

Abstract
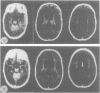
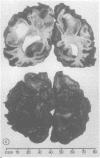
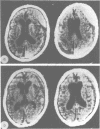
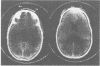
The clinical features and the computed tomographic appearances of the brain in seven children with ornithine carbamoyl transferase deficiency are described. Episodic vomiting and drowsiness, acute encephalopathy, failure to thrive and developmental retardation were common, but focal neurological symptoms and signs were also observed. The CT appearances were non-specific with generalised or focal changes. They were related to the severity, the duration and the age of onset of the hyperammonaemia. Since the CT changes may suggest conditions other than metabolic disease, the emergency investigation of a child with an encephalopathy should include the estimation of plasma ammonium and, if elevated, the appropriate investigations to establish the cause.
Full text
PDF


Images in this article
Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bachmann C., Colombo J. P. Diagnostic value of orotic acid excretion in heritable disorders of the urea cycle and in hyperammonemia due to organic acidurias. Eur J Pediatr. 1980 Aug;134(2):109–113. doi: 10.1007/BF01846026. [DOI] [PubMed] [Google Scholar]
- Batshaw M. L., Roan Y., Jung A. L., Rosenberg L. A., Brusilow S. W. Cerebral dysfunction in asymptomatic carriers of ornithine transcarbamylase deficiency. N Engl J Med. 1980 Feb 28;302(9):482–485. doi: 10.1056/NEJM198002283020902. [DOI] [PubMed] [Google Scholar]
- Bruton C. J., Corsellis J. A., Russell A. Hereditary hyperammonaemia. Brain. 1970;93(2):423–434. doi: 10.1093/brain/93.2.423. [DOI] [PubMed] [Google Scholar]
- Coulter D. L., Allen R. J. Hyperammonemia with valproic acid therapy. J Pediatr. 1981 Aug;99(2):317–319. doi: 10.1016/s0022-3476(81)80489-1. [DOI] [PubMed] [Google Scholar]
- Donn S. M., Swartz R. D., Thoene J. G. Comparison of exchange transfusion, peritoneal dialysis, and hemodialysis for the treatment of hyperammonemia in an anuric newborn infant. J Pediatr. 1979 Jul;95(1):67–70. doi: 10.1016/s0022-3476(79)80085-2. [DOI] [PubMed] [Google Scholar]
- Haan E. A., Danks D. M., Hoogenraad N. J., Rogers J. G. Hereditary hyperammonaemic syndromes--a six year experience. Aust Paediatr J. 1979 Sep;15(3):142–146. doi: 10.1111/j.1440-1754.1979.tb01212.x. [DOI] [PubMed] [Google Scholar]
- Harris M. L., Oberholzer V. G. Conditions affecting the colorimetry of orotic acid and orotidine in urine. Clin Chem. 1980 Mar;26(3):473–479. [PubMed] [Google Scholar]
- Hoogenraad N. J., Mitchell J. D., Don N. A., Sutherland T. M., Mc Leay A. C. Detection of carbamyl phosphate synthetase 1 deficiency using duodenal biopsy samples. Arch Dis Child. 1980 Apr;55(4):292–295. doi: 10.1136/adc.55.4.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley D. P., Kendall B. E. Demyelinating and neuro-degenerative disease in childhood. CT appearances and their differential diagnosis. J Neuroradiol. 1981;8(3):243–255. [PubMed] [Google Scholar]
- Krieger I., Snodgrass P. J., Roskamp J. Atypical clinical course of ornithine transcarbamylase deficiency due to a new mutant (comparison with Reye's disease). J Clin Endocrinol Metab. 1979 Mar;48(3):388–392. doi: 10.1210/jcem-48-3-388. [DOI] [PubMed] [Google Scholar]
- Leonard J. V., Seakins J. W., Griffin N. K. beta-Hydroxy-beta-methyglutaricaciduria presenting as Reye's syndrome. Lancet. 1979 Mar 24;1(8117):680–680. doi: 10.1016/s0140-6736(79)91137-1. [DOI] [PubMed] [Google Scholar]
- Levin B., Dobbs R. H., Burgess E. A., Palmer T. Hyperammonaemia. A variant type of deficiency of liver ornithine transcarbamylase. Arch Dis Child. 1969 Apr;44(234):162–169. doi: 10.1136/adc.44.234.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samtoy B., DeBeukelaer M. M. Ammonia encephalopathy secondary to urinary tract infection with Proteus mirabilis. Pediatrics. 1980 Feb;65(2):294–297. [PubMed] [Google Scholar]
- Saudubray J. M., Cathelineau L., Charpentier C., Boisse J., Allaneau C., Le Bont H., Lesage B. D'eficit hérditaire en ornithine-carbamyl-transférase avec anomalie enzymatique qualitative. Relation d'une forme à révelation néonatale et d'evolution rapidement mortelle chez un garçon. Arch Fr Pediatr. 1973 Jan;30(1):15–27. [PubMed] [Google Scholar]
- Saudubray J. M., Cathelineau L., Laugier J. M., Charpentier C., Lejeune J. A., Mozziconacci P. Hereditary ornithine transcarbamylase deficiency. Report of two male cases with residual enzymatic activity. Acta Paediatr Scand. 1975 May;64(3):464–472. doi: 10.1111/j.1651-2227.1975.tb03866.x. [DOI] [PubMed] [Google Scholar]
- Short E. M., Conn H. O., Snodgrass P. J., Campbell A. G., Rosenberg L. E. Evidence for x-linked dominant inheritance of ornithine transcarbamylase deficiency. N Engl J Med. 1973 Jan 4;288(1):7–12. doi: 10.1056/NEJM197301042880102. [DOI] [PubMed] [Google Scholar]
- Sunshine P., Lindenbaum J. E., Levy H. L., Freeman J. M. Hyperammonemia due to a defect in hepatic ornithine transcarbamylase. Pediatrics. 1972 Jul;50(1):100–111. [PubMed] [Google Scholar]
- Ware A. J., Burton W. C., McGarry J. D., Marks J. F., Weinberg A. G. Systemic carnitine deficiency. Report of a fatal case with multisystemic manifestations. J Pediatr. 1978 Dec;93(6):959–964. doi: 10.1016/s0022-3476(78)81219-0. [DOI] [PubMed] [Google Scholar]
- Wiegand C., Thompson T., Bock G. H., Mathis R. K., Kjellstrand C. M., Mauer S. M. The management of life-threatening hyperammonemia: a comparison of several therapeutic modalities. J Pediatr. 1980 Jan;96(1):142–144. doi: 10.1016/s0022-3476(80)80352-0. [DOI] [PubMed] [Google Scholar]