

Abstract
The increasing availability of molecular genetic testing has changed the landscape of both genetic research and clinical practice. Not only is the pace of discovery of novel disease‐causing genes accelerating but also the phenotypic spectra associated with previously known genes are expanding. These advancements lead to the awareness that some genetic movement disorders may cluster in certain ethnic populations and genetic pleiotropy may result in unique clinical presentations in specific ethnic groups. Thus, the characteristics, genetics and risk factors of movement disorders may differ between populations. Recognition of a particular clinical phenotype, combined with information about the ethnic origin of patients could lead to early and correct diagnosis and assist the development of future personalized medicine for patients with these disorders. Here, the Movement Disorders in Asia Task Force sought to review genetic movement disorders that are commonly seen in Asia, including Wilson's disease, spinocerebellar ataxias (SCA) types 12, 31, and 36, Gerstmann‐Sträussler‐Scheinker disease, PLA2G6‐related parkinsonism, adult‐onset neuronal intranuclear inclusion disease (NIID), and paroxysmal kinesigenic dyskinesia. We also review common disorders seen worldwide with specific mutations or presentations that occur frequently in Asians.
Keywords: movement disorders, genetic, Asia
Starting in the 1980s, advancements in genomics technologies have resulted in the discovery of genetic factors underlying many rare and non‐rare diseases among different populations. 1 Genetic diseases often cluster in different ethnic groups with unique clinical presentations or red flags. Recognition of a particular clinical phenotype, combined with information about the ethnic origin of the patients, could lead to an early and correct diagnosis.
There have been a few studies showing differences in both the clinical phenotypes and genetic causes or risk factors of movement disorders between Asian and Western patients. 2 Some of the disorders may be more common in some populations, or it may be just a specific genetic variant that is more common.
In this article, we review the genetic movement disorders that are considered to be commonly seen in Asians. We also include disorders that are commonly seen worldwide with certain specific variants or presentations occurring frequently in Asians.
Methodology
We searched PubMed from 1969 through March 31, 2022 and used references from relevant articles. Search terms included “Parkinson's disease” (PD), “parkinsonism”, “ataxia”, “dystonia”, “chorea”, “tremor”, “myoclonus”, “movement disorders”, and “Creutzfeldt‐Jakob” with “Asians” or “Asia” without language restrictions. From the search, it was found that comprehensive epidemiological data for non‐PD movement disorders in Asia were largely lacking. Hence, the disorders nominated to be included in this review had to be derived by consensus among experts who are members of the Movement Disorders in Asia Task Force (TF) of the International Parkinson and Movement Disorder Society—Asian and Oceanian Section (MDS‐AOS). This group comprised representatives from most of the major regions in Asia: East Asia (China, Japan, Taiwan, South Korea), the Indian subcontinent (India), South‐East Asia (Thailand, Malaysia, the Philippines), Central Asia (Kyrgyzstan), and the Middle East (Saudi Arabia). Based on their knowledge of the published literature and clinical practice experience, genetic disorders that are widely accepted to be common in Asia, or have been more frequently reported in these populations, were selected. The final reference list was generated by giving priority to the articles directly related to the topic, articles with the latest information, and comprehensive reviews.
Result
A total of 14 genetic movement disorders were found to be common in Asians: Wilson's disease (WD), spinocerebellar ataxias (SCA) types 12, 31, and 36, Gerstmann‐Sträussler‐Scheinker disease (GSS), PLA2G6‐related parkinsonism, adult‐onset neuronal intranuclear inclusion disease (NIID), paroxysmal kinesigenic dyskinesia (PKD), X‐linked dystonia‐parkinsonism (XDP), dentatorubral‐pallidoluysian atrophy (DRPLA), Woodhouse‐Sakati syndrome, benign adult familial myoclonic epilepsy (BAFME), Kufor‐Rakeb disease, and tremulous dystonia associated with variant of the calmodulin‐binding transcription activator 2 (CAMTA2) gene. The latter six conditions have previously been reviewed by the TF (submitted). In this study, we will focus on the first eight disorders listed above. We also discuss the unique presentation of parkinsonism in Asian patients with SCA2 and SCA17, highlight the common genetic variants in PD‐causative genes in Asian patients, and discuss the differences between Asian and Western patients for all the disorders where possible. Key differences in the prevalence, risk factors, and clinical aspects of PD between Asians and Western patients have been previously reviewed. 2
Wilson's Disease
WD is an autosomal recessive (AR) disorder of copper metabolism caused by variants in the ATP7B gene on chromosome 13. 3 Prevalence studies in Asian countries have been done in Chinese (5.9/100,000), 4 Korean (3.8/100,000; allelic variants‐1.3/10,000), 5 Japanese (1:20,000 to 1:30,000; allelic variants‐1.2/10,000) 6 and Taiwanese (1.8/100,000) populations. 7 The disease appears to be especially commonly encountered by physicians in India. WD affected 7.6% of patients in a study of hepatobiliary‐spectrum disorders in North India and about 15–20 new cases are registered annually in a WD clinic in South India. 8 , 9 These relatively large numbers of patients are postulated to be due in part to high rates of consanguinity 8 ; however, systematic epidemiological studies remain lacking.
The clinical presentation of WD is heterogeneous, ranging from asymptomatic to acute or chronic liver involvement and neuropsychiatric illnesses. 10 Previous studies reported that the age of onset in Indian patients may be earlier compared to those from Europe and South America. 11 , 12
Hepatic presentations are common in younger age groups and neuropsychiatric features predominate in later‐onset cases (Video 1). 10 Large cohort studies from Asia, including India, Korea and China, on WD clinical features, show that neuropsychiatric presentations account for 22–77% of the studied populations. 13 , 14 , 15 Kayser–Fleischer rings are reported in 97–100% of neurological cases, 14–87% of hepatic presentations and up to 60% of asymptomatic patients, in the Indian population. 9 Magnetic resonance imaging (MRI) of the brain is a cornerstone for the diagnosis and monitoring of neurological forms of WD. 16 A study in 100 Indian patients showed a variety of MRI features: classical T2‐weighted hyperintensity in the putamen (72%), caudate (61%), thalamus (58%) and/or midbrain (49%); T2‐weighted pallidal hypointensity (34%); “face of the giant panda” sign (12%); central pontine myelinolysis (7%); T1‐weighted striatal hyperintensity (6%); and bright claustral sign (4%) (Fig. 1). 17 Sequential MRI study demonstrated imaging improvement in up to 70% of patients after copper‐chelating therapy. 18
Video 1.
Wilson's Disease. A 33‐year‐old man with a seven‐year history of tremulousness involving bilateral upper limbs and head. He has a past history of jaundice and upper gastrointestinal bleeding. There is a family history of severe liver dysfunction and subsequent demise of his sister. The video shows severe rest and postural tremor of both upper limbs (right more than left) as well as dystonia of the limbs when outstretched. The tremor is a Holmes tremor and is of “wing‐beating” type. Courtesy: Prof. Pramod Kumar Pal.
FIG. 1.
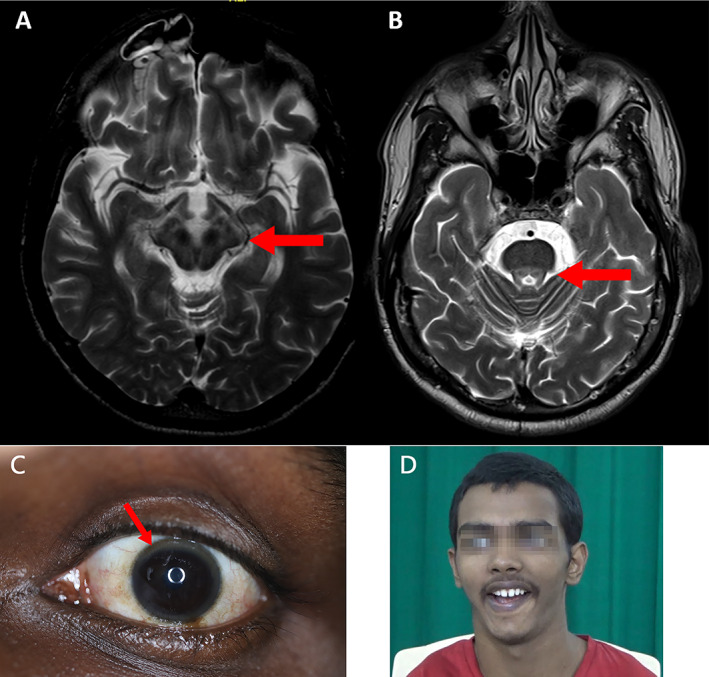
Wilson's disease. Brain MRI T2‐weighted images demonstrating the classic “double panda” sign: (A) Giant panda sign (B) Miniature panda. (C) Kayser‐Fleischer (KF) ring which occurs due to deposition of copper in Descemet's membrane. (D) Classic Wilson's face is shown which is characterized by a facetious smile, pseudo‐laughter, open mouth, dull look, and staring expression in variable combinations. Courtesy: Prof. Pramod Kumar Pal.
The diagnosis of neurologic WD is usually based on clinical presentation, biochemical evaluation, and brain imaging, but sometimes needs genetic confirmation. Variant “hotspots” in ATP7B have been reported to vary by geographic region (Table S1). 19 The p.R778L variant is common in East Asians (30% in China, 20 40% in Taiwan 21 and Korea 22 ) with patients having an earlier onset and predominantly hepatic presentation than those with other genetic variants. 23 The p.P992L variant is the second most common variant in East Asians with a variant frequency of 14.6% in Chinese WD patients. 20 However, no single mutation appears to be dominant in India. 17 In contrast, the p.H1069Q variant has an allelic frequency of 30–70% among Caucasians but is rare in Asians. Most carriers with the p.H1069Q variant have a mean onset age of 20–22 years and a predominantly neurological phenotype. 24 Singh et al. in India have further noted hepatosplenomegaly and extrapyramidal features like bradykinesia, rigidity and dystonia to be associated with truncating variants, and tremors with missense variants. 25
A large Indian cohort showed improvement in clinical symptoms in 76% (176/225) of patients after a mean duration of 46 months of treatment (Video 2). 13 Treatment response and longitudinal tracking can be best done with the Global Assessment Scale (GAS) for WD. 26
Video 2.
Wilson's disease, pre‐ and post‐treatment. Segment 1: Shows a child with Wilson's disease who is confined to the bed and anarthric with generalized choreo‐dystonic movements. Segment 2: The same child after initiation of decoppering therapy shows significant improvement, with ability to speak and walk independently. Some residual generalized chorea and dystonic features are still noted. Courtesy: Dr. Prashanth Lingappa Kukkle.
Spinocerebellar Ataxia Type 12; SCA‐PPP2R2B
SCA12 is a rare autosomal dominant cerebellar ataxia (ADCA) characterized by CAG trinucleotide repeat expansions in the 5′ region of the PPP2R2B gene on chromosome 5q31–5q32, which encodes for a brain‐specific regulatory subunit of the protein phosphatase PP2A. 27 , 28
The gene was first identified in 1999 in a large German kindred. 27 Following the discovery, a study in North India involving 77 families with ADCA phenotype found that SCA12 variants accounted for approximately 7% of the cohort (six patients from five families), and was the third most prevalent SCA, after SCA1 and SCA2. 29 The same group subsequently reported 15 new families. 30 The mean onset age was in the fourth decade, ranging from 26–56 years. 30 These families were from an endogamous population (Agarwal community) originating from Haryana, a Northern Indian region, suggesting a common founder. The mean CAG repeat length in PPP2R2B in the expanded allele was 53.3 ± 6.1 (40–72) with no correlation between the CAG repeat size and the age at symptom onset. 31 Overall, SCA12 accounts for around 16% of ADCA in Northern India, which is considerably higher compared to series from other populations. 30 , 32
Apart from Northern Indian cases, few SCA12 cases have been reported from elsewhere. 33 , 34 , 35 A study among 120 French and 27 Indian families with ADCA without common variants identified one Indian family with SCA12, but none in the French families. 36 In other Asian populations, none of 82 index patients from Thailand, 33 1 of 430 ADCA families from China, 34 and 1 of 204 ataxic patients from Singapore had SCA12. 35
The most common clinical presentations of SCA12 are upper extremity action tremor and gait ataxia (Video 3), followed by varied features including pyramidal dysfunction (hyperreflexia and positive Babinski sign), parkinsonism, dystonia, and cognitive decline. 31 , 32 , 37 Brain MRI usually reveals mild to moderate atrophy of the cerebellum, with more severe atrophy in the vermis compared to the cerebellar hemispheres, as well as in the cerebral cortex with or without subcortical white matter changes. 37
Video 3.
Spinocerebellar Ataxia Type 12; SCA‐PPP2R2B. A 48‐year‐old man with an eight‐year history of shaking of the hands and gait imbalance, with a diagnosis of SCA12. The video shows a coarse amplitude postural tremor of both upper limbs (right more than left) when these are held in front of the chest with shoulders abducted and elbows flexed, rest tremor of the left fourth and fifth fingers, mild head tremor, mild dystonia of the hands (spooning), finger‐nose‐incoordination especially on the left side, inability to perform tandem walking, and tremulousness of both lower limbs while walking. Courtesy: Prof. Pramod Kumar Pal.
The diagnosis of SCA12 requires genetic analysis. Since the majority of patients present with upper extremity action tremor and variable degrees of ataxia, SCA12 should be carefully differentiated from essential tremor (ET) and other late‐onset tremor‐ataxia syndromes, for example, Fragile X‐associated tremor ataxia syndrome (FXTAS). 37 There is no disease‐modifying treatment available for SCA12, but beta‐blockers or GABAergic medications are used for symptomatic relief of the upper limb action tremor.
Spinocerebellar Ataxia Type 31; SCA‐BEAN1
SCA31, caused by a large insertion containing pentanucleotide repeats (TGGAA)n in overlapping introns of the BEAN1 and TK2 genes, is largely restricted to the Nagano district of Japan where it accounts for approximately 42% of ADCA, with a strong founder effect. 38 , 39 , 40 , 41 The abnormal repeat insertion forms abnormal RNA structures, called RNA foci, preferentially in the nuclei of Purkinje cells in affected patients. 38 The length of the SCA31 repeat insertion correlates inversely with the age of onset and shows a pattern of genetic anticipation.
The prevalence of SCA31 ranges between 8–17% of ADCA in other parts of Japan. 40 , 42 , 43 It is rare or absent outside Japan with only one case reported so far in a Chinese patient. 44 , 45 The clinical phenotype of SCA31 is one of late‐onset and relatively pure cerebellar ataxia. A natural history study prospectively enrolling 44 patients with genetically‐proven SCA31 showed that the patients developed ataxic symptoms at the age of 58.5 ± 10.3 years, were confined to wheelchair at 79.4 ± 1.7 years, and died at 88.5 ± 0.7 years. 42 As with other SCAs, therapy is supportive.
Spinocerebellar Ataxia Type 36; SCA‐NOP56
SCA36, another SCA that is rather specifically connected to Asia, is a slowly progressive late‐onset autosomal dominantly (AD) inherited disease that is caused by a hexanucleotide repeat expansion in NOP56. 46 , 47 , 48 This gene encodes a nucleolar protein 56 which is involved in ribosomal RNA methylation and pre‐rRNA processing. 49 This ataxic syndrome was first described in two Japanese patients, 50 and later from the Galicia region in Northwestern Spain. 47 SCA36 is now reported worldwide, but most cases are found in Western Japan and Spain. 51 In Western Japan, SCA36 was initially named “Asidan” ataxia as many patients with SCA36 lived in the Chugoku region near the Asida river, 46 while in Spain, SCA36 was named “Costa de Morte ataxia”. 47 SCA36 is the most frequent spinocerebellar ataxia in the Galicia region, representing 6.3% of adult‐onset ataxia, followed by SCA2 (4.4%), SCA1 (1.9%), SCA3 (1.9%), and SCA7 (1.3%). 47 In Japan, the prevalence of SCA36 is lower than other SCA subtypes and represents 0.6–3.6% of adult‐onset ataxia. 52 SCA36 was reported to contribute to 3% of Italian families with ADCA without one of the commonly‐tested SCAs. Otherwise, SCA36 does not appear to be common in other Asian countries, eg, 0.6% (3/512) of SCA patients in Taiwan, 53 or other world regions, including Greece (none in 98 index patients) and the USA (0.7%, 4/577), 54 , 55 although cases could also be underdiagnosed since this form of ataxia is currently not included in most genetic ataxia panels.
SCA36 is characterized by a late‐onset cerebellar ataxia with a mean onset age in the fourth‐to‐fifth decades, combined with signs of lingual atrophy and fasciculations, and sensorineural hearing loss. 46 , 47 Notably, even though lingual atrophy is prominent in the later stage of the disease, dysphagia is rarely present. 52 Mild cognitive impairment of a fronto‐subcortical pattern has been reported in patients with SCA36. 47 Brain MRI findings range from mild cerebellar vermis atrophy to diffuse cerebellar atrophy. Fluorodeoxyglucose‐positron emission tomography (FDG‐PET) scan can show hypometabolism in the vermis and cerebellar hemispheres. 56
The treatment for SCA36 remains supportive, including speech therapy and communication devices for those with dysarthria.
Gerstmann‐Sträussler‐Scheinker Disease
GSS is an extremely rare, AD‐inherited disease, caused by pathogenic variants in the prion protein (PRNP) gene. 57 GSS was first described in an AD inheritance family with the 25‐year‐old index patient developing cerebellar ataxia and psychosis, eventually dying 6 years after onset. Neuropathological examination revealed prominent cerebellar atrophy with molecular layer “senile” plaques, along with cerebral cortical atrophy. 58 Numerous missense variants have been reported in PRNP with the most prevalent being p.P102L. 59 Octapeptide repeat insertions, particularly longer (>7) insertions in the PRNP gene, can also cause GSS. 60 Although GSS is an AD disorder, up to 30% of patients have no apparent family history. 61
The prevalence of GSS is estimated at 1–10 per 100,000,000 people. 61 One review reported that GSS accounted for approximately 7.9% of genetic prion diseases in European countries and about 10–20% in East Asian countries including Japan, Korea and China. 62 , 63 The p.P102L variant causing GSS is one of the most common PRNP variants in Japan and Korea, but is rare in China. 62 , 63 Another common GSS‐associated variant in Japanese, p.P105L, is also rare in Chinese, 63 while the p.A117V variant is common in Europeans. 61
Clinically, GSS presents with progressive ataxia and lower limb hyporeflexia, followed by cognitive decline and dementia (Video 4). 64 The age of onset is in the fourth‐to‐fifth decades, although two patients with p.P102L were reported to develop symptoms in their twenties. 65 The average disease duration is 40 to 50 months after clinical onset. 66 Some genotype–phenotype correlations are emerging. For example, prominent cognitive decline without cerebellar ataxia as the initial presentation has been reported with p.Q212R. 67 Parkinsonism and psychiatric features such as delusions, paranoia, and hallucinations can be seen in half of the patients with the p.P102L variant. 66 , 68 Less than a quarter of patients with the p.P102L variant developed a sporadic Creutzfeldt–Jakob disease (sCJD)‐like phenotype with rapidly progressive dementia. 69 Recently, a large cohort study enrolling 218 Chinese genetic prion disease patients revealed that GSS p.P102L variant patients had a long survival compared to those with other variants. 63 Patients with the p.P105L variant can present with late‐onset spastic paraparesis. 70 These observations demonstrate that different variants at different positions in PRNP result in different phenotypes of GSS, which vary substantially in their ethnic prevalence and clinical manifestations.
Video 4.
Gerstmann‐Sträussler‐Scheinker (GSS). A 44‐year‐old man with a two‐year history of gradually progressive gait disturbance. His uncle and mother had similar symptoms, and the uncle was clinically diagnosed as having Gerstmann‐Sträussler‐Scheinker disease. On neurological examination, he has saccadic pursuit eye movements in horizontal and vertical directions and dysmetria and decomposition on finger‐nose test and heel–knee test. The deep tendon reflexes are generally decreased, and the plantar reflex extensor on the left. Neither rigidity nor spasticity is seen. Due to severe postural instability, he is not able to walk independently. Genetic analyses revealed the PRNP p.P102L variant, and no pathological variants for SCA1, SCA2, SCA3, SC6, SCA7, SCA10, SCA17, SCA31 and DRPLA. Courtesy: Assoc. Prof. Shinsuke Fujioka.
An integrated evaluation is needed to diagnose GSS, including cerebrospinal fluid (CSF) analysis, electroencephalography (EEG), brain MRI, and genetic analysis. The sensitivity of the real‐time quaking‐induced conversion (RT‐QuIC) test in CSF is approximately 75% in GSS, 71 while it was 92% in sCJD with 100% specificity. 72 EEG periodic synchronous discharges are uncommonly seen (<10%), and there are no specific MRI findings for GSS, although up to 30% of cases show fluid‐attenuated inversion recovery (FLAIR) and diffusion‐weighted imaging (DWI) abnormalities similar to sCJD with increased signal intensity in the cortex (cortical ribboning) and basal ganglia. 73 GSS is often misdiagnosed as other degenerative cerebellar ataxias, especially at the early stage. Genetic analysis of the PRNP gene is needed to confirm the diagnosis. Management is supportive.
DYT/PARK‐PLA2G6 ‐Related Parkinsonism
PLA2G6‐associated neurodegeneration (PLAN) is an AR neurodegenerative disorder caused by variants in PLA2G6. 74 The clinical phenotypes of PLAN are heterogeneous: infantile neuroaxonal dystrophy (INAD), with psychomotor regression or delay between the ages of 6–36 months; atypical neuroaxonal dystrophy (ANAD), with prominent language difficulty and autistic‐like traits between ages 1.5–6.5 years 75 , 76 ; hereditary spastic paraparesis between ages 9–66 years 77 , 78 ; and early‐onset dystonia‐parkinsonism in the second to third decades of life.
A total of 101 PLA2G6‐mutated cases with parkinsonism have been documented worldwide. 74 , 79 , 80 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 The majority of the patients (n = 86) were from Asia, mainly China and Taiwan (n = 36). 89 , 90 Among the Asian cases, 51.8% were males and mean age of onset was 25.1 ± 9.1 years compared to 20.8 ± 9.2 years in Caucasians. The most common manifestations at onset were parkinsonism and dystonia in 62.5% of the Asian patients. Psychiatric features (eg, severe depression or anxiety, psychosis), the second most common manifestations, presented more frequently in Caucasian (40%) than in Asian (21.3%) patients.
The “classical” scenario of early‐onset PLA2G6‐related dystonia‐parkinsonism includes various movement disorders: parkinsonism (100%), dystonia (68.3%), cerebellar ataxia (36.2%), pyramidal signs (63.6%), psychiatric symptoms (76.8%) and cognitive decline (59.7%). 79 , 91 Besides these, autonomic features (including urinary disturbances, constipation, sexual dysfunction and orthostatic hypotension) were observed in 71.9% (23/32) of Asian patients and 75% (3/4) of Caucasian patients. 91 Brain MRI showed cerebral atrophy (especially generalized or frontotemporal lobe) in 52.6% and cerebellar atrophy in 39.1% of Asian cases, whereas only 13.2% of cases reported iron accumulation in the basal ganglia, which is less than in INAD and ANAD patients (26.7%). 92
Pathogenic PLA2G6 variants impair iPLA2β function via a variety of loss‐of‐function mechanisms. 93 The most frequent variant in Chinese is homozygous p.D331Y, 89 , 94 suggesting a common founder effect. The p.R741Q variant is mainly reported in Indian, Saudi Arabian and Pakistani populations, and p.R635Q in Japanese patients. 91 These above‐mentioned variants were rarely reported in Caucasian patients.
The neuropathological findings of patients with PLA2G6 variants are also heterogenous, including Lewy bodies in the substantia nigra and locus ceruleus (similar to idiopathic PD), 95 co‐existing Alzheimer's disease‐like pathology in temporal lobe structures, abundant gliosis, and some may also have excessive iron accumulation in the substantia nigra and basal ganglia. 96
Parkinsonism responded to levodopa in 98.4%, while levodopa‐induced dyskinesias were reported in 86.8% and appeared within the first year of treatment in most cases. Early occurrence of dyskinesias and exacerbation of psychiatric symptoms after levodopa initiation are considered tell‐tale signs of PLA2G6‐related parkinsonism. 79 Motor and non‐motor symptoms and fluctuations responded well to subthalamic nucleus (STN) and globus pallidus internus (GPi) deep brain stimulation (DBS) in a few patients who have received the treatment, in both Asians and Caucasians. 79 , 85 , 97 , 98 , 99
Adult‐Onset NIID and NOTCH2NLC ‐Related Disorders
NIID has been reported since the 1960s, but was rarely diagnosed as this required brain autopsy or invasive (eg, rectal or sural nerve) biopsies. 100 , 101 The recent recognition of cases based on MRI findings and skin biopsy (showing eosinophilic ubiquitin‐positive and p62‐positive intranuclear inclusions in adipocytes, fibroblasts, and sweat glands) paved the way for more widespread recognition of the condition, particularly among Japanese adult patients. 100
In 2019, abnormally increased GGC‐repeat expansions in the 5′ untranslated region (UTR) of the NOTCH2NLC gene were identified to be the cause of NIID, largely among Japanese and other Asian patients. 101 , 102 , 103 Since then, the phenotype has expanded and NOTCH2NLC variants were found in 5.6% of mainland Chinese families with ET 104 ; 1.3% of typical sporadic PD cases in Singapore (all cases were ethnic Chinese) 105 and in China (1.1%, 11/1011) 106 ; and were also reported to be the most frequent genetic cause of adult‐onset leukoencephalopathy in Japan and Taiwan. 107 , 108 In contrast, NOTCH2NLC variants appear to be extremely rare in Caucasians. 101 , 109 , 110 A recent literature search revealed no more than a dozen adult‐onset NIID (and a similar number of juvenile‐onset) cases reported from Europe, North America, and Australia since 2000. 101 Interestingly, a recent analysis of NIID cases of European ancestry (confirmed on post‐mortem brain examination; n = 11) found no case of expanded repeats in NOTCH2NLC, suggesting that NIID may be genetically heterogeneous between Asians and Caucasians. 111
In the “classical” scenario of adult‐onset NIID, onset is usually in mid‐ or later life and features commonly include cognitive decline or dementia, various movement disorders (including parkinsonism, tremor, cerebellar ataxia), muscle weakness, peripheral neuropathy, and autonomic dysfunction, with limb weakness vs. dementia being more prominent in younger‐ vs. older‐onset patients, respectively. 100 Cases may be sporadic or AD in inheritance (sometimes displaying genetic anticipation). 104 Apart from age, variable clinical expressivity may be caused by genetic factors such as the length of the GGC repeats, interruptions (eg, of GGA) in the repeat tracts, or other unknown modifiers. 104 , 112 For example, clinical NIID cases typically have >65 (and up to about 500) repeats (vs. <40 in healthy controls), 105 and typical sporadic PD cases were mostly reported to have intermediate‐length expansions (40–60 GGC repeats). 105 , 106 Interestingly, the Chinese familial ET cases had repeat sizes of 60–250, but did not have other NIID features. 104
Brain MRI could offer diagnostic clues with the characteristic features of high signal intensity in the corticomedullary/gray‐white matter junction on DWI (Fig. 2A,B). FXTAS (also caused by expanded trinucleotide repeats, but in FMR1) has been highlighted as a mimic of NIID (and vice versa), in terms of clinical, radiological (Fig. 2C,D) and also histological findings. 101 , 113 , 114 However, it appears that FXTAS is quite rare in Asian populations (Chinese, Japanese, Koreans, and Singaporeans). 2 , 101
FIG. 2.
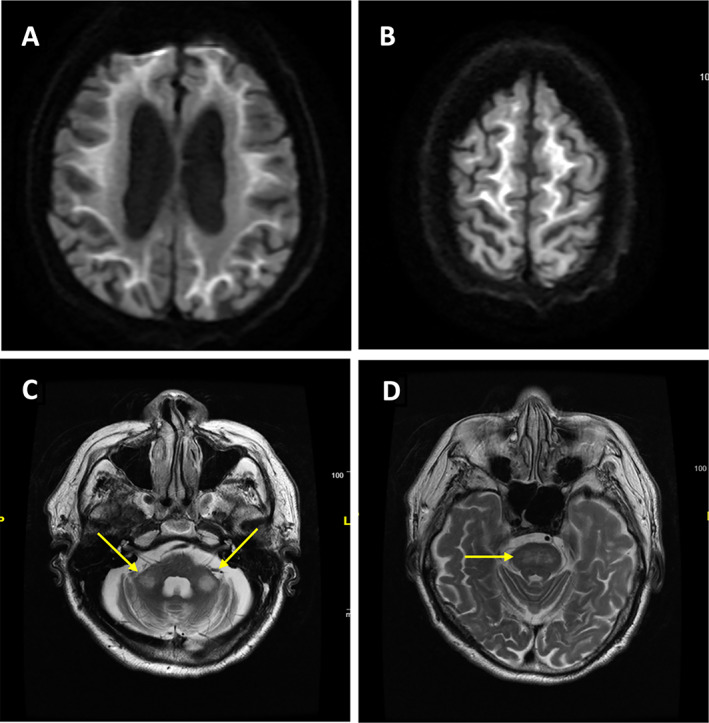
Representative brain MRI from a patient with genetically proven neuronal intranuclear inclusion disease (NIID). (A, B) Characteristic high‐intensity signal along the corticomedullary junction in the cerebral hemispheres on diffusion‐weighted imaging (DWI). High‐intensity signal on T2‐weighted images in bilateral middle cerebellar peduncles (arrows, C) and pons (D), which may mimic the radiological features of FXTAS. This was Malaysian Patient #1 in Lim et al., Ishiura et al. 101 , 102 Courtesy: Prof. Shen‐Yang Lim.
Like most other genetic neurodegenerative disorders, NIID has no specific treatment besides supportive and symptomatic therapy.
Paroxysmal Kinesigenic Dyskinesia; PxMD‐PRRT2 ; PKD‐PRRT2
Paroxysmal dyskinesias are a rare heterogeneous group of conditions. 115 , 116 They are classified into three main subtypes based on triggering factors: PKD, paroxysmal non‐kinesigenic dyskinesia (PNKD) and paroxysmal exercise‐induced dyskinesia (PED). 117 , 118 The prevalence of PKD, which is the most common form of paroxysmal dyskinesias, was estimated to be 1:150,000 in the general population, with a suggestion that it may be more common in Asians, especially Chinese and Japanese, although direct comparisons of prevalence among various ethnic groups are not available. 119 , 120 In studies from Malaysia and Singapore, which are multi‐racial Southeast Asian countries, a preponderance of Chinese (over Malay and Indian) cases has been observed (65–90% Chinese). 121 , 122 , 123 The first multicenter study in Asia was reported by Japanese investigators who analyzed 150 patients with a clinical diagnosis of PKD comprising 53 sporadic cases and 97 affected individuals from 32 pedigrees with a majority compatible with AD inheritance. 124 The mean age of onset was 8.8 years with male predominance (80%). Attacks were precipitated by sudden voluntary movements, startle, or emotional stress, lasted between seconds to 5 min, and upper limbs were most affected. Treatment with carbamazepine or phenytoin was effective in 95% of patients.
The most common genetic variant underlying PKD is c.649dupC (p.Arg217fs) in PRRT2, which was responsible for 76.4% of PRRT2 variant carriers in a Chinese study. 120 , 125 A review of 1444 cases of patients with PPRT2 variants showed that 58.5% of PKD‐PRRT2 were Asians, mainly Chinese, followed by Caucasians (33.9%). 120 PRRT2 variant carriers presented with earlier onset, longer attack duration, and greater complexity (such as having bilateral involvement or a history of infantile convulsions), compared to non‐PRRT2 carriers. The clinical phenotypes include an evolving continuum from benign familial infantile epilepsy, to PKD and paroxysmal headache disorders such as hemiplegic migraine. 117 , 120 The excellent response of PKD to antiepileptic medications, and the recent identification of epilepsy‐related genes (including SCN8A, KCNMA1, DEPDC5, KCNA1, and CHRNA4) in pure or complicated PKD suggest that PKD and epileptic disorders might share similar pathogenesis. 126 , 127 , 128 As such, implications for clinical practice are starting to emerge, with revised clinical diagnostic criteria incorporating genetic diagnosis, and treatment recommendations. 129
Issues of Parkinsonism in SCA2 and SCA17 in Asian Populations
Parkinsonism is not rare in ADCA patients with abnormally expanded trinucleotide repeats. SCA2, caused by abnormal CAG repeat expansion in ATXN2, is the most frequent SCA that can present with parkinsonism, and can mimic PD. 130 , 131 Pathologically, nigral dopaminergic neuronal loss exceeds that seen in PD, 132 with brainstem Lewy body pathology variably found. 132 , 133 Interestingly, midbrain dopaminergic loss appears to be a universal phenomenon in SCA2 regardless of the presence or absence of parkinsonism. 134 Dopamine transporter imaging findings can resemble those of PD with asymmetric and preferential involvement of the posterior putamen, although the dopaminergic denervation is often symmetrical involving the entire striatal regions (Fig. 3A). 134 , 135
FIG. 3.
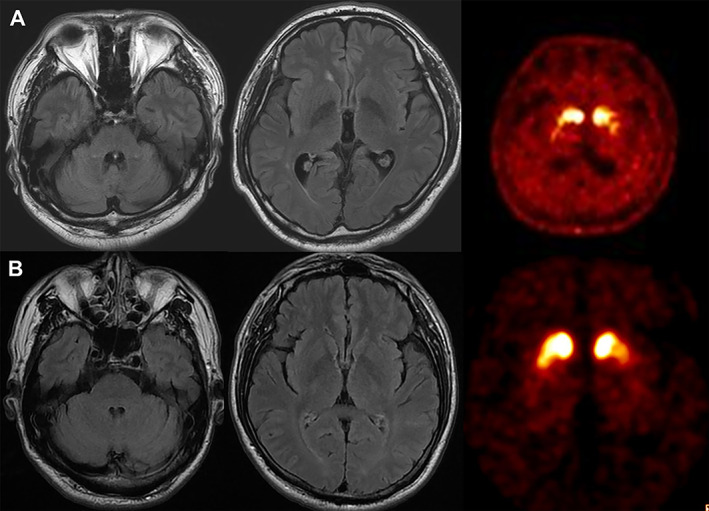
SCA2 and SCA17 presenting with parkinsonism. (A) Brain MRI and 18F‐FP‐CIT PET images of a patient with SCA2 (26/36 expansions) presenting with excellent levodopa‐responsive symmetric parkinsonism with onset at the age of 43 years. There is mild degree pontocerebellar atrophy, but he has no overt ataxia. Dopamine transporter availabilities are reduced bilaterally with anteroposterior gradient. Notably, the patient's family members showed marked clinical heterogeneity. His father and sister manifested severe ataxia and his elder brother has predominantly parkinsonism. (B) Brain MRI and 18F‐FP‐CIT PET images of a patient with SCA17 (36/44 expansions) presenting with levodopa‐responsive parkinsonism with onset at 52 years of age. No clear evidence of cerebellar atrophy is seen, and FP‐CIT bindings are bilaterally reduced in the striatum with anteroposterior gradient. Courtesy: Prof. Jee‐Young Lee and Prof. Beomseok Jeon.
The manifestation of parkinsonism in SCA2 is heterogeneous and it is thought to be more prevalent in Asians compared to other populations, although systematic epidemiological studies are lacking. Studies in sporadic PD populations reported a SCA2 frequency of 2.2% among Singaporean Chinese 136 and 0.4% in Koreans and Taiwanese 134 , 137 ; whereas familial PD populations had frequencies of 1.5–8.7% in mainland China and Taiwan, 138 , 139 2–2.5% in Italy and France, 135 , 140 and 0.9–1.5% in the USA. 131 , 141
Genetic modifications have been suggested as one of the possible mechanisms for the pure parkinsonian phenotype of SCA2. Pathogenic alleles with one or more CAA interruptions and low‐range CAG repeat expansions are reported to be linked to parkinsonism in SCA2. 134 , 135 , 142 A report on quite long‐lasting (up to 34 years) PD phenotype with pure parkinsonism in a Korean SCA2 family without anticipation across generations (40 repeats with four CAA interruptions) supported the hypothesis of CAA interruption in contributing to the pure parkinsonism phenotype. 143 However, CAA interruption was not the only factor determining phenotype, 144 , 145 suggesting other genetic modifiers could play a role in the manifestation of parkinsonism in different populations. Heterogeneous observations suggest that genetic modifiers may be different across populations, which may explain the heterogeneity in the frequency of pure parkinsonism and in the relevant numbers of genetic interruptions and expansions in SCA2 among different ethnic populations. In a large Chinese cohort study, the presence or absence of parkinsonism was independent of the severity of ataxia. 145
In addition to SCA2, levodopa‐responsive parkinsonism has been reported in SCA types 3, 6, 8, and 17. 130 SCA17 is caused by abnormal CAG/CAA repeat expansion in the TATA‐binding protein (TBP) gene. The association with parkinsonism is commonly found in Asian populations. 130 SCA17 patients can present with an atypical parkinsonian syndrome and show poor levodopa response resembling multiple system atrophy or progressive supranuclear palsy. 146 However, SCA17‐pure parkinsonism cases are also reported in Korean, Taiwanese Chinese, and Thai populations. 33 , 147 , 148 , 149 There has been no reported pathologic study of SCA17‐parkinsonism, but dopamine transporter imaging showed heterogeneous features as bilateral severely reduced uptake or diffuse reduction without anterior–posterior gradient, or resembling typical PD (Fig. 3B). Reduced copy number of CAG in the TBP gene may be related to a pure parkinsonian presentation, 147 but the cutoffs remain unclear, and variable movement disorders have been reported with small‐expanded alleles. 150 , 151 One study investigated the frequency of low‐range repeat expansions between parkinsonian patients and normal controls but reported no difference between the groups. 152 Therefore, further studies are required to reveal the exact mechanism of pure parkinsonism in SCA17.
PD‐Causative Genetic Variants in Asian Populations
Approximately 10% of PD can be attributed to a monogenic cause, and the heritable component of PD due to common genetic variability is estimated to be around 22%. 153 LRRK2 variants, the most frequently implicated genetic factor in familial and sporadic PD, display significant ethnic variation. In particular, the LRRK2 p.G2019S variant is very common (up to around 40% of PD cases) in North African Berber Arabs and Ashkenazi Jews and to some extent in Europeans, but very rare in Asians. 154 , 155 In contrast, p.G2385R and p.R1628P (so‐called LRRK2 Asian variants) are common genetic risk factors in Asian PD (Table 1), each being present in around 5–10% of some Asian PD populations such as Han Chinese, vs. approximately half of that in the respective general populations. 2 Although overall the clinical features of LRRK2‐related parkinsonism appear to be comparable to idiopathic PD (Video 5), several studies have reported that patients with the p.G2019S variant are more likely to be women and less likely to have non‐motor symptoms, including olfactory impairment, cognitive dysfunction, and rapid eye movement sleep behavior disorder. 169 , 170 A recent meta‐analysis revealed that patients with the p.G2385R variant have lower motor symptom severity and better cognitive function, but a higher tendency to develop levodopa‐related motor complications than those without this genetic substitution. 171 In addition, heterozygous variants in GBA1 also increase the risk of PD in both Eastern and Western populations, although specific variants (eg, p.L483P which is associated with a more aggressive phenotype (Video 6)) may be over‐represented in Asian populations (Table 2). Interestingly, a recent study suggested that combined LRRK2 p.G2019S and GBA1 variants were not associated with worse disease progression, although no information was available on LRRK2 Asian variants. 197 PINK1 variants have also been commonly reported in Asians. Specifically, the p.L347P variant appears to be a common cause of AR early‐onset PD in Southeast Asia and the Pacific Islands (proposed to reflect ancient migratory patterns of Austronesian races), and displays marked clinical heterogeneity ranging from mild PD (or even dopa‐responsive dystonia without parkinsonism), to cases with extremely severe motor complications (Video 7). 177 , 180 , 198
TABLE 1.
Variant frequency in Parkinson's disease (PD)‐causative genes with autosomal dominant inheritance in Asian patients with PD
| AD inheritance | |||
|---|---|---|---|
| LRRK2 | Heterozygous missense variants |
Similar to sporadic late‐onset PD (Video 5) |
23 of 1402 (1.6%) Japanese PD patients had variants including 7 (0.5%) with p.G2019S, 7 (0.5%) with p.I2020T, 5 (0.4%) with p.R1441H, and 4 (0.3%) with p.R1441G 157 ; 4 of 324 (1.2%) Taiwanese EOPD or FPD patients had variants, including p.R1441H and p.I2012T 149 ; 2 of 662 (0.3%) Chinese EOPD had the p.R1441C variant 158 who, together with other p.R1441C patients in Singapore and Malaysia, were suggested to have a common founder. 156 |
| p.R1628P polymorphism | Similar to sporadic PD | A meta‐analysis revealed that p.R1628P was a risk variant for PD in Asians with an OR of 1.83. 159 | |
| p.G2385R polymorphism | Similar to sporadic PD | A meta‐analysis revealed that p.G2385R was a risk variant for PD in Asians with an OR of 2.27. 159 | |
| Risk variants identified from GWAS | Similar to sporadic PD |
A Japanese GWAS study identified five risk variants (rs1994090, rs7304279, rs4768212, rs2708453 and rs2046932) for PD. 160 A large East Asian study demonstrated that rs1384236 is a risk variant for PD in Asians. 161 |
|
| p.N551K‐p.R1398H haplotype | Similar to sporadic PD | An Asian cohort study showed a protective association in Malays with an OR of 0.45; and meta‐analysis of Chinese patients showed a protective effect (OR: 0.79, 95% CI: 0.67–0.92). 162 | |
| CHCHD2 | Heterozygous missense variants | Similar to sporadic PD | A large Japanese cohort identified two families having p.T61I variant, one family having p.R145Q and another one with a splice‐site variant (300 + 5G > A). 163 A meta‐analysis showed p.P2L to be a risk variant for PD among Chinese. 164 |
| UQCRC1 | Heterozygous missense variants | Early‐onset parkinsonism with polyneuropathy | A large East Asian cohort study identified two Taiwanese families and one Japanese family having UQCRC1 missense or splicing variants. 165 A subsequent large Chinese study identified risk variants in UQCRC1 in sporadic PD. 166 The pathogenic variants of UQCRC1 were not identified in European populations. 167 , 168 |
Abbreviations: AD, autosomal dominant; EOPD, early‐onset Parkinson's disease; FPD, familial Parkinson's disease; OR, odds ratio.
Video 5.
LRRK2 R1441C variant. This Malaysian patient of Chinese ancestry was diagnosed with Parkinson's disease (PD) at the age of 58 years, later found to be associated with the LRRK2 p.R1441C variant. Bilateral subthalamic nucleus (STN) deep brain stimulation (DBS) was performed at the age of 67 years to treat progressively worsening OFF periods (medication effect lasting only 2–3 hr, with disabling OFF symptoms characterized by akinesia, tremors and painful foot dystonia—described further in Lim et al. Patient 2). 156 This video was taken 6 weeks after the DBS surgery, and the patient has come in OFF‐medication for her 1st DBS programming session. The patient is in a wheelchair and is able to take several small steps but requires close supervision. Limb movements are bradykinetic and there is an obvious “striatal toe” on the right side. Courtesy: Prof. Shen‐Yang Lim.
Video 6.
GBA1 p.L483P variant and also the LRRK2 Asian variant p.R1628P. Parkinson's disease in this patient was diagnosed at the age of 44 years. She underwent bilateral subthalamic nucleus (STN) deep brain stimulation (DBS) at the age of 57 years. The video, taken at the age of 59 years, was in the stimulation‐ON, medication‐ON (3.5 hr post‐dose) condition. Parkinsonian signs including bradykinesia can be observed, of mild‐to‐moderate severity. She is able to stand up quickly and walk independently without an aid (albeit slowed, with reduced arm swing bilaterally), and is able to recover during pull test. However, she has difficulty following simple verbal/gestural commands, with Montreal Cognitive Assessment (MoCA) score of only 9/30. The patient was later found to have the GBA1 p.L483P (p.L444P) variant (which could be an explanation for her dementia), and also the LRRK2 “Asian variant” p.R1628P. Courtesy: Prof. Shen‐Yang Lim.
TABLE 2.
Variant frequency in Parkinson's disease (PD)—causative genes with autosomal recessive inheritance and risk gene in Asian patients with PD
| Gene | Variants | Phenotypes | Variant frequency in cohort studies |
|---|---|---|---|
| AR inheritance | |||
| PARK2 | Missense variants or exonic deletions (especially exons 2 to 5) 149 , 172 , 173 | Early to juvenile‐onset levodopa‐responsive PD (average 26.1 yr) 2 , 172 | 5 of 189 (2.6%) Korean patients with EOPD or FPD 174 ; 15 of 324 (4.6%) Taiwanese patients with EOPD or FPD 149 ; 9 of 240 (3.8%) Han Chinese patients with sporadic or familial EOPD, 175 83 of 1676 (5.0%) Han Chinese patients with EOPD or FPD 173 ; 137 of 1204 (11.4%) Japanese FPD patients. 176 |
| PINK1 | Missense variants |
Early‐onset levodopa‐responsive PD (average 30–50 yr) (Video 7) |
2 of 47 (4.3%) Japanese AR‐inheritance families and 1 of 190 (0.5%) sporadic PD patients 178 ; None of 324 Taiwanese patients with EOPD or FPD 149 ; 7 of 1676 (0.4%) Han Chinese patients with EOPD or FPD 173 ; 3 of 289 (1.0%) mixed Asian populations of PD patients 179 ; 6.9% of Malay EOPD patients had homozygous p.L347P variants. 177 7 of 273 (2.6%) EOPD patients from New Zealand had homozygous p.L347P variants. 180 |
| PLA2G6 | Missense variants | Early‐onset parkinsonism and may be combined with atypical features | 3 of 29 (10.3%) Japanese patients with EOPD 91 ; 2 of 324 (0.6%) Taiwanese patients with EOPD or FPD 149 ; 9 of 1676 (0.5%) Han Chinese patients with EOPD or FPD. 173 The p.D331Y variant was almost exclusively found in Chinese patients, suggesting a common founder effect in this population. 89 |
| Risk Gene | |||
| GBA1 | Heterozygous variants | A younger onset age and more severe motor and non‐motor features, 181 , 182 , 183 , 184 , 185 , 186 , 187 for GBA1 p.L483P (old nomenclature p.L444P) variant 184 , 187 , 188 , 189 , 190 (Video 6) |
An international multicenter cohort showed an increased risk for PD in those carrying p.N370S (OR 3.96) and p.L483P (OR 6.73), but p.N370S variant is uncommon among Asians while p.L483P is a pan‐ethnic variant. 191 A meta‐analysis found that p.R159W (old nomenclature p.R120W) increased the risk of PD (OR 14.93) specifically in East Asians. 127 In East Asians, p.L483P (OR 12.43), RecNciI (a recombinant allele containing p.L483P) and A495P (also known as V499V) increased the risk of PD (OR 3.56). 127 The p.L483P variant has been reported to be the most common GBA1 variant in some Asian populations. 173 , 192 , 193 , 194 , 195 , 196 |
Abbreviations: AR, autosomal recessive; EOPD, early‐onset Parkinson's disease; FPD, familial Parkinson's disease.
Video 7.
PINK1 p.L347P variant. A case of young‐onset Parkinson's disease with PINK1 p.L347P variant in an Indonesian Malay woman (Patient 1 in reference Tan et al. 177 ). The patient started having slowness of movement, tremor and stiffness at age 26 years and developed severe motor fluctuations and dyskinesia within months of levodopa therapy initiation. She was dependent on her caregivers by age 34 years. The video was taken 15 min after taking levodopa/benserazide 150 mg/37.5 mg and shows severe generalized choreo‐ballistic movements leading to difficulty in sitting and standing, and tongue protrusion. This troublesome dyskinesia typically lasts for about 2 hours after each dose of medication, during which the patient moves around at home by bottom shuffling. Courtesy: Assoc. Prof. Ai Huey Tan.
In recent years, several novel PD‐causative or related genes have been identified, notably in Asian populations. Variants in the CHCHD2 gene were linked to a late‐onset AD form of PD (PARK22) in a large Japanese cohort. 163 The authors identified two families having the p.T61I variant, one family having the p.R145Q variant, and another with a splice‐site variant (300 + 5G > A). 163 Their phenotypes were similar to idiopathic PD with good levodopa response. A further meta‐analysis showed the p.P2L substitution to be a risk variant for PD. 164 Although the aforementioned variants were not found in a large European cohort of PD patients, three other rare variants (p.A32T, p.P34L, and p.I80V) were identified 199 and a homozygous missense variant (p.A71P) was reported in a young‐onset Caucasian PD patient. 200
Variants in UQCRC1 were reported in a large East Asian PD cohort, including two Taiwanese families (p.Y314S and p.I311L) and one Japanese family (concomitant splicing variant, c.70‐1G4A, and a frameshift insertion, p.Ala25Glyfs*27). 165 The Taiwanese family presented with early‐onset, levodopa‐responsive parkinsonism with polyneuropathy. A subsequent large Chinese study identified risk variants in UQCRC1 in sporadic PD. 166 PD‐related variants in UQCRC1 have not so far been observed in European populations. 167 , 168
Conclusion
Asian patients have unique disease‐causing variants which may come from founder effects, and high rates of consanguinity are also likely to be contributory in specific regions. Furthermore, many patients may present with characteristic phenotypes, in part related to the genetic variants that are more common or almost exclusively found in specific Asian groups. However, access to genetic testing facilities varies vastly between regions in Asia and may skew some of the results. 201
Recent advances in understanding the pathogenic mechanisms of PD and related movement disorders have shed light on the development of disease‐modifying or mechanism‐targeted therapies. Therefore, it is becoming increasingly imperative that clinicians are aware of and have knowledge about genetic disorders that are more commonly encountered in different ethnic groups. Improved recognition of particular phenotypic characteristics, coupled with information about the ethnic origin of patients, would point to specific genetic testing and lead to earlier diagnosis for better prognostication and, potentially, genetics‐based, mechanism‐targeted, therapies. All these issues underscore the need for further improvements in infrastructure and services, and concerted efforts in training and research involving cross‐collaborations between clinicians and researchers in Asia, and the rest of the world.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the first draft, B. Review and Critique.
P.J.: 1A, 1B, 3A, 3B
S.Y.L.: 1A, 1B, 3A, 3B
P.K.P.: 1A, 1B, 3A, 3B
J.Y.L.: 1A, 1B, 3A, 3B
P.L.K.: 1A, 1B, 3A, 3B
S.F.: 1A, 1B, 3A, 3B
H.S.: 1A, 1B, 3A, 3B
O.P.: 1A, 1B, 3A, 3B
R.B.: 1A, 1B, 3A, 3B
N.M.I.: 1A, 1B, 3A, 3B
Y.U.: 1A, 1B, 3A, 3B
Z.A.: 1A, 1B, 3A, 3B
B.J.: 1A, 1B, 3B
C.D.: 1A, 1B, 3B
C.S.: 1A, 1B, 3B
C.H.L.: 1A, 1B, 3A, 3B
Disclosures
Ethical Compliance Statement: The authors confirm that the approval of institutional review board was not required for this review. Written informed consent from patients was obtained for all the supplementary videos. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: S.Y.L. was supported by a grant from the Ministry of Higher Education Malaysia (FRGS/1/2020/SKK0/UM/01/2). All authors report no conflict of interest related to the study.
Financial Disclosures for Previous 12 Months: P.J. has received grants from Ratchadapiseksompotch Fund, Faculty of Medicine, Chulalongkorn University and speaker's honorarium from Boehringer Ingelheim. S.Y.L. has received consultancies honoraria from Michael J. Fox Foundation, Lundbeck International Neuroscience Foundation Editorial Board; speaker's honoraria from International Parkinson and Movement Disorder Society (MDS), International Brain Research Organization (IBRO), Lundbeck, Medtronic; grants from Michael J. Fox Foundation, Malaysian Ministry of Education Fundamental Research Grant Scheme; on Advisory board of Eisai. P.K.P. has received grants from Indian Council of Medical Research (ICMR), Department of Biotechnology (DBT), Department of Science & Technology‐Science and Engineering Research Board (DST‐SERB), Michael J Fox Foundation, USA. J.Y.L. has received NRF grant funded by MSIT of Korea, Research grant from SMU‐SNU BMC. P.L.K. has received consultancy honoraria from Medgenome Labs. R.B. has received consultancies honoraria from Ipsen, Teva‐Lundbeck, Eisai, BL Hua; speaker's honoraria from Teva‐Lundbeck; grants from Thailand Science and Research Innovation Bureau, Thailand Research Fund, Crown Property Bureau, Chulalongkorn University, and the National Science and Technology Development Agency; royalties from Humana Press, Wiley; on advisory board of Ipsen and Britannia Pharmaceuticals; has invented Laser guided walking stick, Parkinson cup, Tremor analysis software; holds intellectual property rights for Laser‐guided walking stick, Nocturnal Hypokinesia Questionnaire, Dopamine lyrics; holds patents for Tremor analysis software, Laser‐guided walking stick, Nocturnal monitoring device. N.M.I. has received grants from Ministry of Science, Technology and Innovation, Malaysia, MJFF GP2 grant, UKM Grant. Y.U. has received honoraria from Takeda Pharmaceutical Company Limited, Eisai Co., Ltd., FP Pharmaceutical Corporation, Otsuka Pharmaceutical Co., Ltd., Elsevier Japan K. K., Kyowa Hakko Kirin Co., Ltd., Dainippon Sumitomo Pharma Co., Ltd., Mitsubishi Tanabe Pharma Corporation, NIHON PHARMACEUTICAL Co., Ltd., and Novartis Pharma K.K.; royalties as journal editor from CHUGAI‐IGAKUSHA, Igaku‐Shoin Ltd, Medical View Co. Ltd., and Blackwell Publishing K.K.; grants (KAKENHI grant numbers 19H01091 and 20 K07866) from Japan Society for the Promotion of Science and grants from the Association of Radio Industries and Business and Tokyo Metropolitan Institute of Medical Science. B.J. has received grants from Seoul National University College of Medicine, Seoul National University Hospital, Sinyang Cultural Foundation, Peptron. C.D. is on the advisory board of Torrent. C.S. has received speaker's honoraria from International Parkinson and Movement Disorders Society, Defeat MSA Alliance; grants from EAN.
Supporting information
TABLE S1: Common variants in ATP7B gene in Asian patients with WD
Acknowledgment
We would like to thank Assoc. Prof. Ai Huey Tan for providing Video 7.
References
- 1. Claussnitzer M, Cho JH, Collins R, et al. A brief history of human disease genetics. Nature 2020;577(7789):179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lim SY, Tan AH, Ahmad‐Annuar A, et al. Parkinson's disease in the Western Pacific region. Lancet Neurol 2019;18(9):865–879. [DOI] [PubMed] [Google Scholar]
- 3. Prashanth LK, Taly AB, Sinha S, Arunodaya GR, Swamy HS. Wilson's disease: diagnostic errors and clinical implications. J Neurol Neurosurg Psychiatry 2004;75(6):907–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng N, Wang K, Hu W, et al. Wilson disease in the south chinese han population. Can J Neurol Sci 2014;41(3):363–367. [DOI] [PubMed] [Google Scholar]
- 5. Choe EJ, Choi JW, Kang M, et al. A population‐based epidemiology of Wilson's disease in South Korea between 2010 and 2016. Sci Rep 2020;10(1):14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamaguchi H, Nagase H, Tokumoto S, et al. Prevalence of Wilson disease based on genome databases in Japan. Pediatr Int 2021;63(8):918–922. [DOI] [PubMed] [Google Scholar]
- 7. Tai CS, Wu JF, Chen HL, Hsu HY, Chang MH, Ni YH. Modality of treatment and potential outcome of Wilson disease in Taiwan: a population‐based longitudinal study. J Formos Med Assoc 2018;117(5):421–426. [DOI] [PubMed] [Google Scholar]
- 8. Taly AB, Prashanth LK, Sinha S. Wilson's disease: an Indian perspective. Neurol India 2009;57(5):528–540. [DOI] [PubMed] [Google Scholar]
- 9. Kumar N, Prashant LK, Goyal V. Wilson's disease update: an Indian perspective. Ann Indian Acad Neurol 2021;24(5):652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poujois A, Woimant F. Wilson's disease: a 2017 update. Clin Res Hepatol Gastroenterol 2018;42(6):512–520. [DOI] [PubMed] [Google Scholar]
- 11. Aggarwal A, Bhatt M. Update on Wilson disease. Int Rev Neurobiol 2013;110:313–348. [DOI] [PubMed] [Google Scholar]
- 12. Aggarwal A, Chandhok G, Todorov T, et al. Wilson disease mutation pattern with genotype‐phenotype correlations from Western India: confirmation of p.C271* as a common Indian mutation and identification of 14 novel mutations. Ann Hum Genet 2013;77(4):299–307. [DOI] [PubMed] [Google Scholar]
- 13. Taly AB, Meenakshi‐Sundaram S, Sinha S, Swamy HS, Arunodaya GR. Wilson disease: description of 282 patients evaluated over 3 decades. Medicine (Baltimore) 2007;86(2):112–121. [DOI] [PubMed] [Google Scholar]
- 14. Lee BH, Kim JH, Lee SY, et al. Distinct clinical courses according to presenting phenotypes and their correlations to ATP7B mutations in a large Wilson's disease cohort. Liver Int 2011;31(6):831–839. [DOI] [PubMed] [Google Scholar]
- 15. Cheng N, Wang H, Wu W, et al. Spectrum of ATP7B mutations and genotype‐phenotype correlation in large‐scale Chinese patients with Wilson disease. Clin Genet 2017;92(1):69–79. [DOI] [PubMed] [Google Scholar]
- 16. Prashanth LK, Sinha S, Taly AB, Vasudev MK. Do MRI features distinguish Wilson's disease from other early onset extrapyramidal disorders? An analysis of 100 cases. Mov Disord 2010;25(6):672–678. [DOI] [PubMed] [Google Scholar]
- 17. Sinha S, Taly AB, Ravishankar S, et al. Wilson's disease: cranial MRI observations and clinical correlation. Neuroradiology 2006;48(9):613–621. [DOI] [PubMed] [Google Scholar]
- 18. Sinha S, Taly AB, Prashanth LK, Ravishankar S, Arunodaya GR, Vasudev MK. Sequential MRI changes in Wilson's disease with de‐coppering therapy: a study of 50 patients. Br J Radiol 2007;80(957):744–749. [DOI] [PubMed] [Google Scholar]
- 19. Gomes A, Dedoussis GV. Geographic distribution of ATP7B mutations in Wilson disease. Ann Hum Biol 2016;43(1):1–8. [DOI] [PubMed] [Google Scholar]
- 20. Dong Y, Ni W, Chen WJ, et al. Spectrum and classification of ATP7B variants in a large cohort of Chinese patients with Wilson's disease guides genetic diagnosis. Theranostics 2016;6(5):638–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen HL, Li HY, Wu JF, Wu SH, Chen HL, Yang YH, et al. Panel‐based next‐generation sequencing for the diagnosis of Cholestatic genetic liver diseases: clinical utility and challenges. J Pediatr 2019;205(153–159):e156. [DOI] [PubMed] [Google Scholar]
- 22. Yoo HW. Identification of novel mutations and the three most common mutations in the human ATP7B gene of Korean patients with Wilson disease. Genet Med 2002;4(6 suppl):43S–48S. [DOI] [PubMed] [Google Scholar]
- 23. Wu ZY, Lin MT, Murong SX, Wang N. Molecular diagnosis and prophylactic therapy for presymptomatic Chinese patients with Wilson disease. Arch Neurol 2003;60(5):737–741. [DOI] [PubMed] [Google Scholar]
- 24. Stapelbroek JM, Bollen CW, van Amstel JK, et al. The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: results of a meta‐analysis. J Hepatol 2004;41(5):758–763. [DOI] [PubMed] [Google Scholar]
- 25. Singh N, Kallollimath P, Shah MH, et al. Genetic analysis of ATP7B in 102 south Indian families with Wilson disease. PLoS One 2019;14(5):e0215779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aggarwal A, Aggarwal N, Nagral A, Jankharia G, Bhatt M. A novel global assessment scale for Wilson's disease (GAS for WD). Mov Disord 2009;24(4):509–518. [DOI] [PubMed] [Google Scholar]
- 27. Holmes SE, O'Hearn EE, McInnis MG, et al. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat Genet 1999;23(4):391–392. [DOI] [PubMed] [Google Scholar]
- 28. Holmes SE, O'Hearn E, Margolis RL. Why is SCA12 different from other SCAs? Cytogenet Genome Res 2003;100(1–4):189–197. [DOI] [PubMed] [Google Scholar]
- 29. Srivastava AK, Choudhry S, Gopinath MS, Roy S, Tripathi M, Brahmachari SK, Jain S. Molecular and clinical correlation in five Indian families with spinocerebellar ataxia 12. Ann Neurol 2001;50(6):796–800. [DOI] [PubMed] [Google Scholar]
- 30. Bahl S, Virdi K, Mittal U, et al. Evidence of a common founder for SCA12 in the Indian population. Ann Hum Genet 2005;69(Pt 5):528–534. [DOI] [PubMed] [Google Scholar]
- 31. Ganaraja VH, Holla VV, Stezin A, et al. Clinical, radiological, and genetic profile of spinocerebellar ataxia 12: a hospital‐based cohort analysis. Tremor Other Hyperkinet Mov (N Y) 2022;12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Prooije T, Ibrahim NM, Azmin S, van de Warrenburg B. Spinocerebellar ataxias in Asia: prevalence, phenotypes and management. Parkinsonism Relat Disord 2021;92:112–118. [DOI] [PubMed] [Google Scholar]
- 33. Choubtum L, Witoonpanich P, Hanchaiphiboolkul S, et al. Analysis of SCA8, SCA10, SCA12, SCA17 and SCA19 in patients with unknown spinocerebellar ataxia: a Thai multicentre study. BMC Neurol 2015;15:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang J, Shen L, Lei L, et al. Spinocerebellar ataxias in mainland China: an updated genetic analysis among a large cohort of familial and sporadic cases. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2011;36(6):482–489. [DOI] [PubMed] [Google Scholar]
- 35. Zhao Y, Tan EK, Law HY, Yoon CS, Wong MC, Ng I. Prevalence and ethnic differences of autosomal‐dominant cerebellar ataxia in Singapore. Clin Genet 2002;62(6):478–481. [DOI] [PubMed] [Google Scholar]
- 36. Fujigasaki H, Verma IC, Camuzat A, et al. SCA12 is a rare locus for autosomal dominant cerebellar ataxia: a study of an Indian family. Ann Neurol 2001;49(1):117–121. [PubMed] [Google Scholar]
- 37. Kumar DSA, Faruq M, Gundluru VR. Spinocerebellar ataxia type 12: an update. Ann Mov Disord 2019;2:48–57. [Google Scholar]
- 38. Sato N, Amino T, Kobayashi K, et al. Spinocerebellar ataxia type 31 is associated with "inserted" penta‐nucleotide repeats containing (TGGAA)n. Am J Hum Genet 2009;85(5):544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Niimi Y, Takahashi M, Sugawara E, et al. Abnormal RNA structures (RNA foci) containing a penta‐nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology 2013;33(6):600–611. [DOI] [PubMed] [Google Scholar]
- 40. Sakai H, Yoshida K, Shimizu Y, Morita H, Ikeda S, Matsumoto N. Analysis of an insertion mutation in a cohort of 94 patients with spinocerebellar ataxia type 31 from Nagano. Japan Neurogenetics 2010;11(4):409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ishikawa K, Nagai Y. Molecular mechanisms and future therapeutics for spinocerebellar ataxia type 31 (SCA31). Neurotherapeutics 2019;16(4):1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakamura K, Yoshida K, Matsushima A, et al. Natural history of spinocerebellar ataxia type 31: a 4‐year prospective study. Cerebellum 2017;16(2):518–524. [DOI] [PubMed] [Google Scholar]
- 43. Nozaki H, Ikeuchi T, Kawakami A, et al. Clinical and genetic characterizations of 16q‐linked autosomal dominant spinocerebellar ataxia (AD‐SCA) and frequency analysis of AD‐SCA in the Japanese population. Mov Disord 2007;22(6):857–862. [DOI] [PubMed] [Google Scholar]
- 44. Lee YC, Liu CS, Lee TY, Lo YC, Lu YC, Soong BW. SCA31 is rare in the Chinese population on Taiwan. Neurobiol Aging 2012;33(2):426 e423–426 e424. [DOI] [PubMed] [Google Scholar]
- 45. Ouyang Y, He Z, Li L, Qin X, Zhao Y, Yuan L. Spinocerebellar ataxia type 31 exists in Northeast China. J Neurol Sci 2012;316(1–2):164–167. [DOI] [PubMed] [Google Scholar]
- 46. Ikeda Y, Ohta Y, Kobayashi H, et al. Clinical features of SCA36: a novel spinocerebellar ataxia with motor neuron involvement (Asidan). Neurology 2012;79(4):333–341. [DOI] [PubMed] [Google Scholar]
- 47. Garcia‐Murias M, Quintans B, Arias M, et al. 'Costa da Morte' ataxia is spinocerebellar ataxia 36: clinical and genetic characterization. Brain 2012;135(Pt 5):1423–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kobayashi H, Abe K, Matsuura T, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 2011;89(1):121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McKeegan KS, Debieux CM, Watkins NJ. Evidence that the AAA+ proteins TIP48 and TIP49 bridge interactions between 15.5K and the related NOP56 and NOP58 proteins during box C/D snoRNP biogenesis. Mol Cell Biol 2009;29(18):4971–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ohta Y, Hayashi T, Nagai M, et al. Two cases of spinocerebellar ataxia accompanied by involvement of the skeletal motor neuron system and bulbar palsy. Intern Med 2007;46(11):751–755. [DOI] [PubMed] [Google Scholar]
- 51. Obayashi M, Stevanin G, Synofzik M, et al. Spinocerebellar ataxia type 36 exists in diverse populations and can be caused by a short hexanucleotide GGCCTG repeat expansion. J Neurol Neurosurg Psychiatry 2015;86(9):986–995. [DOI] [PubMed] [Google Scholar]
- 52. Sugihara K, Maruyama H, Morino H, et al. The clinical characteristics of spinocerebellar ataxia 36: a study of 2121 Japanese ataxia patients. Mov Disord 2012;27(9):1158–1163. [DOI] [PubMed] [Google Scholar]
- 53. Lee YC, Tsai PC, Guo YC, Hsiao CT, Liu GT, Liao YC, Soong BW. Spinocerebellar ataxia type 36 in the Han Chinese. Neurol Genet 2016;2(3):e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Valera JM, Diaz T, Petty LE, et al. Prevalence of spinocerebellar ataxia 36 in a US population. Neurol Genet 2017;3(4):e174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Katsimpouris D, Kartanou C, Breza M, Panas M, Koutsis G, Karadima G. Screening for spinocerebellar ataxia type 36 (SCA36) in the Greek population. J Neurol Sci 2019;402:131–132. [DOI] [PubMed] [Google Scholar]
- 56. Aguiar P, Pardo J, Arias M, et al. PET and MRI detection of early and progressive neurodegeneration in spinocerebellar ataxia type 36. Mov Disord 2017;32(2):264–273. [DOI] [PubMed] [Google Scholar]
- 57. Pirisinu L, Di Bari MA, D'Agostino C, et al. Gerstmann‐Straussler‐Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep 2016;6:20443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gerstmann JSE, Scheinker I. Über eine eigenartige hereditär‐familiäre Erkrankung des Zentralnervensystems. Zugleich ein Beitrag zur Frage des vorzeitigen lokalen Alterns. [about a rare hereditary familial disease of the central nervous system; at the same time a contribution to the problem of premature local aging]. Zeitschr Ges Neurol Psychiatr (Berl) 1936;154:36. [Google Scholar]
- 59. Schmitz M, Dittmar K, Llorens F, Gelpi E, Ferrer I, Schulz‐Schaeffer WJ, Zerr I. Hereditary human prion diseases: an update. Mol Neurobiol 2017;54(6):4138–4149. [DOI] [PubMed] [Google Scholar]
- 60. Kim MO, Takada LT, Wong K, Forner SA, Geschwind MD. Genetic PrP prion diseases. Cold Spring Harb Perspect Biol 2018;10:a0331341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kovacs GG, Puopolo M, Ladogana A, et al. Genetic prion disease: the EUROCJD experience. Hum Genet 2005;118(2):166–174. [DOI] [PubMed] [Google Scholar]
- 62. Jeong BH, Kim YS. Genetic studies in human prion diseases. J Korean Med Sci 2014;29(5):623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shi Q, Chen C, Xiao K, et al. Genetic prion disease: insight from the features and experience of China National Surveillance for Creutzfeldt‐Jakob disease. Neurosci Bull 2021;37(11):1570–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Collins S, McLean CA, Masters CL. Gerstmann‐Straussler‐Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci 2001;8(5):387–397. [DOI] [PubMed] [Google Scholar]
- 65. Nakamaura Y, Ae R, Takumi I, Sanjo N, Kitamoto T, Yamada M, Mizusawa H. Descriptive epidemiology of prion disease in Japan: 1999‐2012. J Epidemiol 2015;25(1):8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tesar A, Matej R, Kukal J, et al. Clinical Variability in P102L Gerstmann‐Straussler‐Scheinker Syndrome. Ann Neurol 2019;86(5):643–652. [DOI] [PubMed] [Google Scholar]
- 67. Piccardo P, Dlouhy SR, Lievens PM, et al. Phenotypic variability of Gerstmann‐Straussler‐Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol 1998;57(10):979–988. [DOI] [PubMed] [Google Scholar]
- 68. Webb TEF, Poulter M, Beck J, et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain 2008;131(10):2632–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Higuma M, Sanjo N, Satoh K, et al. Relationships between clinicopathological features and cerebrospinal fluid biomarkers in Japanese patients with genetic prion diseases. PLoS One 2013;8(3):e60003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yamazaki M, Oyanagi K, Mori O, et al. Variant Gerstmann‐Straussler syndrome with the P105L prion gene mutation: an unusual case with nigral degeneration and widespread neurofibrillary tangles. Acta Neuropathol 1999;98(5):506–511. [DOI] [PubMed] [Google Scholar]
- 71. Bongianni M, Orru C, Groveman BR, et al. Diagnosis of human prion disease using real‐time quaking‐induced conversion testing of olfactory mucosa and cerebrospinal fluid samples. JAMA Neurol 2017;74(2):155–162. [DOI] [PubMed] [Google Scholar]
- 72. Green AJE. RT‐QuIC: a new test for sporadic CJD. Pract Neurol 2019;19(1):49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Geschwind MD. Prion diseases. Continuum (Minneap Minn) 2015;21(6 Neuroinfectious Disease):1612–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Paisan‐Ruiz C, Bhatia KP, Li A, et al. Characterization of PLA2G6 as a locus for dystonia‐parkinsonism. Ann Neurol 2009;65(1):19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gregory A, Westaway SK, Holm IE, et al. Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology 2008;71(18):1402–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Morgan NV, Westaway SK, Morton JE, et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 2006;38(7):752–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ozes B, Karagoz N, Schule R, et al. PLA2G6 mutations associated with a continuous clinical spectrum from neuroaxonal dystrophy to hereditary spastic paraplegia. Clin Genet 2017;92(5):534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Koh K, Ichinose Y, Ishiura H, et al. PLA2G6‐associated neurodegeneration presenting as a complicated form of hereditary spastic paraplegia. J Hum Genet 2019;64(1):55–59. [DOI] [PubMed] [Google Scholar]
- 79. Magrinelli F, Mehta S, Di Lazzaro G, et al. Dissecting the phenotype and genotype of PLA2G6‐related parkinsonism. Mov Disord 2022;37(1):148–161. [DOI] [PubMed] [Google Scholar]
- 80. Li Q, Wu S, Tian J, Guo J, Tang B. Clinical features and mutation analysis of PLA2G6 gene in patients with neurodegeneration with brain iron accumulation. J Apoplexy and Nervous Diseases (in Chinese) 2013;30(4):292–294. [Google Scholar]
- 81. Yang Y, Yuan B, Zhang Z. A case report of juvenile Parkinson's disease and literature review. Chin J of Nerv Ment Dis (in Chinese) 2017;43(7):439–441. [Google Scholar]
- 82. Song B, Yu X, Zhang L, et al. A case of PLA2G6 gene mutation Parkinson's syndrome with onset of psychiatric symptoms. Chin J of Nerv Ment Dis (in Chinese) 2019;45(12):752–754. [Google Scholar]
- 83. Tao Z, Qiao D, Chen L, Han F, Sun L. A case report of traditional Chinese medicine intervention in PLA2G6 Parkinson's disease and literature review. Chin J Integrat Med Cardio‐Cerebrovascular Disease (in Chinese) 2021;19(10):1773–1776. [Google Scholar]
- 84. Xie F, Cen Z, Luo W. A case of early‐onset Parkinson's disease caused by homozygous missense mutation in PLA2G6 gene. Proceedings of the 2014 Zhejiang Provincial Neurology Annual Conference (Abstract: PU‐1285, in Chinese); 2014:149. [Google Scholar]
- 85. Wan Z, Wang H, Su D, Feng T. Clinical features of Parkinson's disease caused by PLA2G6 gene mutation (report of 1 case). J Clin Neuro (in Chinese) 2018;31(4):301–304. [Google Scholar]
- 86. Hu T, Wan G, Zhang J. Early‐onset Parkinson's disease caused by PLA2G6 gene mutation: a case and follow‐up report. Proceedings of the 18th National Neurology Academic Conference of the Chinese Medical Association (Abstract: PU‐0736, in Chinese); 2015:232. [Google Scholar]
- 87. Huang X, Liu X, Ma J, Cao L. Genetic and phenotypic analysis of PLA2G6‐associated neurodegenerative disease (PLAN). Proceedings of the 18th National Neurology Academic Conference of the Chinese Medical Association (Abstract: PU‐1396, in Chinese); 2015:505. [Google Scholar]
- 88. Fan C, Cai W, Sun H, et al. Clinical features of young onset Parkinson's disease caused by PLA2G6 gene mutation (report of one case). J Clin Neurol (in Chinese) 2021;34(5):338–342. [Google Scholar]
- 89. Chu YT, Lin HY, Chen PL, Lin CH. Genotype‐phenotype correlations of adult‐onset PLA2G6‐associated neurodegeneration: case series and literature review. BMC Neurol 2020;20(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Guo YP, Tang BS, Guo JF. PLA2G6‐associated neurodegeneration (PLAN): review of clinical phenotypes and genotypes. Front Neurol 2018;9:1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yoshino H, Tomiyama H, Tachibana N, et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14‐linked parkinsonism. Neurology 2010;75(15):1356–1361. [DOI] [PubMed] [Google Scholar]
- 92. Romani M, Kraoua I, Micalizzi A, et al. Infantile and childhood onset PLA2G6‐associated neurodegeneration in a large north African cohort. Eur J Neurol 2015;22(1):178–186. [DOI] [PubMed] [Google Scholar]
- 93. Engel LA, Jing Z, O'Brien DE, Sun M, Kotzbauer PT. Catalytic function of PLA2G6 is impaired by mutations associated with infantile neuroaxonal dystrophy but not dystonia‐parkinsonism. PLoS One 2010;5(9):e12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shi CH, Tang BS, Wang L, et al. PLA2G6 gene mutation in autosomal recessive early‐onset parkinsonism in a Chinese cohort. Neurology 2011;77(1):75–81. [DOI] [PubMed] [Google Scholar]
- 95. Miki Y, Yoshizawa T, Morohashi S, et al. Neuropathology of PARK14 is identical to idiopathic Parkinson's disease. Mov Disord 2017;32(5):799–800. [DOI] [PubMed] [Google Scholar]
- 96. Klein C, Lochte T, Delamonte SM, et al. PLA2G6 mutations and parkinsonism: long‐term follow‐up of clinical features and neuropathology. Mov Disord 2016;31(12):1927–1929. [DOI] [PubMed] [Google Scholar]
- 97. Yamashita C, Funayama M, Li Y, et al. Mutation screening of PLA2G6 in Japanese patients with early onset dystonia‐parkinsonism. J Neural Transm (Vienna) 2017;124(4):431–435. [DOI] [PubMed] [Google Scholar]
- 98. Choi EG, Lee W‐C, Shin J‐Y, Seo J‐S, Lee CS. Novel compound heterozygous mutations of PLA2G6 in a Korean pedigree of young‐onset Parkinson's disease: a study of whole genome sequencing. Parkinsonism Relat Disord 2016;22:e168–e169. [Google Scholar]
- 99. Wirth T, Weibel S, Montaut S, et al. Severe early‐onset impulsive compulsive behavior and psychosis in PLA2G6‐related juvenile Parkinson's disease. Parkinsonism Relat Disord 2017;41:127–129. [DOI] [PubMed] [Google Scholar]
- 100. Sone J, Mori K, Inagaki T, et al. Clinicopathological features of adult‐onset neuronal intranuclear inclusion disease. Brain 2016;139(Pt 12):3170–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lim SY, Ishiura H, Ramli N, et al. Adult‐onset neuronal intranuclear inclusion disease mimicking fragile X‐associated tremor‐ataxia syndrome in ethnic Chinese patients. Parkinsonism Relat Disord 2020;74:25–27. [DOI] [PubMed] [Google Scholar]
- 102. Ishiura H, Shibata S, Yoshimura J, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet 2019;51(8):1222–1232. [DOI] [PubMed] [Google Scholar]
- 103. Sone J, Mitsuhashi S, Fujita A, et al. Long‐read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 2019;51(8):1215–1221. [DOI] [PubMed] [Google Scholar]
- 104. Sun QY, Xu Q, Tian Y, et al. Expansion of GGC repeat in the human‐specific NOTCH2NLC gene is associated with essential tremor. Brain 2020;143(1):222–233. [DOI] [PubMed] [Google Scholar]
- 105. Ma D, Tan YJ, Ng ASL, et al. Association of NOTCH2NLC repeat expansions with Parkinson disease. JAMA Neurol 2020;77(12):1559–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Shi CH, Fan Y, Yang J, et al. NOTCH2NLC intermediate‐length repeat expansions are associated with Parkinson disease. Ann Neurol 2021;89(1):182–187. [DOI] [PubMed] [Google Scholar]
- 107. Okubo M, Doi H, Fukai R, et al. GGC repeat expansion of NOTCH2NLC in adult patients with leukoencephalopathy. Ann Neurol 2019;86(6):962–968. [DOI] [PubMed] [Google Scholar]
- 108. Liu YH, Chou YT, Chang FP, et al. Neuronal intranuclear inclusion disease in patients with adult‐onset non‐vascular leukoencephalopathy. Brain 2022;145:3010–3021. [DOI] [PubMed] [Google Scholar]
- 109. Yau WY, Vandrovcova J, Sullivan R, et al. Low prevalence of NOTCH2NLC GGC repeat expansion in white patients with movement disorders. Mov Disord 2021;36(1):251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yau WY, Sullivan R, Chen Z, Lynch DS, Vandrovcova J, Wood NW, Houlden H. GGC repeat expansion in NOTCH2NLC is rare in European leukoencephalopathy. Ann Neurol 2020;88(3):641–642. [DOI] [PubMed] [Google Scholar]
- 111. Chen Z, Yan Yau W, Jaunmuktane Z, et al. Neuronal intranuclear inclusion disease is genetically heterogeneous. Ann Clin Transl Neurol 2020;7(9):1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chen Z, Xu Z, Cheng Q, et al. Phenotypic bases of NOTCH2NLC GGC expansion positive neuronal intranuclear inclusion disease in a southeast Asian cohort. Clin Genet 2020;98(3):274–281. [DOI] [PubMed] [Google Scholar]
- 113.Ng ASL, Xu Z, Chen Z, et al. NOTCH2NLC‐linked neuronal intranuclear inclusion body disease and fragile X‐associated tremor/ataxia syndrome. Brain 2020;143(8):e69. [DOI] [PubMed] [Google Scholar]
- 114. Higuchi Y, Ando M, Yoshimura A, et al. Prevalence of fragile X‐associated tremor/ataxia syndrome in patients with cerebellar ataxia in Japan. Cerebellum 2021;21:851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Blakeley J, Jankovic J. Secondary paroxysmal dyskinesias. Mov Disord 2002;17(4):726–734. [DOI] [PubMed] [Google Scholar]
- 116. Erro R, Bhatia KP. Unravelling of the paroxysmal dyskinesias. J Neurol Neurosurg Psychiatry 2019;90(2):227–234. [DOI] [PubMed] [Google Scholar]
- 117. Gardiner AR, Jaffer F, Dale RC, et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain 2015;138(Pt 12):3567–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Demirkiran M, Jankovic J. Paroxysmal dyskinesias: clinical features and classification. Ann Neurol 1995;38(4):571–579. [DOI] [PubMed] [Google Scholar]
- 119. Spacey S, Adams P. Familial paroxysmal Kinesigenic dyskinesia—RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews((R)). Seattle, WA: University of Washington; 1993. [PubMed] [Google Scholar]
- 120. Ebrahimi‐Fakhari D, Saffari A, Westenberger A, Klein C. The evolving spectrum of PRRT2‐associated paroxysmal diseases. Brain 2015;138(Pt 12):3476–3495. [DOI] [PubMed] [Google Scholar]
- 121. Tan LC, Methawasin K, Teng EW, et al. Clinico‐genetic comparisons of paroxysmal kinesigenic dyskinesia patients with and without PRRT2 mutations. Eur J Neurol 2014;21(4):674–678. [DOI] [PubMed] [Google Scholar]
- 122. Tai ML, Lim SY, Tan CT. Idiopathic paroxysmal kinesigenic dyskinesia in Malaysia, a multi‐racial southeast Asian country. J Clin Neurosci 2010;17(8):1089–1090. [DOI] [PubMed] [Google Scholar]
- 123. Tan LC, Tan AK, Tjia H. Paroxysmal kinesigenic choreoathetosis in Singapore and its relationship to epilepsy. Clin Neurol Neurosurg 1998;100(3):187–192. [DOI] [PubMed] [Google Scholar]
- 124. Nagamitsu S, Matsuishi T, Hashimoto K, et al. Multicenter study of paroxysmal dyskinesias in Japan—clinical and pedigree analysis. Mov Disord 1999;14(4):658–663. [DOI] [PubMed] [Google Scholar]
- 125. Huang XJ, Wang SG, Guo XN, et al. The phenotypic and genetic Spectrum of paroxysmal Kinesigenic dyskinesia in China. Mov Disord 2020;35(8):1428–1437. [DOI] [PubMed] [Google Scholar]
- 126. Gardella E, Becker F, Moller RS, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol 2016;79(3):428–436. [DOI] [PubMed] [Google Scholar]
- 127. Tian WT, Huang XJ, Mao X, et al. Proline‐rich transmembrane protein 2‐negative paroxysmal kinesigenic dyskinesia: clinical and genetic analyses of 163 patients. Mov Disord 2018;33(3):459–467. [DOI] [PubMed] [Google Scholar]
- 128. Maini I, Iodice A, Spagnoli C, Salerno GG, Bertani G, Frattini D, Fusco C. Expanding phenotype of PRRT2 gene mutations: a new case with epilepsy and benign myoclonus of early infancy. Eur J Paediatr Neurol 2016;20(3):454–456. [DOI] [PubMed] [Google Scholar]
- 129. Cao L, Huang X, Wang N, et al. Recommendations for the diagnosis and treatment of paroxysmal kinesigenic dyskinesia: an expert consensus in China. Transl Neurodegener 2021;10(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Park H, Kim HJ, Jeon BS. Parkinsonism in spinocerebellar ataxia. Biomed Res Int 2015;2015:125273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Payami H, Nutt J, Gancher S, et al. SCA2 may present as levodopa‐responsive parkinsonism. Mov Disord 2003;18(4):425–429. [DOI] [PubMed] [Google Scholar]
- 132. Schols L, Reimold M, Seidel K, et al. No parkinsonism in SCA2 and SCA3 despite severe neurodegeneration of the dopaminergic substantia nigra. Brain 2015;138(Pt 11):3316–3326. [DOI] [PubMed] [Google Scholar]
- 133. Takao M, Aoyama M, Ishikawa K, et al. Spinocerebellar ataxia type 2 is associated with parkinsonism and Lewy body pathology. BMJ Case Rep 2011;2011:bcr0120113685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kim JM, Hong S, Kim GP, et al. Importance of low‐range CAG expansion and CAA interruption in SCA2 parkinsonism. Arch Neurol 2007;64(10):1510–1518. [DOI] [PubMed] [Google Scholar]
- 135. Charles P, Camuzat A, Benammar N, et al. Are interrupted SCA2 CAG repeat expansions responsible for parkinsonism? Neurology 2007;69(21):1970–1975. [DOI] [PubMed] [Google Scholar]
- 136. Lim SW, Zhao Y, Chua E, et al. Genetic analysis of SCA2, 3 and 17 in idiopathic Parkinson's disease. Neurosci Lett 2006;403(1–2):11–14. [DOI] [PubMed] [Google Scholar]
- 137. Shan DE, Liu RS, Sun CM, Lee SJ, Liao KK, Soong BW. Presence of spinocerebellar ataxia type 2 gene mutation in a patient with apparently sporadic Parkinson's disease: clinical implications. Mov Disord 2004;19(11):1357–1360. [DOI] [PubMed] [Google Scholar]
- 138. Wang JL, Xiao B, Cui XX, et al. Analysis of SCA2 and SCA3/MJD repeats in Parkinson's disease in mainland China: genetic, clinical, and positron emission tomography findings. Mov Disord 2009;24(13):2007–2011. [DOI] [PubMed] [Google Scholar]
- 139. Lu CS, Wu Chou YH, Kuo PC, Chang HC, Weng YH. The parkinsonian phenotype of spinocerebellar ataxia type 2. Arch Neurol 2004;61(1):35–38. [DOI] [PubMed] [Google Scholar]
- 140. Modoni A, Contarino MF, Bentivoglio AR, et al. Prevalence of spinocerebellar ataxia type 2 mutation among Italian parkinsonian patients. Mov Disord 2007;22(3):324–327. [DOI] [PubMed] [Google Scholar]
- 141. Simon‐Sanchez J, Hanson M, Singleton A, et al. Analysis of SCA‐2 and SCA‐3 repeats in parkinsonism: evidence of SCA‐2 expansion in a family with autosomal dominant Parkinson's disease. Neurosci Lett 2005;382(1–2):191–194. [DOI] [PubMed] [Google Scholar]
- 142. Wang C, Xu Y, Feng X, et al. Linkage analysis and whole‐exome sequencing exclude extra mutations responsible for the parkinsonian phenotype of spinocerebellar ataxia‐2. Neurobiol Aging 2015;36(1):545 e541–545 e547. [DOI] [PubMed] [Google Scholar]
- 143. Kim YE, Jeon B, Farrer MJ, et al. SCA2 family presenting as typical Parkinson's disease: 34 year follow up. Parkinsonism Relat Disord 2017;40:69–72. [DOI] [PubMed] [Google Scholar]
- 144. Sun H, Satake W, Zhang C, et al. Genetic and clinical analysis in a Chinese parkinsonism‐predominant spinocerebellar ataxia type 2 family. J Hum Genet 2011;56(4):330–334. [DOI] [PubMed] [Google Scholar]
- 145. Yang L, Dong Y, Ma Y, Ni W, Wu ZY. Genetic profile and clinical characteristics of Chinese patients with spinocerebellar ataxia type 2: a multicenter experience over 10 years. Eur J Neurol 2021;28(3):955–964. [DOI] [PubMed] [Google Scholar]
- 146. Lin IS, Wu RM, Lee‐Chen GJ, Shan DE, Gwinn‐Hardy K. The SCA17 phenotype can include features of MSA‐C, PSP and cognitive impairment. Parkinsonism Relat Disord 2007;13(4):246–249. [DOI] [PubMed] [Google Scholar]
- 147. Kim JY, Kim SY, Kim JM, et al. Spinocerebellar ataxia type 17 mutation as a causative and susceptibility gene in parkinsonism. Neurology 2009;72(16):1385–1389. [DOI] [PubMed] [Google Scholar]
- 148. Wu YR, Lin HY, Chen CM, et al. Genetic testing in spinocerebellar ataxia in Taiwan: expansions of trinucleotide repeats in SCA8 and SCA17 are associated with typical Parkinson's disease. Clin Genet 2004;65(3):209–214. [DOI] [PubMed] [Google Scholar]
- 149. Lin CH, Chen PL, Tai CH, Lin HI, Chen CS, Chen ML, Wu RM . A clinical and genetic study of early‐onset and familial parkinsonism in Taiwan: an integrated approach combining gene dosage analysis and next‐generation sequencing. Mov Disord 2019;34(4):506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Paolini Paoletti F, Prontera P, Nigro P, et al. Small‐expanded allele spinocerebellar ataxia 17: imaging and phenotypic variability. Neurol Sci 2021;42(10):4309–4315. [DOI] [PubMed] [Google Scholar]
- 151. de Oliveira DS, Pedroso JL, Barsottini O, Tomaselli PJ, Marques Junior W, Vale TC. Small‐expanded allele spinocerebellar ataxia type 17 leading to broad movement disorder phenotype in a Brazilian patient. Cerebellum 2022;21(6):1151–1153. [DOI] [PubMed] [Google Scholar]
- 152. Shin JH, Park H, Ehm GH, et al. The pathogenic role of low range repeats in SCA17. PLoS One 2015;10(8):e0135275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson's disease. Lancet Neurol 2020;19(2):170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lesage S, Durr A, Tazir M, et al. LRRK2 G2019S as a cause of Parkinson's disease in north African Arabs. N Engl J Med 2006;354(4):422–423. [DOI] [PubMed] [Google Scholar]
- 155. Ozelius LJ, Senthil G, Saunders‐Pullman R, et al. LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N Engl J Med 2006;354(4):424–425. [DOI] [PubMed] [Google Scholar]
- 156. Lim SY, Lim JL, Ahmad‐Annuar A, et al. Clinical phenotype of LRRK2 R1441C in 2 Chinese sisters. Neurodegener Dis 2020;20(1):39–45. [DOI] [PubMed] [Google Scholar]
- 157. Li Y, Ikeda A, Yoshino H, et al. Clinical characterization of patients with leucine‐rich repeat kinase 2 genetic variants in Japan. J Hum Genet 2020;65(9):771–781. [DOI] [PubMed] [Google Scholar]
- 158. Chen Y, Gu X, Ou R, et al. Evaluating the role of SNCA, LRRK2, and GBA in Chinese patients with early‐onset Parkinson's disease. Mov Disord 2020;35(11):2046–2055. [DOI] [PubMed] [Google Scholar]
- 159. Shu L, Zhang Y, Sun Q, Pan H, Tang B. A comprehensive analysis of population differences in LRRK2 variant distribution in Parkinson's disease. Front Aging Neurosci 2019;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Satake W, Nakabayashi Y, Mizuta I, et al. Genome‐wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet 2009;41(12):1303–1307. [DOI] [PubMed] [Google Scholar]
- 161. Foo JN, Tan LC, Irwan ID, et al. Genome‐wide association study of Parkinson's disease in east Asians. Hum Mol Genet 2017;26(1):226–232. [DOI] [PubMed] [Google Scholar]
- 162. Gopalai AA, Lim JL, Li HH, et al. LRRK2 N551K and R1398H variants are protective in Malays and Chinese in Malaysia: a case‐control association study for Parkinson's disease. Mol Genet Genomic Med 2019;7(11):e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Funayama M, Ohe K, Amo T, et al. CHCHD2 mutations in autosomal dominant late‐onset Parkinson's disease: a genome‐wide linkage and sequencing study. Lancet Neurol 2015;14(3):274–282. [DOI] [PubMed] [Google Scholar]
- 164. Li NN, Wang L, Tan EK, et al. Genetic analysis of CHCHD2 gene in Chinese Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet 2016;171(8):1148–1152. [DOI] [PubMed] [Google Scholar]
- 165. Lin CH, Tsai PI, Lin HY, et al. Mitochondrial UQCRC1 mutations cause autosomal dominant parkinsonism with polyneuropathy. Brain 2020;143(11):3352–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Zhao YW, Pan HX, Wang CY, et al. UQCRC1 variants in Parkinson's disease: a large cohort study in Chinese mainland population. Brain 2021;144(6):e54. [DOI] [PubMed] [Google Scholar]
- 167. Senkevich K, Bandres‐Ciga S, Gan‐Or Z, Krohn L. International Parkinson's disease genomics C. lack of evidence for association of UQCRC1 with Parkinson's disease in Europeans. Neurobiol Aging 2021;101:297 e291–297 e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Courtin T, Tesson C, Corvol JC, Lesage S, Brice A. French Parkinson's disease genetics g. lack of evidence for association of UQCRC1 with autosomal dominant Parkinson's disease in Caucasian families. Neurogenetics 2021;22(4):365–366. [DOI] [PubMed] [Google Scholar]
- 169. Kestenbaum M, Alcalay RN. Clinical features of LRRK2 carriers with Parkinson's disease. Adv Neurobiol 2017;14:31–48. [DOI] [PubMed] [Google Scholar]
- 170. Vilas D, Sharp M, Gelpi E, et al. Clinical and neuropathological features of progressive supranuclear palsy in leucine rich repeat kinase (LRRK2) G2019S mutation carriers. Mov Disord 2018;33(2):335–338. [DOI] [PubMed] [Google Scholar]
- 171. Shu L, Zhang Y, Pan H, Xu Q, Guo J, Tang B, Sun Q. Clinical heterogeneity among LRRK2 variants in Parkinson's disease: a meta‐analysis. Front Aging Neurosci 2018;10:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Hattori N, Mizuno Y. Pathogenetic mechanisms of parkin in Parkinson's disease. Lancet 2004;364(9435):722–724. [DOI] [PubMed] [Google Scholar]
- 173. Zhao Y, Qin L, Pan H, et al. The role of genetics in Parkinson's disease: a large cohort study in Chinese mainland population. Brain 2020;143(7):2220–2234. [DOI] [PubMed] [Google Scholar]
- 174. Chu MK, Kim WC, Choi JM, Hong JH, Kang SY, Ma HI, Kim YJ. Analysis of dosage mutation in PARK2 among Korean patients with early‐onset or familial Parkinson's disease. J Clin Neurol 2014;10(3):244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Li N, Wang L, Zhang J, et al. Whole‐exome sequencing in early‐onset Parkinson's disease among ethnic Chinese. Neurobiol Aging 2020;90:150 e155–150 e111. [DOI] [PubMed] [Google Scholar]
- 176. Yoshino H, Li Y, Nishioka K, et al. Genotype‐phenotype correlation of Parkinson's disease with PRKN variants. Neurobiol Aging 2022;114:117–128. [DOI] [PubMed] [Google Scholar]
- 177. Tan AH, Lohmann K, Tay YW, et al. PINK1 p.Leu347Pro mutations in Malays: prevalence and illustrative cases. Parkinsonism Relat Disord 2020;79:34–39. [DOI] [PubMed] [Google Scholar]
- 178. Kumazawa R, Tomiyama H, Li Y, et al. Mutation analysis of the PINK1 gene in 391 patients with Parkinson disease. Arch Neurol 2008;65(6):802–808. [DOI] [PubMed] [Google Scholar]
- 179. Rogaeva E, Johnson J, Lang AE, et al. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol 2004;61(12):1898–1904. [DOI] [PubMed] [Google Scholar]
- 180. Patel SG, Buchanan CM, Mulroy E, et al. Potential PINK1 founder effect in Polynesia causing early‐onset Parkinson's disease. Mov Disord 2021;36(9):2199–2200. [DOI] [PubMed] [Google Scholar]
- 181. Neumann J, Bras J, Deas E, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 2009;132(Pt 7):1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Winder‐Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community‐based incident cohort. Brain 2013;136(Pt 2):392–399. [DOI] [PubMed] [Google Scholar]
- 183. Brockmann K, Srulijes K, Pflederer S, et al. GBA‐associated Parkinson's disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015;30(3):407–411. [DOI] [PubMed] [Google Scholar]
- 184. Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA‐associated Parkinson's disease: the mutation matters. Ann Neurol 2016;80(5):662–673. [DOI] [PubMed] [Google Scholar]
- 185. Davis MY, Johnson CO, Leverenz JB, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 2016;73(10):1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Creese B, Bell E, Johar I, Francis P, Ballard C, Aarsland D. Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson's disease and Lewy body dementias: review and meta‐analyses. Am J Med Genet B Neuropsychiatr Genet 2018;177(2):232–241. [DOI] [PubMed] [Google Scholar]
- 187. Thaler A, Bregman N, Gurevich T, et al. Parkinson's disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat Disord 2018;55:45–49. [DOI] [PubMed] [Google Scholar]
- 188. Alcalay RN, Caccappolo E, Mejia‐Santana H, et al. Cognitive performance of GBA mutation carriers with early‐onset PD: the CORE‐PD study. Neurology 2012;78(18):1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Liu G, Boot B, Locascio JJ, et al. Specifically neuropathic Gaucher's mutations accelerate cognitive decline in Parkinson's. Ann Neurol 2016;80(5):674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Straniero L, Rimoldi V, Melistaccio G, di Fonzo A, Pezzoli G, Duga S, Asselta R. A rapid and low‐cost test for screening the most common Parkinson's disease‐related GBA variants. Parkinsonism Relat Disord 2020;80:138–141. [DOI] [PubMed] [Google Scholar]
- 191. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361(17):1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Pulkes T, Choubtum L, Chitphuk S, et al. Glucocerebrosidase mutations in Thai patients with Parkinson's disease. Parkinsonism Relat Disord 2014;20(9):986–991. [DOI] [PubMed] [Google Scholar]
- 193. den Heijer JM, Cullen VC, Quadri M, et al. A large‐scale full GBA1 gene screening in Parkinson's disease in The Netherlands. Mov Disord 2020;35(9):1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Lim JL, Lohmann K, Tan AH, et al. Glucocerebrosidase (GBA) gene variants in a multi‐ethnic Asian cohort with Parkinson's disease: mutational spectrum and clinical features. J Neural Transm (Vienna) 2022;129(1):37–48. [DOI] [PubMed] [Google Scholar]
- 195. Wu YR, Chen CM, Chao CY, Ro LS, Lyu RK, Chang KH, Lee‐Chen GJ. Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J Neurol Neurosurg Psychiatry 2007;78(9):977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Tan EK, Tong J, Fook‐Chong S, Yih Y, Wong MC, Pavanni R, Zhao Y. Glucocerebrosidase mutations and risk of Parkinson disease in Chinese patients. Arch Neurol 2007;64(7):1056–1058. [DOI] [PubMed] [Google Scholar]
- 197. Ortega RA, Wang C, Raymond D, et al. Association of Dual LRRK2 G2019S and GBA variations with Parkinson disease progression. JAMA Netw Open 2021;4(4):e215845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Morales‐Briceno H, Ong TL, Duma SR, et al. Recurrent Biallelic p.L347P PINK1 variant in Polynesians with parkinsonism and isolated Dopa‐responsive dystonia. Mov Disord Clin Pract 2022;9(5):696–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Jansen IE, Bras JM, Lesage S, et al. CHCHD2 and Parkinson's disease. Lancet Neurol 2015;14(7):678–679. [DOI] [PubMed] [Google Scholar]
- 200. Lee RG, Sedghi M, Salari M, et al. Early‐onset Parkinson disease caused by a mutation in CHCHD2 and mitochondrial dysfunction. Neurol Genet 2018;4(5):e276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Schumacher‐Schuh AF, Bieger A, Okunoye O, et al. Underrepresented populations in Parkinson's genetics research: current landscape and future directions. Mov Disord 2022;37(8):1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1: Common variants in ATP7B gene in Asian patients with WD