

Abstract
Rationale
Indirect airway hyperresponsiveness (AHR) is a highly specific feature of asthma, but the underlying mechanisms responsible for driving indirect AHR remain incompletely understood.
Objectives
To identify differences in gene expression in epithelial brushings obtained from individuals with asthma who were characterized for indirect AHR in the form of exercise-induced bronchoconstriction (EIB).
Methods
RNA-sequencing analysis was performed on epithelial brushings obtained from individuals with asthma with EIB (n = 11) and without EIB (n = 9). Differentially expressed genes (DEGs) between the groups were correlated with measures of airway physiology, sputum inflammatory markers, and airway wall immunopathology. On the basis of these relationships, we examined the effects of primary airway epithelial cells (AECs) and specific epithelial cell–derived cytokines on both mast cells (MCs) and eosinophils (EOS).
Measurements and Main Results
We identified 120 DEGs in individuals with and without EIB. Network analyses suggested critical roles for IL-33–, IL-18–, and IFN-γ–related signaling among these DEGs. IL1RL1 expression was positively correlated with the density of MCs in the epithelial compartment, and IL1RL1, IL18R1, and IFNG were positively correlated with the density of intraepithelial EOS. Subsequent ex vivo modeling demonstrated that AECs promote sustained type 2 (T2) inflammation in MCs and enhance IL-33–induced T2 gene expression. Furthermore, EOS increase the expression of IFNG and IL13 in response to both IL-18 and IL-33 as well as exposure to AECs.
Conclusions
Circuits involving epithelial interactions with MCs and EOS are closely associated with indirect AHR. Ex vivo modeling indicates that epithelial-dependent regulation of these innate cells may be critical in indirect AHR and modulating T2 and non-T2 inflammation in asthma.
Keywords: asthma, airway hyperresponsiveness, mast cell, eosinophil, airway epithelium
At a Glance Commentary
Scientific Knowledge on the Subject
Despite its specificity for asthma, the precise mechanisms contributing to indirect airway hyperresponsiveness (AHR) are not entirely understood.
What This Study Adds to the Field
Here, we perform a transcriptomic analysis of epithelial brushings obtained from individuals with asthma to identify specific pathways associated with indirect AHR, which demonstrates a critical role for epithelial regulation of mast cells and eosinophils in modulating type-2 and non–type-2 mechanisms of airway inflammation in asthma.
Airway hyperresponsiveness (AHR) is a cardinal feature of asthma and a key driver of symptomatology. Although AHR testing is limited to individuals with sufficient baseline lung function because of safety concerns, it remains a critical feature of asthma at all levels of severity (1). There are two distinct forms of AHR (1): direct or “exogenous” AHR, which occurs when an exogenous stimulus such as methacholine directly stimulates smooth muscle contraction; and (2) indirect or “endogenous” AHR, which occurs after a stimulus such as exercise or allergen challenge that promotes the release of mediators endogenous to the airways and results in bronchoconstriction. It is important to note that direct AHR is not specific to asthma, as it is present in other airway disorders and is related to changes in airway smooth muscle, baseline lung function, and airway geometry (2). In contrast, indirect AHR is a highly specific feature of asthma associated with persistent disease and correlated with disease severity (1, 3, 4), which has prompted our efforts to better understand the pathobiology of indirect AHR using a cohort of individuals with asthma who have undergone detailed assessments of airway physiology, inflammatory markers from induced sputum, and airway wall immunopathology (5, 6). We have found that the epithelial compartment is uniquely infiltrated by both mast cells (MCs) and eosinophils (EOS) in individuals with indirect AHR in the form of exercise-induced bronchoconstriction (EIB). We have also identified a correlation between indirect AHR and airway T2 gene expression, although a significant number of individuals with asthma and indirect AHR do not have evidence of T2 inflammation. This suggests that both T2 and non-T2 mechanisms contribute to indirect AHR, and it raises important questions about the crosstalk between airway epithelial cells (AECs) and these critical innate immune cells in regulating indirect AHR and airway inflammation in asthma.
Here, we performed the RNA sequencing (RNA-seq) of epithelial brushings obtained from this well-characterized cohort of individuals with asthma and identified differentially expressed genes (DEGs) between individuals with and without indirect AHR. We then correlated the expression of these DEGs with measures of airway physiology, induced sputum inflammatory markers, and the precise location of MCs and EOS in the airway wall of endobronchial tissue. On the basis of these results, we conducted ex vivo modeling to examine the effects of primary AECs and epithelial cell–derived cytokines on both MCs and EOS. Overall, we identify several novel pathways by which the epithelium modulates MC and EOS gene expression to support T2 and non-T2 inflammation.
Methods
Study Population
We conducted a cross-sectional study of individuals with asthma who have been described previously (5, 6). Briefly, all participants underwent assessment of direct AHR by means of methacholine challenge testing and endogenous AHR in the form of EIB through a dry-air exercise challenge. Participants were required to have a positive methacholine challenge test (provocation concentration causing a 20% fall in FEV1, <8 mg/ml), have mild to moderately severe asthma, and remain off controller therapy for the duration of the study. Individuals were determined to have indirect AHR in the form of EIB by a ⩾10% decrease in FEV1 after an exercise challenge. Study participants underwent induced sputum testing for sputum cytology analysis. To assess airway T2 gene expression in this cohort, the expression levels of IL4, IL5, and IL13 in induced sputum cells were centered, scaled, and combined into the summary T2 gene mean (T2GM) metric as previously described (5, 7). Using this metric, participants are considered to have T2-high asthma if the T2GM value is >2 SD from the mean of the healthy control population, which has been previously described (5). Finally, participants underwent research bronchoscopy, which included the collection of epithelial brushings and endobronchial biopsies (see Figure E1 in the online supplement). All participants provided informed consent, and the study protocol was approved by the University of Washington Institutional Review Board.
Bronchoscopic Collection of Epithelial Brushings
Research bronchoscopy was conducted, and four epithelial brushings were obtained from second- to fifth-generation airways of the left lower lobe and lingula using a 3-mm nylon cytology brush (8). The four brushings from individual participants were pooled and placed in TRIzol reagent, and then RNA was isolated using the Direct-zol RNA MiniPrep Kit (Zymo Research). Epithelial brushings were available from 20 individuals with asthma (9 individuals without EIB and 11 individuals with EIB).
RNA-Seq of Airway Epithelial Brushings
RNA-seq was performed at the University of Washington’s Northwest Genomics Center (https://nwgc.gs.washington.edu/). Briefly, library generation was performed with the TruSeq Stranded mRNA Library Prep Kit (Illumina), using 1 μg total RNA. Sequencing was performed using an Illumina HiSeq 4000 instrument. Sequences were aligned to GRCh37 using STAR, v2.5.3a. Transcript abundances were quantified using Kallisto, v0.43.1. Genes with a raw count of at least 10 went into further analysis, leaving 26,373 unique genes.
Bioinformatics Analysis
Differential gene expression analysis between asthmatic individuals with and without EIB was performed using the DESeq2 program in R (9). DEGs were defined as those with a log2(fold change) ⩾1.0 or ⩽−1.0 and a false discovery rate ⩽0.1 using the Benjamini-Hochberg procedure. The functional associations and biological significance of DEGs were assessed using WebGestalt (the Web-based Gene Set Analysis Toolkit) (10). Gene Ontology biologic processes, molecular functions, and cellular components, as well as canonical pathways from the Kyoto Encyclopedia of Genes and Genomes and Reactome, were used as databases. Ingenuity Pathway Analysis (QIAGEN; www.qiagen.com/ingenuity) was performed on DEGs and was used to construct regulatory networks (11).
Immunohistochemistry and Design-based Stereology
We previously used design-based stereology to precisely quantify the density and determine the location of both EOS (available in n = 19 individuals, 9 without EIB and 10 with EIB) and MCs (available in n = 18 individuals, 8 without EIB and 10 with EIB) in the epithelium and subepithelial space of endobronchial tissue in this study population (5, 6).
Primary AEC-MC Coculture Studies
AECs were isolated from tracheal segments of individuals without known airway or lung disease as previously described (5, 12). AEC air-liquid interface (ALI) organotypic cultures were differentiated in PneumaCult-ALI Medium (STEMCELL Technologies) on 12-mm (0.4-μm–pore) permeable polyester membrane Transwell inserts (Corning). Human MC (HuMC) cultures included either the Laboratory of Allergic Diseases-2 (LAD2) MC line (13) or HuMCs developed from CD34 + cells isolated from the peripheral blood of human donors and maintained in cell culture for 10–12 weeks as previously described (14). LAD2 MCs or HuMCs were transferred to the basolateral compartment at a concentration of 0.2 × 106 cells per milliliter. Coculture assays were performed in a 1:1 mixture of PneumaCult-ALI Medium and media specific for either LAD2 MCs or HuMCs. Human IL-33 (Peprotech) and dexamethasone (Sigma-Aldrich) were added to the cell culture media. RNA was isolated from MCs, and quantitative PCR analysis was conducted using TaqMan primer probe sets with quantification of IL4, IL5, and IL13 genes relative to the endogenous control gene HPRT1 using the ΔCt method.
Ex Vivo EOS Stimulation Studies
Blood samples were obtained from individuals with a physician diagnosis of asthma and/or allergic rhinoconjunctivitis. Granulocytes were isolated from peripheral blood by density gradient centrifugation, followed by negative immunomagnetic selection of EOS (6). EOS were treated with human IL-18 (R&D Systems) and/or IL-33 (Peprotech). RNA was isolated from EOS, and qPCR analysis was conducted using TaqMan primer probe sets with quantification of IL13, IL18, and IFNG genes relative to the endogenous control gene HPRT1 using the ΔCt method.
Primary AEC-EOS Coculture Studies
AECs isolated from tracheal segments of individuals without known airway or lung disease were differentiated at the ALI as described earlier. EOS isolated from peripheral blood, as described earlier, were transferred to the basolateral compartment at a concentration of 0.5 × 106 cells per milliliter in a 1:1 mixture of PneumaCult-ALI Medium and RPMI with 2% fetal calf serum.
Statistics
Statistical methods are explicitly discussed in the individual figure legends and are detailed in the online supplement.
Results
Study Population Characteristics
The characteristics of individuals with asthma with EIB (n = 11) and without EIB (n = 9), from whom we obtained epithelial brushings for RNA-seq analysis, are presented in Table 1. There were no significant differences in the age, sex, body mass index, baseline FEV1 percent predicted, direct AHR, sputum eosinophilia, density of MCs or EOS in the subepithelial space, or number of positive allergens on skin prick testing. Individuals with EIB had more airflow obstruction measured by the FEV1/FVC ratio, increased airway expression of T2 genes measured by the sputum T2GM, and increased densities of MCs and EOS within the epithelial compartment of the airway wall relative to individuals without EIB.
Table 1.
Baseline Characteristics of the Study Groups
| Characteristic | Individuals with Asthma |
P Value | |
|---|---|---|---|
| Without EIB (n = 9) | With EIB (n = 11) | ||
| General | |||
| Age, yr, mean ± SD | 25.6 ± 5.3 | 24.8 ± 7.5 | 0.81 |
| Male, n (%) | 2 (22.2) | 3 (27.3) | 0.40 |
| BMI, kg/m2, mean ± SD | 22.7 ± 2.7 | 23.8 ± 3.9 | 0.48 |
| Airway physiology, mean ± SD | |||
| FEV1, % predicted | 93.4 ± 9.7 | 90.2 ± 11.5 | 0.51 |
| FVC, % predicted | 96.0 ± 8.6 | 102.9 ± 6.0 | 0.05 |
| FEV1/FVC ratio | 0.82 ± 0.06 | 0.75 ± 0.09 | 0.04 |
| Methacholine PC20 | 1.97 ± 1.32 | 0.85 ± 1.89 | 0.15 |
| Exercise challenge. mean ± SD | |||
| Maximum fall in FEV1 (%) | 2.7 ± 2.9 | 24.7 ± 8.8 | <0.0001 |
| Severity of EIB (AUC30) | −0.9 ± 71.3 | 544.3 ± 251.2 | <0.0001 |
| Induced sputum markers, mean ± SD | |||
| T2GM | −0.76 ± 0.67 | −0.15 ± 0.37 | 0.04 |
| Sputum eosinophils, ×103/ml | 7.3 ± 6.9 | 11.9 ± 11.3 | 0.30 |
| Sputum eosinophils, % | 1.48 ± 1.47 | 1.46 ± 1.76 | 0.99 |
| Airway wall morphometry, mean ± SD | |||
| Epithelial MCs, no./mm2 | 31.4 ± 43.8 | 262.2 ± 171.4 | 0.004 |
| Subepithelial MCs, no./mm2 | 377.0 ± 276.6 | 559.3 ± 478.4 | 0.35 |
| Epithelial EOS, no./mm2 | 5.4 ± 8.4 | 20.1 ± 23.1 | 0.07 |
| Subepithelial EOS, no./mm2 | 1,795.3 ± 1,962.6 | 3,461.8 ± 5,121.1 | 0.32 |
| Allergen skin prick testing, mean ± SD | |||
| No. of positive allergens | 4.0 ± 5.0 | 7.4 ± 2.8 | 0.11 |
Definition of abbreviations: AUC30 = area under the curve of FEV1 versus time curve during exercise challenge; BMI = body mass index; EIB = exercise-induced bronchoconstriction; EOS = eosinophils; MCs = mast cells; PC20 = provocative concentration required to reduce FEV1 by 20%; T2GM = type-2 gene mean.
Values are expressed as means ± SD for continuous variables and number and relative frequencies for categorical variables. P values represent the result of unpaired t tests.
Significant Differential Gene Expression Identified Between Individuals with Asthma on the Basis of the Presence or Absence of Indirect AHR in the Form of EIB
On comparing gene expression profiles of epithelial brushings obtained from asthmatic individuals with and without EIB, we identified 120 DEGs at a false discovery rate ⩽0.1 and with a log2(fold change) ⩾1.0 or ⩽−1.0 (Figure 1A; see Table E1 in the online supplement). Forty-seven DEGs had relatively decreased expression, and 73 DEGs had relatively increased expression in individuals with EIB. These included increased expression of classic T2 inflammatory genes (CLCA1, POSTN, SERPINB2, and NOS2) (15) and MC-related genes (KIT, CPA3, TPSAB1, TPSG1, and SIGLEC6) (16). IL1RL1 (encoding the IL-33 receptor) was increased in individuals with EIB and has previously been implicated as a central regulator of T2 inflammation in asthma (17, 18). We also identified increased expression of genes encoding the IL-18 receptor (IL18R1) and IFN-γ (IFNG) in individuals with EIB, which have previously been implicated in non-T2 mechanisms of airway inflammation (19). Overall, these DEGs were enriched in pathways for “leukocyte activation,” “immune response,” “cytokine production,” “regulation of JAK-STAT cascade,” “regulation of IL-12 production,” and “regulation of KIT signaling” (Figure 1B). A regulatory network analysis suggested that IL-33, IL-18, and IFN-γ, as well as key T2 cytokines (IL-4, IL-5, and IL-13), serve central roles within this gene set implicated in indirect AHR (Figure 1C).
Figure 1.
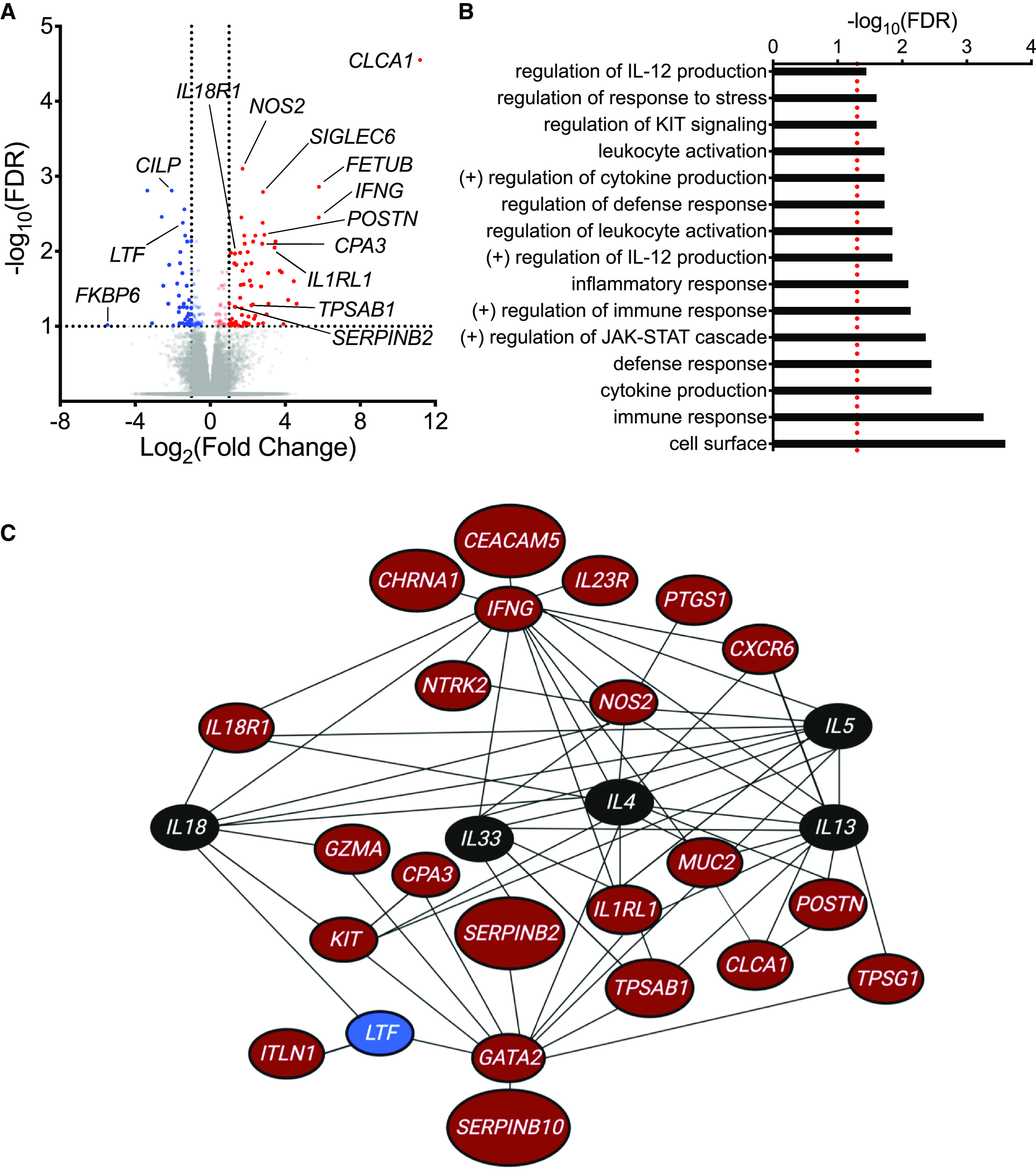
RNA-sequencing analysis of epithelial brushings obtained from individuals with and without exercise-induced bronchoconstriction (EIB). (A) A volcano plot showing the significance and fold-change differences in gene expression between individuals with and without EIB. Red indicates genes that have significantly higher expression, and blue indicates genes that have significantly lower expression in individuals with EIB. False discovery rate (FDR), <0.1; log2(fold change), ⩾1.0 or ⩽−1.0. (B) Gene Ontology biologic processes, cellular components, and molecular functions as well as pathways from the Kyoto Encyclopedia of Genes and Genomes and Reactome databases that were overrepresented among the 120 differentially expressed genes (DEGs) between asthmatic individuals with and without EIB using WebGestalt (FDR <0.05, represented by a red dashed line). (C) Network analysis of DEGs using Ingenuity Pathway Analysis. Genes indicated in red have significantly higher expression, and genes indicated in blue have significantly lower expression in individuals with EIB relative to individuals without EIB. Genes indicated in black are not DEGs identified in our RNA-sequencing analysis but were added to the network to demonstrate interactions between key cytokines of interest (IL4, IL5, IL13, IL18, and IL33) and DEGs.
DEGs Demonstrate Unique Associations with Airway Physiology, Airway Inflammatory Markers, and the Location of MCs and EOS in the Airway Wall
We performed simple linear regression analyses to correlate individual normalized log2 counts of the 120 DEGs with matched measurements of baseline lung function, direct and indirect AHR, airway T2 gene expression, sputum eosinophilia, and the precise location of MCs and EOS within the airway wall (Table 2; see Table E2 in the online supplement). Only two genes were associated with reduced FEV1 (TCN1 and DUOXA2), but several genes were associated with baseline airflow obstruction as measured by the FEV1/FVC ratio, including CD22, CEACAM5, GZMA (encoding granzyme A), NTRK2, CHRNA1, FETUB, and several MC-related genes (CPA3, SIGLEC6, and TPSG1). CEACAM5 and FETUB were also associated with more severe direct AHR to methacholine. Nearly all DEGs with increased expression in individuals with EIB were correlated with the severity of indirect AHR. Multiple DEGs were positively correlated with airway T2 gene expression, including CLCA1, SERPINB2, several MC-related genes (KIT, TPSAB1, CPA3, and SIGLEC6), and the IL-33 receptor (IL1RL1). The density of MCs within the epithelial compartment of the airway wall was positively correlated with multiple MC-related genes (e.g., KIT and CPA3) and genes associated with T2 inflammation (CLCA1, POSTN, SERPINB2, NOS2, and IL1RL1). Finally, 41 DEGs were positively correlated with the density of EOS within the epithelial compartment and included genes associated with MCs (KIT, TPSAB1, TPSG1, CPA3, SIGLEC6), airway mucins (MUC2), prostaglandin synthesis (PTGS1), and T2 inflammation (IL1RL1). However, the density of intraepithelial EOS was also positively correlated with IL18R1 and IFNG expression, suggesting that AEC-EOS interactions may include non-T2 mechanisms.
Table 2.
Correlation of Epithelial Brushing RNA-Sequencing Results with Airway Physiology, Induced Sputum Markers, and Airway Wall Morphometry
| Gene Symbol | Lung Function (FEV1/FVC) | Direct AHR (MCh PC20) | Indirect AHR |
Induced Sputum |
Airway Wall Morphometry |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Max EIB | AUC30 | T2GM | Sputum EOS | Epi MCs | Subepi MCs | Epi EOS | Subepi EOS | |||
| T2 inflammation-associated genes | ||||||||||
| CLCA1 | — | — | 0.48 | 0.41 | 0.32 | — | 0.41 | — | — | — |
| POSTN | — | — | 0.48 | 0.35 | — | — | 0.72 | — | — | — |
| SERPINB2 | — | — | 0.48 | 0.43 | 0.32 | — | 0.40 | — | — | — |
| NOS2 | — | — | 0.42 | 0.37 | — | — | 0.55 | — | — | — |
| Mast cell–related genes | ||||||||||
| CPA3 | 0.20 | — | 0.51 | 0.47 | 0.54 | — | 0.32 | — | 0.36 | — |
| KIT | — | — | 0.31 | 0.28 | 0.32 | — | 0.25 | — | 0.31 | — |
| SIGLEC6 | 0.26 | — | 0.52 | 0.49 | 0.40 | — | 0.25 | — | 0.30 | — |
| TPSAB1 | — | — | 0.44 | 0.38 | 0.58 | — | — | — | 0.45 | — |
| TPSG1 | 0.28 | — | 0.24 | 0.22 | — | — | — | 0.28 | 0.35 | — |
| Cytokines/inflammatory receptors | ||||||||||
| CXCR6 | — | — | — | — | — | — | — | 0.35 | 0.44 | — |
| IFNG | — | — | — | — | — | 0.24 | — | 0.40 | 0.58 | — |
| IL18R1 | — | — | 0.29 | 0.29 | — | — | — | 0.25 | 0.29 | — |
| IL1RL1 | — | — | 0.49 | 0.46 | 0.47 | — | 0.41 | — | 0.30 | — |
| IL23R | — | — | 0.46 | 0.44 | — | — | 0.43 | — | — | — |
| P2RY10 | — | — | — | — | — | — | — | 0.31 | 0.56 | — |
Definition of abbreviations: AHR = airway hyperresponsiveness; AUC30 = area under the curve of FEV1 versus time curve during exercise challenge; EIB = exercise-induced bronchoconstriction; EOS = eosinophils; Epi = the density of a specific cell population within the epithelial compartment of the airway wall (no./mm2); Max EIB = maximum percentage decline in FEV1 from baseline after exercise challenge; MCh PC20 = provocative concentration of methacholine required to reduce FEV1 by 20%; MCs = mast cells; N/A = not applicable; Sputum EOS = the concentration of EOS identified in induced sputum (×103 cells per milliliter); Subepi = the density of a specific cell population in the subepithelial compartment of the airway wall (no./mm2); T2 = type 2; T2GM = type-2 gene mean.
This heatmap shows P values from a simple linear regression analysis of 15 selected differentially expressed genes of interest (genes implicated in type-2 inflammation, mast cell gene expression, and inflammatory cytokines and their receptors) that had relatively increased expression in individuals who had asthma with exercise-induced bronchoconstriction versus individuals who had asthma without exercise-induced bronchoconstriction with airway physiology, induced sputum markers, and airway wall morphometry. Blue indicates a negative relationship, and red indicates a positive relationship, between gene expression and the selected parameter. Correlations highlighted in blue or red in the heatmap have a P value <0.05, and the numerical values listed within the boxes represent the coefficients of determination (r2) obtained from a simple linear regression analysis. Dashes in gray boxes indicate no significant correlation.
AECs Induce Sustained T2 Inflammation in MCs
Given the correlations between intraepithelial MCs and IL1RL1 expression (Figure 2A), we were interested in exploring the relationship between MCs and AECs in the context of treatment with IL-33. LAD2 MCs exposed to primary AECs for 48 hours demonstrated increased expression of genes encoding critical T2 cytokines, primarily IL4 and IL5, but also modest increases in IL13 expression (Figures 2B–2D). Additionally, IL-33 stimulation of MCs alone did not alter MC T2 gene expression at 48 hours, but IL-33 stimulation in the presence of AECs significantly enhanced MC T2 gene expression. This phenomenon of epithelial-dependent, sustained T2 gene expression in MCs after treatment with IL-33 was only partly attenuated by adding maximal ex vivo doses of dexamethasone (Figure 2E), which we also validated in HuMCs (Figure 2F) (20). Overall, these findings suggest that AECs and IL-33, a critical epithelial cell–derived alarmin and central regulator of T2 inflammation, promote sustained T2 gene expression in MCs that is only partially suppressed by corticosteroids.
Figure 2.
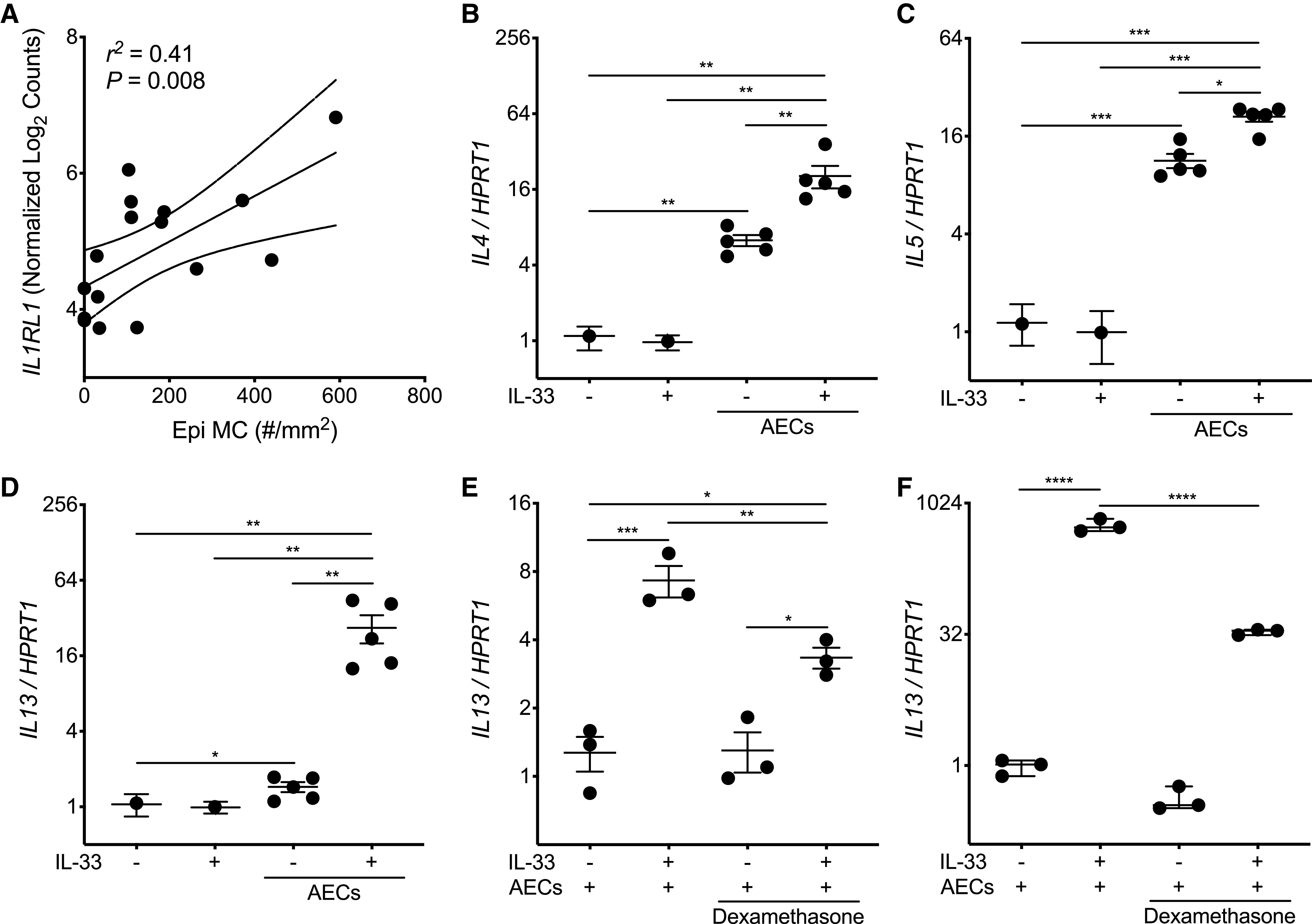
Airway epithelial cells (AECs) promote mast cell (MC) type-2 (T2) gene expression at baseline and enhance sustained T2 gene expression in response to IL-33. (A) Simple linear regression analysis of the density of MCs in the epithelial compartment (Epi MC) versus IL1RL1 expression identified from the epithelial brushings of matched study participants. A solid linear regression line is shown, with the 95% confidence interval indicated with dotted lines. (B–D) Quantitative PCR analysis for expression of IL4 (B), IL5 (C), and IL13 (D) (in comparison with the housekeeping gene HPRT1) in the Laboratory of Allergic Diseases-2 (LAD2) MCs cultured for 48 hours in the presence or absence of AECs obtained from five healthy adult AEC donors and/or IL-33 (10 ng/ml). Individual data points represent the mean value of four LAD2 MC replicates, and each replicate was the result of three PCR reactions. Thus, data columns with AECs have five individual data points, each representing four LAD2 MC replicates cocultured with a single AEC donor. Mean values are indicated, with error bars representing the SEM. P values represent the result of one-way ANOVAs with repeated measures. (E) Quantitative PCR of LAD2 MCs (n = 4; each replicate was the result of three PCR reactions) cocultured with a single healthy adult AEC donor in the presence of IL-33 (10 ng/ml) or dexamethasone (1 μM). Mean values are indicated, with error bars representing the SEM. P values represent the result of a one-way ANOVA. (F) Human MCs derived from CD34+ cells isolated from the peripheral blood of a single donor were cocultured with primary AECs from a single healthy adult donor for 48 hours in the presence or absence of IL-33 (10 ng/ml) and/or dexamethasone (1 μM). n = 3 per condition, and each data point is the result of three PCR reactions. Mean values are indicated, with error bars representing the SEM. P values represent the result of a one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
IL-18, IL-33, and AECs Induce Expression of IL-13 and IFN-γ in EOS
IL1RL1 expression was also positively correlated with the density of intraepithelial EOS, suggesting that IL-33 and AECs may similarly induce T2 gene expression in EOS (Figure 3A). However, the density of intraepithelial EOS was also positively correlated with expression of the IL-18 receptor and IFN-γ (Figures 3B–3C), and IL1RL1 and IL18R1 expression were each positively correlated with IFNG expression (Figures 3D–3E). IL-18 is known to induce IFN-γ through the IL-18 receptor (21), which has been implicated in non-T2 mechanisms of airway inflammation in asthma but has not previously been investigated in EOS. Similarly, IL-33 has been shown to induce IFN-γ production in natural killer cells and γδ T cells (22), but this also has not been demonstrated in EOS. Given that airway eosinophilia has classically been associated with T2 inflammation, we assessed the effects of IL-33, IL-18, and coculture with primary AECs on T2 and non-T2 gene expression by EOS. IL-18 and IL-33 both increased the expression of IL13, IFNG, and IL18 in EOS (Figures 4A–4C), and we found that primary AECs also induce IFNG expression in EOS (Figure 4D). IL13 expression increased in two EOS donors and decreased in one EOS donor cocultured with AECs, suggesting that AEC effects on EOS T2 gene expression may be heterogeneous across individual donors (Figure 4E). Overall, these findings implicate EOS as sources of T2 and non-T2 cytokines in the airways, particularly EOS within the epithelial compartment where they are in immediate proximity to AECs and epithelial cell–derived cytokines.
Figure 3.
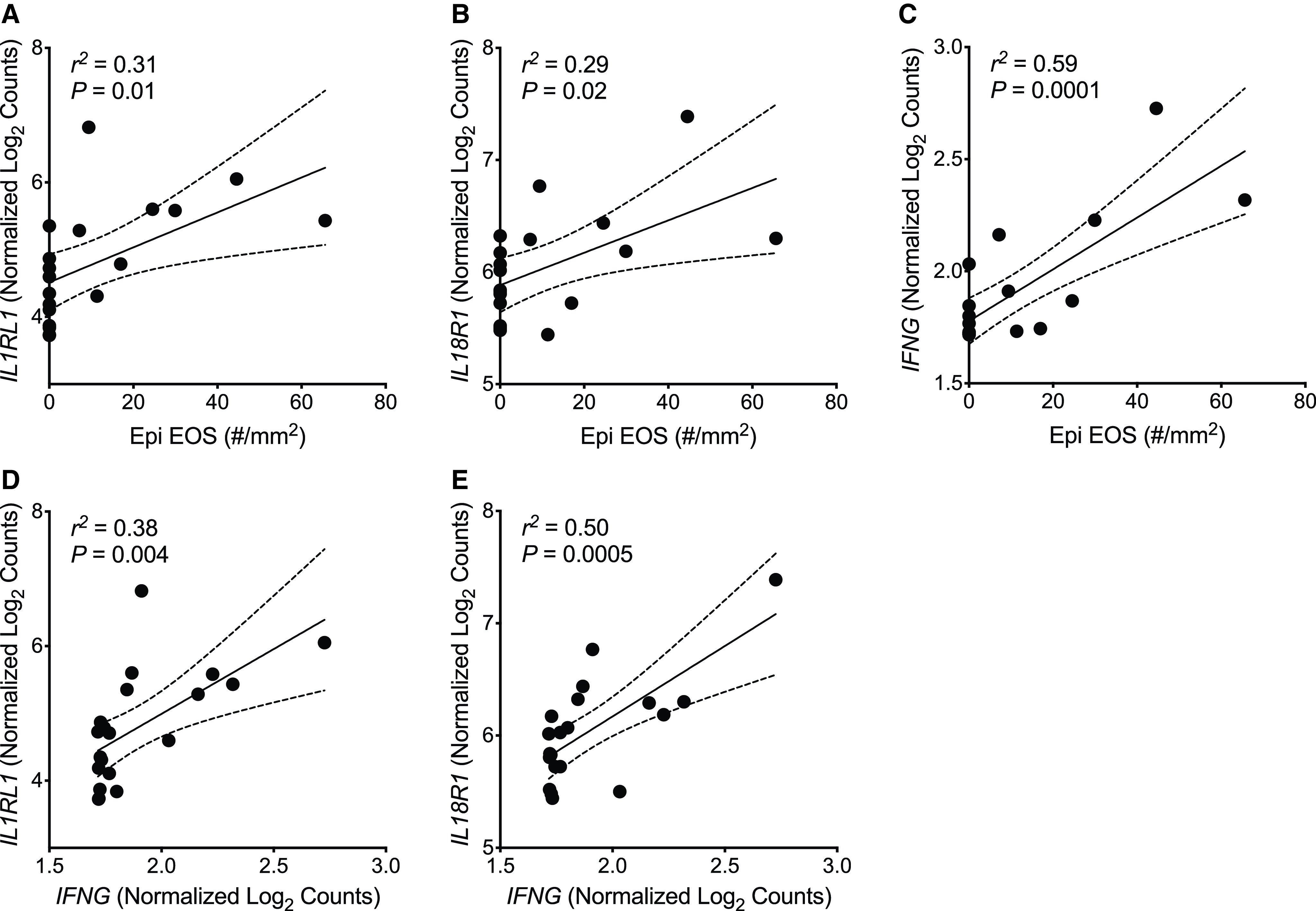
Eosinophils (EOS) within the epithelial compartment are positively correlated with expression of IFN-γ and the receptors for IL-33 and IL-18. (A–C) Simple linear regression analyses of the density of EOS within the epithelial compartment (Epi EOS) and expression of IL1RL1 (A), IL18R1 (B), and IFNG (C) identified from the epithelial brushings of matched study participants. (D and E) Simple linear regression analyses of IFNG expression and expression of IL1RL1 (D) and IL18R1 (E) identified from the epithelial brushings of matched study participants. A solid linear regression line is shown, with the 95% confidence interval indicated with dotted lines.
Figure 4.
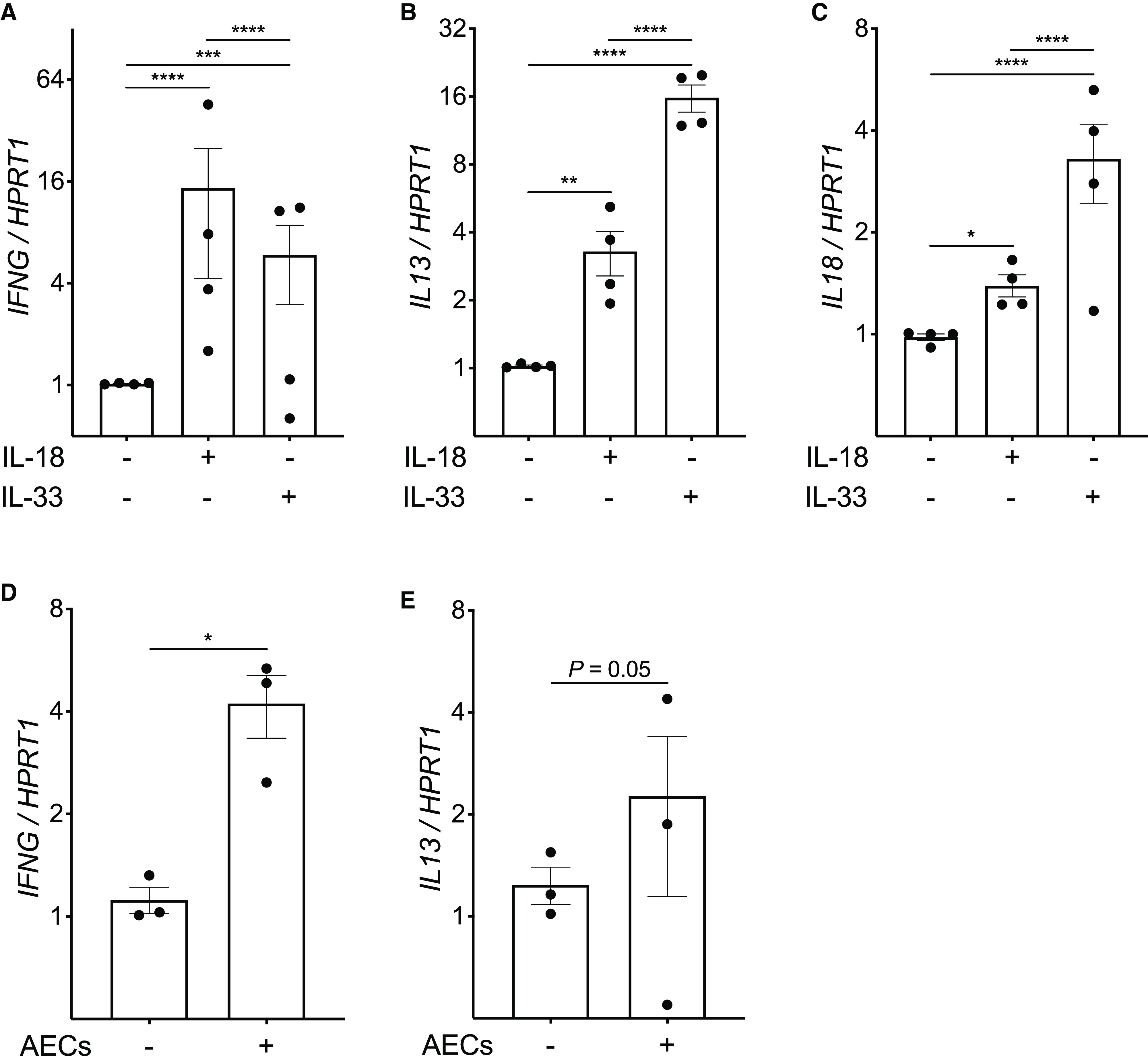
IL-18, IL-33, and airway epithelial cells (AECs) induce IFN-γ and type-2 (T2) gene expression in eosinophils (EOS). (A–C) Quantitative PCR analysis for expression of IFNG (A), IL13 (B), and IL18 (C) (in comparison with the housekeeping gene HPRT1) in EOS isolated from the peripheral blood of four human donors cultured for 2 hours in the presence or absence of IL-18 (10 ng/ml) or IL-33 (10 ng/ml). Individual data points represent the mean value of four EOS replicates per EOS donor; each replicate was the result of three PCR reactions. Mean values are indicated, with error bars representing the SEM. (D and E) Quantitative PCR analysis for expression of IFNG (D) and IL13 (E) in EOS isolated from the peripheral blood of three human donors cultured for 8 hours in the presence or absence of primary AECs from a single healthy adult donor. Individual data points represent the mean value of three EOS replicates per EOS donor; each replicate was the result of three PCR reactions. Mean values are indicated, with error bars representing the SEM. P values represent the result of two-way ANOVAs. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Discussion
Here, we aimed to identify the key mechanisms and intercellular signaling pathways responsible for indirect AHR and inflammation in asthma by performing transcriptomic analyses of epithelial brushings obtained from a well-characterized cohort of individuals with asthma. We identified 120 DEGs between individuals with asthma with and without indirect AHR in the form of EIB, which included several novel genes of interest in addition to genes that have previously been implicated in asthma, and used regression analyses to correlate expression of individual genes with measures of airway physiology, airway inflammatory markers, and airway immunopathology. Our analysis implicates IL-33 as a key regulator of MC interactions with AECs. This prompted further ex vivo coculture modeling that demonstrated that AECs promote basal T2 gene expression in MCs and a marked epithelial-dependent increase in MC T2 gene expression after treatment with IL-33. Our analysis also implicated IL-33 and IL-18 in promoting T2 and non-T2 responses in EOS. Subsequent ex vivo modeling unexpectedly revealed that both IL-33 and IL-18 induce EOS IL13 and IFNG expression and that AECs also induce this response in EOS. Together, these results identify specific pathways associated with indirect AHR in asthma and reveal a critical role for epithelial regulation of MCs and EOS in modulating T2 and non-T2 mechanisms of airway inflammation in asthma.
Despite the specificity of indirect AHR for asthma, the precise mechanisms contributing to indirect AHR are not entirely understood (1). We have recently shown that the epithelial compartment of the airway wall is uniquely infiltrated by MCs and EOS in asthmatic individuals with indirect AHR and that the presence of intraepithelial MCs and EOS is associated with airway T2 gene expression (5, 6). We have also demonstrated that indirect AHR is overall associated with airway T2 gene expression, but several individuals in our cohort with indirect AHR do not have an elevated sputum T2GM, suggesting that non-T2 mechanisms may play a role in indirect AHR either alone or in addition to T2 mechanisms. Here, our sequencing analysis identified increased expression of genes associated with T2 inflammation (CLCA1, POSTN, SERPINB2, and NOS2) in individuals with EIB. However, we also found increased expression of several genes associated with non-T2 inflammation, such as IL18R1 and IFNG, implicating specific non-T2 mechanisms in indirect AHR. Additionally, we identified increased expression of MC-related genes in individuals with EIB, including KIT, CPA3, TPSAB1, TPSG1, and SIGLEC6. Of note, increased expression of the genes encoding the MC proteases carboxypeptidase 3 (CPA3) and tryptase is consistent with prior studies that identified an MC population with high expression of CPA3, persistent expression of tryptase, and loss of chymase expression (MCT/CPA3) as a unique subpopulation found in individuals with T2-high asthma (5, 23). We did not identify increased expression of EOS-specific genes (such as IL5RA, CCR3, CLC, MBP, or ECP) in our analysis, which is potentially due to the smaller difference in the density of intraepithelial EOS between individuals with and without EIB that were included in the transcriptomics analysis for this study. Overall, our sequencing analysis implicates both T2 and non-T2 mechanisms of airway inflammation in indirect AHR, is consistent with prior studies demonstrating the importance of intraepithelial MCs in indirect AHR, and supports the role of unique MC subpopulations in the airways of individuals with asthma.
Given the heterogeneous composition of the epithelial compartment of individuals with asthma, we performed regression analyses between the expression of individual DEGs and the density of intraepithelial MCs and EOS to guide further investigations into the role of the epithelium and epithelial cell–derived cytokines in regulating MC- and EOS-specific immune responses relevant to indirect AHR. We found strong correlations between expression of the gene encoding the IL-33 receptor (IL1RL1) and intraepithelial MCs, which is highly relevant to asthma pathogenesis, as IL-33 is a critical epithelial cell–derived alarmin released in response to proteolytic allergens and respiratory viral infections (17, 24). This prompted further ex vivo coculture studies demonstrating that AECs increase basal MC T2 gene expression. Additionally, although prior studies have shown that IL-33 induces MC T2 responses (5), we show that this effect does not persist at 48 hours. However, IL-33 stimulation of MCs cocultured in the presence of AECs demonstrates a sustained increase in expression of T2 cytokines. Finally, we found that this novel epithelial cell–dependent, persistent T2 response in MCs after IL-33 stimulation is only partially suppressed by maximal dose corticosteroids, implicating this pathway as a potentially critical source of persistent T2 inflammation in individuals with asthma that fail to respond to inhaled corticosteroids (25, 26). These findings also add to the growing evidence of the important role that AECs play in regulating MC function, including reducing degranulation (27) and promoting distinct changes in cell surface markers and gene expression profiles (28, 29).
We also identified novel associations between the density of intraepithelial EOS and the expression of IL1RL1, IL18R1, and IFNG, suggesting that interactions between the epithelium and EOS are critical to indirect AHR and are mediated through both T2 and non-T2 mechanisms. EOS have long been implicated in T2-high asthma as sources of T2 cytokines but are also known to produce non-T2 mediators such as IFN-γ, although the specific role of EOS-derived IFN-γ in asthma has not previously been studied in humans (30, 31). Prior studies have also investigated the effects of epithelial cells on EOS using coculture systems, demonstrating EOS activation after exposure to necrotic epithelial cells (32) and modulating EOS inflammatory responses to antimicrobial peptides (33). More recently, an esophageal epithelial cell line was shown to enhance EOS survival, promote EOS migration, and alter EOS gene expression profiles (34). Here, we show that both AECs and IL-33 not only stimulate IL13 expression in EOS but can also induce IFNG expression, suggesting that IL-33 serves as a pleiotropic proinflammatory alarmin capable of inducing both T2 and non-T2 responses in EOS. We also demonstrate that IL-18 induces EOS expression of both IL13 and IFNG, again implicating EOS in T2 and non-T2 mechanisms in asthma and further suggesting that specific non-T2 mechanisms are critical for indirect AHR. IL-18 is a pleiotropic proinflammatory cytokine that signals through the IL-18 receptor and is capable of inducing IFN-γ in multiple cell types and T2 cytokines in basophils and MCs (21, 35). Multiple studies have previously implicated IL-18 and IFN-γ in asthma, but their precise roles and cellular sources are not well understood. Higher serum IL-18 levels are present in individuals with asthma (36–39), IL18 and IL18R1 gene polymorphisms have been associated with asthma (40–42), and airway biopsies of individuals with fatal asthma have increased immunostaining of IL-18 and the IL-18 receptor within the airway wall (43). A recent study also identified IL-18–mediated signaling as a critical mechanism for a subset of individuals with severe asthma and mixed T1/T2 inflammation (19). In murine models, IFN-γ has been shown to play a key role in promoting AHR (44, 45). In humans, IFN-γ has most frequently been implicated in individuals with severe asthma receiving corticosteroid therapy, who have higher expression of IFNG and numbers of IFN-γ+ staining cells in BAL (43) and airway biopsy samples (46). In contrast, our study population had mild to moderately severe asthma and were not exposed to corticosteroids for the duration of the study, suggesting that IFN-γ plays a critical role in indirect AHR and that EOS may be a novel source of this classical non-T2 mediator.
In summary, we performed transcriptomic analyses on epithelial brushings obtained from individuals with asthma who were further characterized for indirect AHR in the form of EIB. Individuals with EIB had higher expression of genes associated with both T2 and non-T2 mechanisms of airway inflammation, as well as specific MC genes previously implicated in indirect AHR and T2-high asthma. We correlated the expression of individual DEGs increased in individuals with EIB with airway wall morphometry, which implicated specific mechanisms by which the airway epithelium and epithelial cell–derived cytokines could modulate inflammatory signaling in both MCs and EOS. Finally, we modeled these potential mechanisms using ex vivo coculture systems, revealing that AECs sustain T2 gene expression in MCs after IL-33 stimulation, which is only partially suppressed by corticosteroids. We further discovered that AECs, IL-33, and IL-18 each promote both IL13 and IFNG expression in EOS. However, here, we utilized AECs obtained from previously healthy donors rather than AECs obtained from individuals with asthma phenotyped for EIB. We have recently characterized the differential effects of AECs obtained from children with asthma in comparison with AECs obtained from healthy children on LAD2 MC gene expression (47) and suspect that there may also be phenotype-specific changes in epithelial signaling in adult populations with asthma that further modulate interactions with MCs and EOS, which should be the focus of future studies. Collectively, these findings represent novel and potentially targetable mechanisms through which the airway epithelium regulates MCs and EOS within the epithelial compartment to promote indirect AHR and drive both T2 and non-T2 airway inflammation in asthma.
Acknowledgments
Acknowledgment
The authors thank Patrick Heagerty from the University of Washington Department of Biostatistics and Institute for Translational Health Sciences for assistance with the statistical analysis presented in this article.
Footnotes
Supported by NIH grants U19AI125378 (to S.F.Z.), K24AI150991 (to J.S.D.), K24AI130263 and R01HL153979 (to T.S.H.), and F32 HL159889 (to R.C.M.).
Author Contributions: R.C.M., T.S.H., J.S.D., S.A.G., and C.W.F. designed the studies. R.C.M., Y.L., and M.L. performed the experiments. R.C.M. and S.A.G. performed the RNA-sequencing analysis. R.C.M. and T.S.H. wrote the manuscript. R.C.M., M.C.A., T.A.-S., W.A.A., J.S.D., S.F.Z., A.M.P., S.A.G., and T.S.H. edited and revised the manuscript. All authors reviewed and approved the manuscript before submission.
This article has an online supplement, which is accessible from this issue’s table of contents online at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202209-1707OC on March 2, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Hallstrand TS, Leuppi JD, Joos G, Hall GL, Carlsen KH, Kaminsky DA, et al. American Thoracic Society (ATS)/European Respiratory Society (ERS) Bronchoprovocation Testing Task Force ERS technical standard on bronchial challenge testing: pathophysiology and methodology of indirect airway challenge testing. Eur Respir J . 2018;52:1801033. doi: 10.1183/13993003.01033-2018. [DOI] [PubMed] [Google Scholar]
- 2. Coates AL, Wanger J, Cockcroft DW, Culver BH, Carlsen K-H, Diamant Z, et al. Bronchoprovocation Testing Task Force ERS technical standard on bronchial challenge testing: general considerations and performance of methacholine challenge tests. Eur Respir J . 2017;49:1601526. doi: 10.1183/13993003.01526-2016. [DOI] [PubMed] [Google Scholar]
- 3. Stern DA, Morgan WJ, Halonen M, Wright AL, Martinez FD. Wheezing and bronchial hyper-responsiveness in early childhood as predictors of newly diagnosed asthma in early adulthood: a longitudinal birth-cohort study. Lancet . 2008;372:1058–1064. doi: 10.1016/S0140-6736(08)61447-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cabral AL, Conceição GM, Fonseca-Guedes CH, Martins MA. Exercise-induced bronchospasm in children: effects of asthma severity. Am J Respir Crit Care Med . 1999;159:1819–1823. doi: 10.1164/ajrccm.159.6.9805093. [DOI] [PubMed] [Google Scholar]
- 5. Altman MC, Lai Y, Nolin JD, Long S, Chen CC, Piliponsky AM, et al. Airway epithelium-shifted mast cell infiltration regulates asthmatic inflammation via IL-33 signaling. J Clin Invest . 2019;129:4979–4991. doi: 10.1172/JCI126402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Al-Shaikhly T, Murphy RC, Parker A, Lai Y, Altman MC, Larmore M, et al. Location of eosinophils in the airway wall is critical for specific features of airway hyperresponsiveness and T2 inflammation in asthma. Eur Respir J . 2022;60:2101865. doi: 10.1183/13993003.01865-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peters MC, Mekonnen ZK, Yuan S, Bhakta NR, Woodruff PG, Fahy JV. Measures of gene expression in sputum cells can identify TH2-high and TH2-low subtypes of asthma. J Allergy Clin Immunol . 2014;133:388–394. doi: 10.1016/j.jaci.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Busse WW, Wanner A, Adams K, Reynolds HY, Castro M, Chowdhury B, et al. Investigative bronchoprovocation and bronchoscopy in airway diseases. Am J Respir Crit Care Med . 2005;172:807–816. doi: 10.1164/rccm.200407-966WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol . 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res . 2019;47:W199–W205. doi: 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krämer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics . 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol Med . 2005;107:183–206. doi: 10.1385/1-59259-861-7:183. [DOI] [PubMed] [Google Scholar]
- 13. Kirshenbaum AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD. Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase N (CD13) Blood . 1999;94:2333–2342. [PubMed] [Google Scholar]
- 14. Folkerts J, Gaudenzio N, Maurer M, Hendriks RW, Stadhouders R, Tam SY, et al. Rapid identification of human mast cell degranulation regulators using functional genomics coupled to high-resolution confocal microscopy. Nat Protoc . 2020;15:1285–1310. doi: 10.1038/s41596-019-0288-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med . 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robida PA, Rische CH, Morgenstern NB, Janarthanam R, Cao Y, Krier-Burris RA, et al. Functional and phenotypic characterization of Siglec-6 on human mast cells. Cells . 2022;11:1138. doi: 10.3390/cells11071138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gordon ED, Palandra J, Wesolowska-Andersen A, Ringel L, Rios CL, Lachowicz-Scroggins ME, et al. IL1RL1 asthma risk variants regulate airway type 2 inflammation. JCI Insight . 2016;1:e87871. doi: 10.1172/jci.insight.87871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan BCL, Lam CWK, Tam LS, Wong CK. IL33: roles in allergic inflammation and therapeutic perspectives. Front Immunol . 2019;10:364. doi: 10.3389/fimmu.2019.00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Camiolo MJ, Zhou X, Wei Q, Trejo Bittar HE, Kaminski N, Ray A, et al. Machine learning implicates the IL-18 signaling axis in severe asthma. JCI Insight . 2021;6:e149945. doi: 10.1172/jci.insight.149945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rider CF, King EM, Holden NS, Giembycz MA, Newton R. Inflammatory stimuli inhibit glucocorticoid-dependent transactivation in human pulmonary epithelial cells: rescue by long-acting beta2-adrenoceptor agonists. J Pharmacol Exp Ther . 2011;338:860–869. doi: 10.1124/jpet.111.181016. [DOI] [PubMed] [Google Scholar]
- 21. Nakanishi K. Unique action of interleukin-18 on T cells and other immune cells. Front Immunol . 2018;9:763. doi: 10.3389/fimmu.2018.00763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liang Y, Jie Z, Hou L, Yi P, Wang W, Kwota Z, et al. IL-33 promotes innate IFN-γ production and modulates dendritic cell response in LCMV-induced hepatitis in mice. Eur J Immunol . 2015;45:3052–3063. doi: 10.1002/eji.201545696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dougherty RH, Sidhu SS, Raman K, Solon M, Solberg OD, Caughey GH, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J Allergy Clin Immunol . 2010;125:1046–1053.e8. doi: 10.1016/j.jaci.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med . 2014;190:1373–1382. doi: 10.1164/rccm.201406-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peters MC, Kerr S, Dunican EM, Woodruff PG, Fajt ML, Levy BD, et al. National Heart, Lung and Blood Institute Severe Asthma Research Program 3 Refractory airway type 2 inflammation in a large subgroup of asthmatic patients treated with inhaled corticosteroids. J Allergy Clin Immunol . 2019;143:104–113.e14. doi: 10.1016/j.jaci.2017.12.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kraft M, Martin RJ, Lazarus SC, Fahy JV, Boushey HA, Lemanske RF, Jr, et al. Asthma Clinical Research Network Airway tissue mast cells in persistent asthma: predictor of treatment failure when patients discontinue inhaled corticosteroids. Chest . 2003;124:42–50. doi: 10.1378/chest.124.1.42. [DOI] [PubMed] [Google Scholar]
- 27. Martin N, Ruddick A, Arthur GK, Wan H, Woodman L, Brightling CE, et al. Primary human airway epithelial cell-dependent inhibition of human lung mast cell degranulation. PLoS One . 2012;7:e43545. doi: 10.1371/journal.pone.0043545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Derakhshan T, Samuchiwal SK, Hallen N, Bankova LG, Boyce JA, Barrett NA, et al. Lineage-specific regulation of inducible and constitutive mast cells in allergic airway inflammation. J Exp Med . 2021;218:e20200321. doi: 10.1084/jem.20200321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dwyer DF, Ordovas-Montanes J, Allon SJ, Buchheit KM, Vukovic M, Derakhshan T, et al. Human airway mast cells proliferate and acquire distinct inflammation-driven phenotypes during type 2 inflammation. Sci Immunol . 2021;6:eabb7221. doi: 10.1126/sciimmunol.abb7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bousquet J, Chanez P, Lacoste JY, Barnéon G, Ghavanian N, Enander I, et al. Eosinophilic inflammation in asthma. N Engl J Med . 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 31. Spencer LA, Szela CT, Perez SA, Kirchhoffer CL, Neves JS, Radke AL, et al. Human eosinophils constitutively express multiple Th1, Th2, and immunoregulatory cytokines that are secreted rapidly and differentially. J Leukoc Biol . 2009;85:117–123. doi: 10.1189/jlb.0108058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stenfeldt AL, Wennerås C. Danger signals derived from stressed and necrotic epithelial cells activate human eosinophils. Immunology . 2004;112:605–614. doi: 10.1111/j.1365-2567.2004.01906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiao D, Wong CK, Tsang MS, Chu IM, Liu D, Zhu J, et al. Activation of eosinophils interacting with bronchial epithelial cells by antimicrobial peptide LL-37: implications in allergic asthma. Sci Rep . 2017;7:1848. doi: 10.1038/s41598-017-02085-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dunn JLM, Caldwell JM, Ballaban A, Ben-Baruch Morgenstern N, Rochman M, Rothenberg ME. Bidirectional crosstalk between eosinophils and esophageal epithelial cells regulates inflammatory and remodeling processes. Mucosal Immunol . 2021;14:1133–1143. doi: 10.1038/s41385-021-00400-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang W, Tanaka T, Okamura H, Sugita M, Higa S, Kishimoto T, et al. Interleukin-18 enhances the production of interleukin-8 by eosinophils. Eur J Immunol . 2001;31:1010–1016. doi: 10.1002/1521-4141(200104)31:4<1010::aid-immu1010>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 36. Wong CK, Ho CY, Ko FW, Chan CH, Ho AS, Hui DS, et al. Proinflammatory cytokines (IL-17, IL-6, IL-18 and IL-12) and Th cytokines (IFN-gamma, IL-4, IL-10 and IL-13) in patients with allergic asthma. Clin Exp Immunol . 2001;125:177–183. doi: 10.1046/j.1365-2249.2001.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanaka H, Miyazaki N, Oashi K, Teramoto S, Shiratori M, Hashimoto M, et al. IL-18 might reflect disease activity in mild and moderate asthma exacerbation. J Allergy Clin Immunol . 2001;107:331–336. doi: 10.1067/mai.2001.112275. [DOI] [PubMed] [Google Scholar]
- 38. Harada M, Obara K, Hirota T, Yoshimoto T, Hitomi Y, Sakashita M, et al. A functional polymorphism in IL-18 is associated with severity of bronchial asthma. Am J Respir Crit Care Med . 2009;180:1048–1055. doi: 10.1164/rccm.200905-0652OC. [DOI] [PubMed] [Google Scholar]
- 39. Imaoka H, Gauvreau GM, Watson RM, Smith SG, Dua B, Baatjes AJ, et al. Interleukin-18 and interleukin-18 receptor-α expression in allergic asthma. Eur Respir J . 2011;38:981–983. doi: 10.1183/09031936.00033811. [DOI] [PubMed] [Google Scholar]
- 40. Higa S, Hirano T, Mayumi M, Hiraoka M, Ohshima Y, Nambu M, et al. Association between interleukin-18 gene polymorphism 105A/C and asthma. Clin Exp Allergy . 2003;33:1097–1102. doi: 10.1046/j.1365-2222.2003.01739.x. [DOI] [PubMed] [Google Scholar]
- 41. Lee CC, Lin WY, Wan L, Tsai Y, Tsai CH, Huang CM, et al. Association of interleukin-18 gene polymorphism with asthma in Chinese patients. J Clin Lab Anal . 2008;22:39–44. doi: 10.1002/jcla.20218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheng D, Hao Y, Zhou W, Ma Y. The relationship between interleukin-18 polymorphisms and allergic disease: a meta-analysis. BioMed Res Int . 2014;2014:290687. doi: 10.1155/2014/290687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oda H, Kawayama T, Imaoka H, Sakazaki Y, Kaku Y, Okamoto M, et al. Interleukin-18 expression, CD8(+) T cells, and eosinophils in lungs of nonsmokers with fatal asthma. Ann Allergy Asthma Immunol . 2014;112:23–28.e1. doi: 10.1016/j.anai.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 44. Raundhal M, Morse C, Khare A, Oriss TB, Milosevic J, Trudeau J, et al. High IFN-γ and low SLPI mark severe asthma in mice and humans. J Clin Invest . 2015;125:3037–3050. doi: 10.1172/JCI80911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu M, Eckart MR, Morgan AA, Mukai K, Butte AJ, Tsai M, et al. Identification of an IFN-γ/mast cell axis in a mouse model of chronic asthma. J Clin Invest . 2011;121:3133–3143. doi: 10.1172/JCI43598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shannon J, Ernst P, Yamauchi Y, Olivenstein R, Lemiere C, Foley S, et al. Differences in airway cytokine profile in severe asthma compared to moderate asthma. Chest . 2008;133:420–426. doi: 10.1378/chest.07-1881. [DOI] [PubMed] [Google Scholar]
- 47.Murphy RC, Lai Y, Altman MC, Barrow KA, Dill-McFarland KA, Liu M, et al. Rhinovirus infection of the airway epithelium enhances mast cell immune responses via epithelial-derived interferons. J Allergy Clin Immunol . 2023. [DOI] [PMC free article] [PubMed]