

Abstract
CRISPR gene editing holds great promise to modify DNA sequences in somatic cells to treat disease. However, standard computational and biochemical methods to predict off-target potential focus on reference genomes. We developed an efficient tool called CRISPRme that considers SNP and indel genetic variants to nominate and prioritize off-target sites. We tested the software with a BCL11A enhancer targeting guide RNA showing promise in clinical trials for sickle cell disease and β-thalassemia and found that the top candidate off-target is produced by an allele common in African-ancestry populations (MAF 4.5%) that introduces a protospacer adjacent motif (PAM) sequence. We validated that SpCas9 generates strictly allele-specific indels and pericentric inversions in CD34+ HSPCs, although high-fidelity Cas9 mitigates this off-target. This report illustrates how genetic variants should be considered as modifiers of gene editing outcomes. We expect that variant-aware off-target assessment will become integral to therapeutic genome editing evaluation and provide a powerful approach for comprehensive off-target nomination.
INTRODUCTION
CRISPR genome editing offers extraordinary opportunities to develop novel therapeutics by introducing targeted genetic or epigenetic modifications to genomic regions of interest. Briefly, CRISPR offers a simple and programmable platform that couples binding to a genomic target sequence of choice with diverse effector proteins through RNA:DNA (spacer:protospacer) complementary sequence interactions mediated by a guide RNA (gRNA) spacer sequence matching a genomic protospacer sequence restricted by protospacer adjacent motif (PAM) sequences. Editing effectors may consist of nucleases to introduce targeted double strand breaks leading to short insertions/deletions (indels) and templated repairs (e.g. Cas9), deaminases for precise substitutions (base editors), or chromatin regulators for transcriptional interference or activation (CRISPRi/a) among others to achieve a range of desired biological outcomes2.
CRISPR-based systems may create unintended off-target modifications posing potential genotoxicity for therapeutic use. Several experimental assays and computational methods are available to uncover or forecast these off-targets3. Off-target sites are partially predictable based on homology to the spacer and PAM sequence. Beyond the number of mismatches or bulges, a variety of sequence features, like position of mismatch or bulge with respect to PAM or specific base changes, contribute to off-target potential3–6. Computational models can complement experimental approaches to off-target nomination in several respects: to triage gRNAs prior to experiments by predicting the number and cleavage potential of off-target sites and to prioritize target sites for experimental scrutiny. Genetic variants may alter protospacer and PAM sequences and therefore may influence both on-target and off-target potential. Gene editing strategies designed to specifically recognize patient mutations may increase the likelihood of editing mutant alleles, whereas variants that reduce homology to the anticipated target may decrease the efficiency of the desired genetic modification. Although a variety of in vitro and cell-based experimental methods can be used to empirically nominate off-target sites, these methods either use homology to the reference genome as a criterion to define the search space and/or use a limited set of human donor genomes to evaluate off-target potential4,7. Therefore, computational methods may be especially useful to predict the impact of off-target sequences not found in reference genomes.
Prior studies considering gRNAs targeting therapeutically relevant genes using population-based variant databases like the 1000 Genomes Project (1000G) and the Exome Aggregation Consortium have highlighted how genetic variants can significantly alter the off-target landscape by creating novel and personal off-target sites not present in a single reference genome8,9. Although these prior studies provide code to reproduce analyses, implementation choices make these tools not suitable to analyze large variant datasets and to consider higher numbers of mismatches. In addition, these methods ignore bulges between RNA:DNA hybrids, cannot efficiently model alternative haplotypes and indels, and require extensive computational skills to utilize.
Several user-friendly websites have been developed to aid the design of gRNAs and to assess their potential off-targets10–13. Even though variant-aware prediction is an important problem for genome editing interventions, these scalable graphical user interface (GUI) based tools do not account for genetic variants. In addition, these tools artificially limit the number of mismatches for the search and/or do not support DNA/RNA bulges. Therefore, designing gRNAs for therapeutic intervention using current widely available tools could miss important off-target sites that may lead to unwanted genotoxicity. A complete and exhaustive off-target search with an arbitrary number of mismatches, bulges, and genetic variants that is haplotype-aware is a computationally challenging problem that requires specialized and efficient data structures.
We have recently developed a command line tool that partially solves these challenges called CRISPRitz14. This tool uses optimized data structures to efficiently account for single variants, mismatches and bulges but with significant limitations14. Here we substantially extend this work by developing CRISPRme, a tool to aid gRNA design with added support for haplotype-aware off-target enumeration, short indel variants and a flexible number of mismatches and bulges. CRISPRme is a unified, user-friendly web-based application that provides several reports to prioritize putative off-targets based on their risk in a population or individuals.
CRISPRme is flexible to accept user-defined genomic annotations, which could include empirically identified off-target sites or cell type specific chromatin features. It can integrate population genetic variants from sets of phased individual variants (like those from 1000G15), unphased individual variants (like those from the Human Genome Diversity Project, HGDP16) and population-level variants (like those from the Genome Aggregation Database, gnomAD17). Furthermore, it can accept personal genomes from individual subjects to identify and prioritize private off-targets due to variants specific to a single individual.
Here we demonstrate the utility of CRISPRme by analyzing the off-target potential of a gRNA currently being tested in clinical trials for SCD and β-thalassemia1,18,19. We identify possible off-targets introduced by genetic variants included within and extending beyond 1000G. We predict that the most likely off-target site, overlooked by prior analyses, is introduced by a variant common in African-ancestry individuals (rs114518452, minor allele frequency (MAF)=4.5%) and provide experimental evidence of its off-target potential in gene edited human CD34+ hematopoietic stem and progenitor cells. Furthermore, we demonstrate that allele-specific off-target potential is widespread across various nucleic acid targeting therapeutic strategies.
RESULTS
Computational tool for variant-aware off-target nomination
CRISPRme is a web-based tool to predict off-target potential of CRISPR gene editing that accounts for genetic variation. It is available online at http://crisprme.di.univr.it. CRISPRme can also be deployed to local, protected and isolated environments as a web app or command line utility, neither of which transfer nor store data online, therefore respecting genomic privacy and regulations. CRISPRme takes as input a Cas protein, gRNA spacer sequence(s) and PAM, genome build, sets of variants (VCF files for populations or individuals), user-defined thresholds of mismatches and bulges, and optional user-defined genomic annotations to produce comprehensive and personalized reports (Fig. 1a, Supplementary Notes 1–3).
Figure 1. CRISPRme provides web-based analysis of CRISPR-Cas gene editing off-target potential reflecting population genetic diversity.
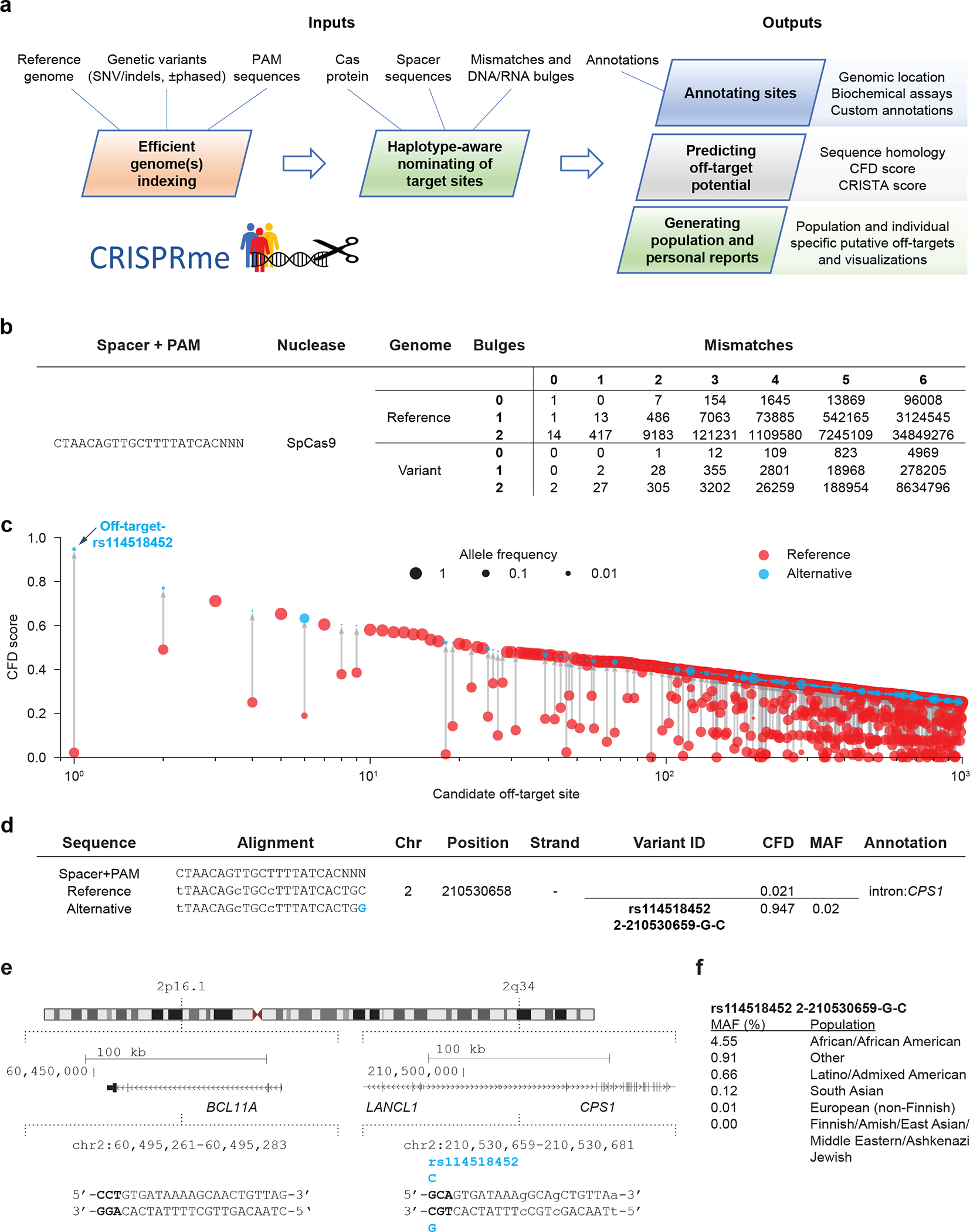
a) CRISPRme software takes as input a reference genome, genetic variants, PAM sequence, Cas protein type, spacer sequence, homology threshold and genomic annotations and provides comprehensive, target-focused and individual-focused analyses of off-target potential. It is available as an online webtool and can be deployed locally or used offline as command-line software. b) Analysis of the BCL11A-1617 spacer targeting the +58 erythroid enhancer with SpCas9, NNN PAM, 1000G variants, up to 6 mismatches and up to 2 bulges. c) Top 1000 predicted off-target sites ranked by CFD score, indicating the CFD score of the reference and alternative allele if applicable, with allele frequency indicated by circle size. d) The off-target site with the highest CFD score is created by the minor allele of rs114518452. Coordinates are for hg38 and 0-start for the potential off-target and 1-start for the variant-ID. MAF is based on 1000G. e) The top predicted off-target site from CRISPRme is an allele-specific off-target with 3 mismatches to the BCL11A-1617 spacer sequence, where the rs114518452-C minor allele produces a de novo NGG PAM sequence. PAM sequence shown in bold and mismatches to BCL11A-1617 shown as lowercase. Coordinates are for hg38 and 1-start. f) rs114518452 allele frequencies based on gnomAD v3.1. Coordinates are for hg38 and 1-start. Spacer shown as DNA sequence for ease of visual alignment.
We have designed CRISPRme to be flexible with support for new gene editors with variable and extremely relaxed PAM requirements20. Thanks to a PAM encoding based on Aho-Corasick automata and an index based on a ternary search tree, CRISPRme can perform genome-wide exhaustive searches efficiently even with an NNN PAM, extensive mismatches (tested with up to 7) and RNA:DNA bulges (tested with up to 2) (Supplementary Note 4).
Notably, a comprehensive search performed with up to 6 mismatches, 2 DNA/RNA bulges and a fully non-restrictive PAM (NNN) on a small computational cluster node using 20 CPU cores and 128 GB RAM (Intel Xeon CPU E5-2609 v4 clocked at 2.2 GHz) takes ~34 hours of real time and ~152 hours of CPU time (including both user and system times). All the 1000G variants, including both SNVs and indels, can be included in the search together with all the available metadata for each individual (sex, super-population and age), and the search operation takes into account observed haplotypes (Supplementary Note 5). Importantly, off-target sites that represent alternative alignments to a given genomic region are merged to avoid inflating the number of reported sites. Although several tools exist to enumerate off-targets, to our knowledge only two command line tools8,21 incorporate genetic variants in the search. However, they have several limitations in terms of scalability to large searches, number of mismatches, bulges, haplotypes, and variant file formats supported and do not provide an easy-to-use graphical user interface (Supplementary Note 6).
CRISPRme generates several reports (Supplementary Note 2). First, it summarizes for each gRNA all the potential off-targets found in the reference or variant genomes based on their mismatches and bulges (Fig. 1b) and generates a file with detailed information on each of these candidate off-targets (Supplementary Data 1, Supplementary Table 1). Second, it compares gRNAs to customizable annotations. By default, it classifies possible off-target sites based on GENCODE22 (genomic features) and ENCODE23 (candidate cis-regulatory elements, cCREs) annotations. It can also incorporate user-defined annotations in BED format, such as empiric off-target scores or cell type specific chromatin features (Extended Data Fig. 1, Supplementary Note 5). Third, using 1000G and/or HGDP16 variants, CRISPRme reports the cumulative distribution of homologous sites based on the reference genome or super-population. These global reports could be used to compare a set of gRNAs based on how genetic variation impacts their predicted on- and off-target cleavage potential using cutting frequency determination (CFD) or CRISPR Target Assessment (CRISTA)24 scores (Extended Data Fig. 2). CRISPRme includes multiple scoring metrics and can be easily extended with new ones, including scores tailored for different editors. Finally, CRISPRme can generate personal genome focused reports called personal risk cards (Supplementary Note 3). These reports highlight private off-target sites due to unique genetic variants.
A common allele-specific off-target for a gRNA in the clinic
We tested CRISPRme with a gRNA (#1617) targeting a GATA1 binding motif at the +58 erythroid enhancer of BCL11A18,19. A recent clinical report described two patients, one with SCD and one with β-thalassemia, each treated with autologous gene modified hematopoietic stem and progenitor cells (HSPCs) edited with Cas9 and this gRNA, who showed sustained increases in fetal hemoglobin, transfusion-independence and absence of vaso-occlusive episodes (in the SCD patient) following therapy. This study as well as prior pre-clinical studies with the same gRNA (#1617) did not reveal evidence of off-target editing in treated cells when considering off-target sites nominated by bioinformatic analysis of the human reference genome and empiric analysis of in vitro genomic cleavage potential (Supplementary Table 2, Supplementary Note 7)1,19,25. CRISPRme analysis found that the predicted off-target site with both the greatest CFD score and the greatest increase in CFD score from the reference to alternative allele was at an intronic sequence of CPS1 (Fig. 1c,d), a genomic target subject to common genetic variation (modified by a SNP with MAF ≥ 1%). CFD scores range from 0 to 1, where the on-target site has a score of 1. The alternative allele rs114518452-C generates a TGG PAM sequence (that is, the optimal PAM for SpCas9) for a potential off-target site with 3 mismatches and a CFD score (CFDalt 0.95) approaching that of the on-target site (Fig. 1e). In contrast, the reference allele rs114518452-G disrupts the PAM to TGC, which markedly reduces predicted cleavage potential (CFDref 0.02). rs114518452-C has an overall MAF of 1.33% in gnomAD v3.1, with MAF of 4.55% in African/African-American, 0.91% in Other, 0.66% in Latino/Admixed American, 0.12% in South Asian, 0.01% in European (non-Finnish) and 0.00% in all other populations represented in gnomAD (Fig. 1f, Supplementary Table 3). To consider the off-target potential that could be introduced by personal genetic variation that would not be predicted by 1000G variants, we analyzed HGDP variants identified from whole genome sequences of 929 individuals from 54 diverse human populations. We observed 249 candidate off-targets for gRNA #1617 with CFD ≥0.2 for which the CFD score in HGDP exceeded that found for either the reference genome or 1000G variants by at least 0.1. (Fig. 2a, Extended Data Fig. 3). These additional variant off-targets not found from 1000G were observed in each super-population, with the greatest frequency in the African super-population (Fig. 2b). 229 (92.0%) of these variant off-targets were unique to a super-population and 172 (69.1%) of these were private to just one individual (Fig. 2c). Furthermore, single individual focused searches, for example an analysis of HGDP01211, an individual of the Oroqen population within the East Asian super-population, showed that most variant off-targets (with higher CFD score than reference) were due to variants also found in 1000G (n=32369, 90.4%), a subset were due to variants shared with other individuals from HGDP but absent from 1000G (n=3177, 8.9%), and a small fraction were private to the individual (n=234, 0.7%) (Fig. 2d). Among these private off-targets was one generated by a variant (rs1191022522, 3-99137613-A-G, gnomAD v3.1 MAF 0.0053%) where the alternative allele produces a canonical NGG PAM that increases the CFD score from 0.14 to 0.54 (Fig. 2d,e).
Figure 2. CRISPRme provides analysis of off-target potential of CRISPR-Cas gene editing reflecting population and private genetic diversity.
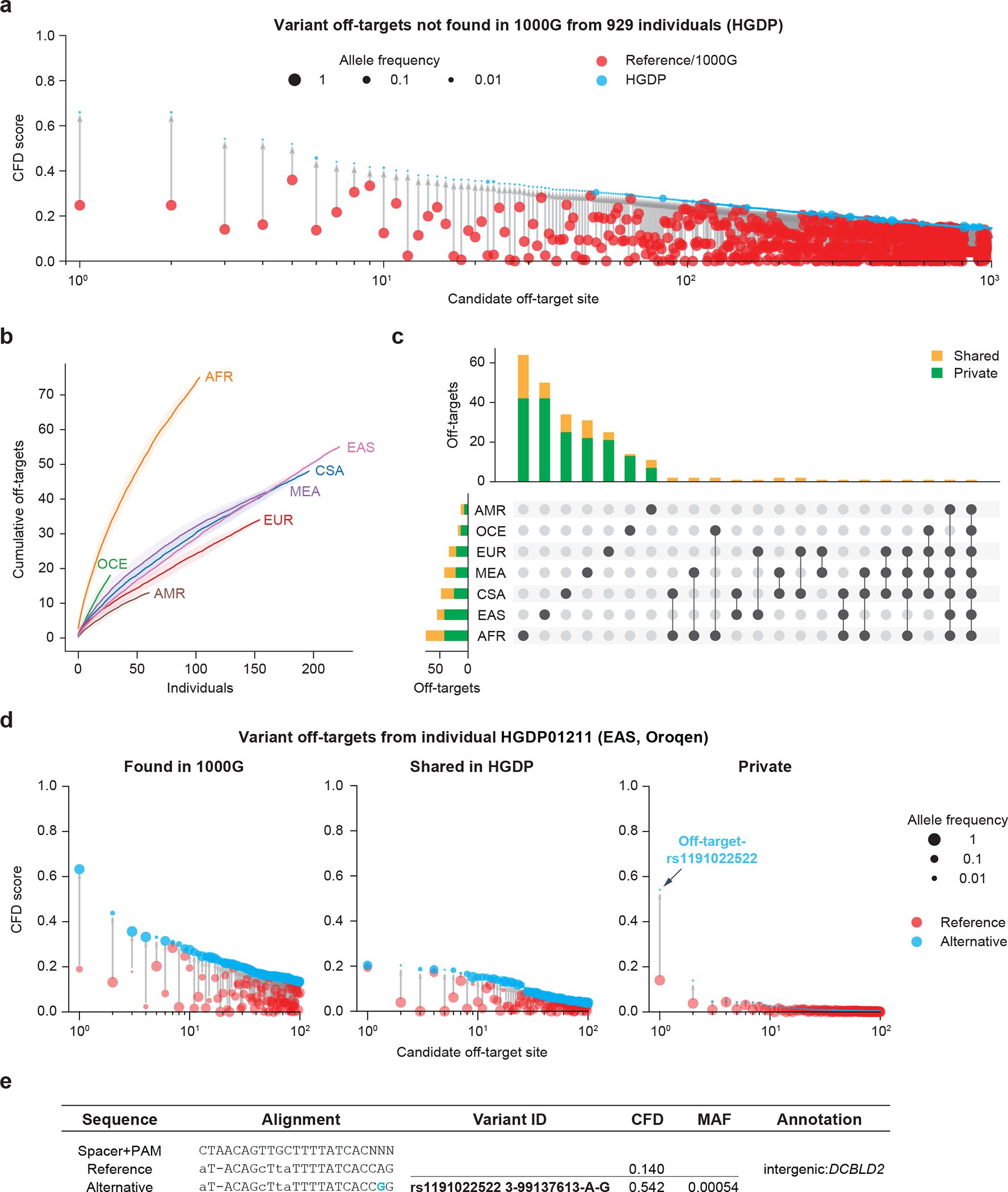
a) CRISPRme analysis was conducted with variants from HGDP comprising whole genome sequencing of 929 individuals from 54 diverse human populations. HGDP variant off-targets with greater CFD scores than the reference genome or 1000G were plotted and sorted by CFD score, with HGDP variant off-targets shown in blue and reference or 1000G variant off-targets shown in red. b) Cumulative distribution plot of HGDP variant off-targets with CFD≥0.2 and increase in CFD of ≥0.1 per super-population. Individual samples from each of the seven super-populations were shuffled 100 times to calculate the mean and 95% confidence interval (shading around lines). c) Intersection analysis of HGDP variant off-targets with CFD≥0.2 and increase in CFD of ≥0.1. Shared variants (orange) were found in 2 or more HGDP samples while private variants (green) were limited to a single sample. d) CRISPRme analysis of a single individual (HGDP01211) showing the top 100 variant off-targets from each of the following three categories: shared with 1000G variant off-targets (left panel), higher CFD score compared to reference genome and 1000G but shared with other HGDP individuals (center panel), and higher CFD score compared to reference genome and 1000G with variant not found in other HGDP individuals (right panel). For the center and right panels, reference refers to CFD score from reference genome or 1000G variants. e) The top predicted private off-target site from HGDP01211 is an allele-specific off-target where the rs1191022522-G minor allele produces a canonical NGG PAM sequence in place of a noncanonical NAG PAM sequence. Spacer shown as DNA sequence for ease of visual alignment.
To experimentally test the top predicted off-target from CRISPRme, we identified a CD34+ HSPC donor of African ancestry heterozygous for rs114518452-C, the variant predicted to introduce the greatest increase in off-target cleavage potential (Fig. 1c–f). We performed RNP electroporation using a gene editing protocol that preserves engrafting HSC function. Amplicon sequencing analysis showed 92.0 ± 0.5% indels at the on-target site and 4.8 ± 0.5% indels at the off-target site. For reads spanning the variant position, indels were strictly found at the alternative PAM-creation allele without indels observed at the reference allele (Fig. 3a–c), suggesting 9.6 ± 1.0% off-target editing of the alternative allele. In an additional 6 HSPC donors homozygous for the reference allele rs114518452-G/G, 0.00 ± 0.00% indels were observed at the off-target site, suggesting strict restriction of off-target editing to the alternative allele (Fig. 3d).
Figure 3. Allele-specific off-target editing by a BCL11A enhancer targeting gRNA in clinical trials associated with a common variant in African-ancestry populations.
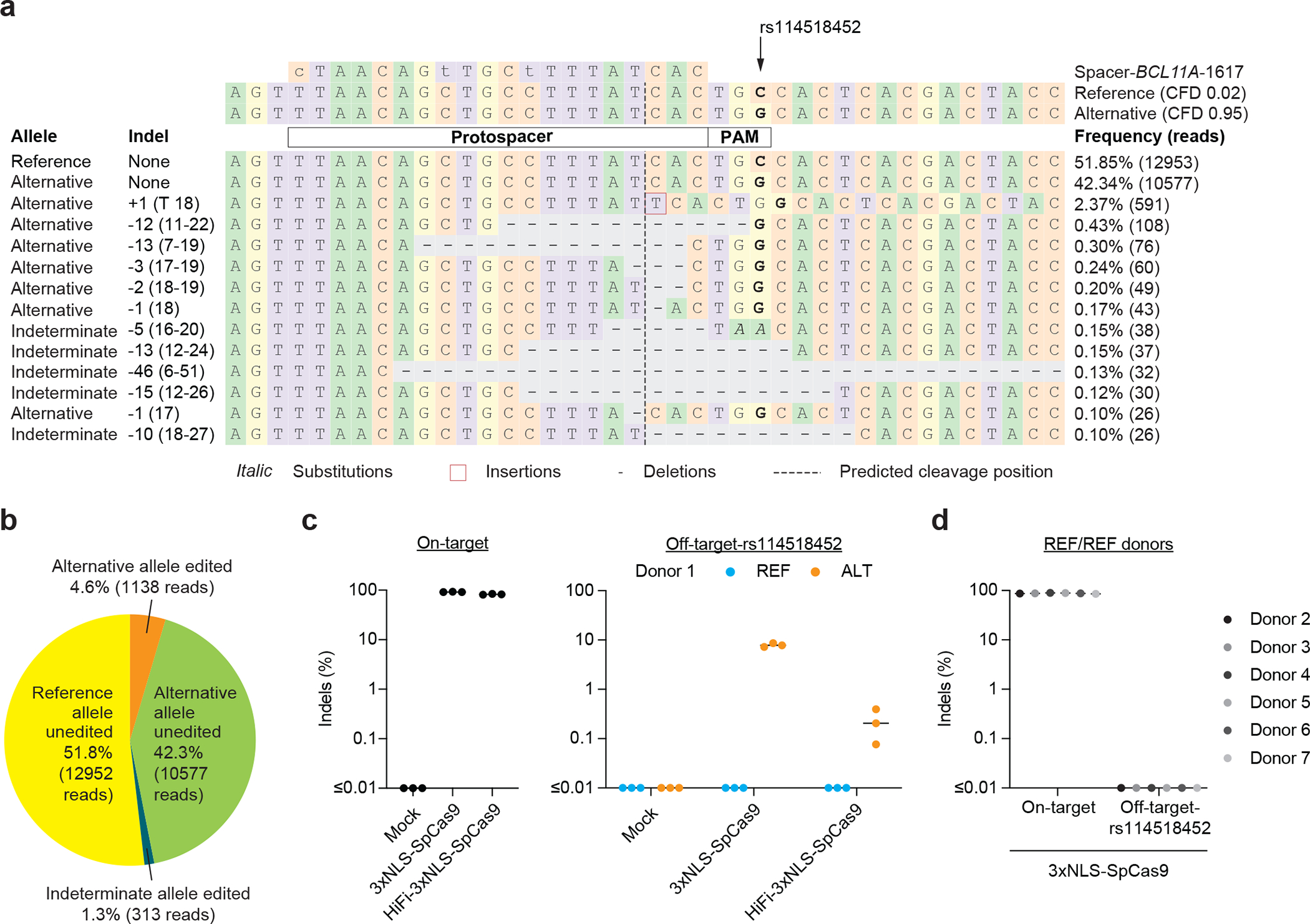
a) Human CD34+ HSPCs from a donor heterozygous for rs114518452-G/C (Donor 1, REF/ALT) were subject to 3xNLS-SpCas9:sg1617 RNP electroporation followed by amplicon sequencing of the off-target site around chr2:210,530,659–210,530,681 (off-target-rs114518452 in 1-start hg38 coordinates). CFD scores for the reference and alternative alleles are indicated and representative aligned reads are shown. Spacer shown as DNA sequence for ease of visual alignment, with mismatches indicated by lowercase and the rs114518452 position shown in bold. b) Reads classified based on allele (indeterminate if the rs114518452 position is deleted) and presence or absence of indels (edits). c) Human CD34+ HSPCs from a donor heterozygous for rs114518452-G/C (Donor 1) were subject to 3xNLS-SpCas9:sg1617 RNP electroporation, HiFi-3xNLS-SpCas9:sg1617 RNP electroporation, or no electroporation (mock) followed by amplicon sequencing of the on-target and off-target-rs114518452 sites. Each dot represents an independent biological replicate (n = 3), lines represent medians. Indel frequency was quantified for reads aligning to either the reference or alternative allele. d) Human CD34+ HSPCs from 6 donors homozygous for rs114518452-G/G (Donors 2–7, REF/REF) were subject to 3xNLS-SpCas9:sg1617 RNP electroporation with 1 biological replicate per donor followed by amplicon sequencing of the on-target and off-target-rs114518452 sites.
The on-target BCL11A intronic enhancer site is on chr2p while the off-target-rs114518452 site is on chr2q within an intron of a non-canonical transcript of CPS1. Inversion PCR demonstrated inversion junctions consistent with the presence of ~150 Mb pericentric inversions between BCL11A and the off-target site only in edited HSPCs carrying the alternative allele (Fig. 4a,b). Deep sequencing of the inversion junction showed that inversions were restricted to the alternative allele in the heterozygous cells (Fig. 4c,d). Droplet digital PCR revealed these inversions to be present at 0.16 ± 0.04% allele frequency (Fig. 4e).
Figure 4. Allele-specific pericentric inversion following BCL11A enhancer editing due to off-target cleavage.
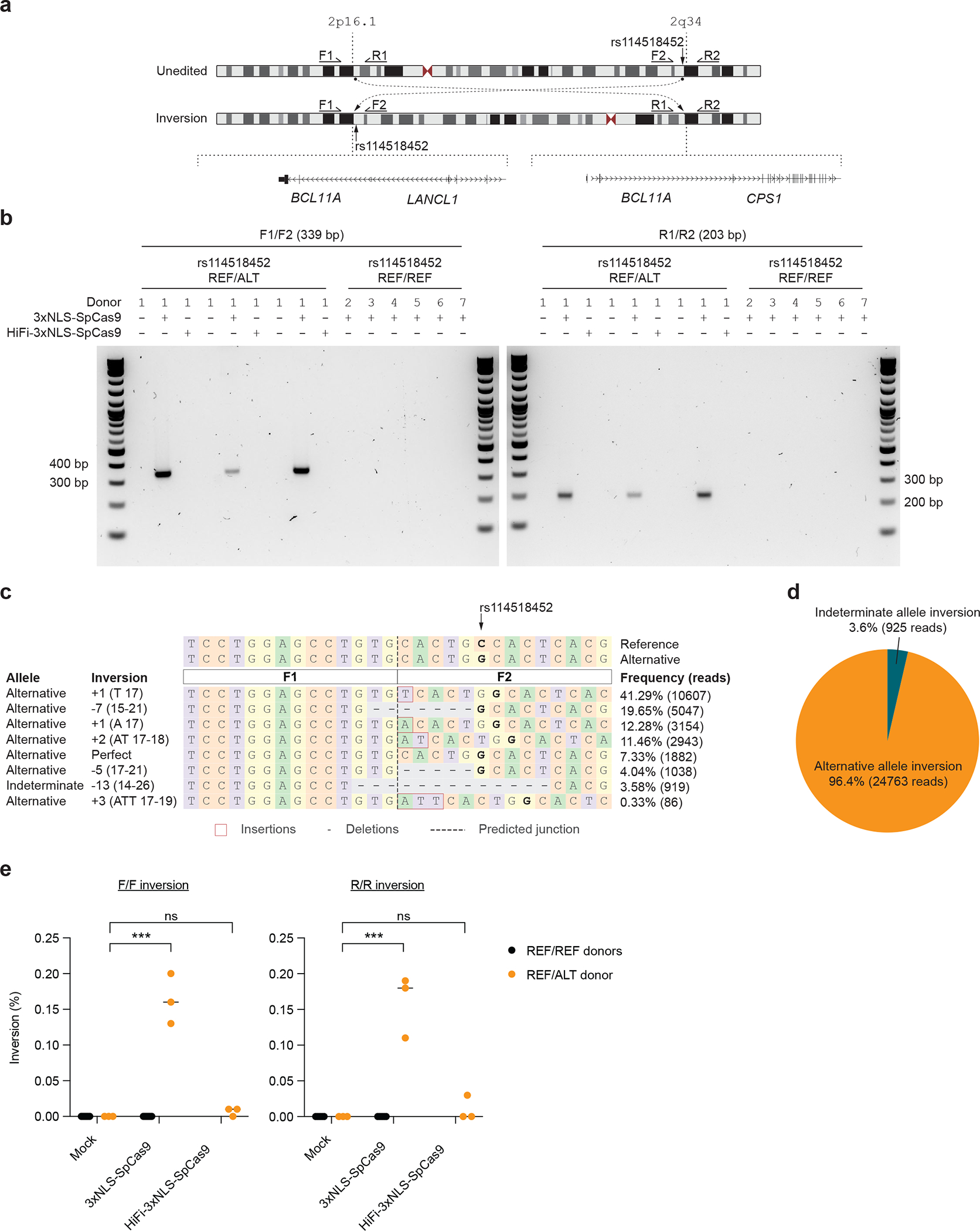
a) Concurrent cleavage of the on-target and off-target-rs114518452 sites could lead to pericentric inversion of chr2 as depicted. PCR primers F1, R1, F2, and R2 were designed to detect potential inversions. b) Human CD34+ HSPCs from a donor heterozygous for rs114518452-G/C (Donor 1) were subject to 3xNLS-SpCas9:sg1617 RNP electroporation, HiFi-3xNLS-SpCas9:sg1617 RNP electroporation, or no electroporation with 3 biological replicates. Human CD34+ HSPCs from 6 donors homozygous for rs114518452-G/G (Donors 2–7, REF/REF) were subject to 3xNLS-SpCas9:sg1617 RNP electroporation with 1 biological replicate per donor. Gel electrophoresis for inversion PCR was performed with F1/F2 and R1/R2 primer pairs on left and right respectively with expected sizes of precise inversion PCR products indicated. c) Reads from amplicon sequencing of the F1/F2 product (expected to include the rs114518452 position) from 3xNLS-SpCas9:sg1617 RNP treatment were aligned to reference and alternative inversion templates. The rs114518452 position is shown in bold. d) Reads classified based on allele (indeterminate if the rs114518452 position deleted). e) Inversion frequency by ddPCR from same samples as in (b) with three replicates from the single REF/ALT donor and one replicate each from the six REF/REF donors. F/F indicates forward and R/R reverse inversion junctions as depicted in (a).
Various high-fidelity Cas9 variants may improve the specificity of gene editing, although at the possible cost of reduced efficiency26. Gene editing following the same electroporation protocol using a HiFi variant 3xNLS-SpCas9 (R691A)27 in heterozygous cells revealed 82.3 ± 1.6% on-target indels with only 0.1 ± 0.1% indels at the rs114518452-C off-target site, i.e. a ~48-fold reduction compared to SpCas9 (Fig. 3c). Inversions were not detected following HiFi-3xNLS-SpCas9 editing (Fig. 4b,e).
Allele-specific off-target potential of additional gRNAs
To examine the pervasiveness of alternative allele off-target potential, we evaluated an additional 13 gRNAs in clinical development or otherwise widely used for SpCas9-based nuclease or base editing28–37 and 6 gRNAs for non-SpCas9-based editing such as for SaCas9 and Cas12a33,38–41 (Supplementary Table 4, Supplementary Data 2–3, Supplementary Note 8). CRISPRme analysis including the 1000G and HGDP genetic variant datasets showed 18% (95% confidence interval 13–23%) of the total nominated off-targets were due to alternative allele-specific off-targets. Most alternative allele-specific off-targets were associated with rare variants (MAF <1%), although candidate off-targets associated with common variants were identified for each gRNA (Fig. 5a). None of these alternative allele-specific off-target sites were described in the original manuscripts reporting the editing strategies and off-target analyses.
Figure 5. CRISPRme illustrates prevalent off-target potential due to genetic variation.
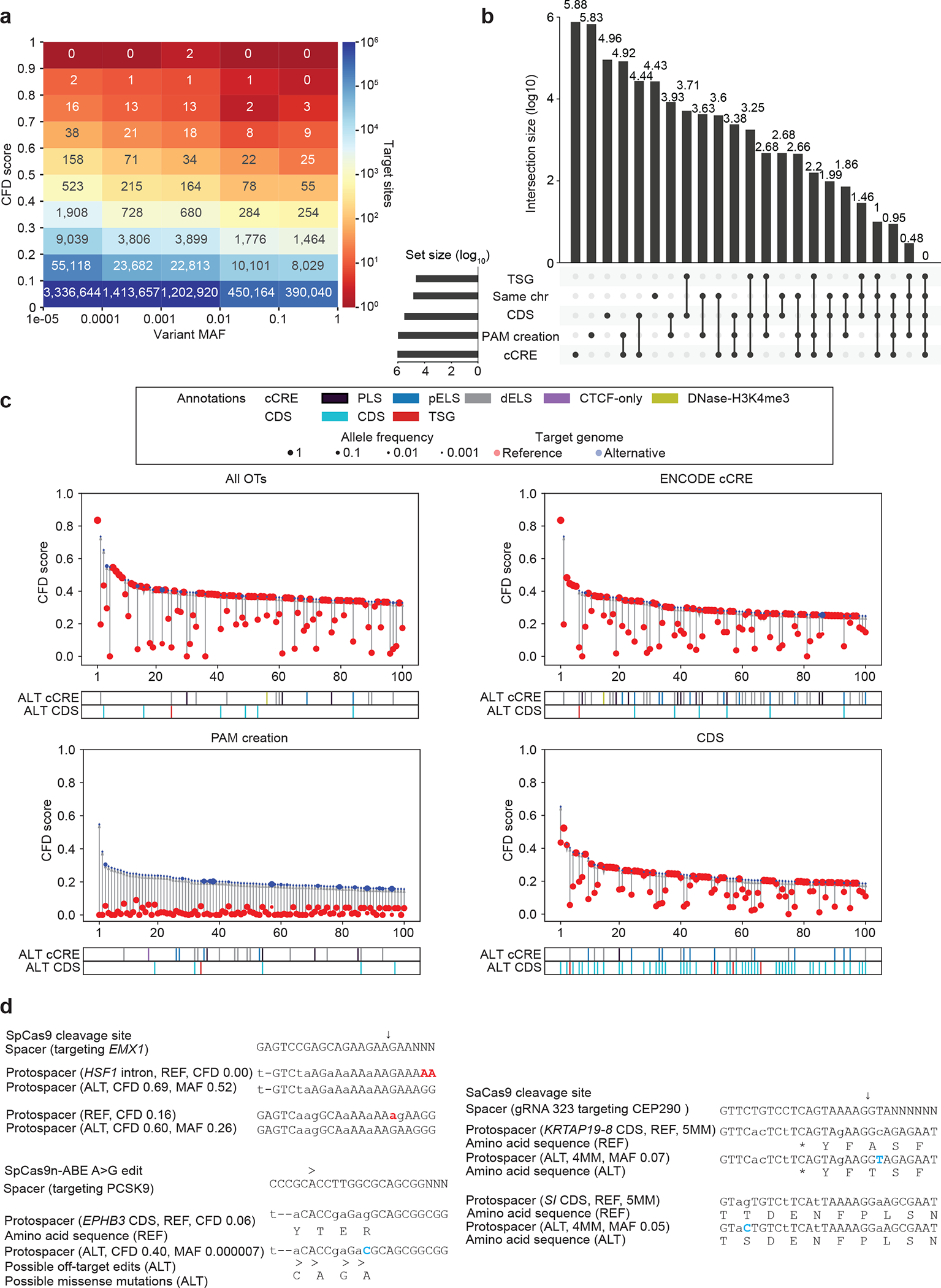
a) Heatmap showing the distribution of alternative allele nominated off-targets for SpCas9 guides by CFD score and MAF. b) UpSet plot showing overlapping annotation categories for candidate off-targets (TSG, tumor suppressor gene; candidate off-targets on the same chromosome as the on-target; CDS regions; cCRE from ENCODE and PAM creation events). c) Top 100 predicted off-target sites ranked by CFD score for the gRNA targeting PCSK9 with no filter, found in cCREs, corresponding to PAM creation events, and in CDS regions) d) Top left: Candidate off-target sites with increased predicted cleavage potential introduced by common (MAF 52% and 26%) indel variants for a SpCas9 gRNA targeting EMX1. Right: Candidate off-target cleavage sites within coding sequences with increased homology to a lead gRNA for SaCas9 targeting of CEP290 to treat congenital blindness in current clinical trials due to common SNPs. Bottom: Potential missense mutations in the EPHB3 tumor suppressor resulting from candidate off-target A-to-G base editing by a preclinical lead gRNA targeting PCSK9 to reduce LDL cholesterol levels. Deletions shown in red, SNPs shown in blue.
CRISPRme produces visualizations to specifically highlight alternative allele-specific candidate off-target sites overlapping candidate cis-regulatory elements and protein coding sequences (including putative tumor suppressor genes42) and/or which involve PAM creation events (Fig. 5b–c, Supplementary Fig. 1). For example, within the top 20 candidate off-targets nominated by CRISPRme for a SpCas9 gRNA targeting EMX135, two sites involve genetic variants with high MAF (52% and 26%) and are associated with substantial increases in CFD score from REF to ALT (+0.69 and +0.44). The first is an intronic PAM creation variant, while the second introduces two PAM-proximal matches to the gRNA (Fig. 5d). Notably, both of these candidate off-targets involve indel variants, underscoring the utility of CRISPRme to account for variants beyond SNPs.
In addition to visualizing candidate off-target sites by predictive score rank (such as CFD or CRISTA) for SpCas9 derived editors, CRISPRme can also visualize candidate off-targets by number of mismatches and bulges, which may be especially useful for Cas proteins with distinct PAMs for which predictive scores are not readily available. For example, SaCas9 is a clinically relevant nuclease whose small size favors packaging to AAV. For a SaCas9-associated gRNA targeting CEP29040 currently being evaluated in clinical trials to treat a form of congenital blindness (NCT03872479), CRISPRme nominated two candidate off-targets associated with common SNPs (MAF 7% and 5%) that reduced mismatches from 5 (REF) to 4 (ALT) which are predicted to produce cleavages within coding sequences (Fig. 5d).
CRISPRme can nominate variant off-targets for base editors and evaluate their base editing susceptibility within a user-defined editing window. For a gRNA targeting PCSK937 that has been used with SpCas9-nickase adenine base editor in vivo in preclinical studies to reduce LDL cholesterol levels, 4 of the top 5 candidate off-target sites involve alternative alleles, including one with CFDref 0.2 and CFDalt 0.75 found in an ENCODE candidate enhancer element. CRISPRme nominated a candidate off-target associated with a rare variant (MAF 0.0007%) that increased the CFD score from 0.06 (REF) to 0.40 (ALT) which would be predicted to produce missense mutations in EPHB3, a putative tumor suppressor gene (Fig. 5d).
The underlying computational challenge that CRISPRme addresses extends beyond CRISPR-based applications to other technologies based on nucleic acid sequence recognition. For example, CRISPRme can nominate off-targets for RNA-targeting strategies, whether RNA-guided gene editors or even oligonucleotide sequences used as RNA interference (RNAi) or antisense oligo (ASO) therapies (Extended Data Fig. 4). We performed a variant-aware search (without PAM restriction) for the FDA-approved antisense oligonucleotide Nusinersen43,44, which targets SMN2 pre-mRNA to treat spinal muscular atrophy. Using CRISPRme, we identified a potential off-target site within a coding region wherein a common SNP (MAF 2%) reduces the number of mismatches from 3 (REF) to 2 (ALT). Similarly, analysis of the FDA-approved RNAi therapy Inclisiran45, which targets PCSK9 mRNA to treat hypercholesterolemia, revealed that its antisense strand has a candidate off-target in the 3’ UTR of the ribosomal gene RPP14 for which a common insertion variant (MAF 36%) reduces the number of mismatches and bulges from 7 (REF) to 4 (ALT).
DISCUSSION
These results demonstrate how personal genetic variation may influence the off-target potential of sequence-based therapies like genome editing. Increased availability of haplotype-resolved genomes of diverse ancestry would enhance ability to nominate variant-associated off-target sites present in human populations. A limitation of current tools including CRISPRme is that potential off-targets cannot be enumerated based on structural variants or other complex genetic events such as combinations of indels and SNPs. Future extensions of CRISPRme based on new data structures such as graph genomes46,47 could enable these complex searches and improve their efficiency.
The practical implications of allele-specific off-target editing need to be considered on a case-by-case basis (also see Supplementary Note 7). In the case of BCL11A enhancer editing, up to ~10% of SCD patients with African ancestry would be expected to carry at least one rs114518452-C allele, leading to ~10% cleavage at an off-target site that was not identified in prior studies of this gRNA using currently available tools (Supplementary Table 2). Our results highlight that allele-specific off-target editing potential is not equally distributed across all ancestral groups, but especially concentrated in those of African ancestry where genomic variation is most pronounced. Therefore, gene editing efforts that include subjects of African ancestry (like those targeting sickle cell disease) might pay particular attention to this issue. Gene editing efforts that focus on a specific patient population should consider genetic variants enriched in that population during off-target evaluation. However, our analysis also shows that variant off-targets may be private to a given individual, so all humans could potentially be susceptible to such an effect.
Implementing off-target analysis and testing into therapeutic genome editing protocols in practice is an important issue that is broader in scope than our report. Fundamentally, variant-aware off-target analysis may identify off-target potential that would be overlooked by conventional analysis. Of note, as is true for off-target genetic changes in general, the mere possibility of somatic genetic alteration does not imply functional consequence. Although in principle ex vivo edited patient cells could be tested by sequencing prior to infusion, the functional importance of off-target edits may range from likely functional to likely neutral, so the mere presence of off-target editing in a cell product may not necessarily preclude its clinical use, and this testing could deplete precious material and delay therapy. We recommend several steps to minimize risk of unintended allele-specific off-target effects during therapeutic genome editing, consistent with regulatory guidance to consider effects of genetic variation48. First, prioritize use of genome editing methods that maximize specificity, such as high-fidelity editors and pulse delivery. Second, nominate off-targets in a variant-aware manner, with particular attention toward genetic variants found in relevant patient populations, using a tool like CRISPRme. Third, employ off-target detection assays that are variant-aware to empirically evaluate the likelihood of off-target editing, although these may imperfectly reflect editing in a therapeutic context (see Supplementary Note 7). When possible, allele-specific off-target editing potential should be validated in primary cells of relevant genotype by sequencing. However, it may be difficult to obtain such primary cells to perform biological validation in a relevant therapeutic context. Fourth, perform a risk assessment of variant off-target editing given predicted genomic annotations, mechanisms of DNA repair, delivery to target cells and disease context. For example, off-target edits within tumor suppressor loci might carry greater risk than those targeting unannotated noncoding sequences. Fifth, if excess allele-specific genome editing risks are identified, consider including genotype among the subject inclusion/exclusion criteria. Finally, for therapeutic genome editing indications in which it is feasible (such as hematopoietic cell targeting), prospectively monitor somatic modifications in patient samples to gather information about the frequency and consequence of such events to help assess patient-specific risk and provide valuable information for the broader field as to the frequency and in vivo dynamics of off-target edits if present.
CRISPRme offers a simple-to-use tool to comprehensively evaluate off-target potential across diverse populations and within individuals. CRISPRme is available at http://crisprme.di.univr.it and may also be deployed locally to preserve privacy (Supplementary Note 9).
METHODS
This research complies with all relevant ethical regulations. Studies were reviewed by the Boston Children’s Hospital Institutional Review Board (P00004546). CD34+ HSPCs were deidentified and the study was determined by IRB not to constitute human subjects research.
Statistics & Reproducibility
The correlation values and p-values(two-sided) were calculated using standard functions from the Python scipy library (v1.7.3). Sample size for the public data analyzed was chosen by the original studies. All the code and data to reproduce the analyses presented in this manuscript were deposited in public repositories (see data and code availability sections).
Cell culture
Fresh G-CSF mobilized peripheral blood cells from healthy donor 1 were obtained from Miltenyi Biotec (Auburn, CA). CD34+ HSPCs were isolated using CliniMACS® CD34 reagent (Miltenyi, 130-017-501). Cryopreserved human CD34+ HSPCs from mobilized peripheral blood of deidentified healthy donors 2–7 were obtained from the Fred Hutchinson Cancer Research Center (Seattle, Washington). CD34+ HSPCs were cultured into Stem Cell Growth Medium (SCGM) (CellGenix, 20806–0500) supplemented with 100 ng ml −1 human Stem Cell Growth Factor (SCF) (CellGenix, 1418–050), 100 ng ml −1 human thrombopoietin (TPO) (CellGenix, 1417–050) and 100 ng ml −1 recombinant human FMS-like Tyrosine Kinase 3 Ligand (Flt3-L) (CellGenix cat# 1415–050). HSPCs were electroporated with 3xNLS-SpCas9:sg1617 RNP or HiFi-3xNLS-SpCas9:sg1617 RNP 24 h after thawing. Twenty-four hours after electroporation, HSPCs were transferred into erythroid differentiation medium (EDM) consisting of IMDM (LIFE, 12440061) supplemented with 330 μg ml −1 holo-human transferrin (Sigma, T0665-1G), 10 μg ml −1 recombinant human insulin (Sigma, 19278-5ML), 2 IU ml−1 heparin (Sigma, H3149), 5% human solvent detergent pooled plasma AB (Rhode Island Blood Center), and 3 IU ml −1 erythropoietin (Pharmacy). Five days after electroporation, cells were harvested for gDNA extraction.
Protein purification
3xNLS-SpCas9 plasmid was constructed in the pET21a expression plasmid (Addgene, 114365). The recombinant Streptococcus pyogenes Cas9 with a 6xHis tag and c-Myc-like NLS at the N terminus, SV40, and nucleoplasmin NLS at the C terminus was expressed in Escherichia coli Rosetta (DE3) pLysS cells (Sigma-Aldrich, 70956). Cells were grown at 37 °C to an OD600 of ~0.2, then shifted to 18 °C and induced at an OD600 of ~0.4 for 16 h with IPTG (1 mM final concentration). Following induction, cells were resuspended with Nickel-NTA buffer (20 mM Tris, 500 mM NaCl, 20 mM imidazole, 1 mM TCEP, pH 8.0) supplemented with HALT protease inhibitor and lysed with M-110s Microfluidizer (Microfluidics) following the manufacturer’s instructions. The protein was purified with Ni-NTA resin and eluted with elution buffer (20 mM Tris, 250 mM NaCl, 250 mM imidazole, 10% glycerol, pH 8.0). Subsequently, 3xNLS-SpCas9 protein was further purified by cation exchange chromatography (column, 5 ml HiTrap-S; buffer A, 20 mM HEPES pH 7.5, 1 mM TCEP; buffer B, 20 mM HEPES pH 7.5, 1 M NaCl, 1 mM TCEP; flow rate, 5 ml min−1; column volume, 5 ml) and size-exclusion chromatography on Hiload 16/600 Superdex 200 pg column (isocratic size-exclusion running buffer: 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP), then reconstituted in a formulation of 20 mM HEPES and 150 mM NaCl, pH 7.4. HiFi-3xNLS-SpCas9 plasmids were transformed into BL21 (DE3) competent cells (MilliporeSigma, 702353) and grown in Terrific Broth (TB) media at 37°C until OD600 2.4–2.8. Cells were induced with 0.5 mM isopropyl ß-d-1-thiogalactopyranoside (IPTG) per liter for 20 hours at 20°C. Pellets were lysed in 25 mM Tris, pH 7.6, 500 mM NaCl, 5% glycerol, passed through homogenizer twice and centrifuged at 20,000 × g for 1 hour at 4°C. Proteins were purified by Nickel-NTA resin and treated with TEV protease (1 mg lab made TEV per 40 mg of protein) and benzonase (100 units ml −1, Novagen 70664-3) overnight at 4°C. Subsequently, the proteins were purified by size exclusion column (Amersham Biosciences HiLoad 26/60 Superdex 200 17-1071-01) and ion exchange with a 5 ml SP HP column (GE 17-1151-01) according to the manufacturer’s instructions. Proteins were dialyzed in 20 mM Hepes buffer pH 7.5 containing 400 mM KCl, 10% glycerol, and 1 mM TCEP buffer, and contaminants were removed by Toxin Sensor Chromogenic LAL Endotoxin Assay Kit (GenScript, L00350). Purified proteins were concentrated and filtered using Amicon ultra filter units – 30k NMWL (MilliporeSigma, UFC903008) and ultrafree-MC centrifugal filter (MilliporeSigma, UFC30GV0S). Protein fractions were further assessed on TGX stain free 4–20% SDS-PAGE (Biorad, 5678093) and quantified by BCA assay.
RNP electroporation
Electroporation was performed using Lonza 4D Nucleofector (V4XP-3032 for 20 μl as the manufacturer’s instructions). CD34+ HSPCs were thawed 24 h before electroporation. For 20 μl Nucleocuvette Strips, the RNP complex was prepared by mixing 3xNLS-SpCas9 protein7 (100 pmol) or HiFi-3xNLS-SpCas9 protein (100 pmol) and sgRNA-1617: CTAACAGTTGCTTTTATCAC (300 pmol, IDT) with glycerol (2% of final concentration, Sigma, G2025) and P3 solution up to 10 μl and incubating for 15 min at room temperature immediately before electroporation. 50K HSPCs resuspended in 10 μl P3 solution were mixed with RNP and transferred to a cuvette for electroporation with program EO-100. The P3 solution was removed after 15 min of room temperature rest. The electroporated cells were resuspended with SCGM medium with cytokines and changed into EDM 24 h after electroporation.
Measurement of +58 BCL11A enhancer on-target and OT40 off-target indel and inversion
Editing frequencies were measured with cells cultured in EDM 5 days after electroporation. Briefly, genomic DNA was extracted using the Qiagen DNeasy Blood and Tissue kit (Qiagen, 69506). The BCL11A enhancer DHS +58 on-target site was amplified using forward primer AGAGAGCCTTCCGAAAGAGG (F1) and reverse primer GCCAGAAAAGAGATATGGCATC (R1). The off-target-rs114518452 site was amplified using forward primer TAAGATTCTTTTGGTTCTGGCT (F2) and reverse primer AGAGAGGCAGTATTTACGATGC (R2). The inversion junction was amplified using +58 forward primer (F1) and off-target-rs114518452 forward primer (F2), or +58 reverse primer (R1) and off-target-rs114518452 reverse primer (R2). KOD Hot Start DNA Polymerase (EMD-Millipore, 71086-31) was used for PCR and followed cycling conditions: 95 degrees for 3 min; 30 cycles of 95 degrees for 20 s, 60 degrees for 10 s, and 70 degrees for 10 s; 70 degrees for 5 min. 1 μl of locus specific PCR product was used for indexing PCR with KOD Hot Start DNA Polymerase and index primers following cycling conditions: 95 degrees for 3 min; 10 cycles of 95 degrees for 20 s, 60 degrees for 10 s, and 70 degrees for 10 s; 70 degrees for 5 min. Resulting PCR products were evaluated by TapeStation using High Sensitivity D1000 Reagents (Agilent, # 5067–5585) and High Sensitivity D1000 ScreenTape (Agilent, 5067–5584), KAPA Universal qPCR Master Mix (KAPA Biosystems, # KK4824 / Roche 07960140001) and Qubit® dsDNA HS Assay Kit (Thermo Fisher, # Q32854), pooled as equimolar products and subjected to deep sequencing using MiniSeq (Illumina).
Amplicon deep sequencing and analysis
Amplicons were sequenced using paired-end 150 bp reads on an Illumina MiniSeq system with >18,000X coverage per sample for the off-target-rs114518452 site and >3,800X coverage per sample for the on-target site. Reads were trimmed for adapters and quality using Trimmomatic v0.36 in paired-end mode for the off-target-rs114518452 site and in single-end mode for the on-target site due to a nearby difficult-to-sequence homopolymer region. Editing outcomes were analyzed using CRISPResso v2.1.0 by aligning to the expected reference and/or alternative allele amplicons. A Needleman-Wunsch gap opening penalty of −30 (CRISPResso2 default: −20) was used to ensure more accurate alignment of reads to the reference vs. alternative allele amplicons for off-target-rs114518452 since they only differ by a single nucleotide. Only indels overlapping the expected SpCas9 cleavage site (3 bp upstream of the PAM) were counted as gene edits. The median observed indel frequency is reported for samples for which technical replicates were performed (n = 4), which includes all amplicon sequencing at the off-target-rs114518452 site for the donor heterozygous for rs114518452. Representative reads collapsed by allele identity and indel type are presented in the plots.
Inversion PCR
Nested PCR was performed to amplify the inversion junction. First step PCR was amplified using the outer primers on-target +58 forward, CACACGGCATGGCATACAAA, and off-target-rs114518452 forward, AATAGCCAAACTACTGAGCATTGTG; or the outer primers on-target +58 reverse, CACCCTGGAAAACAGCCTGA, and off-target-rs114518452 reverse, ACTAAGGCAATTGTTGTCCAAGC. KOD Hot Start DNA Polymerase was used for PCR and followed cycling conditions: 95 degrees for 3 min; 30 cycles of 95 degrees for 20 s, 60 degrees for 10 s, and 70 degrees for 10 s; 70 degrees for 5 min. 1 μl of PCR1 product was used for the second step PCR amplifying with inner primers on-target +58 forward (F1) and off-target-rs114518452 forward (F2), or on-target +58 reverse (R1) and off-target-rs114518452 reverse (R2) with cycling conditions: 95 degrees for 3 min; 10 cycles of 95 degrees for 20 s, 60 degrees for 10 s, and 70 degrees for 10 s; 70 degrees for 5 min. Resulting PCR products were loaded on a 2% agarose (VWR, 97062–250) gel. Images were captured by the BioRad ChemiDocTM MP Imaging System.
Droplet digital PCR
100 ng of gDNA was used for ddPCR with the ddPCR supermix (no dUTP, Bio-Rad, 1863024). See primer and probe sequences in Supplementary Table 5. The premixed samples were placed into the Automated Droplet Generator (Bio-Rad, 1864101) that utilized Automated Droplet Generation Oil for Probes (Bio-Rad, 1864110) for droplet generation prior to PCR. The cycling conditions were: 1 cycle of 95°C for 10 min, 50 cycles of 94°C for 1 min sec (2°C/s ramp) and 56°C for 1 min (2°C/s ramp), 1 cycle of 98°C for 10 min, hold at 4°C. After thermal cycling, plate was placed in the QX200 Droplet Reader and plate layout set-up using QuantaSoft Software (Bio-Rad, 10031906).
Extended Data
Extended Data Figure 1. Top 100 predicted off-target sites for BCL11A-1617 spacer by CFD score.
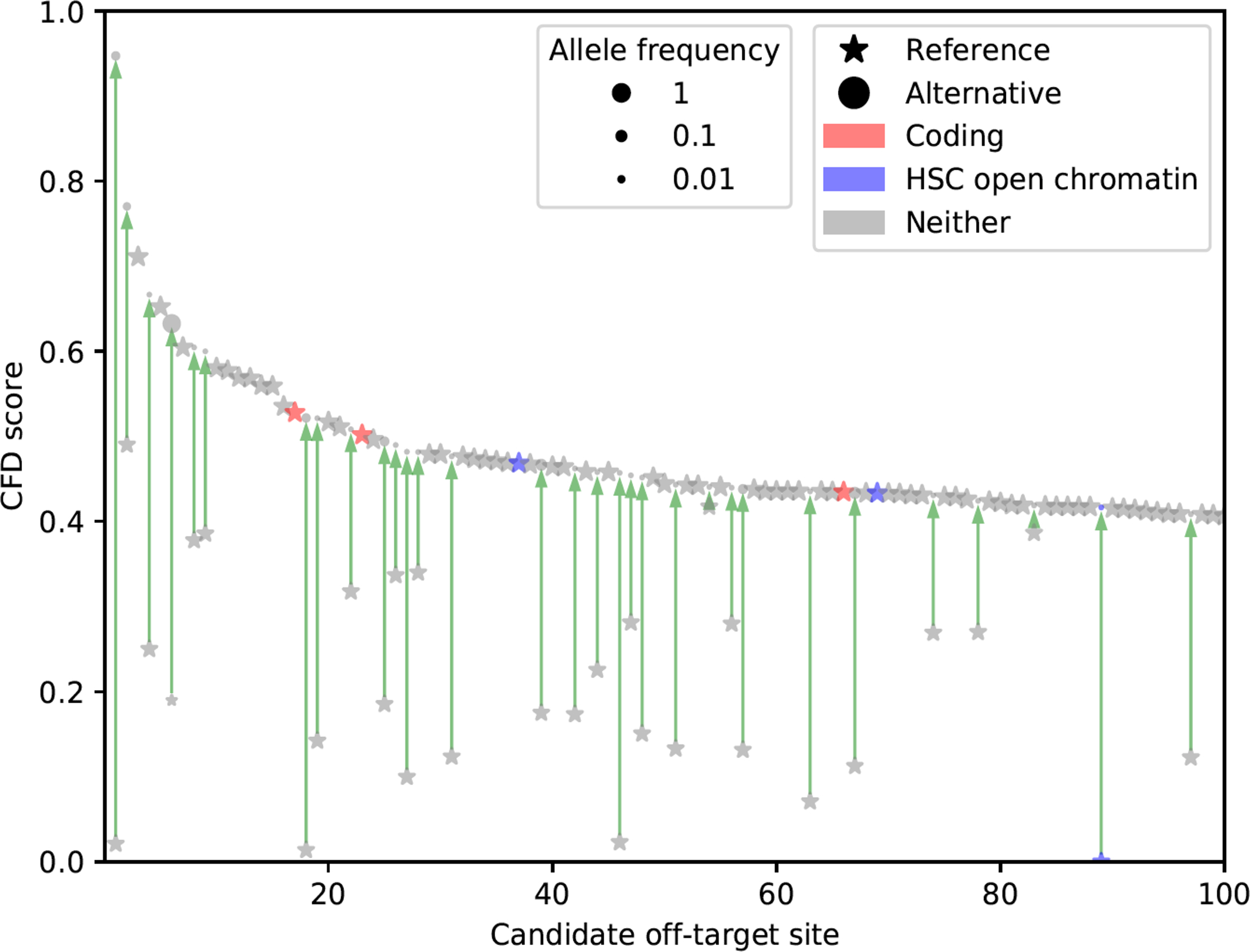
CRISPRme search as in Fig. 1. Candidate off-target sites within coding regions based on GENCODE annotations and ATAC-seq peaks in HSCs based on user-provided annotations (data from Corces et al. 2016) are highlighted.
Extended Data Figure 2. Plots with rank ordered correlation between CFD and CRISTA reported targets.
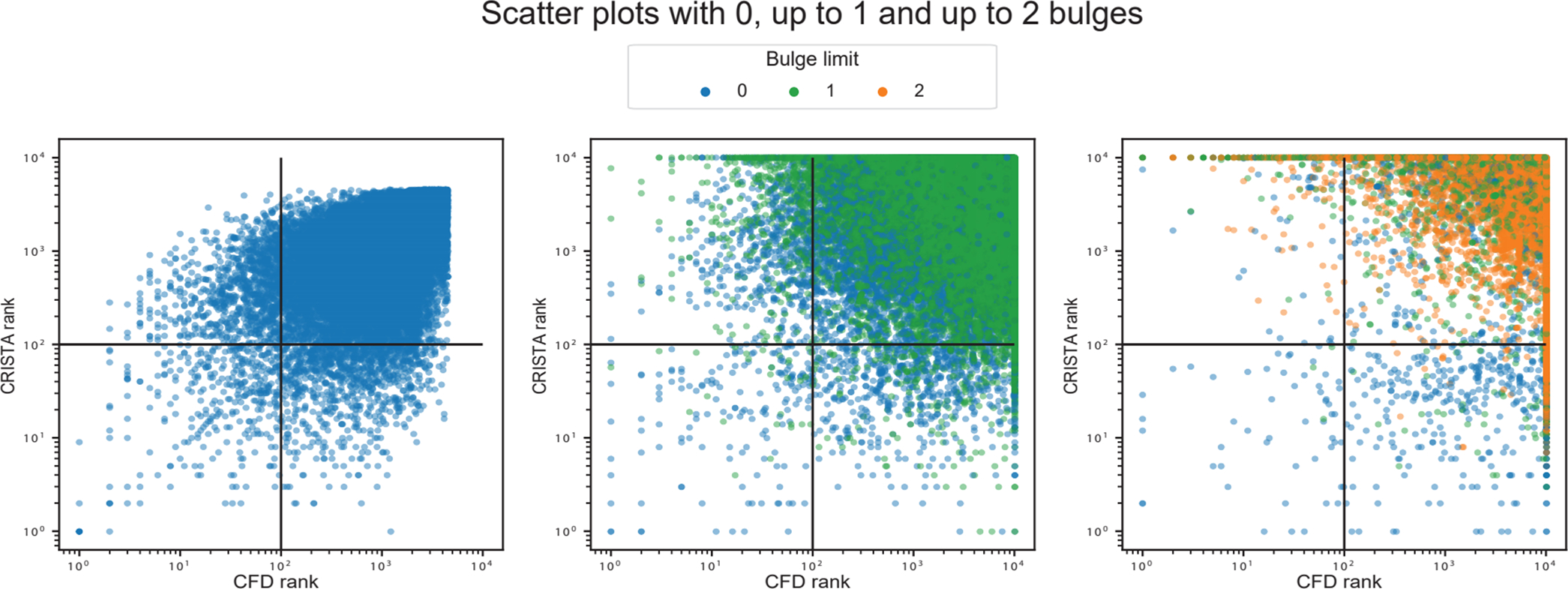
Scatter plots show from left to right, the correlation of ranked targets, extracted by selecting top 10000 targets ordered by CFD and CRISTA score, respectively. The left plot shows the rank correlation of targets with 0 bulges (Pearson’s correlation: 0.57, p < 1e-10, Spearman’s correlation: 0.55, p < 1e-10), the center plot shows rank correlation of targets with 1 bulge (Pearson’s correlation: −0.16, p < 1 e-10, Spearman’s correlation: −0.33, p < 1e-10) and the right plot shows the rank correlation of targets with 2 bulges (Pearson’s correlation: −0.55, p < 1e-10, Spearman’s correlation: −0.80, p < 1e-10). The correlation values and p-values(two-sided) were calculated using standard functions from the Python scipy library. The colors represent the lowest count of bulges for each target, since the two scoring methods may prioritize different alignments and thus different number of mismatches and bulges of the same genomic target.
Extended Data Figure 3. HGDP super-population distribution plots.
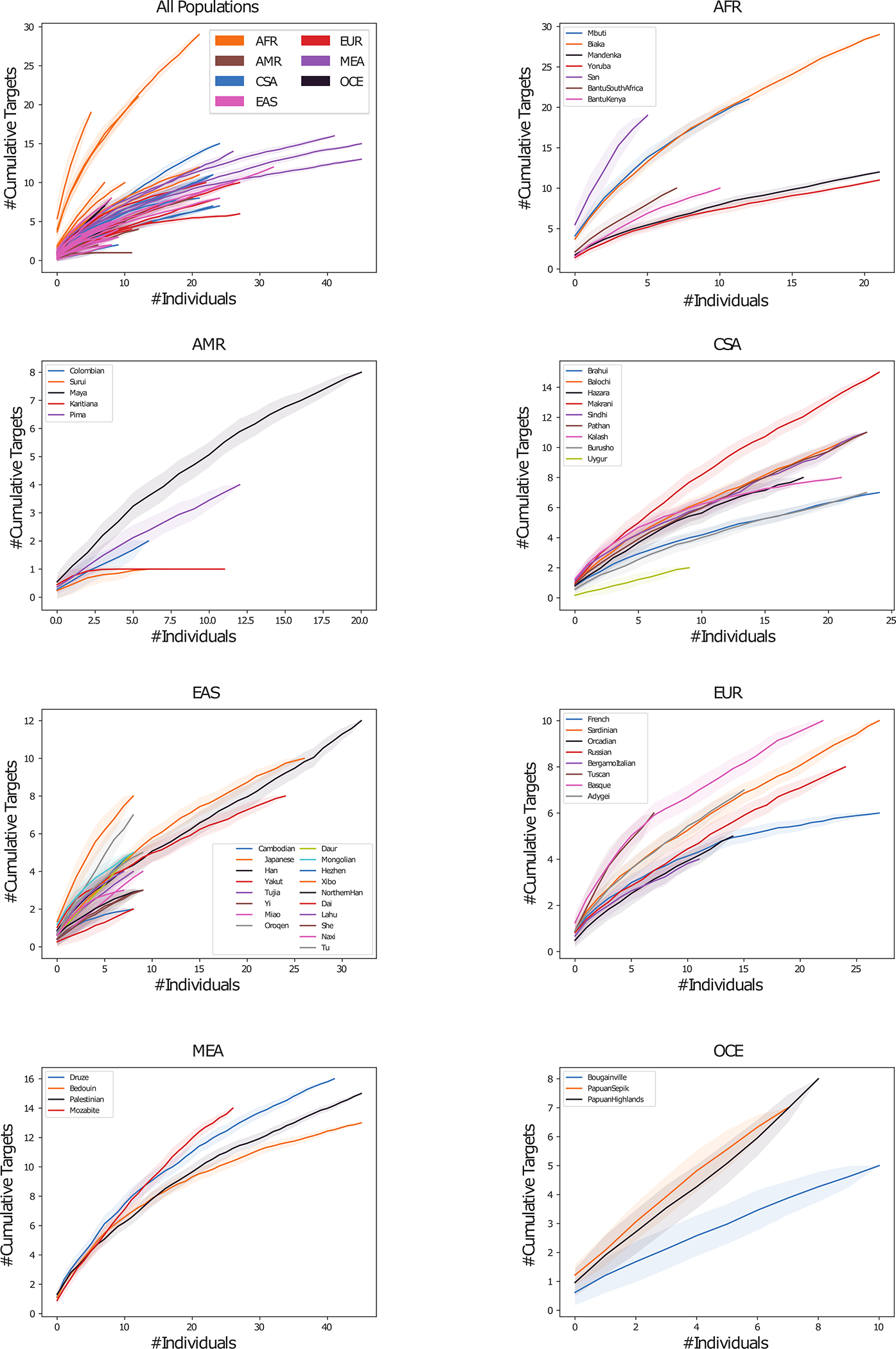
Cumulative distribution plot of HGDP variant off-targets with CFD≥0.2 and increase in CFD of ≥0.1 per super-population. Individual samples from each of the seven super-populations were shuffled 100 times to calculate the mean and 95% confidence interval (shading around lines). First panel shows distribution within all 54 discrete populations, colored by super-population. Additional seven panels show distribution of discrete populations within each listed super-population.
Extended Data Figure 4. Candidate transcript off-targets introduced by common genetic variants for non-CRISPR sequence-based RNA-targeting therapeutic strategies.
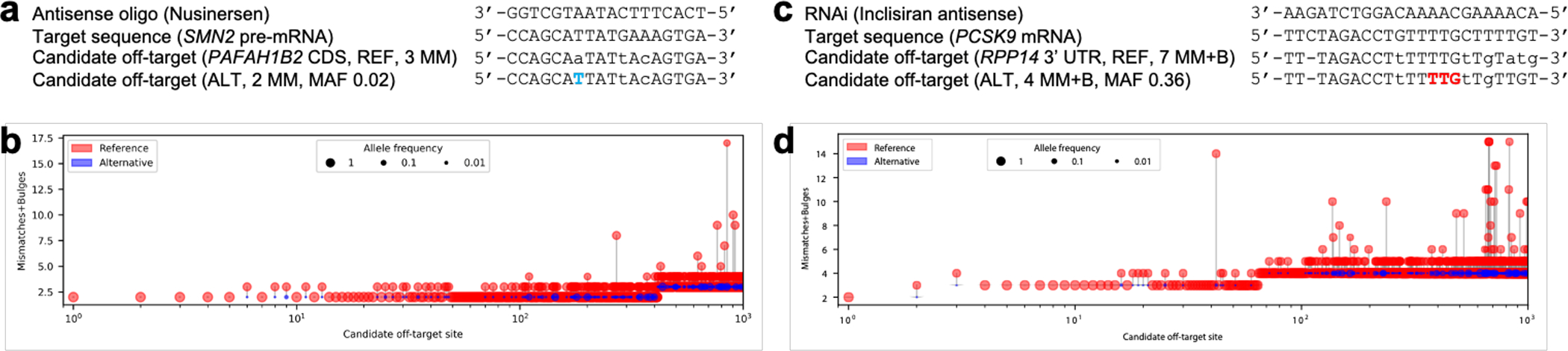
a) A common SNP (in blue) introduces a candidate CDS off-target site with 2 mismatches for the FDA-approved antisense oligo Nusinersen. b) Top 1000 candidate transcript off-targets ranked by mismatches and bulges for Nusinersen from a search performed with the 1000G and HGDP genetic variant datasets. c) A common insertion variant (in red) introduces a candidate 3’UTR off-target site with 4 mismatches + bulges for the FDA-approved RNAi therapy Inclisiran. d) Top 1000 candidate transcript off-targets ranked by mismatches and bulges for Inclisiran from a search performed with the 1000G and HGDP genetic variant datasets.
Supplementary Material
Acknowledgements
L.P. received support from the U.S. NIH (R35 HG010717 and RM1 HG009490). D.E.B. was supported by the National Heart, Lung, and Blood Institute (OT2HL154984, P01HL053749), Burroughs Wellcome Fund, Doris Duke Charitable Foundation and the St. Jude Children’s Research Hospital Collaborative Research Consortium. R.G received support from European Union’s ERA-NET JPCOFUND2 (JPND2019-466-037). We thank Stuart H. Orkin, Guillaume Lettre, J. Keith Joung, Vikram Pattanayak, Karl Petri, Anne H. Shen, Elia Dirupo and Francesco Masillo for helpful input.
Footnotes
Competing financial interests statement
L.P. has financial interests in Edilytics, Inc., Excelsior Genomics, and SeQure Dx, Inc. L.P.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. The remaining authors declare no competing interests.
Code availability
CRISPRme source code is available at https://github.com/pinellolab/crisprme and https://github.com/InfOmics/CRISPRme. The webapp is available online at http://crisprme.di.univr.it. The versions of CRISPRme (1.8.8 & v1.7.7) used to generate the results presented in this manuscript have been deposited on Zenodo: https://doi.org/10.5281/zenodo.5047489. CRISPRitz v2.6.5 used to generate the data presented in this manuscript have been deposited on Zenodo: https://doi.org/10.5281/zenodo.7078220.
The scripts to generate the plots presented in the manuscript have been deposited on Zenodo: https://doi.org/10.5281/zenodo.7193131.
Data availability
Sequencing data is deposited in the NCBI Sequence Read Archive database under accession number PRJNA733110. The data for the 1000 Genomes project was downloaded from DOI: 10.12688/wellcomeopenres.15126.2. The data for the Human Genome Diversity Project was downloaded from DOI: 10.1126/science.aay5012. The full CRISPRme results for NGG PAM gRNAs (including sg1617) are available at https://doi.org/10.5281/zenodo.7195706.
REFERENCES
- 1.Frangoul H et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 384, 252–260 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Clement K, Hsu JY, Canver MC, Joung JK & Pinello L Technologies and Computational Analysis Strategies for CRISPR Applications. Mol. Cell 79, 11–29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bao XR, Pan Y, Lee CM, Davis TH & Bao G Tools for experimental and computational analyses of off-target editing by programmable nucleases. Nat. Protoc. 16, 10–26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu PD et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaudhari HG et al. Evaluation of Homology-Independent CRISPR-Cas9 Off-Target Assessment Methods. CRISPR J 3, 440–453 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lessard S et al. Human genetic variation alters CRISPR-Cas9 on- and off-targeting specificity at therapeutically implicated loci. Proc. Natl. Acad. Sci. U. S. A. 114, E11257–E11266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott DA & Zhang F Implications of human genetic variation in CRISPR-based therapeutic genome editing. Nat. Med. 23, 1095–1101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Concordet J-P & Haeussler M CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Listgarten J et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat Biomed Eng 2, 38–47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Labun K et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park J, Bae S & Kim J-S Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics 31, 4014–4016 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Cancellieri S, Canver MC, Bombieri N, Giugno R & Pinello L CRISPRitz: rapid, high-throughput and variant-aware in silico off-target site identification for CRISPR genome editing. Bioinformatics 36, 2001–2008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowy-Gallego E et al. Variant calling on the GRCh38 assembly with the data from phase three of the 1000 Genomes Project. Wellcome Open Res 4, 50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergström A et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 367, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canver MC et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nature Medicine vol. 25 776–783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walton RT, Christie KA, Whittaker MN & Kleinstiver BP Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fennell T et al. CALITAS: A CRISPR-Cas-aware ALigner for In silico off-TArget Search. CRISPR j. 4, 264–274 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Frankish A et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.ENCODE Project Consortium et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abadi S, Yan WX, Amar D & Mayrose I A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 13, e1005807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demirci S et al. Durable and robust fetal globin induction without Anemia in rhesus monkeys following autologous hematopoietic stem cell transplant with BCL11A Erythroid enhancer editing. (2019).
- 26.Schmid-Burgk JL et al. Highly parallel profiling of Cas9 variant specificity. Mol. Cell 78, 794–800.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vakulskas CA et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 24, 1216–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu L et al. CRISPR/Cas9-mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV-1 resistance in vivo. Mol. Ther. 25, 1782–1789 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L et al. CRISPR-edited stem cells in a patient with HIV and acute Lymphocytic leukemia. N. Engl. J. Med. 381, 1240–1247 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Stadtmauer EA et al. CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillmore JD et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502 (2021). [DOI] [PubMed] [Google Scholar]
- 32.DeWitt MA et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 8, 360ra134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu S et al. Editing aberrant splice sites efficiently restores β-globin expression in β-thalassemia. Blood 133, 2255–2262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Métais J-Y et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 3, 3379–3392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng J et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 26, 535–541 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Musunuru K et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 593, 429–434 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Chu SH et al. Rationally designed base editors for precise editing of the sickle cell disease mutation. CRISPR j. 4, 169–177 (2021). [DOI] [PubMed] [Google Scholar]
- 39.Newby GA et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 595, 295–302 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeder ML et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 25, 229–233 (2019). [DOI] [PubMed] [Google Scholar]
- 41.De Dreuzy E et al. EDIT-301: An experimental autologous cell therapy comprising Cas12a-RNP modified mPB-CD34+ cells for the potential treatment of SCD. Blood 134, 4636–4636 (2019). [Google Scholar]
- 42.Zhao M, Kim P, Mitra R, Zhao J & Zhao Z TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 44, D1023–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finkel RS et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Mercuri E et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 378, 625–635 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Raal FJ et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N. Engl. J. Med. 382, 1520–1530 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Hickey G et al. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 21, 35 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ameur A Goodbye reference, hello genome graphs. Nat. Biotechnol. 37, 866–868 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Center for Biologics Evaluation & Research. Human gene therapy products incorporating human genome editing. U.S. Food and Drug Administration; https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-products-incorporating-human-genome-editing. [Google Scholar]
Methods References
- 49.Corces MR et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Gen. 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data is deposited in the NCBI Sequence Read Archive database under accession number PRJNA733110. The data for the 1000 Genomes project was downloaded from DOI: 10.12688/wellcomeopenres.15126.2. The data for the Human Genome Diversity Project was downloaded from DOI: 10.1126/science.aay5012. The full CRISPRme results for NGG PAM gRNAs (including sg1617) are available at https://doi.org/10.5281/zenodo.7195706.