

Abstract
A wide divergence has been detected in the telomeric sequences among budding yeast species. Despite their length and homogeneity differences, all these yeast telomeric sequences show a conserved core which closely matches the consensus RAP1-binding sequence. We demonstrate that the RAP1 protein binds this sequence core, without involving the diverged sequences outside the core. In Saccharomyces castellii and Saccharomyces dairensis specific classes of interspersed variant repeats are present. We show here that a RAP1-binding site is formed in these species by connecting two consecutive 8 bp telomeric repeats. DNase I footprint analyses specify the binding site as the 13 bp sequence CTGGGTGTCTGGG. The RAP1 protein also binds the variant repeats, although with a lowered affinity. However, a split footprint is produced when RAP1 binds a variant repeat where the two half-sites of the binding site are separated by an additional 6 nt. This is probably caused by the intervening sequence looping out of the RAP1–DNA complex. We suggest that the bipartite subdomain structure of the RAP1 protein allows it to remodel telomeric chromatin, a feature which may be of great relevance for telomeric chromatin assembly and structure in vivo.
INTRODUCTION
Telomeres are the nucleoprotein complexes defining the ends of, and contributing stability to, eukaryotic chromosomes. An important function of this complex is to enable DNA damage repair mechanisms to distinguish natural ends from internal DNA breakage sites. Telomeric DNA consists of tandemly arranged unidirectional repeats which are species specific. Separate telomere-binding proteins bind to the double-stranded telomeric DNA as well as to the single-stranded protruding DNA (1). Proteins binding to double-stranded DNA arrays have been identified in several different organisms. While the overall amino acid sequence similarity between these proteins is usually low, the DNA-binding motifs in Saccharomyces cerevisiae RAP1, Schizosaccharomyces pombe TAZ1 and human TRF1 and TRF2 show similarities to each other and to the DNA-binding motif in the c-Myb family of transcriptional activators (2). Whereas the c-Myb proteins typically consist of three tandem repeats of the Myb DNA-binding motif, where at least two are required for sequence-specific DNA recognition (3), TRF1, TRF2 and TAZ1 only contain one Myb-like domain each (4,5). Although the single Myb-like domain of a TRF1 monomer can interact with telomeric DNA, TRF1 predominantly binds as a preformed homodimer (6). RAP1 binds as a monomer, but is dimeric in nature, with two structurally similar Myb-like subdomains connected by a loosely folded linker (7). The two domains are tandemly arranged on the DNA, so that each interacts with the tandemly repeated GGTGT motif in the telomeric RAP1-binding site 5′-GGTGTGTGGGTGT (2). The RAP1-binding domains are classified as homeodomains, because of the characteristic feature of a short N-terminal arm that interacts with bases in the minor groove (7).
The telomeric DNA of S.cerevisiae consists of ~300 bp of a heterogeneous sequence that is usually abbreviated to TG2–3(TG)1–6 (8). The RAP1-binding site has by in vitro studies been estimated to be present at an average of one per 18 bp (9). Due to the heterogeneous sequences, however, the site spacing is irregular and it was shown that some sites may even overlap. A direct consequence of this is that the spacing between individual RAP1 molecules bound to these repeats varies between different telomeres. This could be an explanation for the length differences found between the telomeres in one cell. RAP1 is suggested to play a role in a protein counting mechanism, where the cell is able to measure and adjust the total number of RAP1 molecules bound to the telomere (10,11). When artificial RAP1 sites were placed internal of the native telomeric sequences, a decrease in the length of telomeric sequences was seen. This indicated that the number of RAP1 molecules, and not the DNA sequences, served as the metric for telomere length regulation. When longer spacer regions were inserted between the internal artificial sites, an increase in the total length of RAP1-bound DNA was observed (11). These results suggest that a longer spacer between Rap1p sites would result in longer telomeres.
In Kluyveromyces lactis the RAP1 homolog has been demonstrated to play a role in telomere length regulation (12,13). The K.lactis homolog is smaller (666 compared to 827 amino acids) but the DNA-binding domain is virtually identical between the two RAP1 homologs. The K.lactis telomeric repeats are 25 bp long, which is in sharp contrast to the short, typically 5–8 bp, telomeric repeats known from other eukaryotes. Moreover, cloning and sequencing of telomeric repeat units from several budding yeast species has shown a length variation between 8 and 26 bp (14,15). Despite the sequence divergence, all the telomeric sequences contain a conserved core showing a close match to a consensus RAP1-binding site (Fig. 1). In contrast to the S.cerevisiae sequences, most of the other yeast telomeric sequences examined are totally homogeneous. However, variant repeats exist in the two species Saccharomyces castellii and Saccharomyces dairensis (26 and 36%, respectively) (14). Although they have the same 8 bp telomeric repeats, TCTGGGTG, the variant repeats differ dramatically between the two species. The major class of variant repeats housed by S.castellii contains additional TG dinucleotides [TCTGGGTG(TG)1–3]. Such variants have been attributed to a tendency for slippage in telomerase polymerization and resemble the irregular sequences present in S.cerevisiae telomeres (16). However, in S.dairensis the major variant repeat is the shorter, uniform sequence TCTGGG, which is not detected in S.castellii. The homogeneous telomeric repeats present in most yeasts would predict equally spaced RAP1 molecules with a species-specific spacing. In S.castellii and S.dairensis the interspersed variant repeats of longer and shorter sizes would introduce irregularities. Whether this would severely affect the spacing of RAP1 molecules along the telomere array would be largely dependant on whether the protein is able to interact with the variants or not.
Figure 1.

Conservation of a potential RAP1-binding site within yeast telomeric sequences. The sequences are aligned to the RAP1 protein consensus binding site derived from S.cerevisiae (13). The respective positions in the 12 bp consensus sequence are numbered at the top. Matches to this site are indicated with enlarged, bold letters. R indicates A or G, Y indicates C or T. The consensus sequence used here closely matches (11/13 positions) the previously published consensus RRTGNNTGGGT(T/G)Y (here translated to its TG-rich strand), which, however, is extended by 2 nt at the 3′-end (27). The species involved in this study are underlined, belonging to the genera Saccharomyces (S.), Candida (C.) and Kluyveromyces (K.).
In the present work we have determined that the conserved region in the telomeric sequences of several budding yeasts is able to function as a binding site for the RAP1 protein. We show that the RAP1 protein needs two consecutive 8 bp S.castellii telomeric repeats to form the 13 bp binding site. In our footprint analyses the cleavage pattern of DNase I reflects the bipartite structure of the RAP1 protein, by allowing access for DNase I between the two half-site sequences of the binding site. Furthermore, we demonstrate that the RAP1 protein is able to bind the different S.castellii and S.dairensis variant repeats, although at a somewhat lowered affinity. In order to determine the effect of variant repeats on the spacing of RAP1 molecules, we performed footprinting analyses on a DNA fragment containing a variant repeat. This revealed a remarkable flexibility of the RAP1 protein in its binding to telomeric DNA. The RAP1 molecule is able to span the variant telomeric repeat, thus encompassing the two half-sites of the binding site which are separated by an additional 6 bp of intervening sequence.
MATERIALS AND METHODS
Preparation of Escherichia coli extract
The RAP1 protein was overexpressed in E.coli BL21(DE3) from a pET vector containing the cloned RAP1 gene (17). Extracts were prepared according to standard protocols in The QIAexpressionist Handbook (Qiagen) from 40 ml cell cultures. Cells were harvested after 3 h of IPTG induction and resuspended in 1 ml of lysis buffer (50 mM HEPES, pH 7.6, 1% Triton X-100, 2 mM EDTA, 10% glycerol, 500 mM NaCl, 5 mM DTT). A control extract not containing the RAP1 construct was also prepared. This extract did not produce any bands in electrophoretic mobility shift assays (EMSAs).
Electrophoretic mobility shift assays
32P-labeled oligonucleotide was annealed with a 5-fold excess of the complementary strand. An aliquot of 20 fmol double-stranded oligonucleotide was mixed with binding buffer (10 mM Tris–HCl, pH 7.5, 7 mM MgCl2, 8% glycerol) and non-specific competitors [0.3 µg yeast tRNA, 0.3 µg poly(dI·dC), 0.3 µg salmon sperm and 0.5 µg E.coli DNA]. An aliquot of 0.05–0.1 µg E.coli expressed recombinant RAP1 protein was added to give a total volume of 15 µl. The reaction was incubated at 25°C for 15 min and then loaded on non-denaturing polyacrylamide gels (4–5%). The gels were run at 150 V at 4°C for ~1 h using 1× TBE as buffer, then dried in a gel drier at 80°C. The gels were analyzed using a Bio-Rad phosphorimager with the software Quantity One. All oligonucleotides used in this study were double stranded. The 32 bp oligonucleotide GGACTTAAAATGGCGTGGCAGAACTAACTCTT was used as the non-specific competitor where indicated in the figures.
Quantitation of the shifted band signals was done with the Bio-Rad Quantity One software using volume density measurements from the phosphorimager scanned gel. The intensities were compared relative to each other.
DNase I footprinting analysis
32P-labeled double-stranded oligonucleotide (0.1 pmol), binding buffer, non-specific competitors [0.3 µg yeast tRNA, 0.3 µg poly(dI·dC), 0.3 µg salmon sperm and 1 µg yeast E.coli DNA] and RAP1 protein-containing E.coli extract (6 µg) were mixed in 50 µl reactions and incubated as for the mobility shift assays. No-protein control reactions were incubated for 1 min with 0.5 U DNase I. The G strand-labeled reactions containing protein were incubated for 1 (Fig. 9) or 5 (Fig. 6) min with 0.5 U DNase I and the C strand-labeled reactions (Fig. 10) were incubated for 1 min with 0.5 or 1.5 U DNase I. The reactions were stopped by addition of 100 µl stop solution (200 mM NaCl, 30 mM EDTA, 1% SDS, 100 µg/ml yeast tRNA). A phenol/chloroform extraction was performed, followed by ethanol precipitation and washing. The pellet was dried and resolved in loading buffer and the samples were loaded on 10% polyacrylamide gels. The gels were analyzed using a phosphorimager.
Figure 9.

DNase I footprinting of the RAP1 protein bound to a S.castellii telomeric repeat variant. Reactions of the probe containing a variant repeat having three extra TGs [(TG)3, lanes 5–8] were run in parallel with reactions of the normal repeats (lanes 1–4). The additional sequence (TG)3 of the variant repeat is indicated by the filled box. Footprint regions are indicated by the bars denoted I and II in the normal probe and I′ and II′ in the variant repeat probe.
Figure 6.
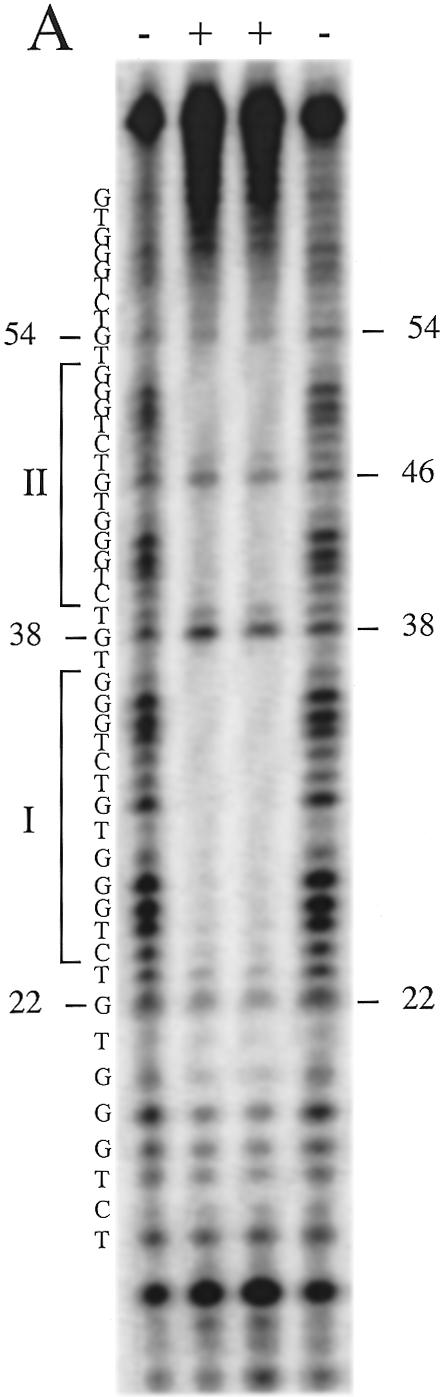

DNase I footprinting of the RAP1 protein bound to the telomeric repeats of S.castellii. (A) Lanes 1 and 4, control cleavage pattern of the naked G-rich strand; lanes 2 and 3, the sample protein–DNA complex. Footprint regions are indicated by the bars denoted I and II. Protected positions were assessed from Maxam–Gilbert G sequencing reactions run in parallel. (B) Schematic illustration of RAP1 protein binding as interpreted from the footprinting results in (A). The telomeric repeats start at base number 15. +/–, with and without protein, respectively.
Figure 10.
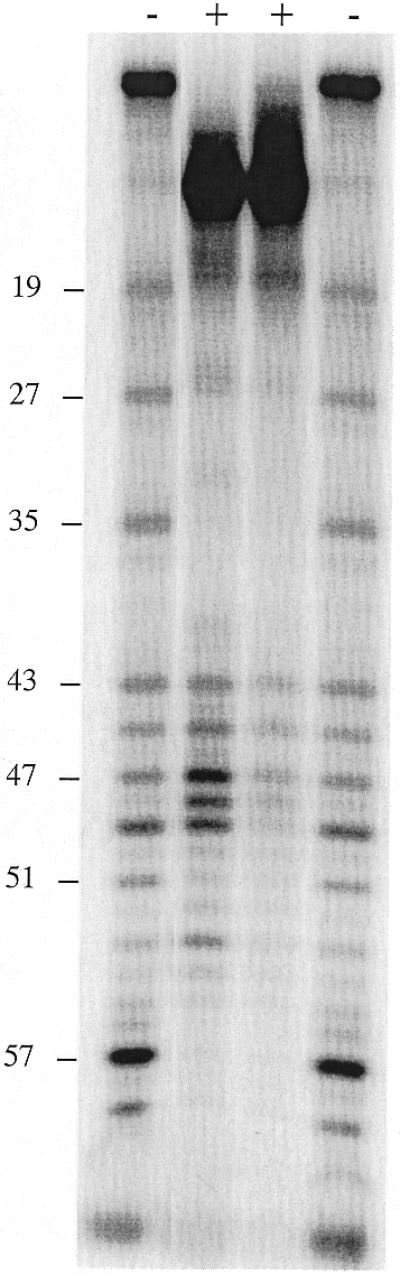
DNase I footprinting of the complementary C-rich strand of the probe containing a variant (TG)3 repeat. The base numbers at the left are the same as in Figure 9. +/–, with and without protein, respectively. Protein-containing samples were treated with 1.5 or 0.5 U DNase I, from left to right.
DMS footprinting analysis
Binding reactions were performed as described for DNase I footprinting above, but using double the volumes and adding 20 µg yeast tRNA. After incubation, 0.5 µl dimethyl sulfate (DMS) was added followed by a 2 min incubation at room temperature. The rest of the methylation steps were performed as described previously (18) and the piperidine cleavage was performed as for a standard Maxam–Gilbert sequencing reaction. The reactions were loaded on 10% polyacrylamide gels and analyzed using a phosphorimager.
RESULTS
RAP1 binds the telomeric sequences of various yeast species
A conserved region with similarity to the RAP1 consensus was previously recognized in the DNA sequences of telomeric DNA repeats from several yeast species (14). To determine whether the S.cerevisiae RAP1 protein is able to bind the 8 bp telomeric DNA repeats in S.castellii and S.dairensis (Fig. 1), we performed EMSAs. It was previously shown that S.cerevisiae RAP1 binds to the 25 bp repeats of K.lactis (12). In this study we used bacterially expressed recombinant RAP1 protein, which has the same binding features as the protein extracted from yeast cells (17). When reacted with a 19 bp oligonucleotide containing the S.cerevisiae RAP1 consensus site, Scer19 (Table 1), a band shift was produced with drastically lowered mobility as compared to the free DNA probe (Fig. 2). The specificity of this binding was analyzed by competition analyses, where unlabeled oligonucleotides were added to the reaction mixes prior to addition of protein extract. The shifted signal was competed with increasing amounts of specific competitor, but not with a 32 bp non-specific competitor.
Table 1. Oligonucleotides used for EMSA.

At the top the RAP1 protein-binding site consensus is indicated (13). In the wild-type sequences the predicted fit to the RAP1 consensus is indicated by bold letters. Scer19, wild-type telomeric sequence from S.cerevisiae; Scas20 and Scas19-2, wild-type telomeric sequences from S.castellii/S.dairensis having different permutations of the repeats; Scas19-1, two S.castellii repeats, one of which has mutations disrupting the fit to the RAP1 consensus; Sklu1-26 and Sklu21-20, different permutations of the S.kluyveri repeat, with one preserved and one split RAP1-binding site, respectively; Sklu26-18 and Sklu8-26, shortened versions of the S.kluyveri repeat containing or not containing the RAP1-binding site, respectively; Klac25 and Cgui20, wild-type telomeric sequences from K.lactis and C.guilliermondii, respectively; (TG)1, (TG)2 and (TG)3, S.castellii variants containing extra TG dinucleotides as indicated; ΔTG-1, containing one shortened S.dairensis variant.
Figure 2.
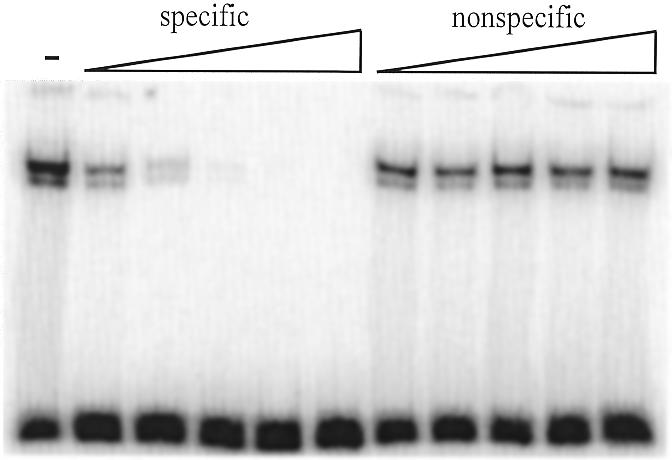
The recombinant S.cerevisiae RAP1 protein binds with high specificity to the telomeric sequences. EMSA of RAP1 protein incubated with the labeled Scer19 oligonucleotide and increasing amounts of unlabeled specific and non-specific competitors. The specific competitor was Scer19 and an unrelated 32 bp sequence was used as the non-specific competitor. Competitors were added in 5, 25, 125, 625 and 2500 times molar excess. The – lane contains no competitors.
To determine the relative affinity of the S.cerevisiae RAP1 protein for the telomeric sequences of different yeast species, the binding of RAP1 to the labeled S.cerevisiae consensus probe was challenged with unlabeled oligonucleotides of the telomeric sequences of S.cerevisiae, S.castellii/S.dairensis, Saccharomyces kluyveri, K.lactis and Candida guilliermondii (Fig. 3). The RAP1 protein binds to the S.castellii/S.dairensis telomeric sequences with a similarly high affinity as to the S.cerevisiae consensus sequence. It binds to the telomeric repeats of the other species with affinities in the order: K.lactis > C.guilliermondii > S.kluyveri. Approximately three and four times less binding was seen to the K.lactis and the C.guilliermondii oligonucleotides, respectively, as compared to the S.cerevisiae sequence. The relatively poor affinity seen for the S.kluyveri sequence (Fig. 3A) might reflect the fact that the S.kluyveri sequence has a mismatch to the RAP1 consensus sequence in its fourth position (Fig. 1). According to the consensus, this position is a conserved T, which is in fact conserved in eight of the 10 species examined. In S.kluyveri this T is substituted by a G. A substitution of this T for a C was shown to greatly reduce binding of the K.lactis RAP1 protein to its telomeric sequence (12). Moreover, the S.kluyveri sequence has an A in the third consensus position, while all the other species have a G (Fig. 1). Similarly, the lowered affinity for the C.guilliermondii repeats may reflect the substitution of a G for a T in the ninth position of the consensus. In all the other species a G occupies this position.
Figure 3.
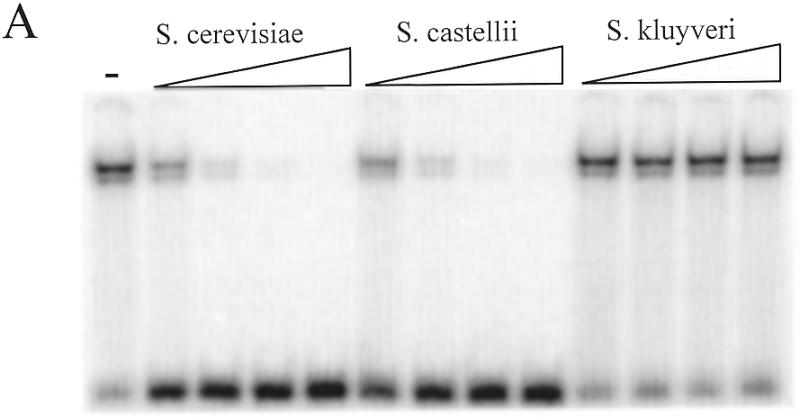
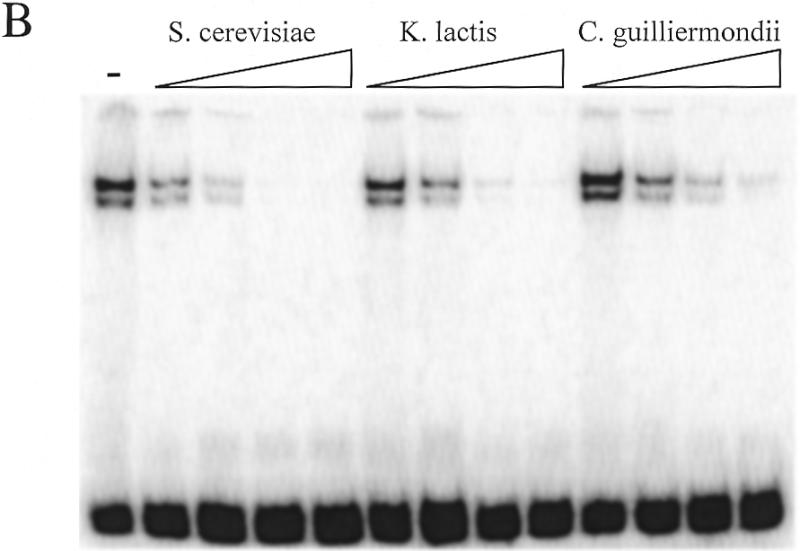
Assay of the relative affinity of RAP1 protein binding to telomeric DNA from various yeast species. The labeled Scer19 probe was competed with unlabeled oligonucleotides consisting of telomeric sequences from (A) S.cerevisiae (Scer19), S.castellii (Scas19-2) and S.kluyveri (Sklu1-26) and (B) S.cerevisiae (Scer19), K.lactis (Klac25) and C.guilliermondii (Cgui20). Competitors were added to the EMSA reactions at 10, 50, 250 and 1250 times molar excess. The – lane contains no competitor.
RAP1 binds specifically to the conserved RAP1-binding site region of diverged telomeric repeats
We wanted to determine whether the conserved tentative binding site found in the telomeric sequences of various yeast species is indeed the crucial motif supporting RAP1 protein binding. We reasoned that the longer S.kluyveri telomeric repeat would be helpful to evaluate this proposition. The S.kluyveri repeat is 26 bp long and is predicted to have a spacing of 14 bp between RAP1-binding sites (Fig. 1). We therefore designed an oligonucleotide having a permutation splitting the consensus motif between positions seven and eight (Sklu21-20, Table 1). The EMSA of this oligonucleotide was compared to an oligonucleotide containing the intact consensus region (Sklu1-26, Table 1). Binding was detected to Sklu1-26 (Fig. 4, lane 2); however, even after extended exposure times, no binding was detected to Sklu21-20, having a permutation splitting the potential binding site into halves (Fig. 4, lane 1). Additionally, two shortened versions of the S.kluyveri repeat were designed. One (Sklu8-26) having a permutation including the potential binding site and the other (Sklu26-18) having a permutation lacking the second half of the consensus region (Table 1). The former oligo supports binding (Fig. 4, lane 3) while the latter does not (Fig. 4, lane 4). These results confirm that the conserved region indicated in Figure 1 is indeed a functional RAP1-binding site and that the diverged sequences outside the core sequence are not involved in binding.
Figure 4.
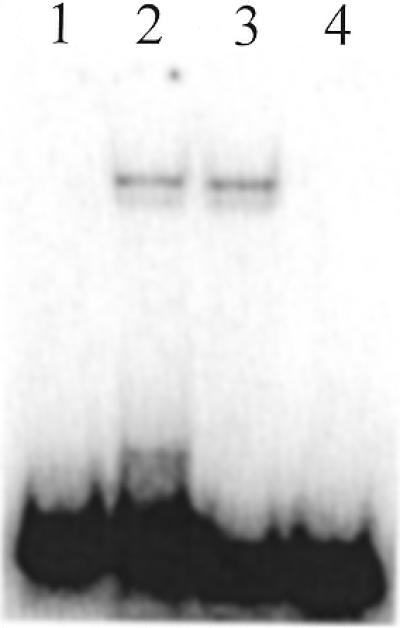
The RAP1 protein only produces mobility shifts when reacted with S.kluyveri telomeric repeats containing an intact putative binding site motif (lane 2, Sklu1-26; lane 3, Sklu8-26). When presented to an oligonucleotide containing a split motif (lane 1, Sklu21-20) or missing half of the motif (lane 4, Sklu26-18) no shift is seen in the EMSA.
The RAP1-binding site in S.castellii and S.dairensis telomeres is formed by connecting two consecutive telomeric repeats
In order to form a S.castellii/S.dairensis telomeric sequence motif which is consistent with the RAP1-binding site consensus, one needs to align two consecutive telomeric 8 bp repeats. To elucidate whether two repeats are actually needed for binding of the protein, we designed a 19 bp oligonucleotide (Scas19-1) where one of the telomeric repeats is replaced by a sequence different from the RAP1 consensus (Table 1). This oligonucleotide was unable to support any RAP1 binding in EMSAs (Fig. 5). We conclude that the RAP1-binding site extends over two consecutive telomeric repeats. This should apply to C.guilliermondii as well, which also has 8 bp telomeric repeats (Fig. 1).
Figure 5.
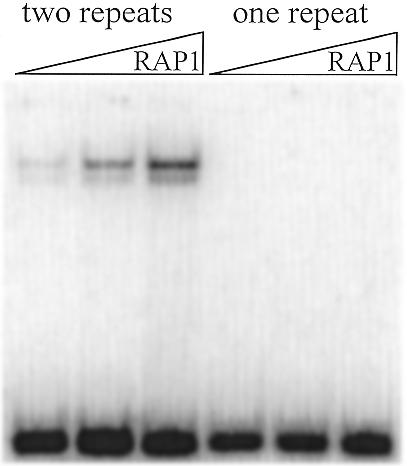
Two 8 bp repeats are needed in order to form a full RAP1 protein-binding site in S.castellii telomeric sequences. The expected shifted band is seen with a probe containing two telomeric repeats (Scas19-2, lanes 1–3). However, when one of the telomeric repeats is replaced by an unrelated sequence, there is no support for RAP1 binding (Scas19-1, lanes 4–6). The protein was added in a dilution range of 2-fold steps.
Footprint analysis of S.castellii telomeric sequences reveals the bipartite structure of the RAP1 protein
To determine the phase and sequence of the RAP1-binding site in the homogeneous S.castellii telomeric repeats, we performed DNase I footprinting analyses. In addition to determining the binding site region, this analysis gives us an estimate of the possible density of bound RAP1 molecules. We used a 65 bp oligonucleotide containing three putative binding sites (Fig. 6). The result was a distinct and very convincing footprinting pattern with two large protected regions, I and II (Fig. 6A). As schematically depicted in Figure 6B, this footprint shows that the oligonucleotide is bound by two RAP1 molecules positioned next to each other, apparently binding the middle repeats, and leaving one unbound telomeric repeat on either side (Fig. 6B). It is evident that these are the positions in which the majority of the protein molecules are bound. Such a preference for RAP1 to bind to adjacent sites has been previously observed (9).
Each protected region, I and II, spans the 13 bp sequence 5′-CTGGGTGTCTGGG-3′. Between the two large protected regions, 3 nt were exposed to DNase I digestion, corresponding to the TGT sequence at positions 37–39 (Fig. 6). In experiments where the no protein control band at position 37 is stronger, the sample band is also stronger. This outcome was unexpected, because of the presence of these base pairs in the GGTGT motif, which was previously determined to be involved in RAP1 binding (2,7). The GGTGT motif, with the same 8 bp spacing as in S.cerevisiae, is also present in the S.castellii sequence. The RAP1 footprint indicates that the phase of binding of the RAP1 protein to the S.castellii sequences is shifted 3 bp to the side, thus excluding the TGT triplet.
Moreover, in the upper protected region (II), the G at position 46 is cleaved by DNase I (Fig. 6A). This G is positioned exactly in the middle of the 13 bp region, but nevertheless is not protected by the bound protein. Most probably, it is exposed to DNase I cleavage because of its location in the region between the two DNA-binding subdomains of the RAP1 molecule (Fig. 6B). The footprint can thus be considered as being split into two halves, each centered on the sequence CTGGG. Thus, the size of the binding site and the 8 bp centre-to-centre spacing of the two domains remained on RAP1 protein binding to these homogeneous sequences. The main structural features thus seem to be in accordance with binding of the irregular S.cerevisiae sequences. Although we have not here determined which bases are actually physically contacting the protein, we prefer to simplify the discussion below by referring to the CTGGG sequences as constituting the two half-sites of the observed binding site region.
RAP1 binds the variant telomeric repeats with lowered affinity
In S.cerevisiae in vitro studies it has been shown that telomeric RAP1 sites occur about once every 18 bp (9). The heterogeneous telomeric sequences, however, make the spacing irregular, so that some sites even overlap (9). In most of the other budding yeasts examined the telomeric sequences consist of totally homogeneous repeats (14,15). This predicts an even distribution of RAP1 sites and a more homogeneous chromatin structure. However, in S.castellii and S.dairensis quite distinct variants of repeats are frequently scattered throughout the telomere (14). Since the optimal fit to the RAP1 consensus only appears when two basic repeats are connected, the variant repeats are predicted to cause irregularities in the distribution of RAP1 sites. This would of course only be of great consequence if the RAP1 protein was unable to bind to these sites. Therefore, we wanted to determine whether the RAP1 protein would be able to bind the various variants in vitro.
In S.dairensis a shortened telomeric repeat variant, TCTGGG, constitutes a prominent amount of the total number of repeats (25%) (14). We designed a 20 bp oligonucleotide containing a combination of one short variant repeat followed by a normal repeat, ΔTG-1 (Table 1). ΔTG-1 has a 6 bp centre-to-centre spacing between the half-sites of the binding site, instead of the normal 8 bp. The RAP1 protein shows a binding affinity for this variant oligonucleotide which is only slightly lower than for the wild-type S.castellii oligonucleotide (Fig. 7). From these results we can conclude that shortening of the distance between the half-site motifs by 2 nt does not cause any steric problems in binding of the protein to this variant. Alternatively, the protein is not strictly using the half-sites for binding, but instead realigning on the total stretch of the oligonucleotide, in which case it would be able to find eight fits to the consensus in total. There have been previous reports on RAP1 allowing some loose stringency in the binding of one of the subdomains, as well as some smaller changes in the allowable spacing of subdomain binding (19,20).
Figure 7.
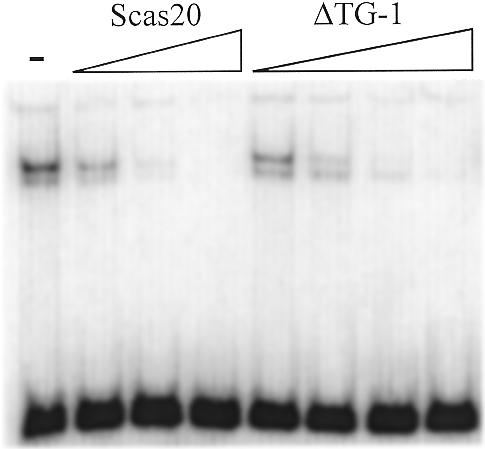
The RAP1 protein binds a S.dairensis variant repeat sequence with only slightly lowered affinity. The relative affinity of RAP1 for a variant (ΔTG-1) sequence containing a shorter variant telomeric repeat was compared to the normal sequence (Scas20). These oligonucleotides were added as cold competitors for the labeled Scer19 probe. Scas20 was added in 10, 100 and 1000 and ΔTG-1 in 10, 100, 1000 and 10 000 times molar excess. The – lane contains no competitor.
The major class of variant telomeric repeats in S.castellii contains additional TG dinucleotides [TCTGGGTG(TG)1–3]. If the half-site motifs (CTGGG) are directing binding, it would mean that when a potential binding site has additional TG bases inserted between the motifs, the 8 bp centre-to-centre spacing between the motifs will be changed. The separate (TG)1, (TG)2 and (TG)3 additions extend the spacing between the motifs to 10, 12 and 14 bp, respectively (Table 1). Because the RAP1 protein binds in a bimodular way, we imagined that this might provide it with a flexibility to extend over these prolonged sequences. Moreover, RAP1 has previously been described to encompass conformational changes (21).
We designed oligonucleotides that contain these different variants with additional TG dinucleotides located in between the half-site motifs. The RAP1 protein is indeed able to bind all three variants (Fig. 8). The relative affinity of the RAP1 protein for the different variants decreased only slightly with a longer intervening distance: (TG)1 > (TG)2 > (TG)3 (Fig. 8). We interpret this result to mean that there is a variation in tolerance of the different half-site distances.
Figure 8.
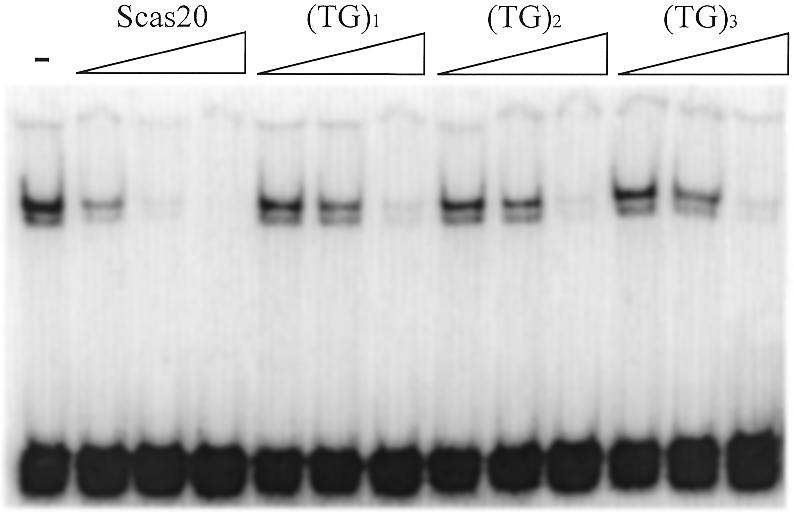
The variant S.castellii repeats are bound by RAP1 with a lower affinity. Different naturally occurring telomeric repeat variants containing one, two and three extra TG dinucleotides (see Table 1) were added as specific EMSA competitors against the labeled probe Scer19. The affinity was compared to the normal Scas20 oligonucleotide. Competitors were added in 10, 100 and 2000 times molar excess. The – lane contains no competitor.
A variant telomeric repeat produces a split footprint
The extra bases in the S.castellii (TG)1, (TG)2 and (TG)3 variant repeats will more or less fit into the consensus and thus could theoretically be recognized by the RAP1 protein. To determine whether they are bound or not, we performed a DNase I footprint analysis on an oligonucleotide containing one such (TG)3 variant repeat (Fig. 9). This oligonucleotide is identical to the one used in the experiment in Figure 6, except for the extra (TG)3 bases inserted at position 46 in the middle of protected region II. In Figure 9 the footprint of the variant-containing oligonucleotide (lanes 5–8) has been run side by side with the original wild-type oligonucleotide (lanes 1–4). The footprints on regions I and I′ are identical in the two experiments. However, a split footprint is produced in the II′ region, where the lower half-site is protected to the same extent as in II, but the consecutive (TG)3 bases are not protected. Instead, protection can be distinguished in the half-site (CTGGG) motif following the (TG)3 stretch in region II′. This suggests that RAP1 binds two half-sites separated by intervening sequences.
To further confirm this finding, we performed a DNase I footprint analysis on the complementary strand of the variant-containing oligonucleotide (Fig. 10). Unfortunately, DNase I does not efficiently cleave this highly C-rich strand. The result nevertheless confirms that the previously detected protected regions span bases 25–59 and that the intervening (TG)3 region is accessible for DNase I cleavage (Fig. 10). To further resolve the protection pattern of the II′ region, we performed a DMS methylation protection footprint analysis. Because DMS modifies G bases that are not involved in binding, this analysis provides information on which G bases make contacts with the protein. In the normal oligonucleotide the G bases in positions 42–44, 51 and 52 are protected from DMS methylation (Fig. 11A). In the variant oligonucleotide the G base positions 42–44 are similarly protected (Fig. 11B). Moreover, the G positions 56–58 are also strongly protected. Thus, the two flanking half-site motifs are firmly protected from DMS modification, whereas the intervening (TG)3 sequence is readily modified, leading to prominent cleavage bands at positions 46, 48, 50 and 52.
Figure 11.
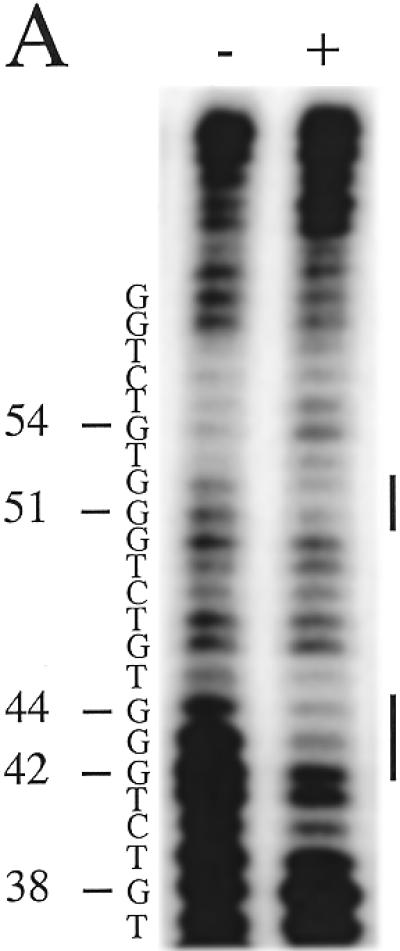
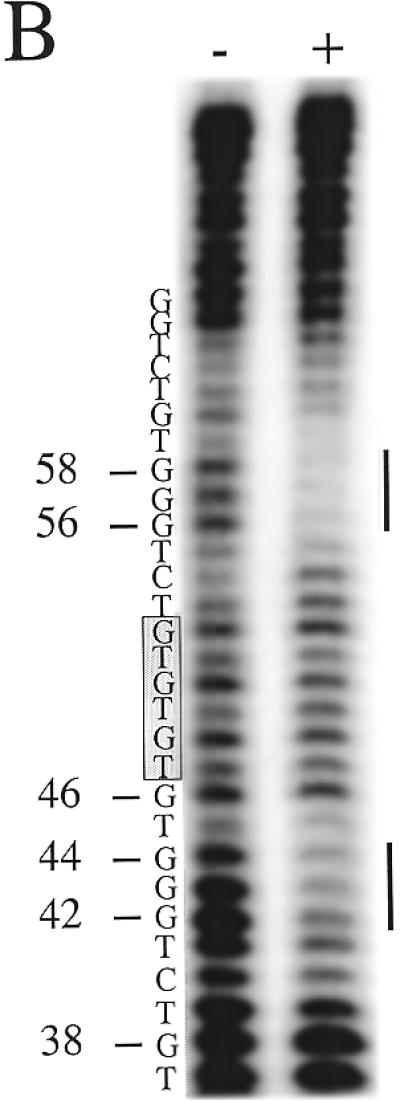
DMS protection footprint analyses. The labeled probes were incubated with or without (+/–) RAP1 protein and then subjected to DMS treatment. (A) The normal sequence probe. (B) The (TG)3 variant-containing probe. The bars indicate the relevant protected G positions. The base numbers at the left are the same as in Figure 9.
The results presented above demonstrate that the RAP1 protein is able to bind to the two half-sites having a centre-to-centre spacing of 14 bp instead of the usual 8 bp. As schematically depicted in Figure 12, this may be enabled by the conformational flexibility of the RAP1 molecule. The two DNA-binding subdomains are connected by a loosely folded linker that may be able to expand and allow the protein to stretch over a longer distance. However, recently it was demonstrated that TRF1 is able to bind to widely separated half-sites by looping out the intervening sequence (6). The lack of protection seen here in the intervening (TG)3 region suggests that they are readily exposed to DNase I attack, which is an indication that the sequences protrude out from the DNA–protein complex. Thus, we suggest that the RAP1 protein is able to remodel the DNA in a similar way to TRF1 in order to bring the two half-sites closer together (Fig. 12C).
Figure 12.

Two different models explaining the observed flexible interaction of the RAP1 protein with the variant repeats containing extra nucleotides inserted between the two half-site motifs of the binding site. In (A) the proteins bind to two normal sites. In the first model (B) the flexible region between the two binding domains is able to stretch out and extend over the elongated intervening sequence. In the second model (C) the RAP1 molecule brings the two half-sites closer together by pushing the intervening sequence out of the protein–DNA complex. We propose that a mechanism including a combination of these two models could be occurring, where both the structural flexibility of the RAP1 molecule and its capacity to remodel DNA add to its ability to bind different kinds of variants.
DISCUSSION
Here we show that the S.cerevisiae RAP1 protein binds the telomeric repeats of several yeast species and that it specifically binds a region with a previously noticed conservation of the RAP1-binding site (14). We demonstrate, using EMSA and DNase I footprint analyses, that the RAP1 protein binds two consecutive 8 bp telomeric repeats in the S.castellii and S.dairensis telomeric DNA sequences. The DNase I footprinting shows a two-part protection pattern, which most likely reflects the bipartite structure of the RAP1 protein. Surprisingly, binding is centered on the CTGGG half-sites present in the full binding site sequence CTGGGTGTCTGGG. These half-sites have the same 8 bp centre-to-centre spacing within the binding site as the GGTGT motif previously identified in S.cerevisiae telomeric sequences. The 3 bp shift in binding site phase is highly unexpected, since the possibility of binding the TGT part of the motif is still offered by the sequence. The observed shift could be due to a tilted complex protecting a region to the side of the bound GGTGT sequence. However, the bands at positions 38 and 46 make this less likely (Fig. 6). Moreover, the DMS footprint data do not support any binding to the underlined G in the GGTGT motif, as seen in positions 46 and 54 of the normal oligonucleotide (Fig. 11A). It would certainly be interesting to obtain conclusive evidence on the specific contacts made, something which is probably best done by resolving the DNA–protein contacts by X-ray crystallography, as was done for the S.cerevisiae sequence (7).
Rearrangements of side-chains have been demonstrated to enable DNA-binding proteins to make alternative base contacts when binding non-consensus sequences (22). Such rearrangements could possibly allow the RAP1 protein to adopt a different structure more suited to bind the 3 bp shifted site observed here. The RAP1 protein was recently demonstrated to form structurally different complexes with the sequences found in the promoters of ribosomal protein genes and telomeres, respectively, although they are two quite similar versions of its consensus (21). It was suggested that RAP1 is able to establish different sets of contacts with different versions of its consensus DNA binding sequence. The function of RAP1 as an activator versus repressor has been reported to be dependent on the DNA context within which it binds. Thus, there is now growing evidence that when binding to different versions of its target sequence, RAP1 is assembled into structurally different complexes having different functionality in vivo (reviewed in 23).
We have also demonstrated the flexibility of the S.cerevisiae RAP1 protein in binding to various naturally occurring variant versions of the telomeric repeats. This capacity to adjust itself to such different targets is most probably due to the subdomain structure of the protein and that the relative positions of the two binding domains with respect to each other are somewhat flexible. However, the affinity for the variants is lowered, which may be of some importance in the context of a longer telomeric sequence stretch. The sequences flanking a potential binding site might affect the ability of a RAP1 molecule to bind the site. A consequence of a lower affinity site might therefore be that the higher affinity sites surrounding it are highly preferred. The reverse could also be argued, that the high affinity sites allow binding of proteins to nearby low affinity sites, as has been demonstrated for the heat shock factors (24). Exactly how profound the in vivo effects of this lowered affinity are may depend on the mechanism of telomere chromatin assembly; whether the proteins are bound consecutively from one end to the other or whether the binding is initially randomly distributed and then adjusted by sliding of the proteins. Another aspect is whether the phasing of the variant irregularities within the telomeric DNA array is optimal for an even distribution of RAP1 molecules. In an in vivo scenario where low affinity sites are not bound a possible outcome of a high number of variants could be an increase in the total length of the telomere. Moreover, if the frequency of variants differs between telomeres, this could possibly lead to different telomeres having different total lengths.
Our footprinting analyses demonstrate a clear and distinct DNase I footprint produced by RAP1 protein interaction with S.castellii telomeric DNA sequences. Here we have shown that the RAP1 protein binds preferentially to sites that are separated by only 3 nt, even though the probe offers other possibilities. It was previously observed that RAP1 molecules preferentially occupy two adjacent sites, rather than two randomly spaced sites, and that this behavior is not caused by any cooperativity in binding (9). One explanation for the extra DNase I cleavage at position 46 in region II, as compared to the totally protected region I, might be a distortion due to the closeness of the sites (Fig. 6A). In fact, steric clashes could be one possible way of explaining the fact that only two molecules are bound, instead of the three possible. The most common site spacing in S.cerevisiae was found to be 18 bp (9). The closeness of the sites in S.castellii/S.dairensis would mean that, if every site is bound, RAP1 protein binding conveys a much more dense structure. The variant repeats may in fact be necessary providers of extra space between sites, so as to decrease the density and thus prevent such steric clashes. It will be necessary to determine whether the structures and sizes of the homologous proteins in these species are comparable to that of S.cerevisiae RAP1 in order to solve this question. The functionally analogous protein of human telomeres, TRF1, is smaller and contains only one DNA-binding domain (25). Thus, TRF1 is predicted to conveniently bind consecutive 6 bp telomeric repeats without any steric clashes (2). However, no structural information is yet available on its interaction with DNA and so the phase of binding of TRF1 along the repeated telomeric sequences is presently unknown. It will be of interest to find out whether S.castellii and S.dairensis have RAP1 proteins of similar structure to that found in S.cerevisiae or whether they have single binding domains like TRF1. The similarities between the S.cerevisiae and K.lactis RAP1 proteins make the first alternative rather likely. Moreover, the RAP1 DNA-binding regions of Saccharomyces unisporus and Zygosaccharomyces rouxii show a high degree of sequence and length conservation when compared to S.cerevisiae (26).
The RAP1 protein functions as a transcriptional activator for many genes encoding glycolytic enzymes and ribosomal proteins. In a study aimed at understanding how RAP1 can regulate expression of this large number of genes, many of them with different expression patterns, it was proposed that structural alterations in RAP1–DNA complexes, both in the DNA and in the protein, affect the transcriptional potential of the complex in an allosteric manner (19). It was further proposed that the dimeric nature of the RAP1 DNA-binding domain is a key structural feature that explains the disparate functions of its DNA-binding sites in vivo (19). A role of the RAP1 protein in opening chromatin to allow activator access to weak binding sites has also been suggested (24). Here we have shown that a split footprint is produced when the RAP1 protein binds a telomeric repeat variant where the two half-sites are separated by an additional 6 bp as compared to the wild-type binding site. The 6 bp of intervening sequence are exposed to DNase I cleavage, which probably indicates that they are looped out from the complex. Thus, this observed split footprint is probably the product of the chromatin remodeling capacity of RAP1 that has been demonstrated at promoters (24). Moreover, this capacity has recently been described for TRF1, where the two Myb domains in a dimerized TRF1 complex are able to bind to widely spaced half-sites by looping out the intervening sequence (6). It was suggested that this spatial flexibility of the TRF1 dimer is enabled by free rotation facilitated by a flexible connection between the two Myb domains. Although intramolecularly localized, a similar flexibility can be predicted to be present between the two Myb domains of the RAP1 protein. Thus, when binding to homogeneous sequences RAP1 appears to be interacting with DNA in a similar manner to the TRF1 protein. The ability of RAP1 molecules to bind the variant repeats in this unpredictable manner in vitro opens up speculation on the possibility of the same in vivo behavior. If this DNA remodeling has a propensity to take place in vivo, it would have significant consequences for the packaging of telomeric DNA in the telomeric arrays of S.castellii, where variants are scattered throughout the array. Moreover, it is predicted to have a pronounced effect on the irregular S.cerevisiae sequences where RAP1-binding sites frequently overlap. The RAP1 protein has a role in recruiting other proteins to the telomeric DNA (the SIR and RIF proteins). Our results suggest that similarly to its function at promoters, the RAP1 protein acts by modulating the conformation of the telomeric DNA and possibly its accessibility to other proteins.
Still, we have much to learn about how telomeric DNA is recognized and packaged and what intricate mechanisms are involved in the regulation of telomere length. The RAP1 protein is a major determinant of telomere chromatin structure in S.cerevisiae. The conservation of a RAP1-binding site, despite the remarkable diversity between the repeats in different yeast species, certainly indicates that it is a well-conserved feature. Since it has been proposed that telomere length is governed by the number of bound RAP1 molecules in S.cerevisiae it will be interesting to find out whether RAP1 protein plays the same important role in the regulation of telomere length in these other budding yeasts.
Acknowledgments
ACKNOWLEDGEMENTS
We are indebted to David Shore for sharing with us the RAP1-containing E.coli expression vector. We are grateful to Michael McEachern for critical reading of the manuscript and thank Anat Krauskopf, Claes von Wachenfeldt and Allan Rasmusson for helpful advice. Andrea Möllenkvist and Kerstin Jönsson are acknowledged for technical assistance. This work was supported by grants to M.C. from the Swedish Medical Research Foundation and the Swedish Cancer Society. The Erik Philip-Sörenssen foundation, the Nilsson-Ehle foundation and the Schyberg foundation are also acknowledged.
REFERENCES
- 1.Fang G. and Cech,T.R. (1995) In Blackburn,E.H. and Greider,C.W. (eds), Telomeres. Cold Spring Harbor Laboratory Press, Plainview, NY, Monograph 29, pp. 69–105.
- 2.König P. and Rhodes,D. (1997) Trends Biochem. Sci., 22, 43–47. [DOI] [PubMed] [Google Scholar]
- 3.Tanikawa J., Yasukawa,T., Enari,M., Ogata,K., Nishimura,Y., Ishii,S. and Sarai,A. (1993) Proc. Natl Acad. Sci. USA, 90, 9320–9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper J.P., Nimmo,E.R., Allshire,R.C. and Cech,T.R. (1997) Nature, 385, 744–747. [DOI] [PubMed] [Google Scholar]
- 5.Broccoli D., Smogorzewska,A., Chong,L. and de Lange,T. (1997) Nature Genet., 17, 231–235. [DOI] [PubMed] [Google Scholar]
- 6.Bianchi A., Stansel,R.M., Fairall,L., Griffith,J.D., Rhodes,D. and de Lange,T. (1999) EMBO J., 18, 5735–5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.König P., Giraldo,R., Chapman,L. and Rhodes,D. (1996) Cell, 85, 125–136. [DOI] [PubMed] [Google Scholar]
- 8.Wang S.-S. and Zakian,V. (1990) Mol. Cell. Biol., 10, 4415–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilson E., Roberge,M., Giraldo,R., Rhodes,D. and Gasser,S.M. (1993) J. Mol. Biol., 231, 293–310. [DOI] [PubMed] [Google Scholar]
- 10.Marcand S., Gilson,E. and Shore,D. (1997) Science, 275, 986–990. [DOI] [PubMed] [Google Scholar]
- 11.Ray A. and Runge,K.W. (1999) Mol. Cell. Biol., 19, 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krauskopf A. and Blackburn,E.H. (1996) Nature, 383, 354–357. [DOI] [PubMed] [Google Scholar]
- 13.Larson G.P., Castanotto,D., Rossi,J.J. and Malafa,M.P. (1994) Gene, 150, 35–41. [DOI] [PubMed] [Google Scholar]
- 14.Cohn M., McEachern,M.J. and Blackburn,E.H. (1998) Curr. Genet., 33, 83–91. [DOI] [PubMed] [Google Scholar]
- 15.McEachern M.J. and Blackburn,E.H. (1994) Proc. Natl Acad. Sci. USA, 91, 3453–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohn M. and Blackburn,E.H. (1995) Science, 269, 396–400. [DOI] [PubMed] [Google Scholar]
- 17.Shore D., Stillman,D.J., Brand,A.H. and Nasmyth,K.A. (1987) EMBO J., 6, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wissmann A. and Hillen,W. (1991) Methods Enzymol., 208, 365–379. [DOI] [PubMed] [Google Scholar]
- 19.Idrissi F.Z. and Pina,B. (1999) Biochem. J., 341, 477–482. [PMC free article] [PubMed] [Google Scholar]
- 20.Graham I.R. and Chambers,A. (1994) Nucleic Acids Res., 22, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Idrissi F.Z., Fernandez-Larrea,J.B. and Pina,B. (1998) J. Mol. Biol., 284, 925–935. [DOI] [PubMed] [Google Scholar]
- 22.Schwabe J.W., Chapman,L. and Rhodes,D. (1995) Structure, 3, 201–213. [DOI] [PubMed] [Google Scholar]
- 23.Morse R.H. (2000) Trends Genet., 16, 51–53. [DOI] [PubMed] [Google Scholar]
- 24.Yu L. and Morse,R.H. (1999) Mol. Cell. Biol., 19, 5279–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith S. and de Lange,T. (1997) Trends Genet., 13, 21–26. [DOI] [PubMed] [Google Scholar]
- 26.Graham I.R., Haw,R.A., Spink,K.G., Halden,K.A. and Chambers,A. (1999) Mol. Cell. Biol., 19, 7481–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buchman A.R., Kimmerly,W.J., Rine,J. and Kornberg,R.D. (1988) Mol. Cell. Biol., 8, 210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]