

Abstract
The transcription factor Smad4 binds DNA in response to a TGF-beta ligand-initiated intracellular signaling cascade. SMAD4 is deleted or mutated during tumorigenesis in many human tumors. Some of these mutations occur in the N-terminal portion of the protein, the Mad homology 1 (MH1) region, which exhibits sequence-specific DNA-binding. We used alanine scanning mutagenesis and natural mutations to map the subregion of the MH1 domain necessary for that function. We created 20 individual mutations in the MH1 region of human Smad4 and assayed their effect on DNA-binding in vitro. Mutation of residues in the less conserved N- and C-terminal areas of the MH1 region had no effect on DNA-binding. However, mutations in the domain from L43 to R135 caused a dramatic reduction of the ability of Smad4 to bind DNA. Previous work demonstrated a β-hairpin protein motif within this region to be responsible for DNA-binding, but suggested that the tumorigenic mutations occurring outside this motif may target a separate function of the MH1 domain. Our results demonstrate that the MH1 domain as a whole is very sensitive to changes in overall structure, and that tumorigenic mutations within the region of L43–R135 indeed would target DNA-binding.
INTRODUCTION
Human Smad4 (DPC4/MADH4) is a central mediator of signal transduction by the transforming growth factor-beta (TGF-beta) superfamily of cytokines. These cytokines induce cellular responses such as growth suppression, apoptosis and differentiation by signaling through the Smad family of effector proteins (reviewed in 1,2). Stimulation of a cell by a TGF-beta superfamily member induces autophosphorylation of ligand-specific receptor pairs. These receptors then phosphorylate/activate a set of pathway-restricted Smad proteins. Activated Smads can complex with the common partner, Smad4, translocate to the nucleus and participate in sequence-specific DNA-binding and transcriptional activation through TGF-beta responsive elements (1,2).
Smad proteins have two evolutionarily conserved regions, MH1 and MH2 (Mad homology 1 and 2) separated by a linker region. The N-terminal MH1 domain is responsible for sequence-specific DNA-binding (3–6), while heteromerization and transactivation functions have been ascribed to the MH2 region (7–9). The MH2 region has also been shown to partially interfere with the DNA-binding function of the MH1 region (5,6,9,10).
Mutation or deletion of components of the Smad signaling pathway allows some human tumors to evade the growth suppressive signals of TGF-beta or related ligands. SMAD4 mutations have been found frequently in pancreatic and colorectal cancers, as well as occasionally in other tumor types (11,12). Several point mutations map to the MH1 region (12–15), implying that functions of this domain may serve as targets for mutational inactivation (9).
The Smad MH1 region was shown to bind to a consensus DNA sequence termed the Smad binding element (SBE) (5). The crystal structure of the MH1 region bound to DNA of a highly homologous family member, Smad3, indicates that the protein contacts DNA via an 11-residue β-hairpin motif. The Smad3 β-hairpin extends beyond the bulk of the MH1 structure to lie within the major groove of the DNA (16).
Since the MH1 regions of Smad3 and Smad4 have 71% similarity at the protein level and Smad3 binds the SBE with a dissociation rate similar to Smad4 (5,16), it is reasonable to expect that Smad4 binds DNA in a manner essentially the same as Smad3. In addition to the β-hairpin motif, there are several other structural features of the MH1 region that are highly conserved among Smad family members. These regions include a helix with several basic residues near the C terminal end of the MH1 domain (a sequence resembling canonical nuclear localization signals), and loops L2 and L4 (termed the double loop region) (16). Interestingly, all of the tumor-derived MH1 mutations of Smad4 map to regions of the protein at a distance from the β-hairpin motif. It is unclear from structural considerations alone whether these mutations would disrupt DNA-binding, or instead affect other potential functions of the domain (16). Indeed, evidence has been presented that a tumorigenic mutation may spare the intrinsic DNA-binding function and affect either the intramolecular association of the MH1 and MH2 domains (10), or a macromolecular interaction interface (16). The relationship of the natural mutations to the primary known function of the MH1 domain is thus in question, and is central to the understanding of the role of Smad4 in tumor-suppression.
In the present study, we employed a scanning alanine mutagenesis strategy to map the area of the MH1 region that was necessary for binding to the SBE. Eighteen individual alanine substitutions within the MH1 region of Smad4 were created, and the effects of these mutations on the DNA-binding function of Smad4 were examined in an in vitro DNA-binding assay. The results of these experiments show that the residues of the MH1 domain which are mutated in human tumors play a role in stabilizing the DNA-binding function of this domain. Mutation of each of these residues to alanine dramatically reduced the ability of the Smad4 MH1 domain to bind DNA. Our work defines a region, Smad4 residues L43–R135, that is highly sensitive to structural changes. The integrity of this region, in addition to the β-hairpin DNA contact point, is important for maintaining the DNA-binding function of Smad4. MH1 residues inactivated during tumorigenesis fall within the L43–R135 region suggesting that mutation of these residues targets the DNA-binding activity of Smad4.
MATERIALS AND METHODS
Plasmid constructs and alanine scanning mutagenesis
Full-length SMAD4 cDNA was cloned into the pcDNA3.1 vector (Invitrogen, Carlsbad, CA) to produce pDPC4-WT as described (9). A stop mutation was engineered at codon 515, removing the C-terminal 37 residues that partially interfere with DNA-binding (9,10) yielding the pDPC4-515st plasmid. By creating the alanine mutagenesis constructs in this background, we could eliminate any potential effects between the residues under study and the autoinhibitory domain, thus focusing on the direct effects of the residues on DNA-binding. This increases the sensitivity of our assay to detect small changes in DNA-binding affinity caused by mutation of individual Smad4 residues.
Thirteen residues within the most highly conserved area of the MH1 region of Smad4 (the L43–R135 area) and five residues outside the most highly conserved area were selected for mutation to alanine. In addition to the alanine mutagenesis, a specific SMAD4 mutation found in a human tumor, R100T, was produced (Table 1). Individual mutations were created by use of the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) with the pDPC4-WT or the pDPC4-515st constructs as the parental vector according to the manufacturer’s protocol. The sequences of selected clones were confirmed by full-length automated sequencing.
Table 1. In vitro DNA-binding activities of Smad4 MH1a mutants.
| MH1 domain mutations | DNA-binding observedb | Crystal structure position |
|---|---|---|
| Outside of highly conserved region | ||
| P10A | +++ | absent from structure |
| Q28A | +++ | surface |
| F166A | +++ | absent from structure |
| E193A | +++ | absent from structure |
| P244A | +++ | absent from structure |
| Within highly conserved region | ||
| L43A | – | hydrophobic core |
| K46A | – | partially buried |
| K50A | – | surface |
| G65Ac | + | surface |
| P68A | +++ | surface |
| R81A | – | β-hairpin |
| K88A | – | β-hairpin |
| P91A | +++ | partially buried |
| R97A | ++ | surface |
| R100Ac | – | partially buried |
| R100Tc | – | partially buried |
| F119A | ++ | buried |
| P130Ac | + | buried |
| R135A | – | partially buried |
aDNA-binding activity of various Smad4 mutant proteins measured upon translation in vitro, binding to labeled SBE probe and separation in an EMSA.
b+++, strongest binding to SBE probe; ++, moderate binding; +, weak binding; –, no binding.
cResidues which are mutated in human cancers.
Preparation of Smad4 protein
The mutant and wild-type Smad4 proteins were produced by use of an in vitro rabbit reticulocyte transcription and translation system (T7 Quick TNT, Promega, Madison, WI). For each DNA construct, the TNT synthesis was divided evenly into an unlabeled reaction mixture (1 ml of 1 mM methionine added) and a labeled reaction [1 ml of [35S]-Translabel from ICN (Costa Mesa, CA) added]. The radiolabeled products were quantified by 8% SDS–PAGE and autoradiography, allowing verification of the efficiency of protein synthesis prior to performance of the subsequent electrophoretic mobility shift assay (EMSA).
Electrophoretic mobility shift assay
EMSAs were performed to directly assess the ability of the pDPC4-515st derived alanine scanning mutation series to bind an SBE probe. The 32P-labeled SBE probe, a double-stranded oligonucleotide containing a single repeat of the SBE, was prepared as described (9). The binding reaction was carried out as described in previous studies (5), with 0.1 ng of labeled probe (1 × 104 c.p.m.) being incubated with 2 µl of unlabeled in vitro-translated Smad4 protein. Smad4 bound to the SBE probe was separated from unbound probe in a 4% polyacrylamide/0.5× TBE gel electrophoresed for 2 h at 200 V and 40°C. The shifted complexes were visualized by autoradiography of the dried gels. The intensities of the probes having shifted mobility, as observed for the complex formed with each mutant construct, were compared to those formed with pDPC4-WT and with the wild-type pDPC4-515st construct.
Computational analysis
The molecular coordinates for the crystal structure of the Smad3 MH1 region bound to the SBE were obtained from Yigong Shi (Department of Molecular Biology, Princeton University) (PDB accession code 1MHD). We used the Rasmol and Sculpt imaging programs to identify the positions corresponding to the Smad4 alanine mutations in this structure. (Rasmol: http://www.umass.edu/microbio/rasmol/getras/htm , Sculpt: Interactive Simulations). We also used the online ClustalW sequence alignment program (http://www.genome.ad.jp/SIT/CLUSTALW.html ).
RESULTS
Functional mapping of the DNA-binding function of Smad4
The MH1 region of Smad4 harbors the DNA-binding function of this transcription factor. The crystal structure of the MH1 domain of a highly related protein, Smad3, shows that a novel β-hairpin motif (corresponding to residues 78–89 in Smad4) serves as the point of DNA contact. This motif lies within an area of maximum evolutionary conservation. Within the MH1 region itself there are several other areas that exhibit a high level of conservation between Smad family members. The precise roles of these structural features have not been defined. It is interesting to note that natural mutations of the Smad4 MH1 region that arose during tumorigenesis (i.e., G65V, Y95N, R100T, N129K and P130S), which presumably abolish the function of Smad4, occurred outside of the β-hairpin DNA contact motif.
Mutations of the MH1 domain are selected during tumorigenesis presumably due to their targeting the inactivation of a specific function of the domain. Previous work has suggested that mutations of the MH1 domain may target an autoinhibitory function of the Smad4 protein, an unknown protein–protein interaction, or the DNA-binding function of Smad4 (9,10,16). This work specifically addresses the sufficiency of the DNA-binding activity as the functional target for these tumorigenic mutations.
To functionally map the region of the MH1 region necessary for DNA-binding, we employed a scanning alanine mutagenesis approach. This strategy was chosen to address the questions apropos to cancer genetics. Missense mutations in human tumor-suppressor genes do not characteristically affect strategic structural points in a highly restrictive distribution. Instead, mutations generally affect a wide distribution of evolutionarily conserved residues due to their random origin followed by the operation of selective pressures. We therefore chose not to selectively mutate the key structural points, as this might lead to foregone conclusions, and because the correspondence of Smad4 to the known Smad3 structure is incomplete. Alternatively, we attempted to compensate for the limited number of reported natural mutations in the MH1 region by construction of a mutation panel that might mimic the diversity of conserved residues expected of the natural mutations.
Eighteen residues in the MH1 region of Smad4 that are evolutionarily conserved among Smad family members were selected and changed to alanine via site-directed mutagenesis. We then assessed the ability of these mutants to bind the SBE in vitro. This allowed us to test the effect of an individual residue on overall protein function, while minimizing global structural change. We examined the effect of alanine substitutions (Fig. 1) in several conserved areas of the MH1 domain: the β-hairpin DNA-contact motif, the basic helix region, and the double loop region to which tumorigenic mutations map (Fig. 2).
Figure 1.
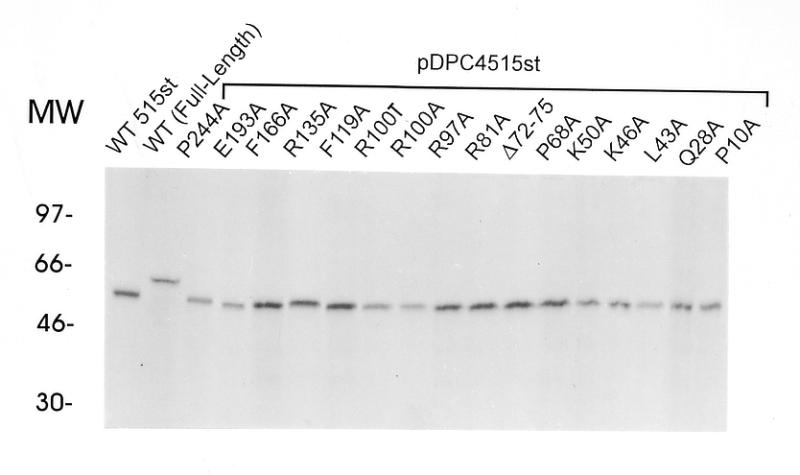
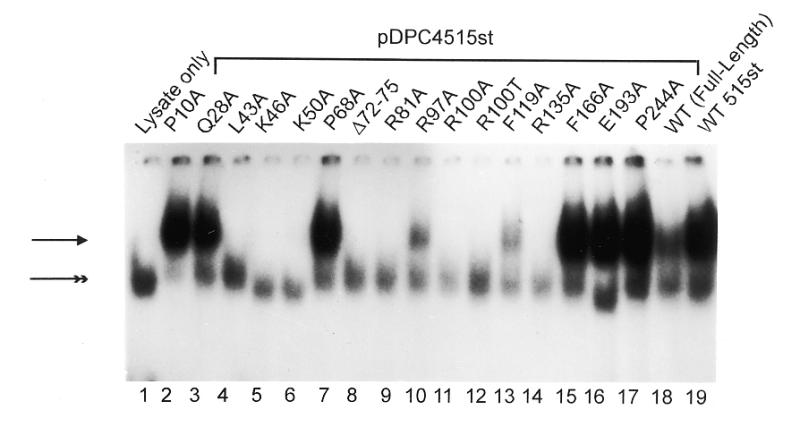
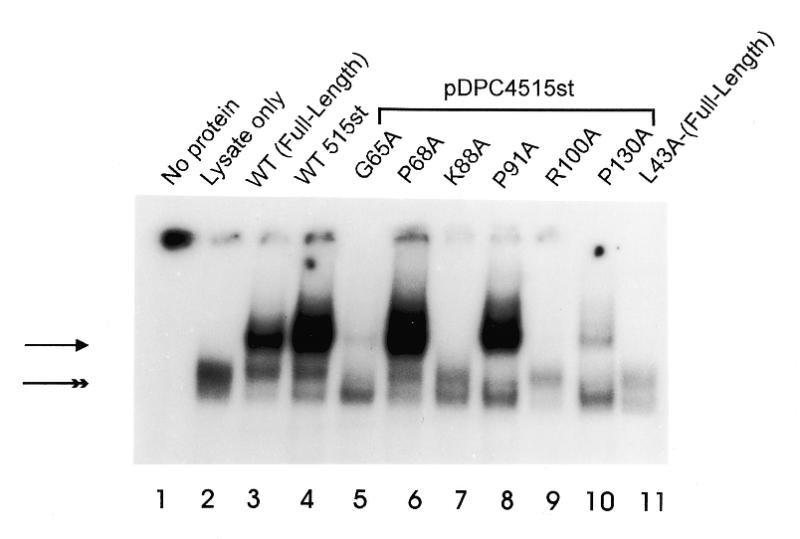
Comparison of the abilities of various mutants of 515st Smad4 protein to bind DNA. (A) Relative quantities of in vitro translated MH1 mutants, 515st and wild-type full-length Smad4 resolved in a denaturing polyacrylamide gel and imaged with autoradiography. (B and C) Each Smad4 mutant protein, in comparison with wild-type, in EMSA with SBE as labeled probe. Arrowhead indicates the shifted complex consisting of Smad4 bound to SBE. Double arrowhead indicates a non-specific complex present in all lanes containing reticulocyte lysate. Mutations created in the 515st background are grouped under a bar. The mutation created in the full-length background (L43A-Full-Length) is indicated.
Figure 2.

Depiction of the distribution and effects of mutation of various Smad4 MH1 residues. (A) Crystal structure of the Smad3 MH1 region bound to DNA. Protein is shown in cyan, DNA in purple. The N-terminus of the structure is noted as well as the distal border of the MH1 region leading to the MH2 region and C-terminus of the protein. Corresponding Smad4 residues selected for alanine mutagenesis are mapped onto this structure. Residues appearing in red, when mutated, severely disrupt the DNA-binding function of this domain while residues in green have no effect. Those residues that have been found to be mutated in human tumors are colored orange. Of these, those tested also disrupted DNA binding. The basic helix, double loop and β-hairpin domains are indicated. (B) Illustration of the similarity between Smad4 and Smad3. The basic helix, double loop and β-hairpin domains are indicated. The same color scheme, depicting residues selected for alanine scanning mutagenesis, is used as detailed above with residues highlighted in orange again signifying residues mutated in cancer. The residues deleted in the mutagenesis construct Δ72–75 are underlined. The area defined here as necessary for wild-type DNA binding is indicated under the magenta bar.
EMSA revealed a shifted complex containing Smad4 (Fig. 1A) bound to the 32P-labeled SBE oligonucleotides (Fig. 1B and C). As expected, the 515st Smad4 protein, containing a wild-type MH1 region but lacking the C-terminal inhibition function, bound the SBE with a much higher affinity than full-length wild-type Smad4 (Fig. 1B, compare lanes 18 and 19; Fig. 1C, lanes 3 and 4). The disruptions of DNA-binding observed by mutations in the 515st background were also apparent when the same mutations were expressed as a full-length protein (e.g., compare lane 4 of Fig. 1B with lane 11 of Fig. 1C).
Single alanine substitutions within the highly conserved region at L43, K46, K50, R81, R100, R135 and K88, as well as a 4-amino acid deletion near the β-hairpin completely abolished detectable DNA-binding in this assay (Fig. 1B, lanes 4–6, 8, 9, 11, 14; Fig. 1C, lane 7). These effects on DNA-binding may be explained by examining the mutations in the context of the Smad3 crystal structure: the conserved residue L43 is buried in the hydrophobic core of the protein, R81 is directly involved in DNA-binding, and K88 may act to stabilize the β-hairpin DNA contact. K46 and K50 seem to be neither structural residues nor involved in direct DNA contact, but their basic character may help the overall domain to associate with DNA. From the Smad3 structure alone, it is unclear why the mutations of R135 and of tumorigenic residue R100 would have such a drastic effect on DNA-binding.
Other mutant proteins within this region (G65A and P130A) showed a severely reduced level of DNA-binding activity (Fig. 1C, lanes 5 and 10). Inspection of the Smad3 crystal structure reveals that P130 is a buried structural residue. Gly65 in Smad4 is substituted by Asp in Smad3, therefore a clear explanation of the role of this residue in the Smad4 structure is not possible.
When alanine substitutions were made outside of the most highly conserved MH1 regions (i.e., P10A, Q28A, F166A, E193A and P244A), no effect was seen on DNA-binding (Fig. 1B, lanes 2, 3 and 15–17). Of these, only Q28 lies within the domain resolved in the Smad3 crystal structure. The corresponding Smad3 residue is located on the surface of the protein and is not likely to be involved in maintaining the overall structure of the domain.
Additionally, several mutations within the region of highest conservation had moderate or no effects on DNA-binding. The mutants R97A and F119A retained a small degree of DNA-binding activity (Fig. 1B, lanes 10 and 13), and mutations at P68 and P91 showed no reduction in the ability to bind the SBE (Fig. 1C, lanes 6 and 8). Analysis of the Smad3 crystal structure shows that R97 is partially solvent-exposed, and would be expected to contribute in a minor way to the stability of the protein as a whole. The negative effect of mutating F119 on DNA-binding is likely due to the fact that it is located near the DNA and packs against residues involved in the protein–DNA contact. As would be expected from the lack of an effect on DNA-binding, the corresponding Smad3 residue for P68 is a surface residue in the crystal structure. P91, however, is partially buried yet apparently has no role in stabilizing the Smad4 structure.
Comparison of the R100A and R100T proteins (Fig. 1B, lanes 11 and 12) showed that DNA-binding was abolished at this residue both by the arginine to threonine substitution found in a human tumor, as well as by the alanine substitution created in this experiment. Interestingly, in all cases, alanine mutagenesis of MH1 residues reported as being mutated in cancer (i.e., G65, R100 and P130) (Fig. 1C, lanes 5, 9 and 10) showed a severe reduction in DNA-binding activity. In the case of G65, an alanine substitution at this residue abrogated DNA-binding, whereas the mutation of a nearby surface residue, P68, had no effect (Fig. 1C, lanes 5 and 6).
DISCUSSION
Elucidation of the crystal structure of the Smad3 MH1 domain bound to DNA shows that the Smad MH1 region exists as a compact globular fold and contacts DNA via a β-hairpin motif (16). In addition to the β-hairpin motif, several other structural features of the MH1 domain exhibit marked conservation between the Smad family members. Whether the MH1 domain of Smad4 has an additional functional activity separate from the DNA-binding ability is unknown. Here we performed scanning alanine mutagenesis of the conserved region of Smad4 to test the effect of various mutants on intrinsic DNA-binding.
The crystal structure of Smad3 predicts that many of the conserved residues are in the hydrophobic core, thus are important for appropriate protein folding and domain stability. Our functional data agree with this prediction: mutation of buried residues L43 and P130 cause the mutant protein to fail to bind DNA. Additionally, the crystal structure predicts that conserved residues R81, K88 and F119 serve to stabilize the β-hairpin DNA contact motif. This was also confirmed by the DNA-binding assay. However, for other residues, such as G65, K46, K50, R100 and R135 the crystal structure was unable to reliably predict the drastic reduction of DNA-binding exhibited by these mutants.
Our results indicate that in addition to the β-hairpin motif, the DNA-binding activity of Smad4 is very sensitive to structural changes in a larger domain from L43 to R135. In this region, 12 out of 14 individual mutations caused a marked reduction or complete failure of the mutant protein to bind DNA. This area encompasses much of the highly conserved portion of the MH1 domain including the basic helix and double loop region. Other mutations tested, outside of this defined area, had no effect on DNA binding.
Human tumors are often able to avert growth suppressive signals from TGF-beta superfamily cytokines by selecting cells that have deleted or mutated components of the Smad family of signaling proteins. Most often, it is SMAD4 that is targeted for such genetic inactivation. A detailed functional comparison and classification of natural mutations in the C-terminal (MH2) region of this protein has been provided (9). Mutations also occur in the N-terminal (MH1) region of SMAD4. The consequence of mutations to this region is disputed. Anecdotal studies of single mutations had suggested that mutations in the MH1 region of Smad4 may serve to increase autoinhibition of the DNA-binding function of the protein, to prevent interaction with other proteins, or to ablate DNA binding (9,10,16). Through consideration of the Smad3 structure it is unclear whether mutation of the tumorigenic residues would cause only a small perturbation of local structure, or global destabilization of the DNA-binding domain (16).
The current study shows that alanine mutagenesis of Smad4 MH1 residues mutated in tumors invariably caused a drastic reduction of the ability of these mutant proteins to bind DNA, presumably through destabilization of the domain as a whole. Furthermore, a newly published report details the discovery of two additional somatic mutations of the SMAD4 MH1 domain in colorectal cancer, Y95N and N129K (15). Both of these residues are located outside of the β-hairpin DNA-contact motif. However, since they are both buried structural residues located within the DNA-binding sensitive area defined here, it is likely that these mutations will disrupt DNA binding.
We previously demonstrated that some MH1 mutations cause a dramatic loss of DNA-binding activity (9). We have now established that the DNA-binding ability of the β-hairpin motif is highly sensitive to small amino acid changes throughout the L43–R135 domain. Therefore, our data favor the suggestion that mutation of these residues may directly cause a disruption of the DNA-binding function of Smad4 and it is this defect that is selected for during tumorigenesis.
In the case of another tumor-suppressor transcription factor, p53, mutations occur in the DNA-binding domain during tumorigenesis either: (i) at residues which directly contact the DNA, or (ii) at more distant residues which destabilize the DNA-binding domain as a whole (17). A similar mechanism of inactivation may operate for Smad4. To date, all known mutations of the DNA-binding function of Smad4 fall into the latter category. In the future we may discover mutations that alter the residues of the β-hairpin DNA contact, since mutations of either class would serve the same purpose, to disrupt the binding of Smad4 to the SBE.
In conclusion, we suggest that the Smad DNA-binding domain has stringent requirements in the region from L43–R135. It is probably the DNA-binding function of this domain that is inactivated by mutation of this region during tumorigenesis. Additionally, as more mutations are found in the MH1 domain of Smad family members, this functional map of Smad4 DNA-binding will aid in the characterization of these mutations.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr Yigong Shi for helpful suggestions and insight, Dr Jiale Dai for provision of subclones and technical advice, Dr Deborah McClellan for her reading of the manuscript, and Dr Cynthia Wolberger for her consultation on structural analysis. This research was supported by NIH grant CA68228. J.B.J. was supported in part by NIH training grant GM07814 as part of the Predoctoral Training Program in Human Genetics.
REFERENCES
- 1.Heldin C.H., Miyazono,K. and ten Dijke,P. (1997) Nature, 390, 465–471. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y., Musci,T. and Derynck,R. (1997) Curr. Biol., 7, 270–276. [DOI] [PubMed] [Google Scholar]
- 3.Yingling J.M., Datto,M.B., Wong,C., Frederick,J.P., Liberati,M.T. and Wang,X.-F. (1997) Mol. Cell. Biol., 17, 7019–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dennler S., Itoh,S., Vivien,D., ten Dijke,P., Huet,S. and Gauthier,J.-M. (1998) EMBO J., 17, 3091–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zawel L., Dai,J.L., Buckhaults,P., Zhou,S., Kinzler,K.W., Vogelstein,B. and Kern,S.E. (1998) Mol. Cell, 1, 611–617. [DOI] [PubMed] [Google Scholar]
- 6.Kim J., Johnson,K., Chen,H.J., Carroll,S. and Laughon,A. (1997) Nature, 388, 304–308. [DOI] [PubMed] [Google Scholar]
- 7.Liu F., Hata,A., Baker,J.C., Doody,J., Carcamo,J., Harland,R.M. and Massague,J. (1996) Nature, 381, 620–623. [DOI] [PubMed] [Google Scholar]
- 8.Shi Y., Hata,A., Lo,R.S., Massague,J. and Pavletich,N.P. (1997) Nature, 388, 87–93. [DOI] [PubMed] [Google Scholar]
- 9.Dai J.L., Turnacioglu,K., Schutte,M., Sugar,A.Y. and Kern,S.E. (1998) Cancer Res., 58, 4592–4597. [PubMed] [Google Scholar]
- 10.Hata A., Lo,R.S., Wotton,D., Lagna,G. and Massague,J. (1997) Nature, 388, 82–87. [DOI] [PubMed] [Google Scholar]
- 11.Hahn S.A., Schutte,M., Hoque,A.T., Moskaluk,C.A., da Costa,L.T., Rozenblum,E., Weinstein,C.L., Fischer,A., Yeo,C.J., Hruban,T.H. and Kern,S.E. (1996) Science, 271, 350–353. [DOI] [PubMed] [Google Scholar]
- 12.Schutte M., Hruban,R.H., Hedrick,L., Cho,K.R., Nadasdy,G.M., Weinstein,C.L., Bova,G.S., Isaacs,W.B., Cairns,P., Nawroz,H., Sidransky,D., Casero,R., Meltzer,P.S., Hahn,S.A. and Kern,S.E. (1996) Cancer Res., 56, 2527–2530. [PubMed] [Google Scholar]
- 13.MacGrogan D., Pegram,M., Slamon,D. and Bookstein,R. (1997) Oncogene, 15, 1111–1114. [DOI] [PubMed] [Google Scholar]
- 14.Thiagalingam S., Lengauer,C., Leach,F.S., Schutte,M., Hahn,S.A., Overhauser,J., Willson,J.K., Markowitz,S., Hamilton,S.R., Kern,S.E., Kinzler,K.W. and Vogelstein,B. (1996) Nature Genet., 13, 343–346. [DOI] [PubMed] [Google Scholar]
- 15.Koyama M., Ito,M., Nagai,H., Emi,M. and Moriyama,Y. (1999) Mutat. Res., 406, 71–77. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y., Wang,Y.F., Jayaraman,L., Yang,H., Massague,J. and Paveltich,N. (1998) Cell, 94, 585–594. [DOI] [PubMed] [Google Scholar]
- 17.Cho Y., Gorina,S., Jeffrey,P.D. and Paveltich,N.P. (1994) Science, 265, 346–355. [DOI] [PubMed] [Google Scholar]