

Abstract
Proper mitochondrial performance is imperative for the maintenance of normal neuronal function to prevent the development of neurodegenerative diseases. Persistent accumulation of damaged mitochondria plays a role in prion disease pathogenesis, which involves a chain of events that culminate in the generation of reactive oxygen species and neuronal death. Our previous studies have demonstrated that PINK1/Parkin-mediated mitophagy induced by PrP106−126 is defective and leads to an accumulation of damaged mitochondria after PrP106−126 treatment. Externalized cardiolipin (CL), a mitochondria-specific phospholipid, has been reported to play a role in mitophagy by directly interacting with LC3II at the outer mitochondrial membrane. The involvement of CL externalization in PrP106−126-induced mitophagy and its significance in other physiological processes of N2a cells treated with PrP106−126 remain unknown. We demonstrate that the PrP106−126 peptide caused a temporal course of mitophagy in N2a cells, which gradually increased and subsequently decreased. A similar trend in CL externalization to the mitochondrial surface was seen, resulting in a gradual decrease in CL content at the cellular level. Inhibition of CL externalization by knockdown of CL synthase, responsible for de novo synthesis of CL, or phospholipid scramblase-3 and NDPK-D, responsible for CL translocation to the mitochondrial surface, significantly decreased PrP106−126-induced mitophagy in N2a cells. Meanwhile, the inhibition of CL redistribution significantly decreased PINK1 and DRP1 recruitment in PrP106−126 treatment but had no significant decrease in Parkin recruitment. Furthermore, the inhibition of CL externalization resulted in impaired oxidative phosphorylation and severe oxidative stress, which led to mitochondrial dysfunction. Our results indicate that CL externalization induced by PrP106−126 on N2a cells plays a positive role in the initiation of mitophagy, leading to the stabilization of mitochondrial function.
Keywords: mitophagy, autophagy, cardiolipin, prion disease, neurodegenerative disease
Highlights
- PrP106−126 caused a temporal course of mitophagy, which gradually increased and subsequently decreased.
- PrP106−126 caused a temporal course of CL externalization, which gradually increased and subsequently decreased.
- Inhibition of CL externalization significantly decreased PrP106−126-induced mitophagy.
- Inhibition of CL redistribution significantly decreased PINK1 and DRP1 recruitment on mitochondria in PrP106−126 treatment but had no significant decrease in Parkin recruitment in mitochondria.
- Inhibition of CL externalization resulted in impaired oxidative phosphorylation and severe oxidative stress, which caused further mitochondrial dysfunction.
Introduction
Prion diseases are a class of devastating and uniformly fatal neurodegenerative diseases whose pathogen can infect both humans and other animal species, and are based on the aggregation of abnormal protein assemblies that results in a cascade of neurodegenerative pathways (Mead et al., 2019; Maddox et al., 2020). Prion diseases are caused by the accumulation of protease-resistant misfolded form (PrPSc) rich in the beta-sheet structure of cellular prion protein (PrPC), which results in astrogliosis and neuronal loss and vacuolation, eventually leading to brain dysfunction and inevitable death (Gu et al., 2002; Minikel et al., 2020; Sevillano et al., 2020). Despite intense studies in recent decades, both the underlying pathogenic mechanism and effective treatment regimen remain unavailable (Takada et al., 2018). The use of PrP106−126 in a prion disease model was first reported in 1993 and has gradually been demonstrated to create the best neurotoxic effect (Tagliavini et al., 1991, 1993; Selvaggini et al., 1993; Singh et al., 2002; Forloni et al., 2019). The advantages of PrP106−126 are as follows: (1) although PrP106−126, such as PrPSc, is highly fibrillogenic, it is soluble in water at micromolar concentrations; (2) this short PrP sequence recapitulates many of the known features of PrPSc including neurotoxicity, gliotrophic activity, proteinase-K resistance, and a β-sheet structure; and (3) the peptide induces neuronal cell death by way of an apoptotic mechanism, a new paradigm in the field of neurodegeneration (Forloni et al., 2019). Additionally, the effective PrP106−126 concentration has also been explored in detail and is reported to be sublethal at 5–50 μM and lethal at 100–200 μM (Tagliavini et al., 1993; White et al., 2001), which our lab has demonstrated for over 20 years.
Healthy mitochondria are imperative for the maintenance of normal neuronal function. Given the non-regenerative nature of neurons, aberrant mitochondrial dynamics and metabolism can cause severe damage to the brain and even the entire nervous system. Numerous studies have uncovered an essential role for mitochondria in the pathogenesis of neurological diseases via different pathways and mechanisms (Kim and Lemasters, 2011; Lou et al., 2020; Lin et al., 2021; Mito et al., 2022). Accordingly, eukaryotic cells, including neural cells, have developed complex mechanisms to eliminate and degrade damaged mitochondria, with the most efficient being mitophagy (Kanki et al., 2009; Harper et al., 2018), which is activated by either ubiquitination or receptor pathways (Palikaras et al., 2018; Pfanner et al., 2019; Zhao et al., 2021). The PINK1/Parkin pathway is the best-characterized ubiquitination-mediated mitophagy mechanism to date; however, defective non-ubiquitination-mediated mitophagy is also recognized to play a role in several neurodegenerative diseases. Cardiolipin (CL), a specific phospholipid found only in the mitochondrial inner membrane, plays a vital and complex role in maintaining mitochondrial homeostasis from fusion and fission to mitophagy and apoptosis (Kagan et al., 2005; Ji et al., 2012; Chu et al., 2013; Liu and Chan, 2017; Ban et al., 2018; Kojima et al., 2019; Falabella et al., 2021). Although the potentially specific role of CL in mitophagy requires further investigation, the discovery of a direct interaction between CL and LC3II uncovered a novel molecular signal that distinguishes CL-mediated mitophagy from ubiquitination-mediated mitophagy (Kagan et al., 2009; Chu et al., 2013, 2014).
A plethora of studies into neurodegeneration have demonstrated that the persistent accumulation of damaged mitochondria in neurons is involved in aging and neurodegenerative disorders, including prion disease (Norat et al., 2020). Our previous study has demonstrated that PrP106−126 peptide can cause mitochondrial damage and dysfunction, in turn exacerbating the course of damage induced by PrP106−126 based on cellular and animal models (Yang et al., 2008a; Li et al., 2018, 2022; Wu et al., 2019; Zhang et al., 2020). Specifically, mitochondrial homeostasis is gradually disrupted in N2a cells stimulated by PrP106−126, causing aggressive fission, reduced fusion, and intensified apoptosis. Furthermore, PrP106−126 initiates mitophagy through a PINK1/Parkin-related ubiquitination pathway, which exacerbates mitochondria-mediated apoptosis (Li et al., 2022), indicating that ubiquitination-mediated mitophagy may play a vital role in PrP106−126-induced neural damage. Other studies have also suggested that ubiquitination-mediated mitophagy may play a protective role against prion disease or PrP106−126 neural damage at the animal level (Lasmézas and Gabizon, 2018; Gao et al., 2020). Although ubiquitination-mediated mitophagy accounts for a large percentage of cellular mitophagic processes, the molecular signals arising from the direct interaction of LC3II with mitophagic factors, such as CL, remain to be elucidated and may provide potential targets for disease treatment.
In the present study, we demonstrate that PrP106−126 caused a temporal course of mitophagy, which significantly increased and subsequently decreased using PrP106−126 on N2a cells. Moreover, we show that the CL content decreased and that mitophagy was dependent on the externalization of CL to the outer membrane of mitochondria following induction with PrP106−126 on N2a cells. Furthermore, we reveal that the suppression of CL-mediated mitophagy resulted in a worse outcome of PrP106−126-induced neural damage on N2a cells due to mitochondrial dysfunction. Taken together, our data indicate that mitophagy caused by PrP106−126 on N2a cells, and mediated by CL externalization as a marker for the elimination of damaged mitochondria, is beneficial as a result of stabilizing normal mitochondrial function and attenuating neuronal damage.
Materials and methods
Cell culture and PrP106−126 peptide
Mouse neuroblastoma N2a cells were cultured in Dulbecco's modified Eagle medium (DMEM) (Hyclone, Logan, UT, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco, Grand Island, NY, USA) at 37°C with 5% CO2 in a humidified incubator. The PrP106−126 peptide was synthesized at >95% purity by Sangon Bio-Tech (Beijing, China) (sequence: KTNMKHMAGAAAAGAVVGGLG), PrP106−126 scrambled sequence was: MEVGWYRSPFSRVVHLYRNGK referred from Benoit Schneider's article (Yang et al., 2008b). The peptide was dissolved in 0.1 M phosphate-buffered saline (PBS) to a stock concentration of 1 mM and then shaken at 4°C for 24 h to allow for aggregation. All procedures were performed under sterile conditions, and all experiments were conducted using a final peptide concentration of 150 μM.
siRNA administration and transfection
The siRNAs targeting murine CL synthase (CLS), phospholipid scramblase-3 (PLS3), and nucleoside diphosphate kinase (NDPK-D), in addition to the negative control siRNA, were synthesized by GenePharm (GENEPHARM). The sequence details are as follows: siCLS: (mouse, 5′-GGGCUACCUGAUUCUUGAATTdTdT-3′, 5′-CCACUCACUUACAUGAUAATTdTdT-3′, 5′-GCUAAGUACUUCAAUCCUUTTdTdT-3′, pooled); siPLS3: (mouse, 5′-GCUGGGAGACCUGUAAUAUTTdTdT-3′, 5′-GGUGAAGACUAAGGAUGAATTdTdT-3′, 5′-CCACGUUCCUCAUCGAUUATTdTdT-3′, pooled); siNDPK-D (mouse, 5′-AGAGCAGCGAACUGUUGAACUdTdT-3′, 5′-GCUCUUAUCAGCUACAUGAGCdTdT-3′, pooled); and siRNA negative control: (mouse, 5′-UUCUCCGAACGUGUCACGUTTACGUGACACGUUCGGAGAATT-3′). The Mito-GFP and DsRed-Mito plasmids were obtained from Clontech (Mountain View, CA USA), and the pSLenti-CMV-mt-mKeima-PGK-Puro-WPRE vector was procured from Obio Technology. Cultured N2a cells were transfected with siRNA or the relative plasmid at a density of 1 × 105 in a 24-well plate using Lipofectamine™ 3000 (Invitrogen, USA) in Opti-MEM (Invitrogen, USA) for 48 h, according to the manufacturer's instructions.
Mitophagy measured by mt-Keima lentivirus
Mitophagy was directly measured by a fluorescence-based imaging method using mt-Keima, since this novel method was published in Nature Protocols in 2017 (Sun et al., 2017) and improved in 2020 (Wang, 2020). Briefly, the mt-Keima lentivirus (also known as the pSLenti-CMV-mt-mKeima-PGK-Puro-WPRE) titers were determined (5 × 108). N2a cells were seeded on 12-well plates and transduced with the mt-Keima virus according to the manufacturer's protocol. After 3 days, green fluorescence was observed in the neurons, indicating successful transfection: green puncta represented normal mitochondria, while red puncta represented mitochondria fused with lysosomes. Data were collected using flow cytometry (BD Calibur). For flow cytometry analysis, harvested cells were washed three times and centrifuged at 0.3 × g for 3 min at 4°C, and resuspended in PBS prior to their analysis on FACSCalibur (BD Bioscience) with 405 and 561 nm lasers and 610/20 filters. Measurement of lysosomal mitochondrially targeted mKeima was made using a dual-excitation ratiometric pH measurement where pH 7 was detected through the excitation at 405 nm and pH 4 at 561 nm. For each sample, 50,000 events were collected and single, Gating for the mKeima treated group, negative control group. Data were analyzed using FlowJo v10.1 (Tree Star).
Immunoblotting
Extracts for immunoblotting were acquired from N2a cells homogenized in RIPA buffer (radioimmunoprecipitation assay buffer, Thermo Fisher Scientific, Catalog Numbers 89900). Briefly, proteins were separated by electrophoresis and subsequently transferred to the PVDF membrane by electroblotting using standard protocols. The primary antibodies used were anti-LC3II (Cell Signaling Technology, catalog #2775s), anti-COX IV (Proteintech, catalog no. 11242–1-AP), anti-TOM40 (Proteintech, catalog no. 18409–1-AP), anti-MnSOD (Proteintech, catalog no. 24127–1-AP), anti-CLS (Proteintech, catalog no.14845–1-AP), anti-PLS3 (Proteintech, catalog no.28028–1-AP), anti-NDPK-D (Bioss, bs-11902R), anti-PINK1 (Novus Biologicals, catalog no. BC100–494), anti-Parkin (Abcam, catalog no. ab77924), anti-Drp1 (Cell Signaling Technology, catalog #8570), anti-Lamp2 (Cell Signaling Technology, catalog #49067), PGC1a monoclonal antibody (Proteintech, catalog no. 66369–1-Ig), recombinant anti-mtTFA antibody (Abcam, ab252432), GAPDH monoclonal antibody (Proteintech, catalog no. 60004–1-Ig), and anti-Tubulin (Proteintech, catalog no. 11224–1-AP).
Assessment of CL externalization and content at the mitochondrial surface
Evaluation of CL externalization to the mitochondrial surface was performed using the flow cytometry annexin V-binding assay. Briefly, as previously described (Chu et al., 2013; Kagan et al., 2016; Chao et al., 2019), MitoTracker Red CMXRos (M7512, Life Technologies) was applied to mark mitochondria before harvesting, and isolated crude mitochondria were subsequently incubated with FITC-labeled annexin V to stain surface-exposed CL prior to immediate analysis by flow cytometry (BD Calibur). The FITC fluorescence from gated red fluorescent mitochondria (MitoTracker Red) was measured to evaluate the binding of annexin V to mitochondria. Data are presented as the relative FITC fluorescence intensity in comparison with that of mitochondria isolated from control samples. The dye 10-N-nonyl acridine orange (NAO), which specifically binds with high affinity to cardiolipin but not to zwitterionic or other anionic phospholipids, was used to assess the mitochondrial cardiolipin content in living cells (Mileykovskaya et al., 2001; Garcia Fernandez et al., 2004; Kaewsuya et al., 2007). Briefly, cultured cells or isolated mitochondria were exposed to 1 μM NAO for 15–20 min in high-glucose DMEM and subsequently washed three times with PBS. Here, two methods were used to detect NAO fluorescence intensity to overcome method drawbacks: (1) confocal microscopy (Nikon, Tokyo) as described below (Kirkland et al., 2002), with NAO excitation at 488 nm; and (2) measurement using a GloMax® 96 Microplate Luminometer at 485/530 nm (Hsu et al., 2015).
Confocal microscopy
N2a cells were cultured on 24-well coverslips and subsequently transfected with Mito-GFP, DsRed-Mito, Thioflavin T, or siRNA before treatment with PrP106−126 or the control peptide. The specific processes were carried out in accordance with our previous studies (Song et al., 2022). Cells were washed twice with PBS before being fixed with 4% paraformaldehyde for 30 min. After washing twice with PBS, the cells were treated with immunostaining permeabilization buffer containing X-100 (Beyotime biotechnology, P0096) for 5–10 min at room temperature to permeabilize them. The cells were then blocked using an immunostaining blocking buffer (Beyotime Biotechnology, P0102) for 1 h at room temperature, followed by overnight incubation with specific primary antibodies (as described in the immunoblotting section) at 4°C (West et al., 2015). Following the PBS rinsing step, the cells were incubated with secondary antibodies for 1 h at 37°C and then washed with PBS five times for 5 min each. The coverslips were mounted on microscope glass slides using a fluorescent antifading buffer (Bioworld Technology, BD5014). Images were acquired using a Nikon A1HD25 confocal microscope at a magnification of 100 × (oil immersion lens). Approximately 10–15 unique images were captured at random for each sample. The acquired images were quantified using ImageJ software.
Tunel and cell viability assay
N2a cells were seeded into a 24-well plate before treatment with peptide and then used with The One Step TUNEL Apoptosis Assay Kit (Beyotime, catalog no. C1086) in accordance with the manufacturer's instructions before observation using A1 confocal microscope (Nikon). N2a cells' viability in response to PrP106−126 was measured using Cell Counting Kit-8 (Beyotime). After treatment, the CCK-8 solution was directly added to the cell culture medium and incubated for 30 min at 37°C and 5% CO2. The absorbance of the samples was measured using a microplate reader (Thermo Fisher Scientific, USA) at 450 nm. The cell viability was expressed as a percent of the untreated control.
Transmission electron microscopy
To collect samples for microscopic, N2a cells were harvested to do further certain processes after PrP106−126 infections. Cell samples were cut into small pieces after fixation in 5% glutaraldehyde in 0.1-M sodium cacodylate buffer (pH 7.4) for 4 h at 4°C and then fixed in glutaraldehyde were rinsed with sodium cacodylate buffer and then further fixed in 1% OsO4 in 0.1 M sodium cacodylate buffer on ice for 2 h before dehydration with acetone. After dehydrating with a series of ethanol and acetone, the cell pellets were embedded in resin before polymerization at 60°C for 48 h. Ultrathin sections (70 nm) were mounted onto copper grids and counter-stained with 4% uranyl acetate and lead citrate, and images were finally observed under a transmission electron microscope (HITACHI HT7700, Japan) operating at 120 kV.
Mitochondrial isolation
Mitochondrial and cytoplasmic proteins were extracted from N2a cells using a Mitochondria Isolation Kit for Cultured Cells (89874, Thermo Fisher Scientific). Briefly, cells were harvested and subsequently washed three times with 0.1 M PBS, homogenized in lysis buffer containing protease inhibitors, and centrifuged at different speeds. The cytosolic fraction was collected after centrifugation at 3,000 × g for 15 min, and isolated mitochondria were recovered after further centrifugation at 12,000 × g for 5 min. All procedures were carried out at 4°C following the manufacturer's instructions.
mtDNA content and gene expression and measurement of mitochondrial function
Total DNA was extracted from N2a cells and quantitated by spectrophotometry (NanoDrop 2000). Real-time PCR was performed using the ViiA7 Fast Real-Time PCR System (ABI) and SYBR Green Master Mix (Bio-Rad). Primer sequences are as follows: mtDNA (F: 5′-CCTATCACCCTTGCCATCAT-3′ and R: 5′-GAGGCTGTTGCTTGTGTGAC-3′) and genomic DNA (gDNA) (F: 5′-ATGGAAAGCCTGCCATCATG-3′ and R: 5′-TCCTTGTTGTTCAGCATCAC-3′). Data are expressed as the fold change relative to control samples, calculated using the comparative CT method (2−ΔΔCT).
Gene of PGC-1α and mTfam expression was also measured by real-time PCR. Total cellular RNA was extracted from N2a cells after temporal treatments with PrP106−126 by using a total RNA Extraction kit (Aidlab Biotechnologies, Beijing, China) according to the manufacturer's instructions. The cDNA was then generated using HiScript III First Strand cDNA Synthesis Kit (+gDNA wiper) (Vazyme, Biotech Co. Ltd). Real-time PCR was performed with the cDNA using the ViiA7 Fast Real-Time PCR System (ABI) and SYBR Green Master Mix (Bio-Rad). Primer sequences are as follows mouse PGC1α: (F: 5′-ACAACGCGGACAGAATTGAG-3′ and R: 5′-GTTTCGTTCGACCTGCGTAA-3′), mouse mTfam: (F: 5′-AAGGATGATTCGGCTCAGG-3′ and R: 5′-GGCTTTGAGACCTAACTGG-3′), and mouse Tubulin (F: 5′-ACATGGCTTGCTGCCTATTG-3′ and R: 5′-GACCACAGTGAGGCTGGTAA-3′) as a control due to its stable internal expression. Each cDNA sample was performed for three biological triplicates, and Ct values of each replicate were normalized to Tubulin cDNA using the 2 −ΔΔCt method. For relative expression (fold), internal control samples were centered at 1.
Reactive oxygen species (ROS) in N2a cells were measured using 2′,7′-dichlorodihydrofluorescein diacetate (Beyotime, Shanghai, China). Mitochondrial superoxide was detected using the MitoSOX™ Red mitochondrial superoxide indicator (M36008, Thermo Fisher Scientific). The mitochondrial membrane potential (MMP) was evaluated using a JC-1 Mitochondrial Membrane Potential Assay Kit (Beyotime, Shanghai, China). ATP was measured using an ATP Determination Kit (Beyotime, Shanghai, China). The mitochondrial complex activity was measured using a Micro Mitochondrial Respiratory Chain Complex Activity Assay Kit (Solarbio, China). Luminescence was measured using a GloMax® 96 Microplate Luminometer, and fluorescence signals were analyzed using a BD Calibur Fluorescence-activated Cell Sorting Instrument. All processes were carried out following the manufacturer's instructions.
Thioflavin T assay
Thioflavin T is a fluorescent stain that specifically binds to beta-sheet-rich structures, which was used to detect the misfolded PrP protein. Upon binding, the dye exhibits a red-shift in its emission spectrum and increased fluorescence intensity. Briefly, the following protocol was below and referred from two articles (Florio et al., 2003; Chen et al., 2020). A total of 1 mM stock solution of Thioflavin T (Th T, MCE: HY-D0218) was prepared in deionized water and filtered through a 0.2 μm syringe filter before use. Thioflavin T was then diluted in PBS buffer (pH 7.4) to a final concentration of 25 μM. For N2a cells, they were seeded into a Lumox 96 multiwell black plate and treated with 150 μM PrP106−126 for 24 h. The cells were washed three times with PBS before Thioflavin T was added to each well (100 μL volume) of the plate. Alternatively, for preformed PrP106−126 fibrils, Thioflavin T was diluted in PBS buffer (pH 7.4) to a final concentration of 25 μM in each well (100 μL volume) of a Lumox 96 multiwell plate. A total of 150 μM of preformed PrP106−126 fibrils were added to the appropriate wells, and the contents were mixed by pipetting. The plate was sealed and placed in a shaking incubator (600 rpm) at 37°C. The data were analyzed by a GloMax® 96 Microplate Luminometer at 450/480 nm or confocal.
Statistical analysis
All assays were repeated three times, and the number of replicates is presented by individual data points on each graph. Data are reported as the mean ± SD. All comparisons of parametric data were performed using one-way ANOVA followed by Tukey's post hoc test and an unpaired t-test (two-tailed) in GraphPad Prism version 7.0 (La Jolla, CA, USA). The gray level was analyzed using ImageJ (National Institutes of Health, Bethesda, MD, USA). A P-value of < 0.05 was considered statistically significant.
Results
PrP106−126 elicits a temporal course of mitophagy in N2a cells
Our previous study demonstrated that PrP106−126 peptide can cause mitophagy deficiency in a cellular model during the late stage of PrP106−126 treatment. To further explore the effect of PrP106−126 peptide on host mitophagy, we decided to use 150 μM PrP106−126 in N2a cells to fully observe the character of mitophagy induced by PrP106−126 peptide, since 100 μM PrP106−126 may not significantly induce mitophagy during the early stages of PrP106−126 treatment (Li et al., 2022). Before the experiments, we first detected the cell viability, toxicity data. and the state of 150 μM PrP106−126 on N2a cells (Supplementary material). We took advantage of the coral-derived protein Keima, since the fluorescence of mitochondrial matrix-targeted Keima (COX8-mKeima) changes from green to red within the acidic lysosome (pH 4.0) after mitophagy (Sun et al., 2017). We also evaluated mitophagy by observing the changes in colocalization between the mitochondrial Marker DsRed-Mito and the autophagosome protein LC3II and LAMP2. As shown in Figures 1A, B (mKeima), a gradual increase in mitophagy was observed following PrP106−126 treatment, which reached a peak at 6 h and then slowly decreased until 24 h. Moreover, we observed a similar trend in the colocalization of GFP-LC3II and LAMP2 with mitochondria by confocal microscopy (Figures 1C–F). PrP106−126 treatment enhanced the colocalization of GFP-LC3II-positive puncta and MitoTracker Red, indicating an increase in the number of mitophagosomes. In combination with immunoblotting of LC3 (LC3II) and P62 in the isolated mitochondrial samples, the autophagic trend gradually increased until 6 h and subsequently decreased until 24 h, which is consistent with the general autophagic trend (Figure 1J). Furthermore, we used the ratio of mitochondrial DNA (mtDNA) to genomic DNA (gDNA) to quantitate the mitochondrial level at six timepoints between 1 h and 24 h after treatment with PrP106−126 peptide (Figure 1G). The mtDNA/gDNA ratio exhibited the same trend in the peptide-treated group vs. the control, reaching a trough at 6 h. Combined with the mRNA expression level of PGC-1α and TFAM after the temporal treatment of PrP106−126 (Figures 1H, I), we found the mRNA expression level of PGC-1α and TFAM was also significantly decreased on whole time points and the lowest level was at 6 h after PrP106−126 temporal treatments. Meanwhile, we also detected the mitochondrial biosynthesis-related proteins (PGC-1α and TFAM) to determine the breakdown of mitochondrial biosynthesis level (Figure 1L). We found that both two protein levels were gradually significantly decreased after temporal treatments of PrP peptide. These results show that PrP106−126 causes mitochondrial biosynthesis breakdown and decreased. Additionally, the use of different levels of mitochondrial marker proteins mirrored the mitochondrial dynamics. The levels of TOM40 in the outer mitochondrial membrane, MnSOD in the matrix, and COXIV in the inner mitochondrial membrane were decreased following PrP106−126 treatment until reaching a trough at 6 h, before subsequently increasing until 24 h (Figure 1K). Overall, these data demonstrate in detail the dynamic trend in mitophagy and mitochondrial biosynthesis induced by PrP106−126 peptide in N2a cells.
Figure 1.

Temporal course of mitophagy after the addition of 150 μM PrP106−126 to N2A cells. (A, B) The temporal course of mitophagy induced by PrP106−126 was characterized by changes in the mKeima fluorescence ratio. (C–F) Detection of the temporal course of mitophagy after PrP106−126 stimulated on N2a cells using immunofluorescence colocalization of the mitochondrial marker DsRed-Mito and LC3II or LAMP2. Scale bar: 10 μm. (G) The temporal mtDNA/gDNA ratio was assessed by real-time PCR, reflecting the relative number of mitochondria after PrP106−126 was stimulated on N2a cells. (H, I) The messenger RNA relative expression level of PGC-1α and mitochondrial transcription factor A (TFAM) were detected at temporal time-course after PrP106−126 treatments using quantitative real-time polymerase chain reaction. (J, K) Immunoblotting of temporal mitochondrial and autophagic markers obtained from the N2a cells treated with PrP106−126. (L) Immunoblotting of temporal mitochondrial biosynthesis-related protein expression levels (PGC-1α and TFAM) obtained from N2a cells with PrP106−126 treatment. Tubulin and GAPDH were used as loading controls. Data are presented as the mean ± SD; ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001. All experiments were repeated at least three times and PrP106−126 treatments were a temporal course from 0 to 24 h. Statistical analysis was done using a one-way analysis of variance with Tukey post-tests and unpaired t-test (two-tailed).
CL externalizes to the mitochondrial outer membrane and decreases its content in response to PrP106−126
Cardiolipin is a mitochondrial-specific phospholipid that is only distributed in the inner membrane but has been reported to be redistributed under several disease conditions (Chu et al., 2014; Hsu et al., 2015; Song et al., 2019); however, no studies have demonstrated its externalization or content in prion disease or model of prion disease. To examine the mitochondrial redistribution and content of CL during mitophagy induced by PrP106−126 peptide, the annexin V-FITC externalization assay was used to monitor the externalization of CL to the mitochondrial surface. Stimulation with PrP106−126 caused robust externalization of CL, which peaked at 6 h and then gradually decreased (Figures 2A, B). Moreover, the CL-specific dye NAO was utilized to monitor CL levels in live cells, directly measuring its luminescence using a microplate luminometer, in addition to monitoring its mean fluorescence intensity by confocal microscopy. Both these results indicate that the CL content decreased dramatically from 1 h to 24 h following PrP106−126 stimulation (Figures 2C–E). In comparison with the control group, the PrP106−126 peptide-treated group showed a marked decrease in NAO fluorescence intensity at 6 h, with greater than a 2-fold decrease seen at 24 h. Furthermore, confocal data demonstrate that the CL content in the PrP106−126 peptide-treated group was significantly decreased at 1 h and very significantly reduced at 6 h (Figures 2C, E). Therefore, PrP106−126 treatment caused CL externalization to the outer mitochondrial membrane and a significant decrease in its content. Despite the continuous decrease in the CL content, a peak in CL externalization was reached at 6 h following PrP106−126 peptide treatment, which then subsequently decreased.
Figure 2.

Analysis of mitochondrial CL externalization and content after the addition of 150 μM PrP106−126 to N2A cells. (A, B) The temporal exposure of CL on the mitochondrial surface was detected by Annexin V. (C, E) The temporal course of CL content induced by PrP106−126 stimulated on N2a cells was assessed by NAO staining as detected by confocal microscopy. Scale bar: 10 μm. (D) Relative CL level was assessed by NAO staining as detected by a microplate luminometer. NAO, 10-N-nonyl acridine orange. Data are presented as the mean ± SD; ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001. All experiments were repeated at least three times, and PrP106−126 treatments were a temporal course from 0 to 24 h. Statistical analysis was done using one-way analysis of variance with Tukey post-tests and unpaired t-test (two-tailed).
CL externalization induced by PrP106−126 prompts mitophagy via the recruitment of LC3II
Previous research has uncovered that CL redistribution to the surface of mitochondria acts as a mitophagic signal by specifically interacting with the autophagic protein LC3II (Chu et al., 2013, 2014; Kagan et al., 2014). Based on our previous discovery that CL externalization is induced by PrP106−126 treatment, we speculated that CL externalization is involved in mitophagy in the cellular model of prion disease. To investigate two essential enzymes that catalyze different stages of CL translocation from the inner to the outer mitochondrial membrane (Figure 3A), we knocked down PLS3 and NDPK-D in vitro. Knockdown of these two proteins using siRNA reduced the exposure of CL to the outer mitochondrial membrane following PrP106−126 treatment (Figures 3B, C). Additionally, monitoring of mKeima by flow cytometry revealed that PrP106−126-induced mitophagy was decreased after the knockdown of these two enzymes (Figures 3D, E). Moreover, the ratio of mtDNA/gDNA, which was used to quantitate mitochondrial levels, indicates that the number of mitochondria decreased following the addition of PrP106−126 (Figure 3H). GFP-LC3II colocalization with mitochondria was increased (Figures 3F, G), and the loss of mitochondrial proteins was inhibited (Figures 3J, K) following the addition of PrP106−126. To confirm the role of CL in recognizing injured mitochondria during PrP106−126-induced mitophagy, we knocked down CL synthase in vitro using siRNA (Figure 3A). After injury with PrP106−126, the level of CL externalization to the outer mitochondrial membrane was diminished (Figures 3B, C). Moreover, flow cytometry of mKeima, in combination with the mtDNA/gDNA ratio, demonstrates that PrP106−126-induced mitophagy was decreased after CLS knockdown (Figures 3D, E, H). Furthermore, colocalization of GFP-LC3II puncta with mitochondria and loss of mitochondrial proteins were reduced following the addition of PrP106−126 (Figures 3F, G, I). Therefore, decreased externalization of CL due to the knockdown of PLS3, NDPK-D, and CLS significantly decreased mitophagy by reducing LC3II recruitment to mitochondria, exacerbating the accumulation of damaged mitochondria.
Figure 3.
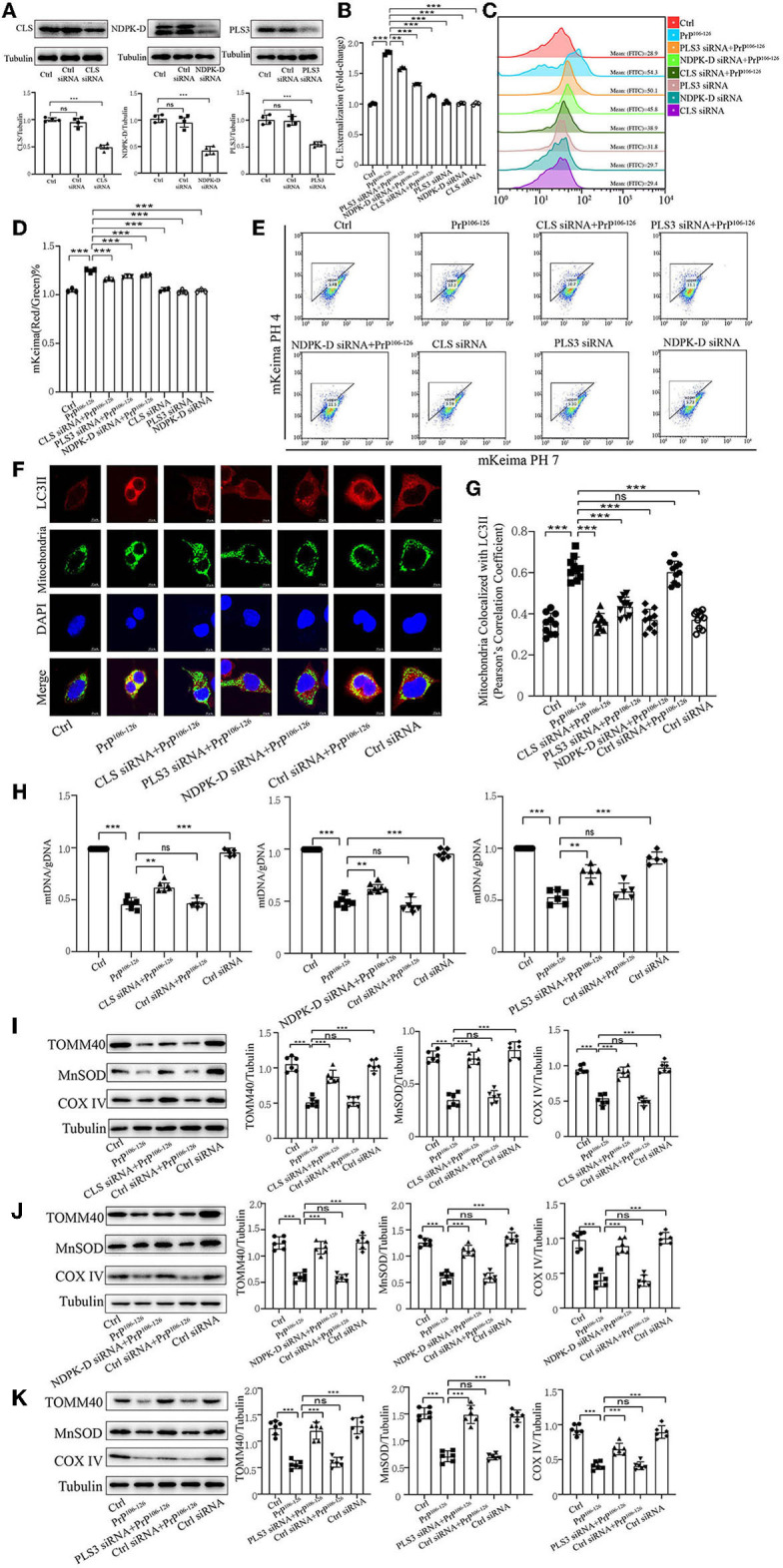
Inhibition of CL externalization dampens 150 μM PrP106−126-induced mitophagy in N2A cells. (A) Immunoblotting of PLS3, CLS, and NDPK-D levels after knockdown. (B, C) Exposure of CL on the mitochondrial surface was detected by Annexin V after the inhibition of CL externalization. (D, E) Detection of mitophagy induced by PrP106−126 was characterized by changes in the mKeima fluorescence ratio after the inhibition of CL externalization. (F, G) Detection of immunofluorescence colocalization of LC3II and the mitochondrial marker mito-GFP after the inhibition of CL externalization after PrP106−126 stimulated on N2a cells. Scale bar: 10 μm. (H) The mtDNA/gDNA ratio was assessed by real-time PCR, reflecting the relative number of mitochondria after the inhibition of CL externalization. (I–K) Immunoblotting of mitochondrial markers obtained from the N2a cells treated with PrP106−126 after the inhibition of CL externalization. Tubulin was used as the loading control. Data are presented as the mean ± SD; ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001. All experiments were repeated at least three times and PrP106−126 treatment was 6 h, causing a peak in mitophagy. Statistical analysis was done using one-way analysis of variance with Tukey post-tests and unpaired t-test (two-tailed).
Decreased CL repartition following PrP106−126 treatment leads to a significant reduction in PINK1 recruitment but has a slight effect on Parkin
Phosphorylation of PARK2/Parkin by PTEN-induced putative kinase 1 (PINK1) plays an essential role in ubiquitin-mediated mitophagy (Geisler et al., 2010a; Lazarou et al., 2015; Gan et al., 2022). Additionally, our previous study has demonstrated that PrP106−126 treatment causes a deficiency in PINK1/Parkin-mediated mitophagy, which aggravates neural apoptosis on the N2a cellular model (Li et al., 2022). Another study has highlighted that CL promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency (Vos et al., 2017); however, the effect of CL externalization on PARK2/Parkin remains unknown. Therefore, we investigated whether the defective CL externalization influences the levels of PINK1 or Parkin recruitment to mitochondria, since depolarized mitochondria induced by PrP106−126 peptide are recognized via a PINK1/Parkin-dependent mechanism. Confocal microscopy and immunoblotting (using isolated mitochondrial samples) demonstrate that the colocalization of PINK1 with mitochondria was significantly reduced after CL repartition, and the levels of PINK1 were also significantly decreased (Figures 4A–C). However, the levels of Parkin recruited to mitochondria were not significantly decreased after CL repartition using both isolated mitochondrial samples (Figure 4D) and confocal microscopy (Figure 4E). Taken together, these results suggest that inhibition on CL repartition induced by PrP106−126 significantly reduces PINK1 recruitment to mitochondria but has no significantly reduced effect on Parkin recruitment.
Figure 4.

CL repartition during 150 μM PrP106−126 treatment in N2A cells leads to a reduction in mitochondrial PINK1 without significantly affecting Parkin levels. (A, B, E, F) Detection of immunofluorescence colocalization of the mitochondrial marker DsRed-Mito and PINK1 or Parkin after the inhibition of CL externalization on N2a cells treated with PrP106−126. Scale bar: 10 μm. (C, D) Immunoblotting of PINK1 and Parkin levels recruiting on mitochondria after the inhibition of CL externalization with PrP106−126 treatment using isolated mitochondrial samples. Cox IV, mitochondrial inner membrane protein, was used as the loading control. Data are presented as the mean ± SD; ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001. All experiments were repeated at least three times and PrP106−126 treatment was 6 h, causing a peak in mitophagy. Statistical analysis was done using one-way analysis of variance with Tukey post-tests and unpaired t-test (two-tailed).
Deficient CL externalization following PrP106−126 treatment downregulates DRP1 translocation to mitochondria
Previous studies have shown that CL externalization to the mitochondrial surface is essential for the activation of fission by directly interacting with DRP1 to enhance its GTPase activity (Ugarte-Uribe et al., 2014; Francy et al., 2017; Gokul, 2018; Rosdah et al., 2020; Mahajan et al., 2021). Moreover, mitochondrial fragmentation plays a vital role in mitophagic processes, in which DRP1 acts as the main regulator of mitochondrial fission and subsequent elimination by autophagy (Twig et al., 2008; Zhang et al., 2013; Lin et al., 2020; Oshima et al., 2021). Our previous study demonstrated that the onset of PrP106−126 stimulation is closely related to DRP1-dependent mitochondrial fragmentation (Li et al., 2018), which plays a pivotal role in PrP106−126-associated mitochondrial dysfunction and neuronal apoptosis. Accordingly, we speculated that deficiency in CL externalization influences DRP1 translocation to mitochondria. Confocal microscopy shows that PrP106−126-induced deficiency in CL externalization significantly decreased DRP1 translocation to mitochondria (Figures 5A, B). Moreover, immunoblotting, using isolated mitochondrial samples, demonstrates that deficient CL redistribution following PrP106−126 treatment reduced DRP1 levels in mitochondria by using mitochondrial samples (Figure 5C). Thus, these results suggest that the inhibition of CL externalization reduces DRP1 recruitment to mitochondria, potentially decreasing mitochondrial fission and inhibiting mitophagy.
Figure 5.

Inhibition of CL externalization downregulates DRP1 translocation to mitochondria during the early stage of mitophagy after 150 μM PrP106−126 treatment in N2A cells. (A, B) Detection of immunofluorescence colocalization of the mitochondrial marker DsRed-Mito and DRP1 after the inhibition of CL externalization on N2a cells treated with PrP106−126. Scale bar: 10 μm. (C) Immunoblotting of DRP1 levels recruiting on mitochondria after the inhibition of CL externalization with PrP106−126 treatment using isolated mitochondrial samples. Cox IV, mitochondrial inner membrane protein, was used as the loading control. Data are presented as the mean ± SD; ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001. All experiments were repeated at least three times and PrP106−126 treatment was 6 h, causing a peak in mitophagy. Statistical analysis was done using one-way analysis of variance with Tukey post-tests and unpaired t-test (two-tailed).
CL externalization ameliorates PrP106−126-induced oxidative stress and mitochondrial dysfunction
Mitophagy caused by PrP106−126 is required for mitochondrial quality control (Keller et al., 2019). As we found that CL externalization increased the mitophagy level to clean damaged mitochondria induced by PrP106−126 and had a positive effect on PINK1 and DRP1 recruitment, the uncertain effects of CL externalization induced by PrP106−126 prompted us to investigate the consequences of oxidative stress on mitochondrial dysfunction, which is widely reported in Prion disease (Milhavet et al., 2000). The results show that the suppression of CL externalization after PrP106−126 stimuli led to more severe oxidative stress and lipid peroxidation (Figures 6A–C) as compared with the PrP106−126 only treatment group. Moreover, deficiency in CL externalization also decreased intracellular ATP levels (Figure 6D) and mitochondrial membrane depolarization (Figures 6E, J) to a greater extent. Furthermore, we detected the activities of the respiratory chain complexes following PrP106−126 treatment. Neurotoxic peptides caused decreased activities of the four complexes to vary degrees (Figures 6F–I), and deficiency in CL externalization intensified these effects on complexes I and III. Thus, the inhibition of CL externalization to the outer mitochondrial membrane aggravates mitochondrial dysfunction induced by the PrP peptide.
Figure 6.

CL externalization ameliorates 150 μM PrP106−126-induced oxidative stress and mitochondrial dysfunction in N2a cells. (A) Malonaldehyde (MDA) levels after the inhibition of CL externalization were detected using a microplate luminometer. (B) ROS production after the inhibition of CL externalization was detected using a microplate luminometer. (C) Mitochondrial superoxide after the inhibition of CL externalization was detected using a microplate luminometer. (D) ATP levels after the inhibition of CL externalization were detected using a microplate luminometer. (E, J) Fluorescence was detected by flow cytometry (FACS) analysis of JC-1 as a marker of mitochondrial membrane potential (MMP) after the inhibition of CL externalization. (F–I) Mitochondrial respiratory chain complexes after the inhibition of CL externalization were detected using a microplate luminometer. Data are presented as the mean ± SD; ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001. All experiments were repeated at least three times, and PrP106−126 treatment was 6 h, causing a peak in mitophagy. Statistical analysis was done using one-way analysis of variance with Tukey post-tests and unpaired t-test (two-tailed).
Discussion
Due to the non-regenerative nature of neurons, mitochondria have gradually become a central therapeutic target in neurodegenerative diseases due to their role in maintaining cell homeostasis (Hao et al., 2022; Lv et al., 2022). Here, we report that mitophagy is involved in the clearance of damaged mitochondria as an early response to PrP106−126 treated with N2a cells. This response was induced by CL translocation to the outer mitochondrial membrane, which stimulated LC3II-mediated autophagy of damaged mitochondria. Moreover, our data targeting PLS, NDPK-D, and CLS in a cellular model strongly support a vital role for CL translocation and repartition in PINK1 and DRP1 recruitment to the mitochondrial membrane. We further demonstrate that the mitochondrial dysfunction of different levels was intensified in PrP106−126 treatment following the prevention of CL externalization, indicating that the mechanisms and pathways of CL translocation may present new targets for drug discovery in prion disease.
Mitophagy is regarded as a regulator in several CNS disorders (Geisler et al., 2010b; Jin and Youle, 2012; Tang et al., 2016; Wang et al., 2019) including prion disease (Forloni et al., 2019). In accordance, we discovered dramatically increased mitophagy induced by PrP106−126 in N2a cells, which decreased for hours. Rescuing damaged mitophagy via PINK1/Parkin signaling, the main ubiquitination-mediated mitophagy pathway would alleviate apoptosis induced by PrP106−126 on N2a cells (Li et al., 2022). These data indicate that mitophagy may play a protective role against neuronal death prior to the induction of apoptosis. To better understand the process of mitophagy in N2a cells stimulated by PrP106−126, we carried out a time-course assessment of mitophagy at different levels. We found that mitophagy induced by PrP106−126 started at 1 h and peaked at 6 h after treatment with PrP106−126, and our previous studies using the TUNEL assay have demonstrated that cells gradually suffer cell death and cytochrome c release after 6 h (Song et al., 2014, 2017a,b; Li et al., 2018). This tendency may provide evidence that cells attempt to clear damaged mitochondria in order to circumvent the necessity to engage in apoptosis after exposure to the PrP106−126 peptide. Moreover, CL also plays a complex role in apoptosis, as previously reported (Kagan et al., 2009; Santucci et al., 2014; Mao et al., 2016; Sparvero et al., 2016); therefore, the potential effects and specific functions of CL in apoptosis induced by PrP106−126 require further investigation.
Furthermore, considering our previous studies on the function of the ubiquitination-mediated PINK1/Parkin pathway, we further determined its potential role in the induction of mitophagy following treatment with PrP106−126. Mitophagy activated by CL externalization has been found to be essential and unique due to the exclusive mitochondrial location of CL, during which externalized CL is rapidly sequestered by autophagosomes and directly binds to LC3II to initiate the recognition of injured mitochondria (Chu et al., 2013; Kagan et al., 2016). A recent study also demonstrated that CL displays different affinities for various members of the LC3 subfamily, which may play disparate roles in CL-mediated mitophagy (Iriondo et al., 2022); therefore, CL can also be regarded as a mitophagic receptor (Chu et al., 2013) that functions as a protective factor in cells, especially neurons. In traumatic brain injury, CL-mediated mitophagy acts as an endogenous neuroprotective process to eliminate damaged mitochondria (Chao et al., 2019). Meanwhile, we also discovered that the CL content was decreased after PrP106−126 peptide treatment via NAO, which specifically binds with high affinity to CL but not to zwitterionic or other anionic phospholipids. Similarly in Parkinson's disease (PD), several studies have reported that a general decrease in the CL content is also detected in different parts of the brain in both patients (Lloyd et al., 2019; Gilmozzi et al., 2020) and mice (Gao et al., 2017). Moreover, further research uncovered that α-Syn, the pathogenic factor of PD, can interact directly with CL to inhibit mitophagy and other cellular processes, which aggravates the progression of PD (Ryan et al., 2018). Several studies have also reported that the CL content displays a similar tendency in Alzheimer's disease (AD) (Guan et al., 1994; Monteiro-Cardoso et al., 2015; Kurokin et al., 2021). Although the potential function of CL in AD is still obscure, CL plays an essential role. Here, we show that non-ubiquitination-related CL-mediated mitophagy also plays a protective role in clearing injured mitochondria to maintain normal neuronal function after PrP106−126 is stimulated on N2a cells. Moreover, we measured the possible contribution of PINK1/Parkin to mitophagy as a result of CL repartition and discovered that PINK1 recruitment was decreased after treatment with PrP106−126 on N2a cells; however, there was no effect on Parkin. It has been reported that the ablation of ALCAT1, an acyltransferase that catalyzes the pathological remodeling of CL, significantly improves mitophagy in PD by promoting the recruitment of Parkin to dysfunctional mitochondria (Song et al., 2019). By contrast, another study found that tafazzin deficiency-induced decreases in the CL content have no significant influence on PINK1/Parkin (Hsu et al., 2015). In the present study, we demonstrate that CL promoted electron transport between ubiquinone and complex I in order to rescue PINK1 deficiency. Furthermore, considering that Parkin is involved in a complex mitophagic activation pathway and is responsible for directing the autophagic clearance of defective mitochondria, which is not only activated by PINK1-mediated phosphorylation but also by other ubiquitinated mitophagic signaling proteins, we suspect that the indifference of Parkin after inhibiting CL externalization may be due to signals other than PINK1 activation (Geisler et al., 2010a; Jin and Youle, 2012; Durcan and Fon, 2015; Seirafi et al., 2015). Therefore, the potential effects of CL externalization on PINK1/Parkin require further exploration.
Cardiolipin externalization can also influence DRP1 since CL provides GTPase activity and other complex functions for the activation of DRP1 in mitochondrial fission (Bustillo-Zabalbeitia et al., 2014; Francy et al., 2017; Kameoka et al., 2018; Mahajan et al., 2021). Here, confocal microscopy and immunoblotting results show that inhibiting CL translocation at its peak decreased DRP1 recruitment to the mitochondrial membrane. Our previous study found that PrP106−126 results in a gradual increase in DRP1 recruitment to mitochondria, but overactive DRP1 eventually causes mitochondrial fragmentation at both cellular and animal levels, indicating that DRP1 recruitment may play a dual role (Li et al., 2018). Our results indicate that the inhibition of CL externalization at its peak decreased DRP1 recruitment, which may influence damaged mitochondrial clearance through fission. It has been previously demonstrated that reduced CL externalization due to ablated ALCAT1 restores mitochondrial dynamics in both PD and coronary artery disease by preventing DRP1 function (Li et al., 2010; Song et al., 2019; Jia et al., 2021). More detailed experiments regarding the effect of CL externalization on DRP1 recruitment are required to explore the underlying mechanisms.
Our study also shows that the deficiency of CL-mediated mitophagy intensifies mitochondrial oxidative stress and dysfunction due to CL externalization. Considering that CL plays a role in the structural integrity and enzymatic activity of all complexes of the respiratory chain to produce ATP (Dudek et al., 2013, 2016), PrP106−126 peptide causes mitochondrial dysfunction and oxidative stress, which decreases mitochondrial ATP production. PrP106−126 peptide-induced externalization of CL from the inner to the outer mitochondrial membrane acts as a signal for the recruitment of LC3II and the initiation of mitophagy to clear damaged mitochondria. Inhibition of CL externalization aggravates mitochondrial oxidative stress and dysfunction, suggesting that the externalization of CL induced by PrP106−126 peptide plays a protective role against the accumulation of damaged mitochondria. Previous studies have demonstrated that CL functions in the respiratory chain to stabilize super-complex formation in the inner mitochondrial membrane (Fry and Green, 1981; Zhang et al., 2002; Arnarez et al., 2016). CL externalization leads to an imbalance in the cycle, which further triggers the dysfunction of different respiratory complexes in diverse diseases (Arias-Cartin et al., 2012; Schwall et al., 2012). Here, we found that the inhibition of CL externalization after PrP106−126 peptide treatment significantly intensified the dysfunction of complexes I and III, aggravating abnormalities in all the respiratory complexes. It has been highlighted that CL can modulate the structure and dynamics of respiratory complex I by specifically binding to certain subunits (Jussupow et al., 2019). Additionally, CL can also affect the function of complex III by preventing its combination with complex IV (Zhang et al., 2005). Moreover, another study showed that CL externalization to the outer mitochondrial membrane in PD can pull α-syn monomers out from preformed fibrils, facilitating their refolding, and effectively buffering synucleinopathy, which may explain the prion-like transmission of α-syn (Ryan et al., 2018). Whether there exists a similar mechanism of CL exposure to the outer mitochondrial membrane upon PrP106−126 protein refolding remains to be explored. The internal regulatory mechanism of CL externalization is complex, and several molecular signals have been uncovered (Kagan et al., 2014). CL translocation and repartition are regulated by SAM50, the depletion of which results in the externalization of CL and mitochondrial outer and inner membrane (including crista membrane) remodeling, triggering Bax mitochondrial recruitment, and mtDNA aggregation and release (Chen et al., 2022). The regulatory mechanism of CL externalization induced by prion disease still needs to be explored. Mitochondrial transplantation is gradually becoming an innovative treatment to cure several neurodegenerative diseases including PD and AD, displaying good efficacy in both cellular and animal models (McCully et al., 2017; Lim, 2020; Liu et al., 2021). Although there is still much work to be done until readiness for clinical application, the importance of mitochondrial homeostasis is recognized.
Conclusion
Taken together, we demonstrate that the activation of CL-mediated mitophagy, and the potential influence of CL translocation to the outer mitochondrial membrane on PINK1 and DRP1, is an essential response elicited by PrP106−126 stimuli on N2a cells. Suppression of CL-mediated mitophagy worsens the overall outcome of N2a cells stimulated by PrP106−126, likely via several different mechanisms. Our findings provide a better understanding of the potential role of mitophagy stimulated by PrP106−126 on N2a cells, in particular non–ubiquitination-mediated mitophagy, laying the foundation for the discovery of new small molecule regulators of mitophagy that could act as potential novel therapeutic targets, not only in prion disease but also in other CNS disorders (Figure 7).
Figure 7.

Schematic diagram summarizing the proposed role of CL in PrP106−126-associated mitophagy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
Material preparation, data collection, and analysis were performed by DY and LY. The first draft of the manuscript was written by DY, and all other authors commented on previous versions of the manuscript. All authors contributed to the study's conception and design. All authors read and approved the final manuscript.
Funding Statement
This study was supported by the Natural Science Foundation of China (Project No. 31972641).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2023.1163981/full#supplementary-material
References
- Arias-Cartin R., Grimaldi S., Arnoux P., Guigliarelli B., Magalon A. (2012). Cardiolipin binding in bacterial respiratory complexes: structural and functional implications. Biochim. Biophys. Acta Bioenerg. 1817, 1937–1949. 10.1016/j.bbabio.2012.04.005 [DOI] [PubMed] [Google Scholar]
- Arnarez C., Marrink S. J., Periole X. (2016). Molecular mechanism of cardiolipin-mediated assembly of respiratory chain supercomplexes. Chem. Sci. 7, 4435–4443. 10.1039/C5SC04664E [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban T., Kohno H., Ishihara T., Ishihara N. (2018). Relationship between OPA1 and cardiolipin in mitochondrial inner-membrane fusion. Biochim. Biophys. Acta Bioenerg. 1859, 951–957. 10.1016/j.bbabio.2018.05.016 [DOI] [PubMed] [Google Scholar]
- Bustillo-Zabalbeitia I., Montessuit S., Raemy E., Basañez G., Terrones O., Martinou J.-C. (2014). Specific interaction with cardiolipin triggers functional activation of dynamin-related protein 1. PLoS ONE 9, e102738. 10.1371/journal.pone.0102738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao H., Lin C., Zuo Q., Liu Y., Xiao M., Xu X., et al. (2019). Cardiolipin-dependent mitophagy guides outcome after traumatic brain injury. J. Neurosci. 39, 1930–1943. 10.1523/JNEUROSCI.3415-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Dong J., Liao S., Wang S., Wu Z., Zuo M., et al. (2022). Loss of Sam50 in hepatocytes induces cardiolipin-dependent mitochondrial membrane remodeling to trigger mtDNA release and liver injury. Hepatology. 76, 1389–1408. 10.1002/hep.32471 [DOI] [PubMed] [Google Scholar]
- Chen M., Li S., Hao M., Chen J., Zhao Z., Hong S., et al. (2020). T-type calcium channel blockade induces apoptosis in C2C12 myotubes and skeletal muscle via endoplasmic reticulum stress activation. FEBS Open Bio. 10, 2122–2136. 10.1002/2211-5463.12965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C. T., Bayir H., Kagan V. E. (2014). LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy 10, 376–378. 10.4161/auto.27191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C. T., Ji J., Dagda R. K., Jiang J. F., Tyurina Y. Y., Kapralov A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. 10.1038/ncb2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek J., Cheng I.-F., Balleininger M., Vaz F. M., Streckfuss-Bömeke K., Hübscher D., et al. (2013). Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 11, 806–819. 10.1016/j.scr.2013.05.005 [DOI] [PubMed] [Google Scholar]
- Dudek J., Cheng I. F., Chowdhury A., Wozny K., Balleininger M., Reinhold R., et al. (2016). Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Molec. Med. 8, 139–154. 10.15252/emmm.201505644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan T. M., Fon E. A. (2015). The three 'P's of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 29, 989–999. 10.1101/gad.262758.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falabella M., Vernon H. J., Hanna M. G., Claypool S. M., Pitceathly R. D. (2021). Cardiolipin, mitochondria, and neurological disease. Trends Endocrinol. Metabol. 32, 224–237. 10.1016/j.tem.2021.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio T., Paludi D., Villa V., Principe D. R., Corsaro A., Millo E., et al. (2003). Contribution of two conserved glycine residues to fibrillogenesis of the 106–126 prion protein fragment. Evidence that a soluble variant of the 106–126 peptide is neurotoxic. J. Neurochemi. 85, 62–72. 10.1046/j.1471-4159.2003.01664.x [DOI] [PubMed] [Google Scholar]
- Forloni G., Chiesa R., Bugiani O., Salmona M., Tagliavini F. (2019). PrP 106-126–25 years after. Neuropathol. Appl. Neurobiol. 45, 430–440. 10.1111/nan.12538 [DOI] [PubMed] [Google Scholar]
- Francy C. A., Clinton R. W., Fröhlich C., Murphy C., Mears J. A. (2017). Cryo-EM Studies of Drp1 Reveal Cardiolipin Interactions that Activate the Helical Oligomer. Scient. Rep. 7, 10744. 10.1038/s41598-017-11008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M., Green D. E. (1981). Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem. 256, 1874–1880. 10.1016/S0021-9258(19)69888-1 [DOI] [PubMed] [Google Scholar]
- Gan Z. Y., Callegari S., Cobbold S. A., Cotton T. R., Mlodzianoski M. J., Schubert A. F., et al. (2022). Activation mechanism of PINK1. Nature 602, 328–335. 10.1038/s41586-021-04340-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G., Wang Z., Lu L., Duan C., Wang X., Yang H. (2017). Morphological analysis of mitochondria for evaluating the toxicity of α-synuclein in transgenic mice and isolated preparations by atomic force microscopy. Biomed. Pharmacother. 96, 1380–1388. 10.1016/j.biopha.2017.11.057 [DOI] [PubMed] [Google Scholar]
- Gao L. P., Xiao K., Wu Y. Z., Chen D. D., Yang X. H., Shi Q., et al. (2020). Enhanced Mitophagy Activity in Prion-Infected Cultured Cells and Prion-Infected Experimental Mice via a Pink1/Parkin-Dependent Mitophagy Pathway. ACS Chem. Neurosci. 11, 814–829. 10.1021/acschemneuro.0c00039 [DOI] [PubMed] [Google Scholar]
- Garcia Fernandez M., Ceccarelli D., Muscatello U. (2004). Use of the fluorescent dye 352 10-N-nonyl acridine orange in quantitative and location assays of cardiolipin: a 353 study on different experimental models. Anal. Biochem. 328, 354. 10.1016/j.ab.2004.01.020 [DOI] [PubMed] [Google Scholar]
- Geisler S., Holmström K. M., Skujat D., Fiesel F. C., Rothfuss O. C., Kahle P. J., et al. (2010a). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- Geisler S., Holmström K. M., Treis A., Skujat D., Weber S. S., Fiesel F. C., et al. (2010b). The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6, 871–878. 10.4161/auto.6.7.13286 [DOI] [PubMed] [Google Scholar]
- Gilmozzi V., Gentile G., Castelo Rueda M. P., Hicks A. A., Pramstaller P. P., Zanon A., et al. (2020). Interaction of alpha-synuclein with lipids: mitochondrial cardiolipin as a critical player in the pathogenesis of Parkinson's disease. Front. Neurosci. 14, 1051. 10.3389/fnins.2020.578993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokul V. (2018). Understanding the role of B-insert in Dynamin related protein 1 (Drp1)-cardiolipin interactions. (Doctoral dissertation) IISER Pune. [Google Scholar]
- Gu Y., Fujioka H., Mishra R. S., Li R., Singh N. (2002). Prion peptide 106–126 modulates the aggregation of cellular prion protein and induces the synthesis of potentially neurotoxic transmembrane PrP. J. Biol. Chem. 277, 2275–2286. 10.1074/jbc.M104345200 [DOI] [PubMed] [Google Scholar]
- Guan Z., Söderberg M., Sindelar P., Edlund C. (1994). Content and fatty acid composition of cardiolipin in the brain of patients with Alzheimer's disease. Neurochem. Int. 25, 295–300. 10.1016/0197-0186(94)90073-6 [DOI] [PubMed] [Google Scholar]
- Hao X., Bu W., Lv G., Xu L., Hou D., Wang J., et al. (2022). Disrupted mitochondrial homeostasis coupled with mitotic arrest generates antineoplastic oxidative stress. Oncogene 41, 427–443. 10.1038/s41388-021-02105-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J. W., Ordureau A., Heo J. M. (2018). Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 19, 93–108. 10.1038/nrm.2017.129 [DOI] [PubMed] [Google Scholar]
- Hsu P., Liu X., Zhang J., Wang H.-G., Ye J.-M., Shi Y. (2015). Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 11, 643–652. 10.1080/15548627.2015.1023984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iriondo M. N., Etxaniz A., Varela Y. R., Ballesteros U., Hervás J. H., Montes L. R., et al. (2022). LC3 subfamily in cardiolipin-mediated mitophagy: a comparison of the LC3A, LC3B and LC3C homologs. Autophagy 18, 2985–3003. 10.1080/15548627.2022.2062111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J., Kline A. E., Amoscato A., Samhan-Arias A. K., Sparvero L. J., Tyurin V. A., et al. (2012). Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nature Neurosci. 15, 1407–1413. 10.1038/nn.3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D., Zhang J., Nie J., Andersen J., Rendon S., Zheng Y., et al. (2021). Cardiolipin remodeling by ALCAT1 links hypoxia to coronary artery disease by promoting mitochondrial dysfunction. Molec. Ther. 29, 3498–3511. 10.1016/j.ymthe.2021.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S. M., Youle R. J. (2012). PINK1-and Parkin-mediated mitophagy at a glance. J. Cell Sci. 125, 795–799. 10.1242/jcs.093849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jussupow A., Di Luca A., Kaila V. R. (2019). How cardiolipin modulates the dynamics of respiratory complex I. Sci. Adv. 5, eaav1850. 10.1126/sciadv.aav1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaewsuya P., Danielson N., Ekhterae D. (2007). Fluorescent determination of cardiolipin using 10-N-nonyl acridine orange. Analyt. Bioanalyt. Chem. 387, 2775–2782. 10.1007/s00216-007-1135-0 [DOI] [PubMed] [Google Scholar]
- Kagan V., Jiang J., Huang Z., Tyurina Y., Desbourdes C., Cottet-Rousselle C., et al. (2016). NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Different. 23, 1140–1151. 10.1038/cdd.2015.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan V. E., Bayir H. A., Belikova N. A., Kapralov O., Tyurina Y. Y., Tyurin V. A., et al. (2009). Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Rad. Biol. Med. 46, 1439–1453. 10.1016/j.freeradbiomed.2009.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan V. E., Chu C. T., Tyurina Y. Y., Cheikhi A., Bayir H. (2014). Cardiolipin asymmetry, oxidation and signaling. Chem. Phys. Lipids 179, 64–69. 10.1016/j.chemphyslip.2013.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan V. E., Tyurin V. A., Jiang J., Tyurina Y. Y., Ritov V. B., Amoscato A. A., et al. (2005). Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem Biol. 1, 223–232. 10.1038/nchembio727 [DOI] [PubMed] [Google Scholar]
- Kameoka S., Adachi Y., Okamoto K., Iijima M., Sesaki H. (2018). Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. 28, 67–76. 10.1016/j.tcb.2017.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T., Wang K., Cao Y., Baba M., Klionsky D. J. (2009). Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Develop. Cell 17, 98–109. 10.1016/j.devcel.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller G., Binyamin O., Frid K., Saada A., Gabizon R. (2019). Mitochondrial dysfunction in preclinical genetic prion disease: A target for preventive treatment? Neurobiol. Dis. 124, 57–66. 10.1016/j.nbd.2018.11.003 [DOI] [PubMed] [Google Scholar]
- Kim I., Lemasters J. J. (2011). Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid. Redox Signal. 14, 1919–1928. 10.1089/ars.2010.3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland R., Adibhatla R., Hatcher J., Franklin J. (2002). Loss of cardiolipin and mitochondria during programmed neuronal death: evidence of a role for lipid peroxidation and autophagy. Neuroscience 115, 587–602. 10.1016/S0306-4522(02)00512-2 [DOI] [PubMed] [Google Scholar]
- Kojima R., Kakimoto Y., Furuta S., Itoh K., Sesaki H., Endo T., et al. (2019). Maintenance of cardiolipin and crista structure requires cooperative functions of mitochondrial dynamics and phospholipid transport. Cell Rep. 26, 518–528.e516. 10.1016/j.celrep.2018.12.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokin I., Lauer A. A., Janitschke D., Winkler J., Theiss E. L., Griebsch L. V., et al. (2021). Targeted lipidomics of mitochondria in a cellular alzheimer's disease model. Biomedicines 9, 1062. 10.3390/biomedicines9081062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasmézas C., Gabizon R. (2018). Identifying therapeutic targets and treatments in model systems. Handbook Clin. Neurol. 153, 409–418. 10.1016/B978-0-444-63945-5.00022-2 [DOI] [PubMed] [Google Scholar]
- Lazarou M., Sliter D. A., Kane L. A., Sarraf S. A., Wang C., Burman J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Wang D., Wu W., Yang W., Ali Shah S. Z., Zhao Y., et al. (2018). DLP 1-dependent mitochondrial fragmentation and redistribution mediate prion-associated mitochondrial dysfunction and neuronal death. Aging Cell 17, e12693. 10.1111/acel.12693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Lai M., Zhang X., Li Z., Yang D., Zhao M., et al. (2022). PINK1-parkin-mediated neuronal mitophagy deficiency in prion disease. Cell Death Dis. 13, 162. 10.1038/s41419-022-04613-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Romestaing C., Han X., Li Y., Hao X., Wu Y., et al. (2010). Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metabol. 12, 154–165. 10.1016/j.cmet.2010.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim G. B. (2020). Intracoronary mitochondrial transplantation. Nat. Rev. Cardiol. 17, 131–131. 10.1038/s41569-020-0340-9 [DOI] [PubMed] [Google Scholar]
- Lin Q., Chen J., Gu L., Dan X., Zhang C., Yang Y. (2021). New insights into mitophagy and stem cells. Stem Cell Res. Ther. 12, 1–14. 10.1186/s13287-021-02520-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X.-H., Qiu B.-Q., Ma M., Zhang R., Hsu S.-J., Liu H.-H., et al. (2020). Suppressing DRP1-mediated mitochondrial fission and mitophagy increases mitochondrial apoptosis of hepatocellular carcinoma cells in the setting of hypoxia. Oncogenesis 9, 1–13. 10.1038/s41389-020-00251-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Gao Y., Liu J., Huang Y., Yin J., Feng Y., et al. (2021). Intercellular mitochondrial transfer as a means of tissue revitalization. Signal Transd. Targeted Ther. 6, 65. 10.1038/s41392-020-00440-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R., Chan D. C. (2017). OPA1 and cardiolipin team up for mitochondrial fusion. Nat. Cell Biol. 19, 760–762. 10.1038/ncb3565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd A. F., Davies C. L., Holloway R. K., Labrak Y., Ireland G., Carradori D., et al. (2019). Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 22, 1046–1052. 10.1038/s41593-019-0418-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou G., Palikaras K., Lautrup S., Scheibye-Knudsen M., Tavernarakis N., Fang E. F. (2020). Mitophagy and Neuroprotection. Trends Mol. Med. 26, 8–20. 10.1016/j.molmed.2019.07.002 [DOI] [PubMed] [Google Scholar]
- Lv J., Yi Y., Qi Y., Yan C., Jin W., Meng L., et al. (2022). Mitochondrial homeostasis regulates definitive endoderm differentiation of human pluripotent stem cells. Cell Death Disc. 8, 69. 10.1038/s41420-022-00867-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox R. A., Person M. K., Blevins J. E., Abrams J. Y., Appleby B. S., Schonberger L. B., et al. (2020). Prion disease incidence in the United States: 2003–2015. Neurology 94, e153–e157. 10.1212/WNL.0000000000008680 [DOI] [PubMed] [Google Scholar]
- Mahajan M., Bharambe N., Shang Y., Lu B., Mandal A., Mohan P. M., et al. (2021). NMR identification of a conserved Drp1 cardiolipin-binding motif essential for stress-induced mitochondrial fission. Proc. Nat. Acad. Sci. 118, e2023079118. 10.1073/pnas.2023079118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao G., Qu F., St. Croix C. M., Tyurina Y. Y., Planas-Iglesias J., Jiang J., et al. (2016). Mitochondrial redox opto-lipidomics reveals mono-oxygenated cardiolipins as pro-apoptotic death signals. ACS Chem. Biol. 11, 530–540. 10.1021/acschembio.5b00737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully J. D., Cowan D. B., Emani S. M., del Nido P. J. (2017). Mitochondrial transplantation: From animal models to clinical use in humans. Mitochondrion 34, 127–134. 10.1016/j.mito.2017.03.004 [DOI] [PubMed] [Google Scholar]
- Mead S., Lloyd S., Collinge J. (2019). Genetic factors in mammalian prion diseases. Ann. Rev. Genet. 53, 117–147. 10.1146/annurev-genet-120213-092352 [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E., Dowhan W., Birke R. L., Zheng D., Lutterodt L., Haines T. H. (2001). Cardiolipin binds nonyl acridine orange by aggregating the dye at exposed hydrophobic domains on bilayer surfaces. FEBS Lett. 507, 187–190. 10.1016/S0014-5793(01)02948-9 [DOI] [PubMed] [Google Scholar]
- Milhavet O., McMahon H. E., Rachidi W., Nishida N., Katamine S., Mangé A., et al. (2000). Prion infection impairs the cellular response to oxidative stress. Proc. Nat. Acad. Sci. 97, 13937–13942. 10.1073/pnas.250289197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minikel E. V., Zhao H. T., Le J., O'Moore J., Pitstick R., Graffam S., et al. (2020). Prion protein lowering is a disease-modifying therapy across prion disease stages, strains and endpoints. Nucl. Acids Res. 48, 10615–10631. 10.1093/nar/gkaa616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito T., Vincent A. E., Faitg J., Taylor R. W., Khan N. A., McWilliams T. G., et al. (2022). Mosaic dysfunction of mitophagy in mitochondrial muscle disease. Cell Metabol. 34, 197–208. 10.1016/j.cmet.2021.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro-Cardoso V. F., Oliveira M. M., Melo T., Domingues M. R., Moreira P. I., Ferreiro E., et al. (2015). Cardiolipin profile changes are associated to the early synaptic mitochondrial dysfunction in Alzheimer's disease. J. Alzheimer's Dis. 43, 1375–1392. 10.3233/JAD-141002 [DOI] [PubMed] [Google Scholar]
- Norat P., Soldozy S., Sokolowski J. D., Gorick C. M., Kumar J. S., Chae Y., et al. (2020). Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. NPJ Regener. Med. 5, 1–9. 10.1038/s41536-020-00107-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima Y., Cartier E., Boyman L., Verhoeven N., Polster B. M., Huang W., et al. (2021). Parkin-independent mitophagy via Drp1-mediated outer membrane severing and inner membrane ubiquitination. J. Cell Biol. 220, e202006043. 10.1083/jcb.202006043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K., Lionaki E., Tavernarakis N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. 10.1038/s41556-018-0176-2 [DOI] [PubMed] [Google Scholar]
- Pfanner N., Warscheid B., Wiedemann N. (2019). Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 20, 267–284. 10.1038/s41580-018-0092-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosdah A. A., Smiles W. J., Oakhill J. S., Scott J. W., Langendorf C. G., Delbridge L. M., et al. (2020). New perspectives on the role of Drp1 isoforms in regulating mitochondrial pathophysiology. Pharmacol. Therapeut. 213, 107594. 10.1016/j.pharmthera.2020.107594 [DOI] [PubMed] [Google Scholar]
- Ryan T., Bamm V. V., Stykel M. G., Coackley C. L., Humphries K. M., Jamieson-Williams R., et al. (2018). Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat. Commun. 9, 1–17. 10.1038/s41467-018-03241-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santucci R., Sinibaldi F., Polticelli F., Fiorucci L. (2014). Role of cardiolipin in mitochondrial diseases and apoptosis. Curr. Med. Chem. 21, 2702–2714. 10.2174/0929867321666140414112156 [DOI] [PubMed] [Google Scholar]
- Schwall C. T., Greenwood V. L., Alder N. N. (2012). The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim. Biophys. Acta Bioener. 1817, 1588–1596. 10.1016/j.bbabio.2012.04.015 [DOI] [PubMed] [Google Scholar]
- Seirafi M., Kozlov G., Gehring K. (2015). Parkin structure and function. FEBS J. 282, 2076–2088. 10.1111/febs.13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaggini C., Degioia L., Cantu L., Ghibaudi E., Diomede L., Passerini F., et al. (1993). Molecular characteristics of a protease-resistant, amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. Biochem. Biophys. Res. Commun. 194, 1380–1386. 10.1006/bbrc.1993.1977 [DOI] [PubMed] [Google Scholar]
- Sevillano A. M., Aguilar-Calvo P., Kurt T. D., Lawrence J. A., Soldau K., Nam T. H., et al. (2020). Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease. J. Clin. Invest. 130, 1350–1362. 10.1172/JCI131564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N., Gu Y., Bose S., Kalepu S., Mishra R. S., Verghese S. (2002). Prion peptide 106–126 as a model for prion replication and neurotoxicity. Front. Biosci. 7, a60–71. 10.2741/A740 [DOI] [PubMed] [Google Scholar]
- Song C., Zhang J., Qi S., Liu Z., Zhang X., Zheng Y., et al. (2019). Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to Parkinson's diseases. Aging Cell 18, e12941. 10.1111/acel.12941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., Ge X., Chen Y., Hussain T., Liang Z., Dong Y., et al. (2022). Mycobacterium bovis induces mitophagy to suppress host xenophagy for its intracellular survival. Autophagy 18, 1401–1415. 10.1080/15548627.2021.1987671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z., Shah S. Z., Yang W., Dong H., Yang L., Zhou X., et al. (2017a). Downregulation of the repressor element 1-silencing transcription factor (REST) is associated with Akt-mTOR and Wnt-β-catenin signaling in prion diseases models. Front. Molec. Neurosci. 10, 128. 10.3389/fnmol.2017.00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z., Yang W., Zhou X., Yang L., Zhao D. (2017b). Lithium alleviates neurotoxic prion peptide-induced synaptic damage and neuronal death partially by the upregulation of nuclear target REST and the restoration of Wnt signaling. Neuropharmacology 123, 332–348. 10.1016/j.neuropharm.2017.05.021 [DOI] [PubMed] [Google Scholar]
- Song Z. Q., Yang L. F., Wang Y. S., Zhu T., Zhou X. M., Yin X. M., et al. (2014). Overexpression of BAT 3 Alleviates Prion Protein Fragment PrP106-126-Induced Neuronal Apoptosis. CNS Neurosci. Therap. 20, 737–747. 10.1111/cns.12243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparvero L. J., Amoscato A. A., Fink A. B., Anthonymuthu T., New L. A., Kochanek P. M., et al. (2016). Imaging mass spectrometry reveals loss of polyunsaturated cardiolipins in the cortical contusion, hippocampus, and thalamus after traumatic brain injury. J. Neurochem. 139, 659–675. 10.1111/jnc.13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N., Malide D., Liu J., Rovira I. I., Combs C. A., Finkel T. (2017). A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Prot. 12, 1576–1587. 10.1038/nprot.2017.060 [DOI] [PubMed] [Google Scholar]
- Tagliavini F., Prelli F., Ghiso J., Bugiani O., Serban D., Prusiner S. B., et al. (1991). Amyloid protein of Gerstmann-Sträussler-Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N-terminal glycine at codon 58. The EMBO J. 10, 513–519. 10.1002/j.1460-2075.1991.tb07977.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliavini F., Prelli F., Verga L., Giaccone G., Sarma R., Gorevic P., et al. (1993). Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc. Nat. Acad. Sci. 90, 9678–9682. 10.1073/pnas.90.20.9678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada L. T., Kim M.-O., Metcalf S., Gala I. I., Geschwind M. D. (2018). Prion disease. Handbook Clin. Neurol. 148, 441–464. 10.1016/B978-0-444-64076-5.00029-6 [DOI] [PubMed] [Google Scholar]
- Tang Y.-C., Tian H.-X., Yi T., Chen H.-B. (2016). The critical roles of mitophagy in cerebral ischemia. Protein Cell 7, 699–713. 10.1007/s13238-016-0307-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. The EMBO J. 27, 433–446. 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte-Uribe B., Müller H.-M., Otsuki M., Nickel W., García-Sáez A. J. (2014). Dynamin-related protein 1 (Drp1) promotes structural intermediates of membrane division. J. Biol. Chem. 289, 30645–30656. 10.1074/jbc.M114.575779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M., Geens A., Böhm C., Deaulmerie L., Swerts J., Rossi M., et al. (2017). Cardiolipin promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency. J. Cell Biol. 216, 695–708. 10.1083/jcb.201511044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. (2020). A sensitive and quantitative mKeima assay for mitophagy via FACS. Curr. Prot. Cell Biol. 86, e99. 10.1002/cpcb.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Liu N., Lu B. (2019). Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 25, 859–875. 10.1111/cns.13140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. P., Khoury-Hanold W., Staron M., Tal M. C., Pineda C. M., Lang S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. 10.1038/nature14156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. R., Guirguis R., Brazier M. W., Jobling M. F., Hill A. F., Beyreuther K., et al. (2001). Sublethal concentrations of prion peptide PrP106–126 or the amyloid beta peptide of Alzheimer's disease activates expression of proapoptotic markers in primary cortical neurons. Neurobiol. Dis. 8, 299–316. 10.1006/nbdi.2001.0386 [DOI] [PubMed] [Google Scholar]
- Wu W., Zhao D., Shah S. Z. A., Zhang X., Lai M., Yang D., et al. (2019). OPA1 overexpression ameliorates mitochondrial cristae remodeling, mitochondrial dysfunction, and neuronal apoptosis in prion diseases. Cell Death Dis. 10, 710. 10.1038/s41419-019-1953-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.-L., Weissman L., Bohr V. A., Mattson M. P. (2008a). Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair 7, 1110–1120. 10.1016/j.dnarep.2008.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Zhou X., Yang J., Yin X., Han L., Zhao D. (2008b). Aspirin inhibits cytotoxicity of prion peptide PrP106–126 to neuronal cells associated with microglia activation in vitro. J. Neuroimmunol. 199, 10–17. 10.1016/j.jneuroim.2008.04.028 [DOI] [PubMed] [Google Scholar]
- Zhang M., Mileykovskaya E., Dowhan W. (2002). Gluing the respiratory chain together: cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556. 10.1074/jbc.C200551200 [DOI] [PubMed] [Google Scholar]
- Zhang M., Mileykovskaya E., Dowhan W. (2005). Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Biol. Chem. 280, 29403–29408. 10.1074/jbc.M504955200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Yan H., Yuan Y., Gao J., Shen Z., Cheng Y., et al. (2013). Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 9, 1321–1333. 10.4161/auto.25132 [DOI] [PubMed] [Google Scholar]
- Zhang X., Zhao D., Wu W., Shah S. Z. A., Lai M., Yang D., et al. (2020). Melatonin regulates mitochondrial dynamics and alleviates neuron damage in prion diseases. Aging 12, 11139. 10.18632/aging.103328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y. G., Codogno P., Zhang H. (2021). Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Molec. Cell Biol. 22, 733–750. 10.1038/s41580-021-00392-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.