

Abstract
Maintenance of genomic integrity is vital to all organisms. A number of human genetic disorders, including Werner Syndrome, Bloom Syndrome and Rothmund–Thomson Syndrome, exhibit genomic instability with some phenotypic characteristics of premature aging and cancer predisposition. Presumably the aberrant cellular and clinical phenotypes in these disorders arise from defects in important DNA metabolic pathways such as replication, recombination or repair. These syndromes are all characterized by defects in a member of the RecQ family of DNA helicases. To obtain a better understanding of how these enzymes function in DNA metabolic pathways that directly influence chromosomal integrity, we have examined the effects of non-covalent DNA modifications on the catalytic activities of purified Werner (WRN) and Bloom (BLM) DNA helicases. A panel of DNA-binding ligands displaying unique properties for interacting with double helical DNA was tested for their effects on the unwinding activity of WRN and BLM helicases on a partial duplex DNA substrate. The levels of inhibition by a number of these compounds were distinct from previously reported values for viral, prokaryotic and eukaryotic helicases. The results demonstrate that BLM and WRN proteins exhibit similar sensitivity profiles to these DNA-binding ligands and are most potently inhibited by the structurally related minor groove binders distamycin A and netropsin (Ki ≤1 µM). The distinct inhibition of WRN and BLM helicases by the minor groove binders suggest that these helicases unwind double-stranded DNA by a related mechanism.
INTRODUCTION
Werner Syndrome (WS) and Bloom Syndrome (BS) are rare human autosomal recessive disorders characterized by chromosomal instability yet distinctly different clinical phenotypes. WS patients display growth retardation and certain phenotypic characteristics associated with premature aging at an early age (1). These symptoms include osteoporosis, atherosclerosis, diabetes mellitus (type II), cataracts, wrinkled skin and gray hair. In addition, cancers, particularly sarcomas, are prevalent in WS patients. In contrast to WS, BS is characterized by an elevated predisposition to a variety of malignant cancers including leukemias and solid tumors (2). Pre- and post-natal growth deficiencies, immunodeficiency and a sun-sensitive facial erythema are also evident in BS.
Despite the differences in clinical symptoms, both WS and BS are characterized as genomic instability disorders. Cells of WS patients display an elevated rate of somatic mutations, chromosomal losses and rearrangements, and large DNA deletions (3,4). Cells of BS patients also display a high mutation frequency characterized by chromatid gaps, breaks and rearrangements (2,5,6). In addition to the chromosomal abnormalities, BS cells, unlike WS cells, display abnormally high sister chromatid exchanges (7). The localization of Bloom (BLM) protein at synaptonemal complexes of homologously paired chromosomes in late meiotic prophase suggests a role of BLM protein in recombination, or possibly in resolution of interlocks between sister chromatids during the first division of meiosis (8). Both WS and BS cells exhibit hyper-recombination (2,9,10) and altered DNA replication (11–13), which likely contribute to the chromosomal instability of the disorders.
While evidence for a molecular function of BLM or Werner (WRN) helicase in human cells remains to be established, the cellular defects suggest that the proteins play important roles in some aspect of DNA metabolism. Consistent with this notion, sequence analysis of the WRN and BLM genes has demonstrated that they share strong sequence homology within a central domain of ~600 amino acids containing the seven ‘signature’ motifs of the RecQ family of DNA helicases and a second region of ~80 amino acids located C-terminal to the helicase domain (10,14,15). At present, five human members of the RecQ family have been identified, including WRN (14), BLM (10), RecQL (16), RecQL4 and RecQL5 (17). Most recently, it was demonstrated that mutations in the RecQL4 gene result in Rothmund–Thomson syndrome (18). It has been suggested that WRN and BLM helicases have a role in regulating genomic stability through suppression of recombination in human cells; however, BLM protein may have an additional function to resume cell cycle progression when S phase is interrupted (19).
The sequence homology between the WRN and BLM proteins suggests that the enzymes may share common biochemical properties. Indeed, both WRN and BLM proteins are DNA-stimulated ATPases and DNA helicases which unwind double-stranded DNA (dsDNA) with a 3′ to 5′ polarity (20–23). However, WRN protein has also been reported to exhibit an exonuclease activity (23–26) whereas BLM protein has not been shown to possess such an activity. Limited studies have addressed the unwinding activity of WRN or BLM helicases on substrates other than the canonical Watson–Crick DNA double helix. It has been shown that WRN helicase unwinds DNA–RNA hybrids (22,26). Recent studies indicate that WRN (27) and BLM (28) helicases are capable of unwinding tetrahelical structures. Future studies are likely to address activities of WRN and BLM enzymes on other alternate DNA structures.
The goal of this study was to investigate the effects of non-covalent DNA modifications on the catalytic function of WRN and BLM helicases. Differences in their function may be an important aspect of the phenotypical differences between the affected individuals. A panel of DNA-binding ligands displaying unique properties for interacting with double helical DNA (Fig. 1) was tested for their effects on unwinding activity of WRN and BLM helicases. A pronounced inhibition of WRN and BLM helicase activities was obtained with the structurally related molecules distamycin A and netropsin, which position themselves in the minor groove of double helical DNA. The results demonstrate that BLM and WRN proteins exhibit very similar sensitivity profiles to these DNA-binding ligands suggesting that the two human helicases are affected similarly by alterations to DNA structure imposed by these ligands. The potent inhibition of WRN and BLM helicases by distamycin A and netropsin, as well as the comparable sensitivity of these enzymes to all of the DNA-binding ligands tested, suggest that the two helicases may unwind dsDNA by a related mechanism. The effect of minor groove modifications on WRN and BLM helicases may be relevant to the biological effects of anti-cancer drugs which perturb DNA structure by binding to the minor groove.
Figure 1.

Chemical structures of DNA-binding compounds.
MATERIALS AND METHODS
Proteins
Recombinant hexa-histidine tagged human WRN protein was overexpressed in Sf9 insect cells and purified by Ni+2 chromatography as described by Brosh et al. (29). Recombinant hexa-histidine tagged human BLM protein was overexpressed in Saccharomyces cerevisiae and purified as described by Karow et al. (30). Human replication protein A (hRPA) containing all three subunits (RPA70, RPA32 and RPA14) was graciously provided by Dr Mark Kenny (Albert Einstein School of Medicine, NY). UvrD (DNA helicase II) was kindly provided by Dr Steven Matson (University of North Carolina at Chapel Hill, NC). T4 polynucleotide kinase was obtained from New England Biolabs, (Beverly, MA).
Nucleotides and DNA
M13mp18 single-stranded DNA (ssDNA) was from New England Biolabs. Poly(dT)~900 was from Midland Certified Reagent Company (Midland, TX). Two 28mer oligonucleotides were purchased from Gibco BRL (Gaithersburg, MD) with the following names and sequences: (i) oligonucleotide A: 5′-TCCCAGTCACGACGTTGTAAAACGACGG-3′, (ii) oligonucleotide B: 5′-ATGCTGATGCAAATCCAATCGCAAGACA-3′. Yeast tRNA was from Boehringer Mannheim, (Indianapolis, IN). [3H]ATP was from Amersham (Arlington Heights, IL) and [γ-32P]ATP was from New England Nuclear (Boston, MA).
DNA-binding ligands
Distamycin A, actinomycin D, camptothecin, Hoescht 33258, mitoxantrone, DAPI, VP16 (Etoposide) and m-AMSA were obtained from Sigma (St Louis, MO). Ethidium bromide was from Research Genetics (Huntsville, AL). Netropsin hydrochloride was obtained from Fluka (Milwaukee, WI). Concentrations of DNA-binding ligands were determined spectrophotometrically according to published extinction coefficients. The solvents used to dissolve the compounds and the extinction coefficients used for spectrophotometric determination of drug concentrations were as follows: distamycin A [dimethyl sulfoxide (DMSO), 237 nm, ɛ 30 000 M–1 cm–1]; mitoxantrone (DMSO, 658 nm, ɛ 20 900 M–1 cm–1); ethidium bromide (H2O, 316 nm, ɛ 50 000 M–1 cm–1); netropsin (DMSO, 304 nm, ɛ 35 000 M–1 cm–1); m-AMSA (methanol, 246 nm, ɛ 46 230 M–1 cm–1); VP16 (methanol, 283 nm, ɛ 4245 M–1 cm–1); camptothecin (DMSO, 370 nm, ɛ 19 900 M–1 cm–1); DAPI (H2O, 340 nm, ɛ 27 000 M–1 cm–1); Hoechst 33258 (5 mM HEPES pH 7.0, 10 mM NaCl, 338 nm, ɛ 42 000 M–1 cm–1); actinomycin D (methanol, 244 nm, ɛ1% 281 cm–1).
DNA helicase substrates
The 28 bp M13mp18 partial duplex substrate constructed with oligonucleotide A complementary to positions 6296–6323 is referred to as M13mp18: A-T[5] because it contains a 5 bp A-T tract. The 28 bp M13mp18 partial duplex substrate constructed with oligonucleotide B complementary to positions 3960–3987 is referred to as M13mp18: A-T[4] because it contains a 4 bp A-T tract. The 28 bp M13mp18 partial duplex substrates were constructed as described by Matson (31) with the following modifications. The 28mer oligonucleotides were labeled at their 5′ ends using T4 polynucleotide kinase and [γ-32P]ATP and annealed to M13mp18 ssDNA circles. Partial duplex DNA substrates were purified by gel filtration column chromatography using Bio-Gel A-5M resin (Bio-Rad, Hercules, CA).
DNA helicase assay
Helicase assay reaction mixtures (20 µl) contained 40 mM Tris (pH 7.4), 4 mM MgCl2, 5 mM dithiothreitol (DTT), 2 mM ATP, 0.5 mg/ml yeast tRNA, 96 nM WRN protein or 19 nM BLM protein, and the indicated amounts of DNA-binding ligand. In those reactions containing hRPA, the concentration of hRPA was 96 nM heterotrimer. The concentration of the 28 bp partial duplex helicase substrate in the reaction mixture was ∼2 µM (nucleotide phosphate). Reactions were initiated by the addition of WRN or BLM protein and incubated at 24°C for 30 min. Reactions were terminated by the addition of 10 µl of 50 mM EDTA–40% glycerol–0.9% sodium dodecyl sulfate (SDS)–0.1% bromophenol blue–0.1% xylene cyanol. The products of helicase reactions were resolved on 12% non-denaturing polyacrylamide gels. Radiolabeled DNA species in polyacrylamide gels were visualized using a PhosphorImager or film autoradiography and quantitated using the ImageQuant software (Molecular Dynamics, Sunnyvale, CA). The percent helicase substrate unwound was calculated by the following formula: % displacement = 100 × P/(S + P) where P is the product and S is the substrate. The values for P and S have been corrected after subtracting background values in the no-enzyme and heat-denatured controls respectively.
ATPase assay
ATPase assay reaction mixtures (20 µl) contained 40 mM Tris pH 7.4, 4 mM MgCl2, 5 mM DTT, M13mp18 ssDNA or poly(dT)~900 (2 µM nucleotide phosphate), 0.8 mM [3H]ATP (42 c.p.m./pmol), 70 nM WRN protein or 11 nM BLM protein, and the indicated amounts of DNA-binding ligand. Reactions were initiated by the addition of WRN protein or BLM protein and incubated for 20 min at 24°C. Samples (5 µl) were removed at the end of incubation and evaluated by thin layer chromatography as previously described (32). Less than 20% of the substrate ATP was consumed in the reaction over the entire time course of the experiment.
RESULTS
Effects of various DNA-binding compounds on WRN and BLM helicase activities
To characterize the effects of non-covalent DNA modifications on the helicase activities catalyzed by WRN and BLM proteins, a series of DNA-binding ligands (Fig. 1) were tested for their effects on the unwinding reaction on a 28 bp M13 partial duplex DNA substrate designated M13mp18: A-T[5] (Table 1). The compounds were tested initially at concentrations of 1, 10 and 100 µM to obtain approximate Ki values and to determine which compounds were effective inhibitors of the WRN and BLM-catalyzed unwinding reactions. The results demonstrated that WRN and BLM helicases exhibit a very similar sensitivity profile to a broad range of DNA-binding ligands including DNA intercalators, compounds which position themselves in the minor or major groove of B-form DNA, and drugs which inhibit topoisomerase activity. Of the compounds tested, distamycin A potently inhibited both WRN and BLM helicase activity at a concentration of 1 µM (Table 1). Other minor groove binders (DAPI, Hoescht 33258 and netropsin) did not inhibit the unwinding activities of WRN or BLM at 1 µM, and required concentrations ≥10 µM to achieve 50% inhibition on the 28 bp M13mp18: A-T[5] partial duplex DNA substrate. All compounds tested in the DNA intercalator class also poorly inhibited WRN and BLM helicase activity at the low dose of 1 µM. Ethidium bromide as well as mitoxantrone, an intercalator which places amino and hydroxyl groups into the major groove, inhibited WRN and BLM helicase activities at concentrations ≥10 µM whereas m-AMSA and actinomycin D inhibited DNA helicase activity at concentrations ≥100 µM. The topoisomerase inhibitors camptothecin and VP16 failed to inhibit WRN and BLM-catalyzed unwinding at the highest concentration tested, 100 µM.
Table 1. Effect of DNA-binding compounds on the unwinding activity of WRN and BLM helicases.
| WRN | BLM | |
|---|---|---|
| Compound | apparent Ki (µM) | apparent Ki (µM) |
| Intercalators | ||
| Ethidium bromide | ~10 | >10 |
| Actinomycin D | ~100 | ~100 |
| m-AMSA | >100 | >100 |
| Mitoxantrone | ~10 | >10 |
| Minor groove binders | ||
| DAPI | ~10 | ~10 |
| Hoescht 33258 | ~10 | ~10 |
| Netropsin | ~10 | >10 |
| Distamycin A | <1 | <1 |
| Topoismerase inhibitors | ||
| Camptothecin | >100 | >100 |
| VP16 (Etoposide) | >100 | >100 |
Helicase reactions were performed under standard reaction conditions as described in Materials and Methods using the M13mp18: A-T[5] partial duplex DNA substrate. In control reactions (no drug), WRN and BLM helicases catalyzed unwinding of ~50% of the 28 bp partial duplex DNA substrate. Final drug concentrations of 1, 10 and 100 µM were tested for inhibition of helicase activity. The apparent Ki value was determined from the drug concentration sufficient to achieve 50 ± 5% inhibition. If <45% inhibition was obtained at the indicated drug concentration, a ‘>’ sign was included in the reported value. If >55% inhibition was obtained at the indicated drug concentration, a ‘<’ sign was included in the reported value. The data represent the average of at least three independent determinations.
Of the DNA-binding compounds tested, the minor groove binder distamycin A was a substantially more effective inhibitor of the unwinding reactions catalyzed by WRN and BLM helicases than any of the other DNA-binding compounds tested. To further explore the inhibition of BLM and WRN helicase activity by the minor groove binder distamycin A, the effects of distamycin A on helicase activity were tested at low concentrations of drug to determine more precisely an apparent Ki for helicase inhibition (Fig. 2). Distamycin effectively inhibited the unwinding activities of WRN (Fig. 2A) and BLM (Fig. 2B) at concentrations of drug <1 µM. Quantitative analysis of the helicase data demonstrates that WRN and BLM are inhibited to ∼40% the level of control (no drug) at a final distamycin A concentration of 0.5 µM (Fig. 2C). At the highest concentration of distamycin A shown (5 µM), WRN and BLM helicase activities were inhibited to 22 and 26% of the level of the control. However, the inhibition curves have not reached a minimum, and further inhibition was achieved at distamycin concentrations ≥10 µM (data not shown). These results contrast sharply with a similar study of UvrD-catalyzed unwinding of the 28 bp M13 partial duplex DNA substrate M13mp18: A-T[5]. Under reaction conditions identical to those used for the WRN/BLM helicase assays, UvrD failed to be inhibited by distamycin A at concentrations of drug up to 100 µM (R.Brosh et al., unpublished data). These results agree well with a previous study demonstrating that distamycin A (100 µM) did not inhibit UvrD-catalyzed unwinding of a 71 bp M13 partial duplex helicase substrate (33) (Table 3). These results suggest that the potent inhibition of WRN and BLM helicases by distamycin A compared to the other compounds tested does not simply reflect a general inhibitory effect of the drug, such as stabilization of duplex DNA, on all helicases. Rather, the failure of distamycin A to inhibit UvrD helicase activity on the same DNA substrate and under identical reaction conditions used for WRN and BLM indicates a specific effect of distamycin on WRN and BLM helicases.
Figure 2.
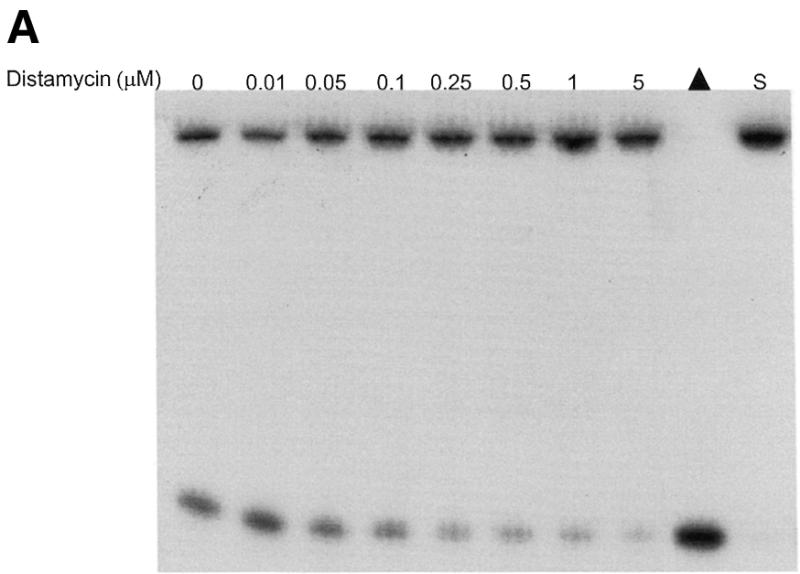
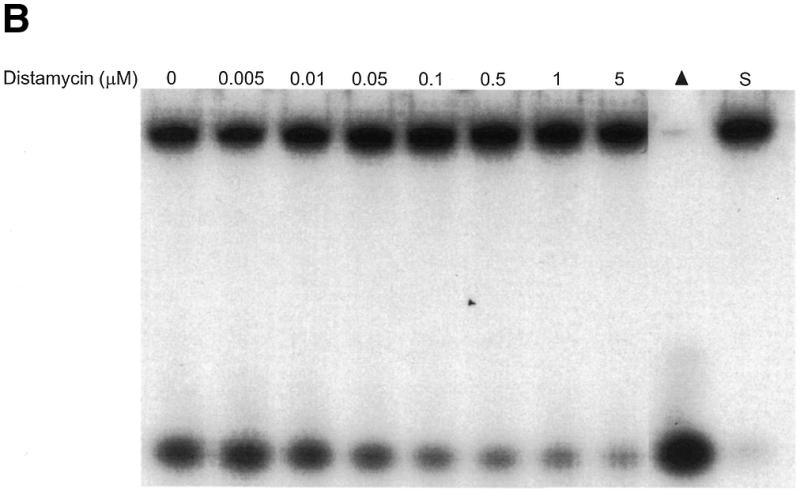

Potent inhibition of WRN and BLM helicase activities on an M13 partial duplex DNA substrate by the minor groove binder distamycin A. WRN protein (96 nM) (A) or BLM protein (19 nM) (B) was incubated with the M13mp18: A-T[5] partial duplex DNA substrate in the presence of the indicated concentrations of distamycin A under the standard helicase reaction conditions as described in Materials and Methods. Incubation was at 24°C for 30 min. Reaction products were analyzed by non-denaturing gel electrophoresis. Representative autoradiographs of WRN and BLM helicase assays are shown. Closed triangle, heat-denatured control; S, no enzyme control. (C) Quantitation of helicase activity (% control activity) as a function of distamycin A concentration. Closed circles, WRN; open circles, BLM. In control reactions, WRN or BLM helicase catalyzed unwinding of ∼50% of the partial duplex substrate. All data points are the average of at least three independent determinations.
Table 3. Inhibition constants of DNA-binding compounds for DNA unwinding by various DNA helicases.
| Apparent Ki (µM) | |||||||
|---|---|---|---|---|---|---|---|
| Drug | WRNa | BLMa | UvrDc | Rad3d | HELA pe | HDHII/Kuf | Large T antigene |
| Intercalator | |||||||
| Ethidium bromide | ~10 | >10 | ~10c ~6d | ~25 | ~12.5 | 8 | ND |
| Mitoxantrone | ~10 | >10 | ~1 | ND | ND | ND | ND |
| m-AMSA | >100 | >100 | >100 | ND | ND | >50 | ND |
| Actinomycin | ~100 | ~100 | >100 | ND | ND | 12 | ~0.8 |
| Minor groove | |||||||
| DAPI | ~10 ~10 | ~10 ~10 | ~10 | ND | ND | ND | ND |
| Hoescht 33258 | ~10 >10 | ~10 >10 | >100 | ND | ND | ND | ND |
| Distamycin | ~0.5a ~2.5b | ~0.5a ~2.5b | >100c 0.02d | ~12 | ~4.5 | ND | ~2 |
| Netropsin | ~10a ~1b | >10a ~1b | >100 | ND | ND | ND | ND |
| Other | |||||||
| Camptothecin | >100 | >100 | >100 | ND | ND | >50 | ND |
| VP16 (Etoposide) | >100 | >100 | ND | ND | >80 | >50 | >80 |
a28 bp M13mp18: A-T[5] partial duplex DNA substrate (this study).
b28 bp M13mp18: A-T[4] partial duplex DNA substrate (this study).
c71 bp M13mp7 partial duplex DNA substrate (33).
d(dU167)-(dA25–30) partial duplex DNA substrate (50).
e17 bp M13mp19 partial duplex DNA substrate (43).
f47 bp M13mp19 partial duplex DNA substrate (42)
Mechanism of inhibition by distamycin A
It has been previously shown that distamycin A binds to the minor groove of B-form dsDNA (34). Interpretation of these structural data suggest that distamycin A blocks WRN or BLM helicase progression through the duplex DNA region (28 bp) in the partial duplex substrate defined by the complementary sequence of the radiolabeled oligonucleotide. Other hypotheses to explain this mechanism are possible. M13 ssDNA circles have been shown to have regions of ssDNA that can adopt a hairpin secondary structure. Distamycin may bind to the minor groove present in the hairpin and prevent the helicase from translocating through the structure. Alternatively, direct interaction of distamycin with ssDNA may prevent the helicase from translocating along the ssDNA to the ssDNA–dsDNA junction. Yet another possibility is that distamycin A binds directly to WRN/BLM helicase and negatively impacts upon the catalytic function of the enzyme and/or prevents the protein from binding to the partial duplex DNA substrate.
To explore the mechanism of distamycin inhibition of these two helicases, we examined helicase-catalyzed ATP hydrolysis in the presence of M13mp18 ssDNA. Both WRN and BLM proteins have been shown to be DNA-stimulated ATPases (20–22) raising the possibility that the mechanism of distamycin inhibition of helicase activity may reflect inhibition of ATP hydrolysis. Thus we tested the helicases for ATP hydrolysis using the M13mp18 ssDNA circle as the DNA effector in the presence of increasing concentrations of distamycin up to 10 µM (Fig. 3). Neither BLM nor WRN ATPase activity was significantly inhibited at concentrations of distamycin that potently inhibited the helicase activity under identical reaction conditions. WRN or BLM-catalyzed ATP hydrolysis was only mildly inhibited (10–15% of the level of control) at a concentration of drug 20-fold higher than the Ki of 0.5 µM for helicase inhibition. These results suggest that distamycin inhibits the helicase activity of WRN and BLM enzymes by a mechanism other than binding directly to BLM or WRN proteins and preventing the enzyme from binding to the DNA effector.
Figure 3.

Effect of distamycin A on the ATPase activities of WRN and BLM helicases in the presence of the M13mp18 ssDNA effector. ATPase reactions containing 0.8 mM [3H]ATP, M13mp18 ssDNA (2 µM nucleotide phosphate) and the indicated concentration of distamycin A were initiated with WRN protein (70 nM) (closed circles) or BLM protein (11 nM) (open circles), as described in Materials and Methods. Reactions were incubated at 24°C for 20 min and analyzed for the production of [3H]ADP. In control reactions, WRN or BLM proteins hydrolyzed <20% of the total ATP in the reaction mixture. The total amount of ATP hydrolyzed by WRN and BLM proteins in the absence of distamycin were 1040 and 1140 pmol, respectively. All data points are the average of at least three independent determinations.
It is possible that distamycin A binds directly to ssDNA and prevents the helicase from translocating along the ssDNA to the ssDNA–dsDNA junction. Using ssDNA effectors of various lengths, a number of DNA helicases have been shown to be better stimulated by long ssDNA effectors compared to short ssDNA effectors (35). These data have been interpreted to suggest that the enzyme translocates with some degree of processivity along ssDNA since the ATPase reaction is dramatically stimulated in the presence of ssDNA. We have tested various DNA effectors in the WRN ATPase reaction (36). The ATPase activity of WRN helicase is more efficiently stimulated by long ssDNA effectors compared to short ssDNA effectors suggesting that the enzyme translocates processively on ssDNA. The ATPase activity of BLM helicase exhibits a similar dependence on length (R.Brosh, unpublished data). The fact that relatively high concentrations of distamycin A only mildly inhibit WRN-catalyzed ATP hydrolysis using M13 ssDNA circles as the effector suggests that the drug does not block translocation of the enzyme along the ssDNA or block the enzyme by binding to a region of secondary structure such as a hairpin in the M13 ssDNA circle. The lack of sensitivity of WRN or BLM enzymes to distamycin A in the ATPase reactions suggests that it is unlikely that the helicase is blocked from translocating to the ssDNA–dsDNA junction of the helicase substrate where the radiolabeled 28mer oligonucleotide is annealed. In addition, we have performed experiments to measure ATP hydrolysis of WRN and BLM proteins in the presence of distamycin using poly(dT)~900 as the DNA effector. Neither WRN nor BLM-catalyzed ATP hydrolysis was affected by the presence of distamycin concentrations up to 10 µM (data not shown). These data provide additional evidence indicating that the mechanism for distamycin inhibition of helicase activity does not involve drug binding to enzyme or dT tracts in the M13 ssDNA circle of the helicase substrate.
Altogether, the results from the DNA-stimulated ATPase assays and the well-documented interaction of distamycin with the minor groove of dsDNA suggest that distamycin blocks progression of WRN or BLM helicases as the enzymes attempt to progress through dsDNA. Importantly, the remarkably similar sensitivity of WRN and BLM proteins to distamycin and the other compounds tested indicate that the non-covalent binding of ligands to the DNA substrate have very similar effects on the unwinding reaction catalyzed by WRN and BLM.
Helicase inhibition using a consecutive 4 bp A-T partial duplex substrate
We found it particularly surprising that distamycin A was very effective in WRN and BLM helicase inhibition whereas the chemically related compound netropsin (Fig. 1) inhibited helicase activity at a 20-fold (WRN) or >20-fold (BLM) higher concentration of drug compared to distamycin (Table 1). Detailed crystal structure data of distamycin A and netropsin bound to oligonucleotides have provided evidence that these compounds insert themselves into the minor groove of B-form double helical DNA (37,38). However, the two compounds exhibit different specificities in their interactions with dsDNA. Distamycin A has been shown to preferably bind to DNA duplex tracts containing a 5 bp A-T tract (39). Netropsin, on the other hand, preferentially binds to a DNA duplex tract containing a 4 bp A-T tract (39). A possible explanation for the lack of inhibition of WRN and BLM helicase activity by netropsin on the M13mp18: A-T[5] partial duplex DNA substrate is that the helicase substrate did not provide the optimal binding site for netropsin.
To address possible effects of nucleotide sequence on the helicase activity inhibition results, we tested the effects of minor groove binders on WRN and BLM helicase activity on M13mp18: A-T[4] partial duplex, a helicase substrate containing a 4 bp A-T tract (Table 2). Netropsin was the most effective inhibitor of WRN and BLM helicase activities on the M13mp18: A-T[4] partial duplex substrate. Concentrations as low as 0.5 µM netropsin significantly inhibited the unwinding reactions catalyzed by WRN and BLM enzymes on the substrate containing the 4 bp A-T tract (Fig. 4). A final netropsin concentration of 1 µM inhibited the BLM and WRN helicase reactions to 51 and 42% of the control (no drug) reaction respectively (Fig. 4). At all concentrations tested, WRN helicase activity was inhibited to a greater extent compared to BLM helicase activity indicating a reproducibly measurable difference between the two enzymes.
Table 2. Effect of minor groove binders on the unwinding activity of WRN and BLM helicases on an M13 partial duplex substrate containing a consecutive 4 bp A-T tract.
| WRN | BLM | |
|---|---|---|
| Compound | apparent Ki (µM) | apparent Ki (µM) |
| DAPI | ~10 | >10 |
| Hoechst 33258 | >10 | >10 |
| Distamycin A | >1 | >1 |
| Netropsin | ~1 | ~1 |
Helicase reactions were performed under standard reaction conditions as described in Materials and Methods using the M13mp18: A-T[4] partial duplex DNA substrate. In control reactions (no drug), WRN or BLM helicases catalyzed unwinding of ~50% of the partial duplex DNA substrate. Final drug concentrations of 1, 10 and 100 µM were tested for inhibition of helicase activity. The apparent Ki value was determined from the drug concentration sufficient to achieve 50 ± 5% inhibition. If <45% inhibition was obtained at the indicated drug concentration, a ‘>’ sign was included in the reported value. If >55% inhibition was obtained at the indicated drug concentration, a ‘<’ sign was included in the reported value. The data represent the average of at least three independent determinations.
Figure 4.

Potent inhibition of WRN and BLM helicase activities on an M13 partial duplex DNA substrate with a 4 bp A-T tract by the minor groove binder netropsin. WRN protein (96 nM) or BLM protein (19 nM) was incubated with the M13mp18: A-T[4] partial duplex DNA substrate in the presence of the indicated concentrations of netropsin under the standard helicase reaction conditions described in Materials and Methods. Helicase activity (% control activity) is expressed as a function of netropsin concentration. Closed circles, WRN; open circles, BLM. In control reactions, WRN or BLM helicase catalyzed unwinding of ∼50% of the partial duplex substrate. All data points are the average of at least three independent determinations.
The potent inhibition of WRN and BLM helicase activity on the M13mp18: A-T[4] partial duplex substrate by netropsin was not observed with the M13mp18: A-T[5] substrate suggesting that the drug had a reduced affinity for the latter substrate. These results suggest that the inhibitory effect of netropsin on WRN/BLM helicase function was not due to the binding of the drug to the protein and altering its function. To further address this issue, we tested the effect of netropsin on UvrD-catalyzed unwinding of the M13mp18: A-T[4] helicase substrate under the identical reaction conditions as those used for the WRN and BLM helicase assays and found no inhibition up to a concentration of 100 µM netropsin (R.Brosh et al., unpublished data). These results concur with a previous study demonstrating that netropsin (100 µM) did not inhibit UvrD-catalyzed unwinding of a 71 bp M13 partial duplex helicase substrate (33) (Table 3). Altogether, these data provide supporting evidence that netropsin exerts its negative effect on the function of WRN and BLM proteins by binding to the M13mp18: A-T[4] DNA substrate and specifically preventing strand displacement by either human helicase.
Both WRN and BLM helicases were similarly inhibited by distamycin A on the M13mp18: A-T[4] helicase substrate (Fig. 5). Using the partial duplex with a 4 bp A-T tract, a final distamycin concentration of 2.5 µM inhibited both WRN and BLM helicase reactions to 40% of the level of the control reaction (Fig. 5). Thus both WRN and BLM helicases exhibit nearly identical sensitivity profiles to distamycin A and netropsin on partial duplex substrates containing either a 4 or 5 bp A-T tract.
Figure 5.

Potent inhibition of WRN and BLM helicase activities on a M13 partial duplex DNA substrate with a 4 bp A-T tract by the minor groove binder distamycin A. WRN protein (96 nM) or BLM protein (19 nM) was incubated with the M13mp18: A-T[4] partial duplex DNA substrate in the presence of the indicated concentrations of netropsin under the standard helicase reaction conditions as described in Materials and Methods. Helicase activity (% control activity) is expressed as a function of distamycin A concentration. Closed circles, WRN; open circles, BLM. In control reactions, WRN or BLM helicase catalyzed unwinding of ∼50% of the partial duplex substrate. All data points are the average of at least three independent determinations.
DAPI and Hoescht 33258 are minor groove binders which require a minimum of four consecutive A-T base pairs (40,41). The requirement for relatively high concentrations of these compounds (≥10 µM) to achieve 50% inhibition of WRN or BLM helicase activity on the M13mp18: A-T[5] substrate raised the question of whether these compounds might behave in a similar manner to that of netropsin and more successfully inhibit WRN/BLM helicase activity on the 4 bp A-T partial duplex substrate. The apparent Ki values for helicase inhibition by Hoechst 33258 and DAPI on M13mp18: A-T[4] were determined to be ≥10 µM (Table 2). Thus, these compounds which bind the minor groove exhibited distinct differences from distamycin or netropsin on partial duplex substrates containing either four or five consecutive A-T base pairs. The results also demonstrate that both BLM and WRN helicases are similarly affected by the two drugs as evidenced by their similar patterns of inhibition.
Effect of distamycin A on hRPA-stimulated WRN or BLM helicase activity
We have recently demonstrated a functional and physical interaction between WRN protein and hRPA (29). WRN helicase activity is stimulated by hRPA whereas heterologous single-strand breaks fail to stimulate WRN helicase activity. A functional interaction has also been detected between BLM helicase and hRPA (R.Brosh et al., unpublished data). The functional interaction between these helicases and hRPA may be important to enable the enzymes to unwind modified DNA substrates or novel DNA structures other than Watson–Crick B-form dsDNA. We investigated the effect of hRPA on the WRN/BLM unwinding reaction in the presence of distamycin on the M13mp18: A-T[5] partial duplex helicase substrate. The helicase reactions were conducted in the presence of sufficient hRPA to coat the helicase substrate and at concentrations of distamycin A up to 10 µM. Despite the presence of hRPA, WRN or BLM helicase activity was inhibited by distamycin A concentrations as low as 0.1–0.25 µM (Fig. 6). Fifty percent inhibition of either WRN or BLM helicase reactions was achieved at a distamycin concentration of 0.5 µM. The overall pattern of distamycin inhibition for WRN or BLM helicases was the same in the presence or absence of hRPA (Fig. 2). These results suggest that hRPA does not alleviate the potent inhibition of WRN or BLM unwinding activity by the minor groove binder distamycin A.
Figure 6.

The presence of hRPA in the WRN or BLM helicase reactions does not alleviate the potent inhibition of unwinding activity by the minor groove binder distamycin A. WRN protein (96 nM) or BLM protein (19 nM) was incubated with the M13mp18: A-T[5] partial duplex DNA substrate in the presence of hRPA (96 nM, heterotrimer) and the indicated concentration of distamycin A. Helicase reactions were conducted as described in Materials and Methods. Helicase activity (% control activity) is expressed as a function of distamycin A concentration. Closed circles, WRN; open circles, BLM. In control reactions, WRN or BLM helicase catalyzed unwinding of ∼50% of the partial duplex substrate. All data points are the average of at least three independent determinations.
DISCUSSION
To better understand the similarities and differences between the unwinding reactions catalyzed by WRN and BLM helicases, the two human enzymes were tested for their sensitivities to DNA-binding compounds using a DNA strand displacement assay. The compounds used in this study belong to three classes of DNA-binding ligands: (i) intercalators, (ii) minor groove binders and (iii) non-intercalating topoisomerase inhibitors. In addition to the distinction by class, the compounds within each group exhibit unique properties defined by sequence specificity, perturbation of DNA structure and interaction of functional groups with the DNA double helix.
A limited number of studies have addressed the effects of DNA-binding ligands on the unwinding activity of DNA helicases. The results of some of these are summarized in Table 3, which provides a comparison of WRN and BLM helicases to viral, bacterial, yeast and human DNA helicases which have been tested using at least some of the same DNA-binding ligands. Inhibition constants (apparent Ki) of DNA-binding compounds for DNA unwinding by the various DNA helicases on specific helicase substrates are shown. By far the most thoroughly characterized helicase with respect to the effect of DNA-binding compounds on unwinding activity is the Escherichia coli enzyme UvrD (33). In control experiments, we performed helicase reactions with UvrD on the M13mp18 28 bp partial duplex substrates used in this study and selected DNA-binding ligands (distamycin A, netropsin, mitoxantrone and ethidium bromide) and found the results to correlate well with those of George et al. on a 71 bp M13mp7 partial duplex DNA substrate (33) (R.Brosh et al., unpublished data). Thus direct comparison of Ki values between helicases on slightly different helicase substrates can be useful, but careful consideration of sequence differences between substrates should be taken into account (see below).
WRN and BLM proteins were relatively insensitive to the DNA intercalators m-AMSA and actinomycin D, and displayed only a mild sensitivity to the DNA intercalators ethidium bromide and mitoxantrone. The Kd values for interaction of these intercalators with DNA (summarized in 33) are at least an order of magnitude less than the respective Ki values for inhibition of WRN/BLM helicase activity determined in this study. Thus the reduced inhibitory activity of these compounds does not reflect a relatively low binding affinity. Previous studies have demonstrated that the helicase activity of human autoantigen Ku complex (HDHII) was also mildly sensitive to ethidium bromide and actinomycin D (42) (Table 3). In contrast, Large T antigen, the SV40-encoded DNA helicase used in mammalian DNA replication of the virus, was substantially inhibited by actinomycin D (Ki ~0.8 µM) (43). These results suggest that eukaryotic DNA helicases exhibit marked differences in their abilities to unwind dsDNA substrates with planar molecules intercalated between the base pairs.
The mild sensitivity of WRN and BLM helicases to mitoxantrone contrasts sharply with the potent inhibition of UvrD-catalyzed unwinding on a 71 bp M13mp7 partial duplex helicase substrate previously reported (33) (Table 3). We tested the effect of mitoxantrone on UvrD helicase activity on the 28 bp helicase substrate used in this study (M13mp18: A-T[5]), and also found mitoxantrone to potently inhibit unwinding at a drug concentration of 1 µM (data not shown). These results suggest that mitoxantrone, and possibly other DNA intercalators, exert markedly different effects on unwinding activity of various DNA helicases. These differences may reflect the mechanism used to unwind dsDNA since the same helicase substrate and identical reaction conditions were used to measure WRN, BLM and UvrD helicase reactions in this study. George et al. reported a similar Ki value of 0.9 µM for inhibition of UvrD-catalyzed helicase and ATPase activities using the M13 partial duplex DNA substrate (33). However, it was shown that the inhibition of the ATPase reaction at low mitoxantrone concentrations is DNA structure-dependent since the drug (at concentrations <2.5 µM) had little effect on the ATPase reaction when poly(dT) was the DNA effector (33). The authors concluded that the inhibition of ATPase activity using the M13 partial duplex substrate most likely reflected mitoxantrone intercalation into the secondary structure of M13 ssDNA. Based on the helicase and ATPase data, George et al. suggested that intercalation of mitoxantrone into dsDNA generates a complex that impedes the movement of UvrD on the DNA helicase substrate (33). The results presented here suggest that WRN and BLM helicases are capable of overcoming the block posed by a mitoxantrone–dsDNA complex during the unwinding reaction. NMR structural studies of mitoxantrone–DNA complexes suggest that the compound intercalates between the base pairs as well as placing functional groups (a positively charged amino group and two hydroxyl groups) into the major groove of B-form double-helical DNA (44). If the positioning of a functional group into the major groove is responsible for mitoxantrone inhibition of UvrD progression through dsDNA, this is not the case for the human BLM and WRN proteins. Further studies will be necessary to address the effects of specific DNA ligands, triplex substrates or DNA-binding proteins that position functional groups into the major groove on the catalytic activities of WRN and BLM enzymes.
A range of sensitivities to minor groove binders was observed for the WRN and BLM helicases. DAPI and Hoescht 33258 failed to inhibit the BLM or WRN unwinding reactions at low concentrations on either helicase substrate, but unwinding was reduced by 50% at 10 µM. The Kd values for the high and low affinity binding sites of DAPI to dsDNA are 0.03 and 3.4 µM, respectively (45). A range of approximate Kd values for the interaction of Hoescht 33258 with dsDNA (0.04–15.1 µM) have been reported, depending on the arrangement of a 4 bp A-T tract in a synthetic DNA fragment (46). Thus the high end of the dissociation constant values for these minor groove binders are of the same approximate magnitude as the inhibition constant values determined here.
Crystallographic structure analysis of Hoechst 33258 bound to B-form DNA indicates that the molecule binds to the minor groove similarly to netropsin (47); however, the bulky and non-planar piperazine ring of Hoechst 33258 (Fig. 1) requires a wider groove that only a G-C region can provide. Thus, in the identical base sequence, the binding site for netropsin is g-A-A-T-T-c (48) whereas that for Hoechst 33258 is g-a-A-T-T-C (47). It is possible that the reduced binding affinity of Hoechst 33258 to DNA or its precise positioning in the minor groove may reduce its ability to inhibit WRN or BLM helicase activity.
The relative insensitivity of WRN/BLM helicase to the minor groove binder DAPI may reflect the small size of the DAPI molecule. DAPI is only 1.4 nm long compared with 2.0 nm for distamycin A (49). However, both compounds require an A-T tract of ≥4 bp, reflecting a requirement for the deep electrostatic potential in the minor groove. Indeed, footprinting results indicate a similar size of protection for distamycin A and DAPI, suggesting that the relatively small DAPI molecule ‘floats’ in the electrostatic well (49). Thus the size and/or nature of the interaction between DAPI and the minor groove may reduce its effectiveness as an inhibitor of WRN or BLM helicases during the unwinding reaction. A possible explanation for the differential effect of distamycin versus DAPI or Hoechst on WRN/BLM helicase activity is the side-by-side binding of two distamycin molecules to the minor groove, not observed for DAPI or Hoechst (37). Alternatively, additional factor(s) other than steric hindrance due to minor groove occupation may be at least partly responsible for the potent inhibition of WRN/BLM unwinding by distamycin. DNA structure perturbation by distamycin, such as widening of the minor groove and/or alteration of helix conformation (37), may be important in the mechanism of inhibition.
Distamycin A effectively inhibited WRN and BLM-catalyzed unwinding of the two helicase substrates used in this study at drug concentrations of ~1 µM. Similar potent inhibition of WRN/BLM helicase activity on the M13mp18: A-T[4] substrate was obtained with netropsin, resulting in a Ki of ~1 µM. The weak inhibition by netropsin of WRN/BLM unwinding on the M13mp18: A-T[5] substrate may reflect DNA sequence preferences of netropsin as reported previously (46). DNase I footprinting of DNA fragments containing different arrangements of AT base pairs has revealed that differences in binding affinity to DNA sequence elements are greater for netropsin than distamycin (46). Since the oligonucleotides used for the helicase substrates are clearly different from one another, it is possible that the sequence context of the A-T tract accounts for the differential effect of netropsin on WRN/BLM helicase activity against the two substrates. Distamycin A is able to bind to 4 or 5 bp A-T tracts (46), and is more tolerant of isolated GC base pairs than netropsin (39), which may explain its ability to inhibit WRN/BLM helicase activity on both partial duplex substrates. Approximate dissociation constants for interaction of distamycin and netropsin to DNA fragments containing different arrangements of four consecutive A-T base pairs were 0.15–0.97 and 0.09–1.25 µM, respectively, depending on the DNA fragment sequence (46). Thus the Ki values of 1 µM for distamycin and netropsin are comparable to the Kd values for the two drugs. The close similarity in chemical structure between distamycin A and netropsin (Fig. 1) suggests that the two compounds interfere with the helicase action of BLM and WRN enzymes by their space occupation properties in the minor groove which block processive movement of the helicases on the DNA strands. Alternatively, distamycin A and netropsin may induce structural or topological changes to the DNA double helix which impede progression of the WRN and BLM enzymes. The inhibitory effect of these minor groove binders on WRN and BLM helicase activity distinctly contrasts with their null effect on UvrD unwinding activity. To our knowledge, the effect of distamycin or netropsin on unwinding activity of RecQ family DNA helicases has not been previously tested. It needs to be investigated whether these minor groove binders would inhibit such RecQ helicases as E.coli RecQ or S.cerevisiae Sgs1.
To our knowledge, WRN and BLM helicases are more potently inhibited by netropsin and distamycin A than any other helicases characterized to date using the M13 partial duplex substrate (Table 3). Netropsin or distamycin failed to inhibit UvrD helicase activity on a 71 bp M13 partial duplex substrate at concentrations up to 100 µM (33) (Table 3). The 71 bp partial duplex DNA substrate contains one tract of three consecutive A-T base pairs, two tracts of six consecutive A-T base pairs and one tract of 10 consecutive A-T base pairs. We also found that distamycin A failed to inhibit UvrD helicase activity on the M13mp18: A-T[5] partial duplex substrate (containing the tract of five consecutive A-T base pairs) at concentrations up to 100 µM drug (data not shown). However, Naegeli et al. reported that distamycin A potently inhibits UvrD helicase activity on a poly(dU)-oligo(dA) partial duplex substrate (50) (Table 3), leading the authors to suggest that the discrepancy between their data and the study of George et al. (33) might reflect the presence of multiple high affinity binding sites for distamycin A in the poly(dU)-oligo(dA) substrate. The unwinding reaction of yeast DNA repair helicase Rad3 is only mildly inhibited by distamycin A on the poly(dU)-oligo(dA) substrate (50) (Table 3), as evidenced by a (5- to 24-fold) greater Ki value compared to either WRN or BLM helicases. The (dU167-dA25–30) duplex substrate used in the Rad3 study should be a preferred substrate for distamycin binding because the drug has been shown to preferably bind to DNA duplex tracts containing a 5 bp A-T tract (48). These notable differences suggest that interactions of distamycin A with the minor groove differentially affect the catalytic function of DNA helicases.
The results presented here suggest that WRN and BLM enzymes are particularly sensitive to distamycin compared to a number of other DNA-binding compounds. The apparent Ki values of distamycin for the human helicase HELAp and eukaryotic mammalian viral SV40 Large T antigen on a 17 bp partial duplex substrate containing three or less consecutive A-T base pairs were determined to be 4.5 and 2 µM, respectively (43) (Table 3). Thus, distamycin A was an effective inhibitor of these helicases, but not quite to the extent observed for WRN and BLM. The difference may reflect the absence of a sufficiently long A-T tract, such as 4 or 5 bp present in the helicase substrates used in this study, or other sequence-specific effects.
Our previously reported stimulation of hRPA on WRN/BLM helicase activity suggested to us the possibility that hRPA may enable the helicases to overcome the block to unwinding imposed by distamycin. Further, it was recently reported that the herpes simplex virus type-1 ssDNA-binding protein ICP8 enhances the ability of the viral DNA helicase-primase to unwind intrastrand cross-linked DNA produced by cisplatin (51). NMR solution structure of the d(GpG) 1,2-intrastrand cross-link indicates that the minor groove opposite the site of platination is widened and flattened (52). The crystal structures of the side-by-side binding of distamycin to AT-containing DNA octamers also demonstrates that the minor groove is widened, and in addition, the helix twist is altered (37). Clearly, hRPA does not stimulate WRN or BLM helicase to overcome the perturbation to DNA structure imposed by distamycin (Fig. 6). Further studies in this area will address the ability of hRPA to impact WRN/BLM helicase function on other types of alternate DNA structure.
Studies using modified substrates containing benzo[α]pyrene adducts have demonstrated that such adducts block transcription by T7 RNA polymerase (53) and translocation by T7 gene 4 protein (54). Molecular modeling of the major adducts of (+)-anti-B[α]PDE demonstrates that four of the eight predominant conformations place a dG moiety in the minor groove (55). Benzo[α]pyrene-induced alterations, and other specific modifications to the minor groove, may pose formidable blocks to certain enzymes which translocate along the DNA lattice.
The anti-tumor drug CC-1065 reacts with duplex DNA to form a N3-adenine DNA adduct which lies in the minor groove of DNA (56,57). The unwinding activities of T4 dda protein and E.coli UvrD (DNA helicase II) are strongly inhibited by CC-1065 modified helicase substrates (58). CC1065 may exert its biological effect by virtue of its covalent bond to dsDNA, perturbation of the minor groove, and interference with some important aspect of DNA metabolism. Replicative DNA helicases, which lead the replication complex to encounter the CC-1065 modified DNA, may be targeted for inhibition. Studies of the effects of specific therapeutic drugs, which interact with DNA on human enzymes such as WRN and BLM helicases, may help to elucidate the mechanism of drug action.
Non-covalent or covalent modifications to the minor groove may influence processes of chromosomal metabolism by blocking complexes involved in replication, transcription or other processes. For example, distamycin A effectively arrests RNA polymerase II transcription by blocking chain elongation (59). However, distamycin A can exert the opposite effect by enhancement of RNA polymerase II transcription at a natural pause site. Thus conformational changes in DNA structure induced by such ligands as the anti-tumor antibiotic distamycin A can profoundly and uniquely affect various biochemical processes in the cell. The findings presented here suggest that specific modifications within the minor groove of genomic DNA may perturb function of the WRN and BLM proteins in vivo. Further studies to address the mechanism of inhibition by specific minor groove binders, and the potency of inhibition of WRN and BLM enzymatic activities by other DNA structural modifications, may yield insight to the differences in molecular functions of these related proteins.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Jimmy Burril for artwork and the Danish Center for Molecular Gerontology for interactions. J.K.K. and I.D.H. are supported by the Imperial Cancer Research Fund. J.K.K. is a Boehringer Ingelheim Fonds Fellow.
REFERENCES
- 1.Martin G.M. (1978) Birth Defects, 14, 5–39. [PubMed] [Google Scholar]
- 2.German J. (1993) Medicine (Baltimore), 72, 393–406. [PubMed] [Google Scholar]
- 3.Salk D., Au,K., Hoehn,H. and Martin,G.M. (1981) Cytogenet. Cell Genet., 30, 92–107. [DOI] [PubMed] [Google Scholar]
- 4.Fukuchi K., Martin,G.M. and Monnat,R.J.J. (1989) Proc. Natl Acad. Sci. USA, 86, 5893–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis N.A. and German,J. (1996) Hum. Mol. Genet., 5, 1457–1463. [DOI] [PubMed] [Google Scholar]
- 6.Watt P.M. and Hickson,I.D. (1996) Curr. Biol., 6, 265–267. [DOI] [PubMed] [Google Scholar]
- 7.Langlois R.G., Bigbee,W.L., Jensen,R.H. and German,J. (1989) Proc. Natl Acad. Sci. USA, 86, 670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walpita D., Plug,A.W., Neff,N.F., German,J. and Ashley,T. (1999) Proc. Natl Acad. Sci. USA, 96, 5622–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng R.Z., Murano,S., Kurz,B. and Shmookler,R.R. (1990) Mutat. Res., 237, 259–269. [DOI] [PubMed] [Google Scholar]
- 10.Ellis N.A., Groden,J., Ye,T.Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- 11.Hanaoka F., Yamada,M., Takeuchi,F., Goto,M., Miyamoto,T. and Hori,T. (1985) Adv. Exp. Med. Biol., 190, 439–457. [DOI] [PubMed] [Google Scholar]
- 12.Gianneli F., Benson,P.F., Pawsey,S.A. and Polani,P.E. (1977) Nature, 265, 466–469. [DOI] [PubMed] [Google Scholar]
- 13.Lonn U., Lonn,S., Nylen,U., Winblad,G. and German,J. (1990) Cancer Res., 50, 3141–3145. [PubMed] [Google Scholar]
- 14.Yu C.E., Oshima,J., Fu,Y.H., Wijsman,E.M., Hisama,F., Alisch,R., Matthews,S., Nakura,J., Miki,T., Ouais,S. et al. (1996) Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 15.Morozov V., Mushegian,A.R., Koonin,E.V. and Bork,P. (1997) Trends. Biochem. Sci., 22, 417–418. [DOI] [PubMed] [Google Scholar]
- 16.Seki M., Miyazawa,H., Tada,S., Yanagisawa,J., Yamaoka,T., Hoshino,S., Ozawa,K., Eki,T., Nogami,M. and Okumura,K. (1994) Nucleic Acids Res., 22, 4566–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitao S., Ohsugi,I., Ichikawa,K., Goto,M., Furuichi,Y. and Shimamoto,A. (1998) Genomics, 54, 443–452. [DOI] [PubMed] [Google Scholar]
- 18.Kitao S., Shimamoto,A., Goto,M., Miller,R.W., Smithson,W.A., Lindor,N.M. and Furuichi,Y. (1999) Nature Genet., 22, 82–84. [DOI] [PubMed] [Google Scholar]
- 19.Yamagata K., Kato,J., Shimamoto,A., Goto,M., Furuichi,Y. and Ikeda,H. (1998) Proc. Natl Acad. Sci. USA, 95, 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray M.D., Shen,J.C., Kamath-Loeb,A.S., Blank,A., Sopher,B.L., Martin,G.M., Oshima,J. and Loeb,L.A. (1997) Nature Genet., 17, 100–103. [DOI] [PubMed] [Google Scholar]
- 21.Karow J.K., Chakraverty,R.K. and Hickson,I.D. (1997) J. Biol. Chem., 272, 30611–30614. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki N., Shimamoto,A., Imamura,O., Kuromitsu,J., Kitao,S., Goto,M. and Furuichi,Y. (1997) Nucleic Acids Res., 25, 2973–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen J.C., Gray,M.D., Oshima,J., Kamath-Loeb,A.S., Fry,M. and Loeb,L.A. (1998) J. Biol. Chem., 273, 34139–34144. [DOI] [PubMed] [Google Scholar]
- 24.Huang S., Li,B., Gray,M.D., Oshima,J., Mian,I.S. and Campisi,J. (1998) Nature Genet., 20, 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamath-Loeb A.S., Shen,J.C., Loeb,L.A. and Fry,M. (1998) J. Biol. Chem., 273, 34145–34150. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki N., Shiratori,M., Goto,M. and Furuichi,Y. (1999) Nucleic Acids Res., 27, 2361–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fry M. and Loeb,L.A. (1999) J. Biol. Chem., 274, 12797–12802. [DOI] [PubMed] [Google Scholar]
- 28.Sun H., Karow,J.K., Hickson,I.D. and Maizels,N. (1998) J. Biol. Chem., 273, 27587–27592. [DOI] [PubMed] [Google Scholar]
- 29.Brosh R.M.J., Orren,D.K., Nehlin,J.O., Ravn,P.H., Kenny,M.K., Machwe,A. and Bohr,V.A. (1999) J. Biol. Chem., 274, 18341–18350. [DOI] [PubMed] [Google Scholar]
- 30.Karow J.K., Newman,R.H., Freemont,P.S. and Hickson,I.D. (1999) Curr. Biol., 9, 597–600. [DOI] [PubMed] [Google Scholar]
- 31.Matson S.W. (1986) J. Biol. Chem., 261, 10169–10175. [PubMed] [Google Scholar]
- 32.Matson S.W. and Richardson,C.C. (1983) J. Biol. Chem., 258, 14009–14016. [PubMed] [Google Scholar]
- 33.George J.W., Ghate,S., Matson,S.W. and Besterman,J.M. (1992) J. Biol. Chem., 267, 10683–10689. [PubMed] [Google Scholar]
- 34.Zimmer C. and Wahnert,U. (1986) Prog. Biophys. Mol. Biol., 47, 31–112. [DOI] [PubMed] [Google Scholar]
- 35.Matson S.W. and George,J.W. (1987) J. Biol. Chem., 262, 2066–2076. [PubMed] [Google Scholar]
- 36.Orren D.K., Brosh,R.M.J., Nehlin,J.O., Machwe,A., Gray,M.D. and Bohr,V.A. (1999) Nucleic Acids Res., 27, 3557–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X., Ramakrishnan,B. and Sundaralingam,M. (1997) J. Mol. Biol., 267, 1157–1170. [DOI] [PubMed] [Google Scholar]
- 38.Nunn C.M., Garman,E. and Neidle,S. (1997) Biochemistry, 36, 4792–4799. [DOI] [PubMed] [Google Scholar]
- 39.Kopka M.L., Yoon,C., Goodsell,D., Pjura,P. and Dickerson,R.E. (1985) Proc. Natl Acad. Sci. USA, 82, 1376–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin R.F. and Holmes,N. (1983) Nature, 302, 452–454. [DOI] [PubMed] [Google Scholar]
- 41.Portugal J. and Waring,M.J. (1988) Biochim. Biophys. Acta, 949, 158–168. [DOI] [PubMed] [Google Scholar]
- 42.Tuteja N., Phan,T.N., Tuteja,R., Ochem,A. and Falaschi,A. (1997) Biochem. Biophys. Res. Commun., 236, 636–640. [DOI] [PubMed] [Google Scholar]
- 43.Bachur N.R., Johnson,R., Yu,F., Hickey,R., Applegren,N. and Malkas,L. (1993) Mol. Pharmacol., 44, 1064–1069. [PubMed] [Google Scholar]
- 44.Lown J.W., Morgan,A.R., Yen,S.F., Wang,Y.H. and Wilson,W.D. (1985) Biochemistry, 24, 4028–4035. [DOI] [PubMed] [Google Scholar]
- 45.Barcellona M.L., Favilla,R., von Berger,J., Avitabile,M., Ragusa,N. and Masotti,L. (1986) Arch. Biochem. Biophys., 250, 48–53. [DOI] [PubMed] [Google Scholar]
- 46.Abu-Daya A., Brown,P.M. and Fox,K.R. (1995) Nucleic Acids Res., 23, 3385–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pjura P.E., Grzeskowiak,K. and Dickerson,R.E. (1987) J. Mol. Biol., 197, 257–271. [DOI] [PubMed] [Google Scholar]
- 48.Kopka M.L., Yoon,C., Goodsell,D., Pjura,P. and Dickerson,R.E. (1985) J. Mol. Biol., 183, 553–563. [DOI] [PubMed] [Google Scholar]
- 49.Jeppesen C. and Nielsen,P.E. (1989) Eur. J. Biochem., 182, 437–444. [DOI] [PubMed] [Google Scholar]
- 50.Naegeli H., Modrich,P. and Friedberg,E.C. (1993) J. Biol. Chem., 268, 10386–10392. [PubMed] [Google Scholar]
- 51.Le Gac N.T., Villani,G. and Boehmer,P.E. (1998) J. Biol. Chem., 273, 13801–13807. [DOI] [PubMed] [Google Scholar]
- 52.Gelasco A. and Lippard,S.J. (1998) Biochemistry, 37, 9230–9239. [DOI] [PubMed] [Google Scholar]
- 53.Choi D.J., Marino-Alessandri,D.J., Geacintov,N.E. and Scicchitano,D.A. (1994) Biochemistry, 33, 780–787. [DOI] [PubMed] [Google Scholar]
- 54.Brown W.C. and Romano,L.J. (1989) J. Biol. Chem., 264, 6748–6754. [PubMed] [Google Scholar]
- 55.Kozack R.E. and Loechler,E.L. (1999) Carcinogenesis, 20, 85–94. [DOI] [PubMed] [Google Scholar]
- 56.Hurley L.H., Reynolds,V.L., Swenson,D.H., Petzold,G.L. and Scahill,T.A. (1984) Science, 226, 843–844. [DOI] [PubMed] [Google Scholar]
- 57.Scahill T.A., Jensen,R.M., Swenson,D.H., Hatzenbuhler,N.T., Petzold,G., Wierenga,W. and Brahme,N.D. (1990) Biochemistry, 29, 2852–2860. [DOI] [PubMed] [Google Scholar]
- 58.Maine I.P., Sun,D., Hurley,L.H. and Kodadek,T. (1992) Biochemistry, 31, 3968–3975. [DOI] [PubMed] [Google Scholar]
- 59.Mote J.J., Ghanouni,P. and Reines,D. (1994) J. Mol. Biol., 236, 725–737. [DOI] [PubMed] [Google Scholar]