

Key Points
-
•
Contact pathway–initiated thrombin generation is increased in C1INH-deficient humans and mice.
-
•
Venous, but not arterial, thrombosis is increased in C1INH-deficient mice.
Visual Abstract
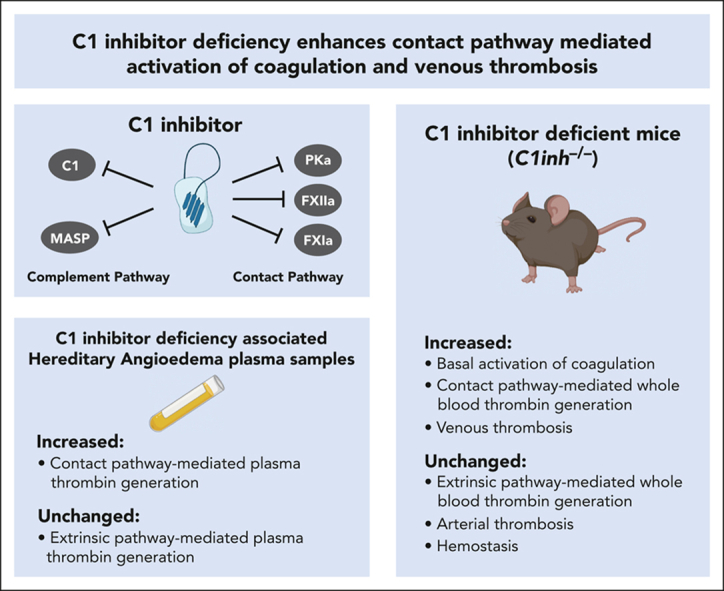
Abstract
C1 inhibitor (C1INH) is a multifunctional serine protease inhibitor that functions as a major negative regulator of several biological pathways, including the contact pathway of blood coagulation. In humans, congenital C1INH deficiency results in a rare episodic bradykinin-mediated swelling disorder called hereditary angioedema (HAE). Patients with C1INH deficiency–associated HAE (C1INH-HAE) have increased circulating markers of activation of coagulation. Furthermore, we recently reported that patients with C1INH-HAE had a moderate but significant increased risk of venous thromboembolism. To further investigate the impact of C1INH deficiency on activation of coagulation and thrombosis, we conducted studies using patient samples and mouse models. Plasmas from patients with C1INH-HAE had significantly increased contact pathway–mediated thrombin generation. C1INH-deficient mice, which have been used as a model of C1INH-HAE, had significantly increased baseline circulating levels of prothrombin fragment 1+2 and thrombin-antithrombin complexes. In addition, whole blood from C1INH-deficient mice supported significantly increased contact pathway–mediated thrombin generation. Importantly, C1INH-deficient mice exhibited significantly enhanced venous, but not arterial, thrombus formation. Furthermore, purified human C1INH normalized contact pathway–mediated thrombin generation and venous thrombosis in C1INH-deficient mice. These findings highlight a key role for endogenous C1INH as a negative regulator of contact pathway–mediated coagulation in humans and mice. Further, this work identifies endogenous C1INH as an important negative regulator of venous thrombus formation in mice, complementing the phenotype associated with C1INH-HAE.
C1 inhibitor (C1INH) is a serine protease inhibitor that is a negative regulator of several pathways, including the contact pathway of coagulation. Congenital absence of C1INH is the major cause of hereditary angioedema (HAE). Grover et al report that patients with HAE have an increased risk of thrombosis, and in this follow-up study, the authors demonstrate that plasma from patients with HAE leads to increased contact-mediated thrombin generation. Furthermore, C1INH-deficient mice have increased venous thrombosis, which is reversible by C1INH administration.
Introduction
C1 inhibitor (C1INH) is a multifunctional serine protease inhibitor that serves as an important endogenous negative regulator of the complement pathway, the contact pathway of coagulation, and the kallikrein-kinin pathway.1,2 C1INH prevents excessive activation of the classical and lectin pathways of complement activation through inhibition of complement component 1 (C1) and mannose-binding lectin-associated serine proteases.3,4 Likewise, C1INH prevents excessive activation of the contact pathway of coagulation by inhibiting activated factor XII (FXIIa) and factor XI (FXIa).5 C1INH is also a key inhibitor of plasma kallikrein (PKa), which is involved in the activation of both the kallikrein-kinin system and the contact pathway of coagulation.6
Congenital C1INH deficiency is the major cause of hereditary angioedema (HAE), a rare autosomal dominant disease with a prevalence of roughly 1 in 50 000 in the general population.7 Most HAE cases are caused by mutations in the gene encoding C1INH, SERPING1, which lead to either a quantitative or qualitative C1INH deficiency.8 In rare cases, HAE can be caused by mutations in other genes, including those that encode FXII, plasminogen, and high molecular weight kininogen.9, 10, 11 C1INH deficiency-associated HAE (C1INH-HAE) presents with recurrent, unpredictable, and potentially life-threatening episodes of swelling.7 Angioedema may involve the subcutaneous tissue (extremities, trunk, face, and genitals) and/or submucosal tissue of the upper airway and gastrointestinal tract.7 Angioedema observed in patients with C1INH-HAE is driven by uncontrolled kallikrein-mediated bradykinin generation.12 Although C1INH-HAE primarily manifests as a swelling disorder, recent evidence suggests that it may also be associated with an increased risk of other comorbidities, including autoimmune and cardiovascular diseases.13
Several prior studies have found that, compared with healthy controls, patients with C1INH-HAE have increased circulating levels of coagulation biomarkers, including prothrombin fragment 1+2 (F1+2), thrombin-antithrombin (TAT) complexes, and D-dimer.14, 15, 16 Patients with C1INH-HAE also have shorter activated partial thromboplastin times (aPTT) compared with healthy controls.16,17 Further, we recently reported an association between C1INH-HAE and an increased risk of venous thromboembolism (VTE).18 Together, these findings suggest that endogenous C1INH plays a physiologically relevant role in regulating coagulation.
To further evaluate the impact of C1INH deficiency on coagulation and thrombosis, we conducted a series of studies using samples from patients with C1INH-HAE and C1INH-deficient mice. Loss of C1INH was found to enhance contact pathway–mediated thrombin generation (TG) in humans and mice. In addition, C1INH deficiency in mice selectively enhanced venous, but not arterial thrombosis, complementing the phenotype recently described in patients with C1INH-HAE.
Methods
Additional methods are available in the supplemental Materials, available on the Blood website.
Human plasma samples
The collection of samples was approved by the institutional review board of Semmelweis University of Budapest, and informed written consent was obtained from each participant in accordance with the Declaration of Helsinki. Fasting citrate anticoagulated blood samples were collected from patients with a confirmed diagnosis of C1INH-HAE during remission (n = 19) and from age- and sex-matched healthy controls (n = 20) (supplemental Table 1). Platelet-poor plasma (PPP) was generated by centrifuging blood at 2500×g for 15 minutes at room temperature and freezing aliquots at −80°C. Plasma levels of C1INH antigen were determined using an in-house radial immunodiffusion assay, and C1INH activity was determined using a commercial assay (MicroVue C1INH Plus, Quidel, San Diego, CA).
Mice
All procedures were approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee and complied with National Institutes of Health guidelines. C1INH-deficient mice (referred to as C1inh−/−) on a pure C57Bl/6J background were obtained from a commercial vendor (Innovative Research, Novi, MI). Heterozygous- and homozygous-deficient breeding colonies were established. All studies were conducted in 8- to 12-week-old male and female mice unless otherwise stated.
Mouse blood collection
Whole blood (WB) was collected from the inferior vena cava (IVC) via 2 methods. For enzyme-linked immunosorbent assays, 200 μL of sodium citrate (3.8%, Ricca Chemicals, Arlington, TX) was injected into the IVC using a 23-gauge needle, and 500 μL of WB was withdrawn. For coagulation experiments, 500 μL of blood was collected directly from the IVC using a 23-gauge needle into 50 μL of sodium citrate (3.8%, Ricca Chemicals) and immediately mixed with corn trypsin inhibitor (CTI, 50 μg/mL final concentration, Haematologic Technologies, Essex, VT). PPP was prepared by centrifugation of WB at 4500×g for 15 minutes at room temperature.
Mouse plasma assays
Plasma levels of F1+2 (EKC37706, Biomatik, Kitchener, ON, Canada), TAT complexes (Enzygnost TAT Micro, Siemens, Munich, Germany), and C5a (DY2150, R&D Systems, Minneapolis, MN) were measured using commercial enzyme-linked immunosorbent assays. PT (Stago, Parsippany, NJ) and aPTT (Stago) assays were conducted using a Start 4 coagulometer (Stago).
Thrombin generation
Human plasma TG was assessed by calibrated automated thrombography, as previously described.19 In brief, per reaction 10 μL of silica (1:45 000 final dilution, Kontact, Pacific Hemostasis, MA) or tissue factor (TF) (1 or 5 pM, Innovin, Siemens) trigger with 4 μM lipids (60% PC, 20% PE, 20% PS, Synapse, Maastricht, The Netherlands) was added to 40 μL of PPP and TG was initiated by automated dispensing of 10 μL of fluorogenic substrate and calcium chloride (FluCa, Stago). For experiments using the TF trigger, CTI (50 μg/mL final concentration, Haematologic Technologies) was added to prevent contact pathway autoactivation.
Mouse WB TG was assessed by calibrated automated thrombography, as previously described.20 In brief, per reaction, 20 μL of silica (1:120 final dilution, Kontact, Pacific Hemostasis) or TF (0.05 pM, Innovin, Siemens) trigger solution containing calcium chloride (16.7 mM) was mixed with 30 μL of citrated WB and 10 μL of fluorogenic substrate (Z-Gly-Gly-Arg-7-amino-4-methylcoumarin, Bachem, Torrance, CA). Fluorogenic substrate cleavage was measured at λex = 355 nm and λem = 460 nm using a Fluoroskan Ascent fluorometer (Thermo Fisher Scientific, Waltham, MA) with Ascent Software (version 2.6, Thermo Fisher Scientific) at 37°C. Thirty-six wells were measured with an integration time of 6 seconds to facilitate continuous mixing.21 In some WB TG experiments, an inhibitory anti-Mouse C5 antibody (10 μg/mL, BB5.1, Antibody Research Corp, Cottleville, MO) or immunoglobulin G (Antibody Research Corp) was added to C1inh−/− mouse WB. In other WB TG experiments, purified human C1INH (0.24-0.96 mg/mL, Berinert, kindly provided by CSL Behring, King of Prussia, PA) or vehicle control was added to C1inh−/− WB.
For mouse PPP TG, 10 μL of silica (1:120 final dilution, Kontact, Pacific Hemostasis) or TF (0.5 pM, Innovin, Siemens) with 4 μM lipids (60% PC, 20% PE, 20% PS, Synapse) was added to 40 μL of 1:3 (plasma:buffer) diluted mouse PPP collected in the presence of CTI (50 μg/mL, Haematologic Technologies). TG was initiated by automated dispensing of 10 μL of fluorogenic substrate and calcium chloride (FluCa, Stago). Substrate cleavage was measured using a fluorescence plate reader (Fluoroscan Ascent, Thermo Fisher Scientific). Data were analyzed using Thrombinoscope software (v5; Thrombinoscope, Maastricht, The Netherlands).
IVC stenosis model
The murine IVC stenosis model of venous thrombosis (VT) was conducted in male mice, as previously described.22 In brief, a midline laparotomy was made, and the bowel was externalized onto wetted gauze. IVC side branches were first ligated using 4 to 0 silk suture. The IVC was then ligated at the level of the left renal vein using a 4 to 0 silk suture with a piece of 5 to 0 prolene suture used as a spacer. The 5 to 0 prolene suture was removed, resulting in stenosis of the IVC. In rescue experiments, C1inh−/− mice were administered human purified C1INH (Berninert, CSL Behring) by intravenous injection at a dose of 15 μg/g or vehicle control 5 minutes before stenosis of the IVC. Thrombus formation was assessed at 24 and 48 hours after induction. The operator was blinded with respect to the genotype and treatment of mice.
Carotid artery ferric chloride model
The murine carotid artery ferric chloride (FeCl3) model was conducted, as previously described.23 Additional details are provided in the supplemental Materials.
Tail tip amputation bleeding model
Mice were anesthetized with a ketamine-xylazine cocktail. The distal 4 mm section of the tail was amputated using a scalpel, and the tail was immediately immersed in prewarmed 37°C normal saline (Mckesson, Irving, TX) in a 50 mL conical centrifuge tube. Bleeding was monitored over a 20-minute period, and active bleeding time was recorded. Blood collected in the 50 mL conical centrifuge tube was mixed 1:1 with ACK lysis buffer (Gibco, Waltham, MA) to lyse erythrocytes. Absorbance was measured at 490 nm (Spectramax M5, Molecular Devices, San Jose, CA), and blood loss was estimated by interpolation from a standard curve generated from known volumes of WB. The operator was blinded with respect to the genotype of mice.
Statistics
Normality of the data was assessed using Shapiro-Wilk tests, and parametric or nonparametric data were selected as appropriate. For two-group analyses, either parametric unpaired Student t tests or nonparametric Mann-Whitney U tests were used. For multigroup analyses, either parametric 1-way analysis of variance with post hoc Bonferroni tests or nonparametric Kruskal-Wallis with post hoc Dunn tests were used. Correlations were assessed using Spearman rank tests. For survival analyses, log-rank tests were used. Differences in thrombus incidence were analyzed using Fischer exact test. P < .05 were considered significant. Data were analyzed using Prism software (Version 9.4, GraphPad, San Diego, CA).
Results
Increased contact pathway–initiated coagulation in human C1INH-deficient plasma
To determine the effect of C1INH deficiency on coagulation in human PPP, TG was measured in samples from patients with C1INH-HAE and matched healthy controls (supplemental Table 1) using calibrated automated thrombography initiated with either silica or TF.
The median silica-initiated TG curves did not differ markedly between plasmas from patients with C1INH-HAE and healthy controls (Figure 1A). There was a nonsignificant trend toward a shorter lag time in C1INH-HAE plasma (Figure 1B). However, peak TG was significantly increased in plasmas from patients with C1INH-HAE compared with those from healthy controls (Figure 1C). Residual plasma levels of C1INH activity varied markedly in C1INH-HAE samples (range: 1%-54% of normal). We therefore subdivided C1INH-HAE samples into those with ≥25% C1INH activity (range: 27%-54% of normal) or those with <25% C1INH activity (range: 1%-24% of normal), resulting in 2 similarly sized subgroups. Subdivision of samples in this manner revealed differences in the median curves of silica-initiated TG between plasmas from patients with C1INH-HAE and healthy controls (Figure 1D). Lag time was significantly reduced (Figure 1E) and peak significantly increased (Figure 1F) in plasmas from patients with C1INH-HAE with <25% C1INH activity compared with those from healthy controls. A trend toward increased endogenous thrombin potential (ETP) in plasmas from patients with C1INH-HAE with <25% C1INH activity compared with those from healthy controls was also observed (supplemental Table 2). Plasmas from patients with C1INH-HAE with ≥25% activity had a trend toward increased peak TG compared with healthy control plasma (Figure 1F). When C1INH activity was considered a continuous variable, a significant positive correlation was observed for lag time (Figure 1G) and a significant negative correlation was observed for peak TG (Figure 1H).
Figure 1.

Increased contact pathway–initiated TG in C1INH-HAE plasma. (A) Median silica-initiated TG curves in plasma from patients with C1INH-HAE (n = 19) or matched controls (n = 20) with quantification of (B) TG lag time and (C) peak TG. (D) Median silica-initiated TG curves in plasma from patients with C1INH-HAE with ≥25% C1INH activity (n = 9), <25% C1INH activity (n = 10), and in matched controls (n = 20) with quantification of TG (E) lag time and (F) peak. Data are represented as individual values with median and interquartile range and analyzed by Kruskal-Wallis with post hoc Dunn’s. Plots of TG (G) lag time and (H) peak vs C1INH activity in plasma from patients with C1INH-HAE and matched controls. Data are represented as individual values with linear regression line. Correlations analyzed using Spearman rank tests. ∗P < .05,
When TG was initiated via the extrinsic pathway using high- (5 pM) or low (1 pM)–dose TF in the presence of the FXIIa inhibitor CTI, no significant difference in lag time, peak, or ETP was observed between plasmas from patients with C1INH-HAE and healthy controls (supplemental Figure 1; supplemental Table 2). The lack of a significant difference was maintained when C1INH-HAE samples were subdivided into those with ≥25% C1INH activity or <25% C1INH activity. However, a nonsignificant trend toward increased peak TG and ETP was noted (supplemental Figure 1; supplemental Table 2). No significant correlations were observed between C1INH activity and TG lag time or peak TG initiated with either 5 or 1 pM TF (supplemental Figure 1).
Characterization of C1INH-deficient mice
C1INH-deficient mice have been developed as a model to study C1INH-HAE, demonstrating significantly increased bradykinin-mediated vascular permeability.24, 25, 26 Consistent with prior reports, heterozygous breeding resulted in the generation of C1inh+/+, C1inh+/−, and C1inh−/− mice at the expected mendelian frequencies (supplemental Table 3).24,25 As expected, C1INH protein levels were markedly reduced in C1inh+/− mice plasma compared with C1inh+/+ mice plasma and undetectable in C1inh−/− mouse plasma (Figure 2A).24,26 Semiquantitative densitometric analysis demonstrated that C1inh+/− mice had a disproportionate ∼80% reduction in plasma C1INH protein compared with C1inh+/+ controls (Figure 2B). Further, at baseline, the ratio of cleaved to total high molecular weight kininogen was significantly increased in the plasma of C1inh−/− mice compared with that of C1inh+/+ controls (Figure 2C; supplemental Figure 2), consistent with increased basal in vivo bradykinin generation.27 Circulating blood cell counts were not significantly different in C1inh+/− and C1inh−/− mice than in C1inh+/+ controls (supplemental Table 4). Histological assessment of organs from C1inh−/− and C1inh+/− mice demonstrated no overt organ pathology compared with those from C1inh+/+ controls (supplemental Figure 3).
Figure 2.

Characterization of C1INH-deficient mice. (A) Western blot of C1INH and transferrin with corresponding (B) densitometric analysis in plasma from C1inh+/+, C1inh+/−, and C1inh−/− mice. (C) Ratio of cleaved to total high molecular weight kininogen in plasma from C1inh+/+, C1inh+/−, and C1inh−/− mice. Data are represented as mean ± standard deviation and analyzed by one-way ANOVA with post hoc Bonferroni. Levels of (D) F1+2 and (E) TAT complexes in plasma from C1inh+/+, C1inh+/−, and C1inh−/− mice. (F) aPTT and (G) prothrombin time assays in plasma from C1inh+/+, C1inh+/−, and C1inh−/− mice. Data are represented as individual values with median and interquartile range and analyzed by Kruskal-Wallis with post hoc Dunn’s. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001 ∗∗∗∗P < .0001. ANOVA, analysis of variance.
Increased basal and contact pathway–initiated coagulation in C1INH-deficient mice
To evaluate basal activation of coagulation in C1INH-deficient mice, levels of plasma markers were measured. Both TAT complexes and F1+2 were significantly increased in the plasma of C1inh−/− mice, but not in C1inh+/− mice, compared with C1inh+/+ littermate controls (Figure 2D-E). By contrast, aPTT and PT clotting times did not differ significantly between plasmas from C1inh−/−, C1inh+/−, and C1inh+/+ mice (Figure 2F-G). To evaluate the effect of C1INH deficiency on contact pathway and extrinsic pathway–mediated coagulation in a more sensitive manner, a WB TG assay was used.20 Representative curves indicated that C1INH-deficient WB supported enhanced silica-initiated TG (Figure 3A). A nonsignificant trend toward shortened TG lag time and a significant increase in peak TG was observed in C1inh−/− mice compared with C1inh+/+ controls (Figure 3B-C). No significant effect of C1INH deficiency on TF-initiated mouse WB TG was observed (Figure 3D-F; supplemental Table 5). Further, C1INH deficiency had no significant effect on silica-initiated or TF-initiated TG in mouse PPP (supplemental Figure 4; supplemental Table 6). Antibody-mediated inhibition of C5a generation had no significant effect on silica-initiated TG in C1inh−/− WB (supplemental Figure 5; supplemental Table 7).
Figure 3.

C1INH deficiency enhances contact pathway–initiated mouse WB TG. (A) Representative silica-initiated TG curves in WB collected from C1inh+/+, C1inh+/−, and C1inh−/− mice with quantification of TG (B) lag time and (C) peak. (D) Representative TF-initiated TG curves in WB collected from C1inh+/+, C1inh+/−, and C1inh−/− mice with quantification of TG (E) lag time and (F) peak. Data are represented as individual values with median and interquartile range and analyzed by Kruskal-Wallis with post hoc Dunn’s. ∗P < .05.
VT is increased in C1INH-deficient mice
To determine the effect of C1INH deficiency on VT, C1inh+/+, C1inh+/−, and C1inh−/− littermate mice were subjected to the IVC stenosis model of VT. Median thrombus weight was significantly increased in C1inh−/− mice compared with C1inh+/+ controls at both 24 hours and 48 hours after induction (Figure 4A-B). A nonsignificant trend toward increased thrombus weight was observed for C1inh+/− mice (Figure 4A-B). Thrombus incidence was not significantly different between C1inh+/+, C1inh+/−, and C1inh−/− mice at 24 hours (6/8 vs 5/9 vs 6/7, respectively) or 48 hours (8/10 vs 9/10 vs 8/9, respectively).
Figure 4.

C1INH deficiency enhances VT in mice. VT was assessed in C1inh+/+, C1inh+/−, and C1inh−/− mice using the IVC stenosis model. Thrombus weight was assessed at (A) 24 hours and (B) 48 hours after IVC stenosis. Data are represented as individual values with median and analyzed by Kruskal-Wallis with post hoc Dunn’s. ∗P < .05.
The composition of venous thrombi was assessed using western blotting of lysates generated from thrombi collected at 48 hours. Levels of fibrin(ogen), the platelet marker CD41, the neutrophil marker Ly6G, the cellular marker β actin, the nucleated cell marker histone H3, and the neutrophil extracellular trap marker citrullinated histone H3 were analyzed (supplemental Figure 6). Semiquantitative densitometric analysis revealed that although there was no significant difference in the relative abundance of these markers in thrombi from C1inh−/− and C1inh+/− mice compared with those from C1inh+/+ controls, there were nonsignificant trends toward increased neutrophil content and reduced neutrophil extracellular trap formation in C1inh−/− mice (supplemental Figure 6). A nonsignificant trend toward increased C5a in the plasma of C1inh−/− mice subjected to the IVC stenosis model compared with those of controls was also observed at 48 hours after induction (supplemental Figure 7).
Arterial thrombosis is not altered in C1INH-deficient mice
To determine the effect of C1INH deficiency on arterial thrombosis, C1inh+/+, C1inh+/−, and C1inh−/− littermate mice were subjected to mild and severe carotid artery FeCl3 injury models. In the severe injury model (8% FeCl3 for 3 minutes), occlusion times in C1inh−/− and C1inh+/− mice did not differ significantly compared with those in C1inh+/+ controls (Figure 5A). Further, no significant difference in carotid artery patency over time was observed between C1inh+/+, C1inh+/−, and C1inh−/− mice (Figure 5B). In the mild injury model (2.5% FeCl3 for 5 minutes), occlusion times in C1inh−/− and C1inh+/− mice did not differ significantly compared with those in C1inh+/+ controls (Figure 5C). Concordantly, carotid artery patency did not differ significantly between C1inh+/+, C1inh+/−, and C1inh−/− mice over time (Figure 5D).
Figure 5.

C1INH deficiency does not enhance arterial thrombosis in mice. Arterial thrombosis was assessed in C1inh+/+, C1inh+/−, and C1inh−/− mice using the carotid artery FeCl3 model under conditions resulting in either severe or mild injury. In the severe model induced with application of 8% FeCl3 for 3 minutes, (A) occlusion time and (B) carotid artery patency were evaluated. Similarly, in the mild model induced with application of 2.5% FeCl3 for 5 minutes, (C) occlusion time and (D) carotid artery patency were evaluated. Data are represented as individual values with median and analyzed using Kruskal-Wallis with post hoc Dunn or log-rank tests.
Tail bleeding is not altered in C1INH-deficient mice
To evaluate the effect of C1INH deficiency on the response to vascular injury, C1inh+/+, C1inh+/−, and C1inh−/− littermate mice were subjected to the tail tip amputation model of hemostasis. No significant difference in the proportion of C1inh+/+, C1inh+/−, and C1inh−/− mice actively bleeding over time was observed (Figure 6A). Further, no significant difference in tail bleeding time (Figure 6B) or blood loss (Figure 6C) was observed.
Figure 6.

C1INH deficiency does not alter hemostasis in mice. Hemostasis was assessed in C1inh+/+, C1inh+/−, and C1inh−/− mice in the tail tip amputation model. (A) The proportion of mice with active bleeding over time, (B) tail bleeding time, and (C) tail bleeding blood loss was assessed. Data are represented as individual values with median and analyzed using Kruskal-Wallis with post hoc Dunn’s or log rank tests.
Purified human C1INH rescues the procoagulant phenotype in C1INH-deficient mice
The effect of exogenous, purified human C1INH on the procoagulant phenotype observed in C1INH-deficient mice was evaluated. The addition of purified human C1INH to C1inh−/− WB ex vivo reduced silica-initiated TG in a dose-dependent manner (Figure 7A). Human purified C1INH significantly prolonged TG lag time (Figure 7B) and decreased the peak TG (Figure 7C) at final concentrations of 0.24 mg/mL and above, with a nonsignificant trend toward decreased ETP also observed (supplemental Table 8). The addition of purified human C1INH to C1inh−/− WB at concentrations between 0.24 and 0.48 mg/mL resulted in peak TG comparable to that observed in C1inh+/+ mice. Finally, we found that administration of human purified C1INH at a dose of 15 μg/g significantly reduced venous thrombus weight, but not incidence, in C1inh−/− mice compared with vehicle control (Figure 7D-F). Thrombus weight in C1inh−/− mice administered human purified C1INH was similar to that observed in C1inh+/+ mice.
Figure 7.

Human purified C1INH dose dependently reduces WB TG and normalizes venous thrombus formation in C1INH-deficient mice. Purified human C1INH was added to WB from C1inh−/− mice at increasing concentrations ex vivo and the effect on silica-initiated WB TG compared with vehicle control. (A) Representative thrombin silica-initiated WB TG curves were plotted and (B) TG lag time and (C) peak TG quantified. Data are represented as individual values with mean ± standard error of the mean and analyzed by one-way ANOVA with post hoc Bonferroni. (D) C1inh−/− mice were administered purified human C1INH or vehicle control and VT induced using the IVC stenosis model. Schematic of C1INH VT rescue experiment. (E) Thrombus weight and (F) thrombus incidence was assessed 24 hours after induction. Thrombus weight data are represented as individual values with median and analyzed using Kruskal-Wallis with post hoc Dunn’s, and thrombus incidence data are analyzed using Fisher exact test. ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001. ANOVA, analysis of variance.
Discussion
The contact pathway has been shown to play an important role in pathological activation of coagulation and thrombosis.28, 29, 30, 31 However, the role of C1INH, the major endogenous inhibitor of the contact pathway, in these processes has been incompletely understood. Here, we demonstrate a key role for endogenous C1INH as a negative regulator of contact pathway–mediated coagulation and VT.
Using plasma samples from patients with C1INH-HAE, we found that C1INH deficiency in humans results in enhanced contact pathway–mediated TG. This phenotype was most apparent in samples from patients with C1INH-HAE with severe C1INH deficiency. It is possible that residual C1INH in patients with a more modest C1INH deficiency may have been sufficient to prevent excess contact pathway–mediated coagulation in this assay. This would be consistent with our finding that contact pathway–mediated TG parameters correlated significantly with residual C1INH activity. It would also be consistent with the reported ability of C1INH to dose-dependently inhibit FXII autoactivation.32 Most prior studies demonstrating increased plasma markers of activation of coagulation in C1INH-HAE samples have not considered the effect of residual C1INH. However, 1 study found that plasma levels of F1+2 were inversely correlated with residual C1INH activity during acute attacks.16
A nonsignificant increase in extrinsic pathway–initiated TG was also observed in samples from patients with C1INH-HAE with severe deficiency. Given that we observed no association between residual C1INH activity and extrinsic pathway–mediated TG parameters, it is unlikely that C1INH is a significant negative regulator of extrinsic pathway–initiated TG. By contrast, a previous study reported that plasmas from patients with C1INH-HAE supported significantly increased extrinsic pathway–initiated TG.33 It should be noted, however, that unlike our study, the previous report evaluated extrinsic pathway–initiated TG in the absence of a contact pathway inhibitor, such as CTI.34,35 A potential contribution of enhanced thrombin-mediated activation of FXI to the observed trend cannot be discounted, given that FXIa is a target of C1INH.36 Consistent with involvement of thrombin-mediated activation of FXI, the trend toward enhanced extrinsic pathway–mediated TG was only apparent at a low TF concentration.36
Our observation of enhanced contact pathway-initiated TG in C1INH-HAE samples, when considered in the context of previous findings demonstrating increased plasma levels of F1+2, TAT complexes, and D-dimer, further supports the notion that C1INH deficiency is associated with a procoagulant state.14, 15, 16 High plasma levels of F1+2, TAT complexes, D-dimer, and plasma TG have all independently been associated with an increased risk of VTE in other populations.37, 38, 39, 40, 41 However, despite evidence of a procoagulant state, C1INH-HAE has not traditionally been associated with an increased risk of thrombotic pathologies.
In a recent retrospective case-control study, we reported that patients with C1INH-HAE had an increased risk of composite VTE but not of composite arterial thromboembolism.18 The observed odds ratio for the association between C1INH-HAE and VTE of 3.6 is in line with bottom-end estimates of other classical hereditary thrombophilias, such as heterozygous factor V Leiden or prothrombin gene mutation G20210A, which confer an ∼2- to 10-fold increased risk of VTE.42, 43, 44, 45, 46 Further supporting a role for plasma C1INH as a modulator of VTE risk, low plasma levels in the general population were associated with a significantly increased risk of VTE in a recent plasma proteomics biomarker discovery study.47 Taken together with our observations, these data suggest that C1INH deficiency is likely to be a moderate risk factor for VTE.
To further investigate the role of endogenous C1INH as a negative regulator of coagulation, we conducted additional experiments in C1INH-deficient mice. Complete C1INH deficiency in mice resulted in elevated basal and contact pathway–mediated activation of coagulation, complementing findings in C1INH-HAE samples. In addition, mice completely deficient in C1INH also demonstrated enhanced VT formation in the IVC stenosis model. This model is physiologically relevant as stenosis results in pathological low blood flow through the vessel but does not cause overt vessel injury.48 We and others have shown that this model is sensitive to deficiencies in the C1INH targets PKa, FXIIa, and FXIa.22,49, 50, 51 Although thrombus formation in this model occurs in a contact pathway–dependent manner, its relevance to VTE in humans is not clear. Patients with FXI deficiency have a reduced risk of VTE, whereas elevated plasma levels of FXI have been associated with an increased risk.52, 53, 54, 55 However, there is no human evidence to date that PK or FXII deficiency is associated with a reduced risk of VTE.56, 57, 58, 59 It is possible that there are species-dependent differences in the contribution of the contact pathway to venous thrombus formation.
Although no significant difference in thrombus composition was observed in C1INH-deficient mice, a nonsignificant trend toward increased thrombus neutrophil content was apparent. Components of the contact pathway and complement system, including PKa and C5a, have been identified as potent neutrophil chemoattractants.60,61 It is possible that the loss of C1INH could support increased PKa activity and C5a generation, contributing to increased neutrophil recruitment. The nonsignificant trend toward increased plasma C5a observed in C1INH-deficient mice could also indicate a minor contribution of complement activation to the observed phenotype. Activation of the complement pathway has been implicated in the pathogenesis of VT in both humans and mice.62, 63, 64, 65 Further, C5a has been shown to induce platelet activation by direct and indirect mechanisms and could provide an increased surface for coagulation reactions.66, 67, 68 Although we did not observe any effect of inhibiting C5a generation on TG in C1INH-deficient mice, the role of other proximal complement factors cannot be discounted.69, 70, 71, 72 Crosstalk between the complement and coagulation systems may also play a role.73, 74, 75 Further work is required to determine the full extent to which modulation of complement activation by C1INH might affect TG and VT.
Interestingly, no significant difference in arterial thrombus formation was evident in the carotid artery ferric chloride model under the conditions of mild injury used to evaluate prothrombotic phenotypes. The lack of an arterial thrombosis phenotype was surprising given that this model is sensitive to the C1INH targets PKa, FXIIa, and FXIa.76, 77, 78, 79 However, this phenotype does recapitulate the findings of our study, in which C1INH-HAE was associated with an increased risk of VTE but not arterial thromboembolism.18 It is possible that the absence of an arterial thrombosis phenotype in C1INH-deficient mice is indicative of redundancy, with other endogenous inhibitors, such as antithrombin, providing compensation in this model.
Our studies indicate that there are some important differences between the phenotypes associated with C1INH deficiency in humans compared with mice. In C1INH-HAE, complete C1INH deficiency is rare given that most cases are associated with a single variant SEPRING1 allele.8 However, our findings indicate that partial C1INH deficiency in patients with C1INH-HAE is sufficient to increase contact pathway–mediated activation of coagulation and VTE risk. By contrast, in mice, complete deficiency was required to demonstrate subtle but significant enhancements in contact pathway–mediated activation of coagulation and VT. It is possible that species-dependent differences in C1INH function or species-dependent differences in assay sensitivity may have contributed to this discordant phenotype. For example, plasma TG in mice, but not humans, requires predilution owing to high endogenous anticoagulant activity.80,81 Further studies are required to better understand the differential effect of C1INH deficiency in humans and mice. However, it remains important that complete C1INH deficiency in mice results in a procoagulant and prothrombotic phenotype.
Development of C1INH replacement therapy represented a significant advance in the management of patients with C1INH-HAE.82 Although initially developed as an acute treatment for swelling attacks, C1INH replacement therapy has increasingly been used as a long-term prophylactic therapy in patients with C1INH-HAE.83 We sought to determine if human purified C1INH could reverse the procoagulant phenotype seen in C1INH-deficient mice. The mean plasma concentration of C1INH in humans is 0.24 mg/mL, but the corresponding value for mice is not known owing to the lack of robust assays.84 Reconstitution of C1INH-deficient mouse WB with human purified C1INH to 0.24 mg/mL corrected the enhanced contact pathway–mediated TG. Interestingly, although TG lag time was not significantly altered by C1INH deficiency in mouse WB, human purified C1INH dose-dependently prolonged this parameter. It is possible that human and mouse C1INH possess different anticoagulant activities. Indeed, there is little cross-species amino acid conservation in the reactive center loop of C1INH, which drives substrate selectivity.1
Administration of human purified C1INH was previously found to rescue the enhanced vascular permeability observed in C1INH-deficient mice.24 Importantly, in our study, administration of purified human C1INH reduced VT formation in C1INH-deficient mice to levels observed in C1INH-sufficient mice. This indicates that the enhanced VT associated with C1INH deficiency in mice is a gene-specific effect and not the result of off-target effects induced during gene deletion. Moreover, it suggests that C1INH replacement therapy could have additional beneficial anticoagulant effects in C1INH-HAE. Indeed, C1INH replacement therapy has been associated with a significant reduction in the incidence of VTE in patients with C1INH-HAE.85 It will be valuable to determine if newer therapies targeting PKa and FXIIa have similar protective effects on VT in the setting of C1INH-HAE.
To conclude, this work demonstrates that C1INH deficiency is associated with selective enhancement of contact pathway–mediated coagulation in both humans and mice. Further, C1INH deficiency in mice results in a selective enhancement of VT, modeling the phenotype recently described in humans with C1INH-HAE.
Conflict-of-interest disclosure: S.P.G. has received research support from CSL Behring and has received honoraria from CSL Behring. H.F. has received honoraria and/or served as a consultant for CSL Behring, Shire/Takeda, Swedish Orphan Biovitrum, Octapharma, Kalvista, and Pharming; and has participated in clinical trials/registries for BioCryst, CSL Behring, Pharming, Pharvaris, and Shire/Takeda. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Ying Zhang for excellent technical assistance.
This study was supported in part by research funding from CSL Behring (Heimburger Award to S.P.G.). This work was supported by grants from the American Heart Association (19POST34370026 to S.P.G.), the American Society of Hematology (Scholar Award to S.P.G.), the National Heart Lung and Blood Institute of the National Institutes of Health (T32HL007149 to S.P.G., R35HL155657 to N.M., R01HL126974 to A.S.W.), and the John C. Parker Professorship (N.M.).
Authorship
Contribution: S.P.G. designed the study, conducted experiments, analyzed data, interpreted data, and wrote the manuscript; N.M. designed the study, interpreted data, and edited the manuscript; T.K., J.W., Z.P., P.T., Y.J.S., S.D., and R.K. conducted experiments and analyzed data; and S.B., O.S., P.K.B., K.R.M., A.S.W., J.-B.H., and H.F. interpreted data and edited the manuscript.
Footnotes
Data are available on request from the corresponding author, Steven P. Grover (steven_grover@med.unc.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Davis AE, 3rd, Mejia P, Lu F. Biological activities of C1 inhibitor. Mol Immunol. 2008;45(16):4057–4063. doi: 10.1016/j.molimm.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grover SP, Mackman N. Anticoagulant SERPINs: endogenous regulators of hemostasis and thrombosis. Front Cardiovasc Med. 2022;9 doi: 10.3389/fcvm.2022.878199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ratnoff OD, Lepow IH. Some properties of an esterase derived from preparations of the first component of complement. J Exp Med. 1957;106(2):327–343. doi: 10.1084/jem.106.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong NK, Kojima M, Dobo J, Ambrus G, Sim RB. Activities of the MBL-associated serine proteases (MASPs) and their regulation by natural inhibitors. Mol Immunol. 1999;36(13-14):853–861. doi: 10.1016/s0161-5890(99)00106-6. [DOI] [PubMed] [Google Scholar]
- 5.Forbes CD, Pensky J, Ratnoff OD. Inhibition of activated Hageman factor and activated plasma thromboplastin antecedent by purified serum C1 inactivator. J Lab Clin Med. 1970;76(5):809–815. [PubMed] [Google Scholar]
- 6.Schapira M, Scott CF, Colman RW. Contribution of plasma protease inhibitors to the inactivation of kallikrein in plasma. J Clin Invest. 1982;69(2):462–468. doi: 10.1172/JCI110470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. 2020;382(12):1136–1148. doi: 10.1056/NEJMra1808012. [DOI] [PubMed] [Google Scholar]
- 8.Ponard D, Gaboriaud C, Charignon D, et al. SERPING1 mutation update: mutation spectrum and C1 inhibitor phenotypes. Hum Mutat. 2020;41(1):38–57. doi: 10.1002/humu.23917. [DOI] [PubMed] [Google Scholar]
- 9.Bjorkqvist J, de Maat S, Lewandrowski U, et al. Defective glycosylation of coagulation factor XII underlies hereditary angioedema type III. J Clin Invest. 2015;125(8):3132–3146. doi: 10.1172/JCI77139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dickeson SK, Kumar S, Sun MF, et al. A mechanism for hereditary angioedema caused by a lysine 311-to-glutamic acid substitution in plasminogen. Blood. 2022;139(18):2816–2829. doi: 10.1182/blood.2021012945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bork K, Wulff K, Rossmann H, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy. 2019;74(12):2479–2481. doi: 10.1111/all.13869. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan AP, Joseph K. Pathogenesis of hereditary angioedema: the role of the bradykinin-forming cascade. Immunol Allergy Clin North Am. 2017;37(3):513–525. doi: 10.1016/j.iac.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Sundler Bjorkman L, Persson B, Aronsson D, Skattum L, Nordenfelt P, Egesten A. Comorbidities in hereditary angioedema-a population-based cohort study. Clin Transl Allergy. 2022;12(3) doi: 10.1002/clt2.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cugno M, Cicardi M, Bottasso B, et al. Activation of the coagulation cascade in C1-inhibitor deficiencies. Blood. 1997;89(9):3213–3218. [PubMed] [Google Scholar]
- 15.Cugno M, Zanichelli A, Bellatorre AG, Griffini S, Cicardi M. Plasma biomarkers of acute attacks in patients with angioedema due to C1-inhibitor deficiency. Allergy. 2009;64(2):254–257. doi: 10.1111/j.1398-9995.2008.01859.x. [DOI] [PubMed] [Google Scholar]
- 16.Csuka D, Veszeli N, Imreh E, et al. Comprehensive study into the activation of the plasma enzyme systems during attacks of hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis. 2015;10:132. doi: 10.1186/s13023-015-0351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bork K, Witzke G. Shortened activated partial thromboplastin time may help in diagnosing hereditary and acquired angioedema. Int Arch Allergy Immunol. 2016;170(2):101–107. doi: 10.1159/000447695. [DOI] [PubMed] [Google Scholar]
- 18.Grover SP, Sundler Bjorkman L, Egesten A, Moll S, Mackman N. Hereditary angioedema is associated with an increased risk of venous thromboembolism. J Thromb Haemost. 2022;20(11):2703–2706. doi: 10.1111/jth.15870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Machlus KR, Colby EA, Wu JR, Koch GG, Key NS, Wolberg AS. Effects of tissue factor, thrombomodulin and elevated clotting factor levels on thrombin generation in the calibrated automated thrombogram. Thromb Haemost. 2009;102(5):936–944. doi: 10.1160/TH09-03-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan J, Tanratana P, Roest M, et al. A novel mouse whole blood thrombin generation assay sensitive to FXI- and FIX-mediated amplification of coagulation. Blood Adv. https://doi.org/10.1182/bloodadvances.2022008720 Published online 30 December 2022. [DOI] [PMC free article] [PubMed]
- 21.Wan J, Konings J, Yan Q, et al. A novel assay for studying the involvement of blood cells in whole blood thrombin generation. J Thromb Haemost. 2020;18(6):1291–1301. doi: 10.1111/jth.14786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grover SP, Olson TM, Cooley BC, Mackman N. Model-dependent contributions of FXII and FXI to venous thrombosis in mice. J Thromb Haemost. 2020;18(11):2899–2909. doi: 10.1111/jth.15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee RH, Kawano T, Grover SP, et al. Genetic deletion of platelet PAR4 results in reduced thrombosis and impaired hemostatic plug stability. J Thromb Haemost. 2022;20(2):422–433. doi: 10.1111/jth.15569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE., 3rd Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. 2002;109(8):1057–1063. doi: 10.1172/JCI14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oschatz C, Maas C, Lecher B, et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity. 2011;34(2):258–268. doi: 10.1016/j.immuni.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Qiu T, Chiuchiolo MJ, Whaley AS, et al. Gene therapy for C1 esterase inhibitor deficiency in a Murine Model of hereditary angioedema. Allergy. 2019;74(6):1081–1089. doi: 10.1111/all.13582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shim YJ, Chatterjee V, Swaidani S, et al. Polyphosphate expression by cancer cell extracellular vesicles mediates binding of factor XII and contact activation. Blood Adv. 2021;5(22):4741–4751. doi: 10.1182/bloodadvances.2021005116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gailani D, Renne T. Intrinsic pathway of coagulation and arterial thrombosis. Arterioscler Thromb Vasc Biol. 2007;27(12):2507–2513. doi: 10.1161/ATVBAHA.107.155952. [DOI] [PubMed] [Google Scholar]
- 29.Grover SP, Mackman N. Intrinsic pathway of coagulation and thrombosis. Arterioscler Thromb Vasc Biol. 2019;39(3):331–338. doi: 10.1161/ATVBAHA.118.312130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maas C, Renne T. Coagulation factor XII in thrombosis and inflammation. Blood. 2018;131(17):1903–1909. doi: 10.1182/blood-2017-04-569111. [DOI] [PubMed] [Google Scholar]
- 31.Raghunathan V, Zilberman-Rudenko J, Olson SR, Lupu F, McCarty OJT, Shatzel JJ. The contact pathway and sepsis. Res Pract Thromb Haemost. 2019;3(3):331–339. doi: 10.1002/rth2.12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaplan AP, Joseph K. Complement, kinins, and hereditary angioedema: mechanisms of plasma instability when C1 inhibitor is absent. Clin Rev Allergy Immunol. 2016;51(2):207–215. doi: 10.1007/s12016-016-8555-6. [DOI] [PubMed] [Google Scholar]
- 33.van Geffen M, Cugno M, Lap P, Loof A, Cicardi M, van Heerde W. Alterations of coagulation and fibrinolysis in patients with angioedema due to C1-inhibitor deficiency. Clin Exp Immunol. 2012;167(3):472–478. doi: 10.1111/j.1365-2249.2011.04541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hojima Y, Pierce JV, Pisano JJ. Hageman factor fragment inhibitor in corn seeds: purification and characterization. Thromb Res. 1980;20(2):149–162. doi: 10.1016/0049-3848(80)90381-3. [DOI] [PubMed] [Google Scholar]
- 35.Hansson KM, Nielsen S, Elg M, Deinum J. The effect of corn trypsin inhibitor and inhibiting antibodies for FXIa and FXIIa on coagulation of plasma and whole blood. J Thromb Haemost. 2014;12(10):1678–1686. doi: 10.1111/jth.12707. [DOI] [PubMed] [Google Scholar]
- 36.Gailani D, Broze GJ., Jr. Factor XI activation in a revised model of blood coagulation. Science. 1991;253(5022):909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 37.Ginsberg JS, Brill-Edwards P, Panju A, et al. Pre-operative plasma levels of thrombin-antithrombin III complexes correlate with the development of venous thrombosis after major hip or knee surgery. Thromb Haemost. 1995;74(2):602–605. [PubMed] [Google Scholar]
- 38.Cofrancesco E, Cortellaro M, Corradi A, Ravasi F, Bertocchi F. Coagulation activation markers in the prediction of venous thrombosis after elective hip surgery. Thromb Haemost. 1997;77(2):267–269. [PubMed] [Google Scholar]
- 39.Andreescu ACM, Cushman M, Rosendaal FR. D-dimer as a risk factor for deep vein thrombosis: the Leiden Thrombophilia study. Thromb Haemost. 2002;87(1):47–51. [PubMed] [Google Scholar]
- 40.Hansen ES, Rinde FB, Edvardsen MS, et al. Elevated plasma D-dimer levels are associated with risk of future incident venous thromboembolism. Thromb Res. 2021;208:121–126. doi: 10.1016/j.thromres.2021.10.020. [DOI] [PubMed] [Google Scholar]
- 41.Lutsey PL, Folsom AR, Heckbert SR, Cushman M. Peak thrombin generation and subsequent venous thromboembolism: the Longitudinal Investigation of Thromboembolism Etiology (LITE) study. J Thromb Haemost. 2009;7(10):1639–1648. doi: 10.1111/j.1538-7836.2009.03561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gohil R, Peck G, Sharma P. The genetics of venous thromboembolism. A meta-analysis involving approximately 120,000 cases and 180,000 controls. Thromb Haemost. 2009;102(2):360–370. doi: 10.1160/TH09-01-0013. [DOI] [PubMed] [Google Scholar]
- 43.Koster T, Rosendaal FR, de Ronde H, Briet E, Vandenbroucke JP, Bertina RM. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet. 1993;342(8886-8887):1503–1506. doi: 10.1016/s0140-6736(05)80081-9. [DOI] [PubMed] [Google Scholar]
- 44.Blom JW, Doggen CJM, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293(6):715–722. doi: 10.1001/jama.293.6.715. [DOI] [PubMed] [Google Scholar]
- 45.Bank I, Libourel EJ, Middeldorp S, et al. Prothrombin 20210A mutation: a mild risk factor for venous thromboembolism but not for arterial thrombotic disease and pregnancy-related complications in a family study. Arch Intern Med. 2004;164(17):1932–1937. doi: 10.1001/archinte.164.17.1932. [DOI] [PubMed] [Google Scholar]
- 46.Emmerich J, Rosendaal FR, Cattaneo M, et al. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism--pooled analysis of 8 case-control studies including 2310 cases and 3204 controls. Study Group for Pooled-Analysis in Venous Thromboembolism. Thromb Haemost. 2001;86(3):809–816. [PubMed] [Google Scholar]
- 47.Jensen SB, Hindberg K, Solomon T, et al. Discovery of novel plasma biomarkers for future incident venous thromboembolism by untargeted synchronous precursor selection mass spectrometry proteomics. J Thromb Haemost. 2018;16(9):1763–1774. doi: 10.1111/jth.14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Diaz JA, Saha P, Cooley B, et al. Choosing a mouse model of venous thrombosis. Arterioscler Thromb Vasc Biol. 2019;39(3):311–318. doi: 10.1161/ATVBAHA.118.311818. [DOI] [PubMed] [Google Scholar]
- 49.Bird JE, Smith PL, Wang X, et al. Effects of plasma kallikrein deficiency on haemostasis and thrombosis in mice: murine ortholog of the Fletcher trait. Thromb Haemost. 2012;107(6):1141–1150. doi: 10.1160/th-11-10-0682. [DOI] [PubMed] [Google Scholar]
- 50.von Bruhl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Revenko AS, Gao D, Crosby JR, et al. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood. 2011;118(19):5302–5311. doi: 10.1182/blood-2011-05-355248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105(2):269–273. doi: 10.1160/TH10-05-0307. [DOI] [PubMed] [Google Scholar]
- 53.Preis M, Hirsch J, Kotler A, et al. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017;129(9):1210–1215. doi: 10.1182/blood-2016-09-742262. [DOI] [PubMed] [Google Scholar]
- 54.Cushman M, O'Meara ES, Folsom AR, Heckbert SR. Coagulation factors IX through XIII and the risk of future venous thrombosis: the longitudinal investigation of thromboembolism etiology. Blood. 2009;114(14):2878–2883. doi: 10.1182/blood-2009-05-219915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342(10):696–701. doi: 10.1056/NEJM200003093421004. [DOI] [PubMed] [Google Scholar]
- 56.Zeerleder S, Schloesser M, Redondo M, et al. Reevaluation of the incidence of thromboembolic complications in congenital factor XII deficiency--a study on 73 subjects from 14 Swiss families. Thromb Haemost. 1999;82(4):1240–1246. [PubMed] [Google Scholar]
- 57.Girolami A, Candeo N, De Marinis GB, Bonamigo E, Girolami B. Comparative incidence of thrombosis in reported cases of deficiencies of factors of the contact phase of blood coagulation. J Thromb Thrombolysis. 2011;31(1):57–63. doi: 10.1007/s11239-010-0495-z. [DOI] [PubMed] [Google Scholar]
- 58.Girolami A, Allemand E, Bertozzi I, Candeo N, Marun S, Girolami B. Thrombotic events in patients with congenital prekallikrein deficiency: a critical evaluation of all reported cases. Acta Haematol. 2010;123(4):210–214. doi: 10.1159/000313361. [DOI] [PubMed] [Google Scholar]
- 59.Koster T, Rosendaal FR, Briet E, Vandenbroucke JP. John Hageman's factor and deep-vein thrombosis: Leiden thrombophilia study. Br J Haematol. 1994;87(2):422–424. doi: 10.1111/j.1365-2141.1994.tb04937.x. [DOI] [PubMed] [Google Scholar]
- 60.Kaplan AP, Kay AB, Austen KF. A prealbumin activator of prekallikrein. 3. Appearance of chemotactic activity for human neutrophils by the conversion of human prekallikrein to kallikrein. J Exp Med. 1972;135(1):81–97. doi: 10.1084/jem.135.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiggins RC, Giclas PC, Henson PM. Chemotactic activity generated from the fifth component of complement by plasma kallikrein of the rabbit. J Exp Med. 1981;153(6):1391–1404. doi: 10.1084/jem.153.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Norgaard I, Nielsen SF, Nordestgaard BG. Complement C3 and high risk of venous thromboembolism: 80517 individuals from the Copenhagen General Population Study. Clin Chem. 2016;62(3):525–534. doi: 10.1373/clinchem.2015.251314. [DOI] [PubMed] [Google Scholar]
- 63.Skjeflo EW, Braekkan SK, Ludviksen JK, et al. Elevated plasma concentration of complement factor C5 is associated with risk of future venous thromboembolism. Blood. 2021;138(21):2129–2137. doi: 10.1182/blood.2021010822. [DOI] [PubMed] [Google Scholar]
- 64.Damoah CE, Snir O, Hindberg K, et al. High levels of complement activating enzyme MASP-2 are associated with the risk of future incident venous thromboembolism. Arterioscler Thromb Vasc Biol. 2022;42(9):1186–1197. doi: 10.1161/ATVBAHA.122.317746. [DOI] [PubMed] [Google Scholar]
- 65.Subramaniam S, Jurk K, Hobohm L, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. 2017;129(16):2291–2302. doi: 10.1182/blood-2016-11-749879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanderson SD, Kirnarsky L, Sherman SA, Ember JA, Finch AM, Taylor SM. Decapeptide agonists of human C5a: the relationship between conformation and spasmogenic and platelet aggregatory activities. J Med Chem. 1994;37(19):3171–3180. doi: 10.1021/jm00045a023. [DOI] [PubMed] [Google Scholar]
- 67.Ferrer-Lopez P, Renesto P, Schattner M, Bassot S, Laurent P, Chignard M. Activation of human platelets by C5a-stimulated neutrophils: a role for cathepsin G. Am J Physiol. 1990;258(6 Pt 1):C1100–1107. doi: 10.1152/ajpcell.1990.258.6.C1100. [DOI] [PubMed] [Google Scholar]
- 68.Aiello S, Gastoldi S, Galbusera M, et al. C5a and C5aR1 are key drivers of microvascular platelet aggregation in clinical entities spanning from aHUS to COVID-19. Blood Adv. 2022;6(3):866–881. doi: 10.1182/bloodadvances.2021005246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grossklaus C, Damerau B, Lemgo E, Vogt W. Induction of platelet aggregation by the complement-derived peptides C3a and C5a. Naunyn Schmiedebergs Arch Pharmacol. 1976;295(1):71–76. doi: 10.1007/BF00509775. [DOI] [PubMed] [Google Scholar]
- 70.Sauter RJ, Sauter M, Reis ES, et al. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation. 2018;138(16):1720–1735. doi: 10.1161/CIRCULATIONAHA.118.034600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krarup A, Wallis R, Presanis JS, Gal P, Sim RB. Simultaneous activation of complement and coagulation by MBL-associated serine protease 2. PLoS One. 2007;2(7):e623. doi: 10.1371/journal.pone.0000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gulla KC, Gupta K, Krarup A, et al. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology. 2010;129(4):482–495. doi: 10.1111/j.1365-2567.2009.03200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Conway EM. Reincarnation of ancient links between coagulation and complement. J Thromb Haemost. 2015;13(Suppl 1):S121–132. doi: 10.1111/jth.12950. [DOI] [PubMed] [Google Scholar]
- 74.Pryzdial ELG, Leatherdale A, Conway EM. Coagulation and complement: key innate defense participants in a seamless web. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.918775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaplan AP, Ghebrehiwet B. The plasma bradykinin-forming pathways and its interrelationships with complement. Mol Immunol. 2010;47(13):2161–2169. doi: 10.1016/j.molimm.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 76.Rosen ED, Gailani D, Castellino FJ. FXI is essential for thrombus formation following FeCl3-induced injury of the carotid artery in the mouse. Thromb Haemost. 2002;87(4):774–776. [PubMed] [Google Scholar]
- 77.Wang X, Cheng Q, Xu L, et al. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3(4):695–702. doi: 10.1111/j.1538-7836.2005.01236.x. [DOI] [PubMed] [Google Scholar]
- 78.Cheng Q, Tucker EI, Pine MS, et al. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116(19):3981–3989. doi: 10.1182/blood-2010-02-270918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kokoye Y, Ivanov I, Cheng Q, et al. A comparison of the effects of factor XII deficiency and prekallikrein deficiency on thrombus formation. Thromb Res. 2016;140:118–124. doi: 10.1016/j.thromres.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tchaikovski SN, VAN Vlijmen BJM, Rosing J, Tans G. Development of a calibrated automated thrombography based thrombin generation test in mouse plasma. J Thromb Haemost. 2007;5(10):2079–2086. doi: 10.1111/j.1538-7836.2007.02719.x. [DOI] [PubMed] [Google Scholar]
- 81.Dargaud Y, Spronk HMH, Leenders P, Hemker HC, Ten Cate H. Monitoring platelet dependent thrombin generation in mice. Thromb Res. 2010;126(5):436–441. doi: 10.1016/j.thromres.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 82.Gadek JE, Hosea SW, Gelfand JA, et al. Replacement therapy in hereditary angioedema: successful treatment of acute episodes of angioedema with partly purified C1 inhibitor. N Engl J Med. 1980;302(10):542–546. doi: 10.1056/NEJM198003063021002. [DOI] [PubMed] [Google Scholar]
- 83.Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363(6):513–522. doi: 10.1056/NEJMoa0805538. [DOI] [PubMed] [Google Scholar]
- 84.Gregorek H, Kokai M, Hidvegi T, Fust G, Sabbouh K, Madaliński K. Concentration of C1 inhibitor in sera of healthy blood donors as studied by immunoenzymatic assay. Complement Inflamm. 1991;8(5-6):310–312. doi: 10.1159/000463201. [DOI] [PubMed] [Google Scholar]
- 85.Farkas H, Kőhalmi KV, Veszeli N, Zotter Z, Varnai K, Varga L. Risk of thromboembolism in patients with hereditary angioedema treated with plasma-derived C1-inhibitor. Allergy Asthma Proc. 2016;37(2):164–170. doi: 10.2500/aap.2016.37.3933. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.