

Abstract
Protein aggregation results in an array of different size soluble oligomers and larger insoluble fibrils. Insoluble fibrils were originally thought to cause neuronal cell deaths in neurodegenerative diseases due to their prevalence in tissue samples and disease models. Despite recent studies demonstrating the toxicity associated with soluble oligomers, many therapeutic strategies still focus on fibrils or consider all types of aggregates as one group. Oligomers and fibrils require different modeling and therapeutic strategies, targeting the toxic species is crucial for successful study and therapeutic development. Here, we review the role of different‐size aggregates in disease, and how factors contributing to aggregation (mutations, metals, post‐translational modifications, and lipid interactions) may promote oligomers opposed to fibrils. We review two different computational modeling strategies (molecular dynamics and kinetic modeling) and how they are used to model both oligomers and fibrils. Finally, we outline the current therapeutic strategies targeting aggregating proteins and their strengths and weaknesses for targeting oligomers versus fibrils. Altogether, we aim to highlight the importance of distinguishing the difference between oligomers and fibrils and determining which species is toxic when modeling and creating therapeutics for protein aggregation in disease.
Keywords: aggregation, fibril, kinetic modeling, molecular dynamics simulation, neurodegeneration, oligomer, post‐translational modification
1. INTRODUCTION
Protein aggregates are any protein species of a higher molecular weight than the expected native species. Unlike external pathogens, protein aggregation diseases are caused by native proteins which have crucial functions within cells. Through mutations, external factors such as toxins or metals, post‐translational modifications (PTMs), crowding, or a combination of effects, these native proteins aggregate into a range of soluble and insoluble species. We will refer to smaller soluble aggregates as oligomers and larger insoluble aggregates as fibrils since the latter species typically form β‐sheet rich strands (Sipe & Cohen, 2000).
In the context of disease, fibrils were originally thought to be the toxic species since they were readily identified in patient samples. However, it has since been shown that fibril concentration does not correlate with disease severity (Kuemmerle et al., 1999; Tiwari & Kepp, 2016). Beginning in the 2000s, soluble oligomers began to be investigated as the potential toxic species in many protein aggregation diseases (A. K. R. Dasari et al., 2019; Ferreira et al., 2007; Haass & Selkoe, 2007; Lasagna‐Reeves et al., 2013; Martinelli et al., 2019; Proctor et al., 2016; C. Wells et al., 2021), whereas fibrils were shown to either have a protective effect or no effect (Congdon & Duff, 2008; Hnath & Dokholyan, 2022; Zhu et al., 2018). Yet, the aggregation field still debates the toxicity of oligomers versus fibrils, with many therapeutic and biomarker strategies still targeting the fibrils (Alam et al., 2019; Cascella et al., 2022; Stefani, 2010; Taneja et al., 2015). Fibrils are visualized in tissue samples using immunostaining, β‐sheet binding dyes such as Congo red and thioflavin T, or just by the naked eye in some cases (Nilsson, 2004; Sipe & Cohen, 2000). Oligomers are difficult to identify in patient samples since they typically cannot be visualized using β‐sheet binding dyes and some oligomer species are structurally independent from the native protein structure and fibril structure, hence most antibodies cannot detect them (Doig et al., 2017; Hnath & Dokholyan, 2022). While the fibril secondary structure is typically composed of β‐sheets, α‐helices have been identified as a prominent structure component in many disease‐associated oligomers, and many disordered proteins adopt helical‐rich structures when in contact with lipids or in specific environments (Figure 1; D. Ghosh et al., 2015; Hnath & Dokholyan, 2022; Pannuzzo et al., 2013). Most protein aggregation diseases involve a toxic gain of function from aggregates developing aberrant interactions that are not present in the normal functional state of the proteins (Hoffner & Djian, 2002; Rajagopalan & Andersen, 2001; Redler & Dokholyan, 2012; Sharma et al., 2016) as opposed to a toxic loss of function in which aggregates sequester the proteins and prevent them from performing crucial jobs (still observed in a few exceptions; Broeck et al., 2014; Choi & Dokholyan, 2021).
FIGURE 1.
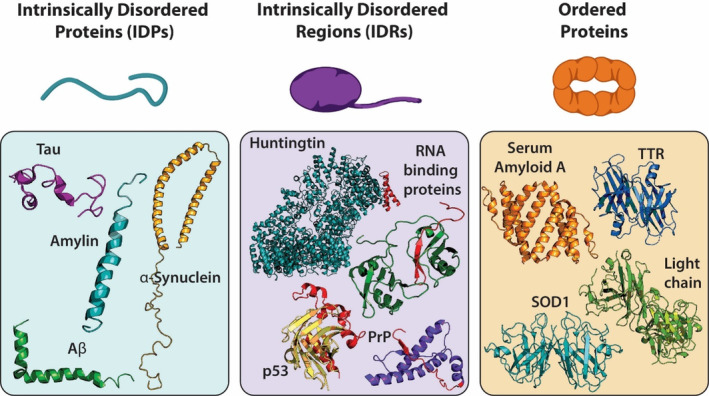
Different types of aggregating proteins. Intrinsically disordered proteins have unstable tertiary or secondary structures such as Tau (PDB 2MZ7), Amylin (PDB 2KB8), Aβ (PDB 1IYT), and α‐synuclein (PDB 1XQ8). Proteins with intrinsically disordered regions have an unstable region that is prone to aggregation, additive mutations typically occur in this region (red region of proteins). Some examples of IDRs are Huntingtin (PDB 6RMH), p53 (PDB 1T4W), PrP (PDB 1QLX), and RNA binding proteins such as FUS and TDP‐43 (PDB 4BS2). Ordered proteins have stable secondary structures which must be destabilized for aggregation to occur such as hexameric Serum Amyloid A (PDB 4IP9, depicts a dimeric piece of the whole structure), tetrameric TTR (PDB 3TFB), dimeric SOD1 (PDB 1SPD), and dimeric antibody light chain (PDB 1YUH).
In this review, we describe how different size protein aggregates (oligomers vs. fibrils) are toxic or protective in many diseases, and explain how factors that promote aggregation (mutations, metals, PTMs, or lipid interactions) may differentially promote oligomers or fibrils. Additionally, we will outline computational modeling and therapeutic strategies targeting different aggregate sizes. Distinguishing whether oligomers or fibrils are being targeted is crucial when modeling aggregation in disease and designing therapeutics.
2. PROTEIN AGGREGATION IN DISEASE
There are three groups of proteins associated with disease progression: intrinsically disordered proteins (IDPs), proteins with intrinsically disordered regions (IDRs), and ordered proteins which aggregate after the native form is destabilized (Figure 1). IDPs are defined as a group of proteins with unstable tertiary or secondary structures under physiological circumstances and are represented by a heterogeneous conformational ensemble (Wright & Jane Dyson, 2015). The disorder may alternatively be limited to specific protein regions, which are called IDRs (R. Lee et al., 2014). The amino acid compositions of IDPs/IDRs are significantly different from those of structured globular proteins or regions in terms of hydrophobicity, charge, sequence complexity, and other properties (Uversky et al., 2008). Although IDPs/IDRs are highly flexible, some of them transition from the disordered to an ordered state when interacting with partners (Uversky, 2013). Misfolded IDPs/IDRs may aggregate and form toxic species that cause neurodegenerative diseases, diabetes, cardiovascular disease, and cancer (Martinelli et al., 2019). In this section, we highlight some IDPs, proteins with IDRs, and misfolded ordered proteins and their aggregation associated with diseases.
α‐Synuclein, an IDP with 140 amino acids commonly studied in association with Parkinson disease (PD), is encoded by the SNCA gene and widely exists in the presynaptic terminals of neurons (Iwai et al., 1995). α‐Synuclein interacts with synaptic vesicle membranes and plays an important role in synaptic vesicle homeostasis and neurotransmitter release (Fortin, 2004). Monomeric α‐synuclein has flexible structures and is characterized by a heterogenous conformational ensemble (J. Chen, Zaer, et al., 2021). Bartels et al. reported that native α‐synuclein isolated from various mammalian cell lines, mouse brain tissue, and living human cells exists primarily as a helically folded tetramer, while monomeric, dimeric, and trimeric forms were detected in small amounts in certain cell types (Bartels et al., 2011). On the contrary, using in‐cell NMR, Binolfi et al. discovered that native α‐synuclein is predominantly monomeric and disordered inside intact Escherichia coli cells (Binolfi et al., 2012). Due to these controversial findings, the most abundant physiologic species of α‐synuclein in vivo are undetermined and need to be investigated thoroughly in the future. α‐Synuclein aggregates including toxic oligomers and fibrils act as precursors in Lewy body formation, a hallmark in synucleinopathies (e.g., PD, multiple system atrophy [MSA], and Lewy body dementia [LBD]; Alam et al., 2019). Although the specific toxic α‐synuclein species is still undetermined and debated, increasing research suggests that soluble α‐synuclein oligomers are cytotoxic, whereas larger Lewy inclusions might be the consequence of a protective response (Martinelli et al., 2019).
Prion protein (PrP) is a glycoprotein abundantly present on the outer surface of neurons (Kovač & Šerbec, 2022). Accumulation of toxic prion aggregates in the brain contributes to prion disease, also named as transmissible spongiform encephalopathies, occurring in several species including humans (C. Chen & Dong, 2016), felines (Colby & Prusiner, 2011), cervids (N. A. Rivera et al., 2019), cattle (G. A. H. Wells et al., 1991), goats and sheep (Acín et al., 2021). PrP consists of an intrinsically disordered N‐terminal region and a structured C‐terminal portion comprised of three α‐helices and two β‐sheets (Kovač & Šerbec, 2022). The protein is localized on the cell membrane through a glycosylphosphatidylinositol anchor at the C‐terminus (Kovač & Šerbec, 2022). The normal form of PrP has been implicated in several important biological processes including myelin maintenance, mitochondrial function, metal ion homeostasis, circadian rhythm, intercellular signaling, and neuroprotection (Castle & Gill, 2017; Fevrier et al., 2004; Zamponi & Stys, 2009). This conformation of prion protein converts into a β‐sheet‐rich structure, which stimulates autocatalytic transformation and protein aggregation (Yamaguchi & Kuwata, 2018). Although prion diseases have been associated with abnormal protein aggregation, the molecular mechanism underlying the conversion of prion protein from normal to pathological forms and the development of prion disease are still poorly understood.
Proteins with prion‐like amino acid composition or prion‐like domains induce aggregation, cause neurotoxicity, spread between cells (Porta et al., 2018), and are associated with many diseases, showing similar features as those of PrP (Feiler et al., 2015; Furukawa et al., 2011; Laferrière et al., 2019; Nonaka et al., 2013). For instance, RNA‐binding proteins containing prion‐like domains, including fused in sarcoma (FUS), Ewing sarcoma breakpoint region 1 (EWSR1), TAR DNA‐binding protein 43 (TDP‐43), TATA‐binding protein associated factor 15 (TAF15), and heterogeneous nuclear ribonucleoproteins A1 and A2 (hnRNPA1 and hnRNPA2), are known to generate pathological inclusions that are related to neurodegenerative diseases, such as frontotemporal disorders (FTD), multisystem proteinopathy (MSP), and amyotrophic lateral sclerosis (ALS; Harrison & Shorter, 2017). These proteins share a similar modular architecture consisting of a disordered prion‐like domain and a structured RNA‐recognition motif or other RNA‐binding domains (Sprunger & Jackrel, 2021). Prion‐like domains assist with the function of RNA‐binding proteins, while also increasing protein misfolding and aggregation, which contributes to neurodegenerative diseases (Harrison & Shorter, 2017).
Human p53 is a G1‐S checkpoint transcription factor playing essential roles in cell proliferation, apoptosis, angiogenesis, senescence, and DNA repair. Pathological aggregation of p53 has been discovered in tumors and is proposed to cause functional alterations that finally influence tumor progression (Oliveira et al., 2020). P53 is composed of 393 residues that can be divided into several domains: an intrinsically disordered N‐terminal transactivation domain, a proline‐rich domain, a structured DNA‐binding domain, and a structurally disordered C‐terminal (Cho et al., 1994; Clore et al., 1995; S. Fields & Jang, 1990; Haupt et al., 1997; Honda et al., 1997; Joerger & Fersht, 2008; W. Lee et al., 1994). The disordered regions mediate interactions with partner proteins and facilitate p53 function. Under physiological conditions, p53 oligomerize through the oligomerization domain at the C‐terminal. Monomer and dimer of p53 distribute in the cytoplasm, while its tetramer localizes in the nucleus and is responsible for transcriptional activation (Stommel, 1999). The most toxic aggregates of p53 are small prefibrillar aggregates and soluble oligomers (Lasagna‐Reeves et al., 2013). In contrast, large p53 amyloid aggregation is non‐toxic and serves biological roles for tumor cells (Ham et al., 2019; Xu et al., 2011). Mechanisms of p53 aggregation are summarized by J. Li et al. (2022).
Amylin, also known as islet amyloid polypeptide, is an IDP with 37 amino acids. It is co‐secreted with insulin from the same pancreatic cells (Cooper et al., 1987; Mosselman et al., 1988; Westermark et al., 1987). Amylin aggregates are the major component of the amyloid deposits in pancreatic islets, a hallmark feature of type 2 diabetes (Hieronymus & Griffin, 2015). Amylin aggregation follows a sigmoidal formation curve (first‐order kinetics) with lag, log, and plateau phases (Rhoades et al., 2000). The lag phase initiates by the interaction of one structurally disordered monomer to another, and further addition of more monomers to produce oligomers (Abedini et al., 2016). These oligomers function as a seed to form higher‐order oligomers and fibrils. The formation of fibrils facilitates the production of new fibrils, and the elongation of fibrils is accelerated in this log phase (Padrick & Miranker, 2002; Patil et al., 2011). Eventually, protein aggregation enters the plateau phase wherein proto‐fibrils and amyloid fibrils rich in β‐sheet structure are formed (Rambaran & Serpell, 2008). At first, amylin fibrils were thought to be the main cause of cytotoxicity. However, it was later found that small oligomers were the most toxic species among the various intermediates. Amylin misfolding and aggregation are associated with a gradual loss of pancreatic cell function and mass in diabetic patients (Bhowmick et al., 2022). It causes cytotoxicity by inducing endoplasmic reticulum stress (C. Huang et al., 2007), oxidative stress (Zraika et al., 2009), mitochondrial malfunction (X.‐L. Li, Chen, et al., 2011), inflammatory cytokine release (Westwell‐Roper et al., 2011), and autophagy pathway disruption (J. F. Rivera et al., 2011).
Alzheimer disease (AD) is the most common neurodegenerative disorder, responsible for 60%–70% of dementia cases, which involves the aggregation of two major kinds of IDPs, extracellular amyloid‐β (Aβ) aggregates and intracellular neurofibrillary Tau tangles. Both types of aggregates form highly ordered cross‐β structures (Bulic et al., 2009). The major component of extracellular aggregates is the Aβ peptide, a cleavage product of the amyloid precursor protein. β‐sheet‐rich structures of Aβ tend to associate with other monomers to aggregate into oligomers and fibrils. Early studies suggested that Aβ fibrils are the toxic agents that cause neuronal cell death and other hallmarks of AD. Further studies have shown that Aβ oligomers rather than Aβ fibrils are toxic to neuronal cells (G. Chen et al., 2017; D. Y. Zhang et al., 2022). Bernstein et al. determined that the aggregation prone form of Aβ, Aβ42, has a different oligomer‐size distribution than the more commonly found form, Aβ40 (Bernstein et al., 2009). The crystal structure of amyloid‐like fibrils revealed that the formation of highly ordered parallel or antiparallel β‐sheets and the steric zipper interface between β‐sheets are two essential elements of amyloid fibril formation (Nelson et al., 2005). The major component of the intracellular neurofibrillary is Tau, which plays important roles in regulating microtubules, neurite outgrowth, and axonal transport. Tau adopts β‐sheet structures resulting in a highly ordered morphology of paired helical filaments.
Huntington disease is an incurable late‐onset and progressive neurodegenerative disorder caused by a mutation in the IT‐15 gene, which encodes a 350 kDa protein of unknown function, huntingtin (Ross & Poirier, 2004). One of the hallmarks of Huntington disease is the aggregation of mutant huntingtin protein (Bonfanti et al., 2019; Khare et al., 2005; Lakhani et al., 2010). The mutant huntingtin protein which contains an IDR of expanded polyglutamine from CAG trinucleotide expansion in the gene is prone to misfolding and aggregating. The amyloid of mutant huntingtin protein, β‐sheet structures tightly held together by hydrogen bonds, is considered a main cause of Huntington disease pathogenesis, causing dysfunction and death of neurons (S. Kim & Kim, 2014). Oligomeric Htt residue in the cytoplasm translocates to the nucleus in neurons as well as in the cytosol in non‐neurons. The huntingtin oligomers affect motor function and cognition (C. Wells et al., 2021). There are over 900 protein interactions involving oligomeric Htt with a major role of interfering with nuclear transport (Woerner et al., 2016). Oligomers may play a pivotal and toxic role in Huntington disease and several studies have shown that fibril formation prevents cell death, similarly to other neurodegenerative diseases (Arrasate et al., 2004; Leitman et al., 2013).
Unlike IDPs, the three main proteins associated with systemic amyloid diseases (amyloidosis; Eisele et al., 2015); transthyretin (TTR), antibody light chain, and serum amyloid A, all have ordered structures that are necessary for the proteins' primary functions. Transthyretin is a tetrameric protein responsible for the transport of retinol and the thyroid hormone thyroxine to the liver (Saelices et al., 2015), antibody light chain is a two‐part small polypeptide subunit of an antibody (Blancas‐Mejia et al., 2018), and serum amyloid A is a hexameric apolipoprotein responsible for the transport of lipids and the recruitment of immune cells (J. Lu et al., 2014). Dissociation of each protein's primary conformation is a necessary first step for aggregation. The location of protein aggregates in the body dictates how the disease presents, with aggregate presentation typically leading to the affected organ failing (Eisele et al., 2015). Soluble oligomers of transthyretin have been shown to be more toxic than fibrils (A. K. R. Dasari et al., 2019), but the toxicity of different size aggregates of antibody light chain and serum amyloid A are still undetermined (Allen et al., 2012; Blancas‐Mejia et al., 2018).
Superoxide dismutase‐1 (SOD1) is a 32‐kDa homodimeric antioxidant enzyme, binding copper and zinc ions (Ding et al., 2012; Khare et al., 2003, 2004, 2005). SOD1 was the first genetic factor associated with ALS, a fatal neurodegenerative disease characterized by the loss of motor neurons, leading to paralysis and eventual death (Redler & Dokholyan, 2012). The main known function of SOD1 is as a dismutase that removes dangerous superoxide radicals by metabolizing them into molecular oxygen and hydrogen peroxide (Nguyen et al., 2021). SOD1 dimer dissociation and loss of metals are necessary precursors for aggregation to occur (Khare et al., 2004; Wilcox et al., 2009). SOD1 apo‐monomers were shown to aggregate into large insoluble fibrils or competing toxic trimers (Hnath & Dokholyan, 2022; Redler et al., 2011, 2014). Toxic trimeric SOD1 is structurally different from other larger aggregates, presenting a new therapeutic target for ALS (Hnath & Dokholyan, 2022; Proctor et al., 2016; Zhu et al., 2018).
Protein aggregation diseases are complex and not fully understood; many lack a known cure (Croce & Yamamoto, 2021). Cell mechanisms for maintaining proteostasis exist, but misfolded proteins take on aberrant functions or loss of function that manifests clinically (Hegde et al., 2023). In many aggregation‐related diseases smaller soluble oligomers are beginning to be identified as the toxic species while larger insoluble fibrils may be protective. IDPs proteins with IDRs are affected by processes promoting aggregation (mutations, metals, or PTMs) in different ways than ordered proteins which misfold prior to aggregation.
3. PROCESSES PROMOTING AGGREGATION
3.1. Mutations
Most mutations that promote aggregation do so by either increasing the length or hydrophobicity of the prion‐like region. Length‐increasing mutations can occur in three main ways; repeats in a specific amino acid (Huntington disease and Ataxias), repeats of a specific domain (Tau and c9orf72), or increasing the length of a tail region (Aβ42). Huntington disease is caused by an expanded CAG trinucleotide repeat in the Huntingtin gene. This trinucleotide repeat region is typically repeated 10–26 times in healthy individuals but is repeated over 40 times in individuals with Huntington disease (Barton et al., 2007; Bates et al., 2015; Budworth & McMurray, 2013; Lakhani et al., 2010). Similarly, spinocerebellar ataxias also occur due to CAG trinucleotide repeats in the long polyglutamine track of ataxin proteins (Fan et al., 2014). Insertion mutations in prion protein cause additional copies of an octapeptide repeat domain at the N‐terminus of the protein; there are typically five copies of the octapeptide repeat, but mutations insert up to eight additional domains (Kovács et al., 2002). Half of the known tau mutations have their primary effect at the RNA level. Some of these mutations increase the splicing of exon 10, overproducing four‐repeat tau which is more prone to aggregation (Goedert, 2005; Panda et al., 2003). A hexanucleotide repeat expansion in chromosome 9 open reading frame 72 (c9orf72), found in 40%–50% of familial ALS patients and 5%–10% of sporadic ALS patients, produces a glycine‐arginine repeat protein that promotes the aggregation of TDP‐43 (Cook et al., 2014; Gitler & Tsuiji, 2016; Umoh et al., 2016). Finally, the aggregation‐prone form of amyloid beta (Aβ42) has a longer tail and is formed due to mutations in the amyloid precursor protein (Finder & Glockshuber, 2007). An increased number of inserted repeats in a protein directly correlates with decreased solubility and rate of aggregation (Adegbuyiro et al., 2017; Yu et al., 2007). Ordway et al. demonstrated that even inserting a glutamine repeat in a protein not associated with disease (hypoxanthine phosphoribosyl transferase) could induce neurological symptoms, shorten the lifespan, and trigger cerebral inclusions in mice (Brais et al., 1998; Ordway et al., 1997; Perutz, 1999). Expanding the length of a protein through insertion mutations promotes aggregation, increasing the formation of oligomers and fibrils.
In addition to insertion mutations, point mutations also promote aggregation by increasing the hydrophobicity of prion regions leading to both mislocalization and aggregation. TDP‐43 (TARDBP), FUS, and other RNA binding proteins naturally contain low complexity domains contributing to liquid–liquid phase separation and the formation of aggregated RNA granules. RNA granules need to be dynamic for normal cell function; the granules must be stable enough to facilitate RNA transport and other cellular processes, but flexible enough to dissociate when necessary. ALS‐related mutations in TARDBP and FUS promote highly stable aggregates which affect the necessary dynamics of RNA granules (Maziuk et al., 2017). Some ALS mutations to TARDBP have even been observed to disrupt RNA granules, forming smaller oligomers instead (Conicella et al., 2016; Fang et al., 2014; B. S. Johnson et al., 2009). TDP‐43 and FUS are both nuclear‐localized proteins, but point mutations to the C‐terminus region affect their nuclear localization signal causing them to enter the cytoplasm (H. Deng et al., 2014; Prasad et al., 2019). Further, point mutations in α‐synuclein (Srinivasan et al., 2021), TARDBP (Prasad et al., 2019), and FUS (H. Deng et al., 2014) all increase the hydrophobicity of the N‐terminus, promoting aggregation. α‐Synuclein point mutations, which result in the most severe dopaminergic loss in the substantia nigra, promote oligomer formation and toxic interactions with membranes (Winner et al., 2011). While mutations in aggregating proteins are associated with diseases, there are other factors that promote aggregation without the need for mutations.
3.2. Metals
Protein‐metal interactions either promote or prevent aggregation. The copper and zinc ions in functional SOD1 are necessary for the superoxide function of the protein, loss of the metals destabilize the dimer interface leading to aggregation (Ding & Dokholyan, 2008; Khare et al., 2004; Khare & Dokholyan, 2006). Similarly, copper and zinc binding to the octapeptide repeat domain of prion protein partially fold the domains preventing aggregation (Inanami et al., 2005; Jackson et al., 2001; Leclerc et al., 2006). Alternatively, metal binding to the prion domains of α‐synuclein (Hillmer et al., 2010; Rasia et al., 2005; Uversky et al., 2001; Yamin et al., 2003), Aβ (Bolognin et al., 2011; Sarell et al., 2010; Tõugu et al., 2011), tau (Yamamoto et al., 2004; Yang et al., 2010), and ataxin (Ricchelli et al., 2007; Stawoska et al., 2009) increase the hydrophobicity of the proteins and promote aggregation. The binding of different metals affects whether soluble oligomers or large insoluble fibrils are formed, the soluble forms are typically more toxic while the large insoluble fibrils are less toxic (Breydo & Uversky, 2011; Drago et al., 2008; T. D. Kim et al., 2000; B. Liu et al., 2011; Miyake et al., 2011). The differing effects of different metals on what type of aggregates are formed (oligomers versus fibrils), along with the high concentrations of zinc and copper at synapses may be a contributing factor to why many aggregating proteins contribute to neurodegeneration (E. P. Huang, 1997; L. Wang et al., 2020).
3.3. Post‐translational modifications
PTMs are a common cause of protein aggregation, specifically in the context of traumatic injuries and age‐related neurodegenerative disorders. PTMs are broadly defined as any covalent attachment of a functional group or targeted cleavage that occurs to a protein following its synthesis. While over 600 PTMs have been described (Bradley, 2022), only a subset have been experimentally confirmed to play a role in protein aggregation. In broad terms, PTMs influence protein aggregation similarly to mutations and metals, primarily by modifying protein folding or altering solubility. Shaffert and Carter have recently reviewed the influence of PTMs on aggregation and their role in neurodegenerative diseases (Schaffert & Carter, 2020). Here we provide a brief overview of the most well‐characterized PTMs and their role in protein aggregation.
Protein phosphorylation is defined as the covalent addition of a phosphate group to a receiving protein, a process typically catalyzed by a kinase. Physiologically, phosphorylation is utilized by cells to transfer and store energy, activate or deactivate a protein through a conformational change, and transmit information through signaling pathways. Aberrant phosphorylation occurs when kinase function becomes dysregulated, frequently due to a genetic error, trauma, or other causes of cellular stress (Gendron & Petrucelli, 2009; Giasson & Mushynski, 1996; Perluigi et al., 2016). Proteins that have been shown to aggregate upon phosphorylation include Aβ (Jamasbi et al., 2017; Kumar & Walter, 2011), tau (Buée et al., 2000; G. V. W. Johnson & Stoothoff, 2004; Lippens et al., 2007), α‐synuclein (Fujiwara et al., 2002; Samuel et al., 2016), TDP‐43 (Carlomagno et al., 2014; Hasegawa et al., 2008), and PrPc (Giannopoulos et al., 2009). However, the phosphorylation of these proteins does not always result in aggregate formation. Numerous groups have demonstrated that the phosphorylation of certain residues in Aβ (Kumar et al., 2016), tau (W. Hu et al., 2016), and TDP‐43 (Brady et al., 2011; H.‐Y. Li, Yeh, et al., 2011) do not promote aggregation. Phosphorylation of different residues could also contribute to whether oligomers or fibrils are formed (Kumar et al., 2016).
While phosphorylation is perhaps the most well‐studied PTM that leads to pathologic protein aggregation, other pathways have been implicated in the formation of aggregates. Oxidation has been shown to promote aggregate formation by altering kinase function (T. Zhang et al., 2015), as well as disrupting the structural conformation of proteins and damaging genetic elements that disrupt protein structure after translation (Butterfield & Kanski, 2001). Specifically, the oxidation of α‐synuclein (El‐Agnaf & Brent Irvine, 2000) and Aβ (Oda et al., 1995) are well characterized. Nitration has also been demonstrated to increase aggregate formation in multiple neurodegenerative disorders (Hyun et al., 2004; Ischiropoulos & Beckman, 2003; Reynolds et al., 2007; Uversky et al., 2005).
There is emerging evidence that methylation (Balmik & Chinnathambi, 2021) and ubiquitination promote aggregation in specific contexts (Berke & Paulson, 2003; Dantuma & Bott, 2014); however, these PTMs may affect other proteins in unique ways. Similarly, multiple groups have described how the addition of long‐chain functional groups result in aggregate formation. Acetylation of tau (S. I. A. Cohen et al., 2013; Cook et al., 2014) and TDP‐43 (T. J. Cohen et al., 2015), glycation of tau (K. Liu et al., 2016), glycosylation of tau (F. Liu et al., 2002), and SUMOylation of tau (X. Chen, Zhang, et al., 2021; Luo et al., 2014) all result in protein aggregation. Glutathionylation of SOD1 destabilizes the dimer leading to aggregation (Redler et al., 2011). While decades of research have dissected individual modifications and their role in the overall aggregate formation, research has only recently begun to investigate how these modifications affect oligomers versus fibrils (Barrett & Timothy Greenamyre, 2015; Ercan‐Herbst et al., 2019; Kumar et al., 2016).
3.4. Lipid interactions
Protein–lipid interactions promote aggregation and contribute to cell death in many aggregation‐related diseases. One of the key factors contributing to Aβ toxicity in AD is the interaction between the cell membrane and Aβ. There are several ways in which the lipid membrane interacts with Aβ (Arispe et al., 1993). For instance, membranes can act as a surface for Aβ to adsorb onto, leading to increased concentration and proximity which affects aggregation (Yanagisawa et al., 2011). Certain lipid components, such as cholesterol and sphingolipids, can promote the formation of Aβ oligomers by altering the physical properties of the lipid membrane, modifying the conformation of Aβ and forming ion channels. Moreover, the membrane can also serve as a nucleation site for Aβ aggregation, promoting the formation of toxic oligomers and fibrils (Vetrivel & Thinakaran, 2010). Aβ peptides can insert themselves into the lipid bilayer and disrupt the normal structure and function of the membrane, leading to increased membrane permeability and cellular dysfunction. In addition, the membrane can facilitate the interaction between Aβ and other proteins on the membrane, such as Apolipoprotein E. The interaction between the membrane and Aβ can also activate intracellular signaling pathways that affect Aβ toxicity (Reed‐Geaghan et al., 2009).
α‐Synuclein and lipid interactions play essential roles in both maintaining normal physiological functions as well as contributing to the pathogenesis of several neurodegenerative diseases, termed as synucleinopathies (Runwal & Edwards, 2021). α‐Synuclein monomers and oligomers bind to lipid membranes through electrostatic interactions between the negatively charged lipid headgroups and the positively charged lysine‐rich N‐terminus of α‐synuclein (Galvagnion, 2017; Grey et al., 2011; Rooijen et al., 2008). Both monomeric and oligomeric forms of α‐synuclein have a high binding affinity to loosely packed membranes and small unilamellar vesicles with high surface curvature (Middleton & Rhoades, 2010; Ouberai et al., 2013). Among all different species of α‐synuclein, the oligomeric forms cause neurotoxicity and are likely responsible for the onset and development of disease (Lashuel et al., 2013). The cytotoxic effects of α‐synuclein oligomers are attributed to the interactions with lipid membranes, deteriorating membrane integrity and impairing cellular homeostasis (Alam et al., 2019; Fusco et al., 2017; Iyer & Claessens, 2019; Killinger et al., 2019). Several scenarios have been proposed to explain the cellular toxicity caused by α‐synuclein oligomer–membrane interactions; including bilayer thinning, detergent‐like solubilization, and pore and nanodisc formation, which have been summarized by Musteikytė et al. (2021). On the other hand, several studies found that lipid membranes could trigger α‐synuclein aggregation and oligomer formation in vivo (Bae et al., 2013; Galvagnion, 2017; Näsström et al., 2011). Assayag et al. reported that alterations in the level of polyunsaturated fatty acids on lipid membrane induced cytotoxic α‐synuclein oligomer formation in the cytoplasm, which preceded Lewy‐like inclusions in dopaminergic cells (Assayag et al., 2007).
Aβ and α‐synuclein lipid interactions are the most widely studied, but there is also evidence that Tau oligomers and misfolded SOD1 can interact with, and may disrupt, mitochondrial membranes (Abbasabadi et al., 2013; Britti et al., 2020; Camilleri et al., 2020; H.‐X. Deng et al., 2006; Lin & Flint Beal, 2006). Mitochondrial dysfunction and energy deficiency are established components of many neurodegenerative diseases (Cozzolino & Carrì, 2012; Johri & Flint Beal, 2012; X. Wang et al., 2014). Overall, lipid interactions contribute to protein aggregation and maybe a central component of cell death in some diseases (Gonzalez‐Garcia et al., 2021).
4. MODELING PROTEIN AGGREGATION
4.1. Molecular dynamics modeling of protein aggregation
Protein aggregation is involved in many neurodegenerative diseases, and research on the mechanism of protein aggregation will aid the development of therapeutic interventions that modulate protein aggregation. Although known aggregated proteins have different sequences, many of them adopt β‐sheet‐rich structures in coincidence for aggregation. One of the hypotheses is that hydrogen‐bond interactions unrelated to amino acid side chains play a key role in the process of protein aggregation. The validation of this hypothesis and the exploration of other mechanisms of protein aggregation rely on wet‐lab experiments. However, many proteins that aggregate are disordered in nature and only form specific three‐dimensional structures transiently, which makes studying them experimentally very hard. An alternative approach is to resort to computational molecular dynamics (MD) simulation. Here we review computational studies of the aggregation of Aβ, α‐synuclein, and SOD1 with all‐atom MD simulations as well as coarse‐grained molecular dynamics. More research on the aggregation of other proteins can be found in the reviews of Kulkarni et al. (2022) and Thirumalai (2003).
Extensive modeling of Aβ monomers, intermediates, and fibrils has been performed by multiple groups. Sanches et al. utilized the energy landscape visualization method (ELViM) to analyze coarse‐grained simulations of normal Aβ‐40 and Aβ‐42 monomers, along with six single‐point mutations associated with early‐onset disease. They found that the aggregation rate is correlated with the β‐strand content of the monomers, and most of the contacts necessary to seed the aggregation have already been formed in Aβ42 monomers and the monomers of less soluble mutants, while the monomers of Aβ40 need to unfold some helical structure before aggregating (Sanches et al., 2022). Baumketner et al. performed both ion mobility mass spectrometry and theoretical modeling to study the conformational states of the monomeric Aβ42 peptide. Their simulations revealed that Aβ42 in aqueous solution adopts both extended chains as well as collapsed‐coil structures. Aβ42 did not display a unique fold like a typical protein, but rather a mixture of rapidly interconverting conformations. They analyzed the secondary structure and found that Aβ42 peptide conformations are dominated by loops and turns but show some helical structure in the C‐terminal hydrophobic tail (Baumketner, 2006). Klimov and Thirumalai conducted a series of MD simulations for Aβ and found that Aβ16–22 peptides form antiparallel β‐sheet structures, and α‐helical intermediates are transiently populated. They proposed that Aβ forms by fibrils maximizing the number of salt bridges and hydrophobic interactions, and the oligomers must have a high α‐helical content (Klimov & Thirumalai, 2003). Studying monomers and fibril intermediates gives some information on the structure of oligomeric intermediates, but further simulations expand on the structure of specific‐size oligomers.
Urbanc et al. studied the formation of Aβ dimer by discrete molecular dynamics (DMD) (Urbanc et al., 2004). They first studied the formation of the Aβ dimer through coarse‐grained simulations and then studied the thermodynamic properties through all‐atom simulation. They found that the free energy of the dimer is generally higher than that of the monomer, and there is no significant difference in the free energy of the Aβ42 dimer and Aβ40 dimer. Barz et al. utilized transition networks derived from all‐atom molecular dynamics to determine that the oligomer size distribution is different between Aβ42 and Aβ40 due to higher solvent exposure of hydrophobic residues in Aβ42. Additionally, dimers and tetramers of Aβ42 and Aβ40 were structurally different which could contribute to their differences in aggregation (Barz et al., 2018). Ma and Nussinov simulated the stabilities of oligomeric AGAAAAGA and AAAAAAAA (A8) with MD simulations and found these two peptides are stable in aggregates of 6–8 monomers, providing insight into the mechanism of seed growth with a multilayer β‐sheet model (Ma & Nussinov, 2009). They further studied the oligomers of Aβ fragments 16–22, 16–35, and 10–35. Their simulations indicated that the antiparallel β‐sheet orientation is the most stable for the Aβ16‐22, in agreement with a solid state NMR‐based model (Ma & Nussinov, 2002). In addition, recent crystal structures of Aβ16‐22 suggest that the region 16–22 can form multiple polymorphs, with variations in their molecular structure, hydrogen‐bonding network, and conformational dynamics (Colvin et al., 2016; S. Dasari & Mallik, 2020; J.‐X. Lu et al., 2013; Qiang et al., 2017; Tycko, 2015; Wälti et al., 2016). The structural diversity of these polymorphs may contribute to the heterogeneity of Alzheimer disease and its clinical subtypes. Zhang et al. have performed MD simulations to computationally investigate the interactions between Aβ and GM1, a member of the ganglion series of gangliosides. They performed 100 ns MD simulations for GM1 membranes and five Aβ42 monomers and found that GM1 tightly binds Aβ42. They found that the fifth residue (arginine, R5) of Aβ is within 1–2 Å from GM1, suggesting that R5 stably binds GM1. They further computationally substituted R5 to glycine, and they found that the average distance between G5 and GM1 increased to >10 Å and was within a binding distance (<2 Å) for as little as 2% of the time, indicating that the R5G mutation disrupted the tight binding interaction between the fifth residue and GM1. Thus, the fifth residue in Aβ42 plays a critical role in the direct binding between AC and GM1 (D. Y. Zhang et al., 2022). The recently‐solved cryo‐EM structures of Aβ have provided valuable insights into the molecular architecture of amyloid fibrils and their role in the pathogenesis of Alzheimer disease (Fitzpatrick et al., 2017; Gremer et al., 2017; Q. Li et al., 2021; R. Zhang et al., 2009). These structures can enable researchers to perform detailed molecular dynamics simulations to better understand the dynamics and stability of the fibrils. It is important to model different size Aβ oligomers to be able to determine the toxicity of different oligomers. More MD research on Aβ can be found in the review by Nasica‐Labouze et al. (2015).
Chen et al. used inter‐dye distance distributions from bulk time‐resolved Förster resonance energy transfer as constraints in DMD simulations to map the conformational space of α‐synuclein monomers (J. Chen, Zaer, et al., 2021). They found that some conformations of α‐synuclein are surprisingly stable, exhibiting conformational transitions in milliseconds. Their comprehensive analysis of conformational ensembles revealed fundamental structural properties and underlying conformations that facilitate their various functions in membrane interactions or oligomer and fibril formation. By integrating cross‐linking mass spectrometry with DMD simulations, the same group utilized cross‐linking data as constraints to simulate the dimer structures of α‐synuclein. They identified one compact and stable dimer structure where tyrosine 39 is sufficiently close to forming a dityrosine covalent bond, which may be involved in α‐synuclein amyloid fibril formation (Zamel et al., 2019).
To computationally propose potential SOD1 trimer structures, DMD was used to simulate several nanoseconds of SOD1 trimer trajectory. To obtain constraints for certain interatomic distances and to narrow the structural search space, Proctor et al. applied limited proteolysis to SOD1 trimers and used mass spectrometry to identify likely solvent‐exposed sites on the surface of the trimer. A custom bias potential was designed iteratively so that the experimentally deduced solvent‐exposed sites were solvent‐exposed in the DMD simulations (Proctor et al., 2016).
DMD has proven to be a useful tool for the investigation of protein aggregation. DMD applies discrete step function potentials to define interatomic interactions rather than continuous potentials widely adopted in traditional molecular dynamics simulations (Brodie et al., 2019; Dokholyan et al., 1998; Proctor et al., 2011; Shirvanyants et al., 2012). This innovative application greatly reduces calculations and therefore enhances sampling and allows DMD to achieve protein folding, especially for smaller oligomers, in a practical time scale (Ding et al., 2008; Dokholyan et al., 1998). For intrinsically disordered proteins, to the best of our knowledge no experimental technique characterizes their conformational ensembles at the atomic level. Therefore, experimentally guided DMD simulations provide the best solution. DMD simulations are combined with various experimental technologies, such as FRET, Mass Spectrometry, which have been demonstrated by previous studies (J. Chen, Zaer, et al., 2021; Proctor et al., 2016; Zamel et al., 2019). Protein structures predicted by DMD simulations that are guided and validated by experimental data are more accurate than those predicted solely by computational methods (J. Chen, Zaer, et al., 2021; Seffernick & Lindert, 2020) making DMD a crucial tool to observe complex conformational changes in the monomeric, oligomeric, and aggregated forms of Aβ, α‐synuclein, and SOD1.
4.2. Kinetic models of protein aggregation
Without detailed knowledge of a protein's structure in the aggregated state, it is still possible to propose systems of ordinary differential equations (ODEs) that model the formation of aggregates of various size (Hall et al., 2005, 2015; Hirota et al., 2019). Toy models of protein aggregation proposed by Knowles, Dobson, Vendruscolo, Hall, Hirota, and others use physical reasoning to propose reasonable systems of ODEs and predict the relative abundances of the various sizes of amyloid as a function of initial conditions, particularly the size of the initial nucleation site (Hirota et al., 2019; Hall et al., 2005, 2015; Knowles & Buehler, 2011; Knowles et al., 2007; 2009). Such modeling often uses simplifying assumptions, such as irreversible misfolding of the protein upon aggregation, and roughly spherical shapes for the protein; however, the results are flexible enough to fit a wide variety of time‐dependent concentration signals.
An extensive accounting of protein aggregation in humans and other organisms is found in the review by Chiti and Dobson (2006). Important factors in aggregation include increased hydrophobicity of amino acid side chains and neutral total protein charge, as aggregates are energetically favored in proteins with these properties. Investigation into Aβ42 aggregates has shown that the major factor for formation and growth is the presence of existing fibrils (S. I. A. Cohen et al., 2013), which is termed a secondary nucleation process. Since Aβ monomer is known to be an intrinsically disordered protein (Uversky et al., 2008), the assumptions made in ODE modeling of aggregation are thought to hold, since there is no preferred native monomer structure that would bias the aggregation pathways. Additionally, metal‐binding may play an important role in aggregate nucleation and growth, since Aβ plaques found in Alzheimer disease are enriched in copper and zinc (Faller et al., 2014).
Proteins can exist in different structural states, including ordered and intrinsically disordered forms. The mechanisms of protein aggregation for these two forms are distinct from each other. In the case of ordered proteins, they need to undergo a destabilization of their unique structures to form amyloidogenic intermediates. This process is typically referred to as denaturation and misfolding, which can eventually lead to the formation of amyloid fibrils. On the other hand, IDPs require at least partial folding to form amyloidogenic or aggregation‐prone species (Uversky & Fink, 2004). It has been shown that IDPs can aggregate in the absence of a well‐defined tertiary structure and that the nature of the disordered regions can influence the kinetics and thermodynamics of the aggregation process (Avni et al., 2019; Uversky, 2014; W. Wang et al., 2010). Therefore, understanding the differences in the mechanisms of protein aggregation for ordered and intrinsically disordered proteins is important for developing effective strategies to prevent or treat protein misfolding diseases.
Recently, it has been determined that the formation of superoxide dismutase‐1 (SOD1) trimers implicated in the development of amyotrophic lateral sclerosis form off‐pathway from the larger aggregates (Khare & Dokholyan, 2006; Proctor et al., 2016; Zhu et al., 2018). The theoretical justification for this result was that the model from Hall and Hirota predicts that off‐pathway trimer pathway aggregation results in a strong correlation between the decrease in trimer with an increase in aggregate (Hirota et al., 2019), which was observed in SOD1 aggregation data (r = −0.78 to 0.98, p < 0.001). Despite the exact structures of the SOD1 trimer and larger aggregates being unknown at present, there is indirect evidence that SOD1 trimers form off‐pathway (Hnath & Dokholyan, 2022).
Efforts to understand the formation of Aβ from both the computation (Auer et al., 2007; Blondelle et al., 1997) and experiments (Fandrich, 2002; X. Hu et al., 2009; Nelson et al., 2005; Serio et al., 2000) point to a nucleated growth mechanism, in which high concentrations of protein are needed for the initial formation of misfolded seeds, before a first‐order kinetic process results in the exponential growth of Aβ fibrils (Ding et al., 2005). However, it is currently unknown how nucleation occurs, since the concentration needed for the misfolded seeds to form are in the mM range (Auer et al., 2007; Ilie & Caflisch, 2019), whereas Aβ is physiologically found at nM to μM concentrations, both in cerebrospinal fluid (Andreasen et al., 1999) and neuronal tissue (Gong et al., 2003; Lazarevic et al., 2017). Recently, it has been proposed that Aβ may interact with the plasma membrane to form seeds (D. Y. Zhang et al., 2022), with the GM1 ganglioside serving as a catalyst that binds and localizes Aβ monomer.
5. THERAPEUTIC STRATEGIES TO TREAT PROTEIN AGGREGATION
Four main therapeutic strategies are used to target different stages of protein aggregation in disease; protein reduction, aggregation inhibition, breaking down aggregates or preventing toxic interactions (Figure 2). For most of these strategies, it is necessary to know the native function, toxic aggregate size and structure, and mechanism of cell death of the aggregating proteins, of which parts remain unknown for most diseases associated with aggregating proteins. Very few therapeutics targeting protein aggregations have been successful in clinical trials (Doig et al., 2017; Lang, 2010; Petrov et al., 2017).
FIGURE 2.
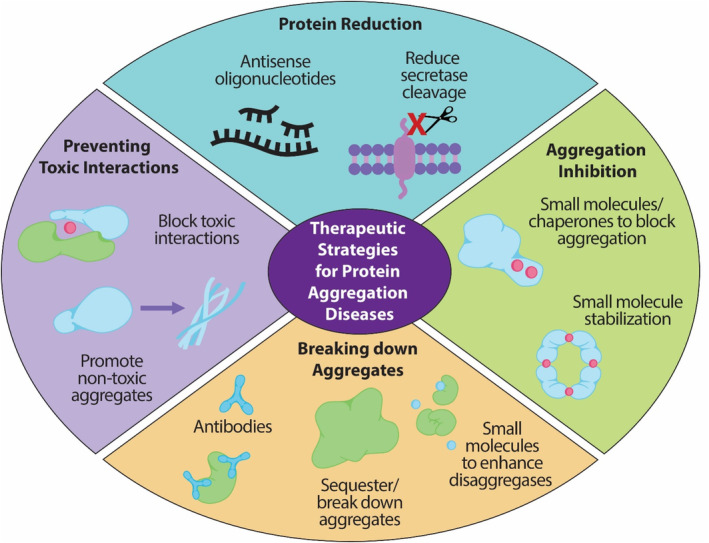
Therapeutic strategies for protein aggregation diseases. The four current strategies for targeting aggregating proteins are through (1) overall protein reduction, (2) inhibiting aggregation using small molecules or chaperones to block protein interactions or stabilizing the native form, (3) breaking down formed aggregates using antibodies or disaggregases, or (4) preventing toxic interactions by blocking the interaction or promoting non‐toxic aggregates.
Crowding and increased protein concentration contribute to protein aggregation (Munishkina et al., 2004); for example, copy number gains of α‐synuclein are regularly seen in Parkinson disease (Mokretar et al., 2018). To combat overexpression and prevent aggregation many different methods have been tested to reduce the level of aggregation‐prone proteins. Antisense oligonucleotide therapy uses short strands of modified nucleotides to target RNA and reduce the protein expression (Kuijper et al., 2021). This therapy is currently being tested to reduce SOD1 (Meyer et al., 2023; Miller et al., 2022), tau (Mummery et al., 2021), huntingtin (Smith & Tabrizi, 2020), amylin (Novials et al., 1998), α‐synuclein (Alarcón‐Arís et al., 2018), FUS (Korobeynikov et al., 2022), and TTR (Teresa Coelho et al., 2013) with various degrees of success in clinical trials. To reduce the amount of Aβ, strategies focus on decreasing the activity of β and γ‐secratases (A. K. Ghosh & Osswald, 2014). Protein reduction is only a feasible therapeutic strategy for protein aggregates with a toxic gain of function. Another issue with this strategy is that many aggregating proteins have crucial native roles in the body, decreasing the overall level of some proteins causes a different phenotype due to a toxic loss of function. Protein reduction strategies need to carefully reduce the expression level of aggregating proteins without losing the native function, and the dosing may not transfer from person to person due to natural differences in protein expression levels (Southwell et al., 2012). Since this strategy is aimed at reducing all aggregation instead of just targeting fibrils it may be helpful against oligomers.
Another strategy for preventing or slowing aggregation is using small molecules or chaperones to inhibit aggregation. For ordered proteins such as SOD1 and TTR, small molecule therapeutics focus on stabilizing the dimer or tetramer interfaces to prevent the native proteins from dissociating (Amporndanai et al., 2020; Capper et al., 2018; T. Coelho et al., 2012; Cotrina et al., 2020; Tojo et al., 2006). For IDPs like amylin (López et al., 2016) and α‐synuclein (C. R. Fields et al., 2019), small molecule therapeutics target the prion‐like region to prevent aggregation. The same strategy of blocking the aggregating region is accomplished using chaperone proteins such as heat shock protein (Klucken et al., 2004; Webster et al., 2019) or other engineered chaperones (Ferretti et al., 2022). While this strategy may be effective against both oligomers and fibrils it relies on treating patients early in disease progression before the aggregates have spread and done extensive damage, which is difficult since most of the above‐mentioned diseases do not have early biomarkers to test for the disease before the symptoms have progressed. More extensive descriptions of small molecule aggregation inhibitors are reviewed by Malik and Wiedau (2020), Jokar et al. (2019), and Pena‐Diaz et al. (2020).
Once aggregates have already formed, antibodies or disaggregases modulate them or direct them to proteostasis machinery for sequestration or degradation. Both active and passive immunotherapy techniques have been tested to treat protein aggregation‐related diseases with mixed results. Vaccines of small surface‐exposed regions of the toxic forms of protein aggregates (active immunotherapy) or monoclonal/ polyclonal antibody treatments which target specific regions of toxic aggregates (passive immunotherapy) have recently begun evaluation in clinical trials (Eisele et al., 2015; Vassilakopoulou et al., 2021). In the past year, two different monoclonal antibody treatments targeting Aβ oligomers were given emergency FDA approval to treat AD (Aducanumab and Lecanemab; Marsool et al., 2023; Perneczky et al., 2023; Rahman et al., 2023; Vitek et al., 2023). The key difference between new immunotherapy methods and past methods is immunotherapy is now being designed to target smaller soluble oligomers of proteins or specific misfolded regions found in larger aggregates, as opposed to the previous strategy of designing antibodies that only detect large insoluble aggregates. In‐depth descriptions of failed and new immunotherapy methods to treat different diseases were reviewed by Reiss et al. (2021), Schwab et al. (2020), Gittings and Sattler (2020), and Villoslada et al. (2008). Disaggregases are a category of chaperone proteins that help break down or remove aggregated proteins (Mogk & Bukau, 2004); Skd3 (Caseinolytic peptidase B protein homolog; Rizo et al., 2019) and heat shock proteins (specifically Hsp70 [Gao et al., 2015; Thackray et al., 2022], Hsp104 [Shorter & Southworth, 2019], and Hsp110 [O'Driscoll et al., 2015]) are the two groups of disaggregases typically studied. Small molecule treatments are also being investigated to promote the cell's own degradation processes to break down aggregates. For example, rapamycin has been shown to increase the clearance of α‐synuclein due to its inhibition of mTOR, inducing autophagy (Maiese et al., 2013; Sarkar et al., 2007). Rapamycin and many of these degradation‐enhancing small molecules have many other off‐target effects (such as acting on other essential pathways involved in immunosuppression; Abraham & Wiederrecht, 1996), for this strategy to be effective different small molecules with fewer off‐target effects need to be tested. Small soluble oligomers are beginning to be identified as the toxic species in many protein aggregation diseases (A. K. R. Dasari et al., 2019; Lasagna‐Reeves et al., 2013; Martinelli et al., 2019; Proctor et al., 2016; C. Wells et al., 2021), while larger aggregates are protective (Ham et al., 2019; Hnath & Dokholyan, 2022; Leitman et al., 2013; Xu et al., 2011; Zhu et al., 2018). Strategies that target and break down larger aggregates may be increasing toxicity by undoing the sequestration of smaller oligomers and increasing the abundance of the toxic species. Antibody strategies have therapeutic potential if they are highly specific for the toxic forms of aggregates, but the debate is ongoing to determine what the primary toxic species are for most protein aggregation diseases.
The last therapeutic strategy in development to treat protein aggregation‐related diseases is in the initial phases of exploration. Most protein aggregation diseases involve a toxic gain of function (Hoffner & Djian, 2002; Rajagopalan & Andersen, 2001; Redler & Dokholyan, 2012; Sharma et al., 2016), meaning the toxic aggregates interact with another protein or membrane in a way they should not be. To combat this, researchers are inhibiting the toxic interaction either by blocking the binding site (Akhtar et al., 2021; Choi & Dokholyan, 2021; with specific antibodies or small molecules) or by using chaperones to promote non‐toxic aggregate formation (promote fibrils). Since the structures and toxic interactions of protein aggregates are largely unknown this strategy is in the early stages.
Many protein aggregation diseases occur in the central nervous system (Alzheimer, Parkinson, and Huntington diseases), so these therapeutic strategies have the added challenge of crossing the blood–brain barrier to be effective. Additionally, these strategies cannot reverse tissue damage, so early diagnosis is crucial for any of the strategies to have any effect. Other therapies are in development that focus on treating cellular damage after it occurs instead of preventing it by targeting the protein aggregation (Poppe et al., 2014; Srivastava et al., 2021). Preventing aggregate formation from the beginning, either through reducing the overall amount of protein or the addition of small molecules/ chaperones, has the potential to prevent both oligomer and fibril formation if biomarkers are discovered which allows patients to be treated earlier in disease progression. Breaking down aggregates is one of the most widely used current therapeutic strategies, despite the dangerous possibility of increasing toxic species by breaking down protective fibrils. Targeting toxic interactions has the potential to be an effective therapeutic once more work is done to determine the proteins or membranes being affected during the toxic gain of function. Many drug design strategies that attack aggregation have failed in the past due to administration too late in disease progression or targeting the wrong size aggregate. Therapeutic strategies become not just ineffective but dangerous by not considering the toxicity of oligomers versus fibrils in different diseases.
6. CONCLUSION
Protein aggregates are still studied as one group, despite soluble oligomers and insoluble fibrils having drastically different toxicities and structures. Aggregation of IDPs, proteins with IDRs, or ordered proteins all contribute to a variety of diseases, the majority form into toxic oligomers that begin a gain of function while insoluble fibrils have protective effects. Modifications to aggregation‐prone proteins by mutations, metal interactions/ loss of metals, PTMs, or lipid interactions contribute to the rate of aggregation and what size aggregates are formed. Due to the span of sizes and structural conformations of aggregates, different computational methods need to be utilized to study oligomers and fibrils. Finally, many different therapeutic strategies have been attempted to target aggregated proteins in disease, but more research needs to be done into the actual toxic interactions instead of just breaking apart aggregates and potentially increasing toxic oligomers. Overall, it is crucial to determine what size aggregate is toxic in each disease. Specifically targeting the toxic size can increase the effectiveness of protein aggregation modeling and therapeutic strategies to treat protein aggregation‐related diseases.
ACKNOWLEDGMENTS
We acknowledge support from the National Institutes for Health (R35 GM134864, 1RF1 AG071675‐01, 1R01 AT012053), National Science Foundation (2210963), and the Passan Foundation.
Hnath B, Chen J, Reynolds J, Choi E, Wang J, Zhang D, et al. Big versus small: The impact of aggregate size in disease. Protein Science. 2023;32(7):e4686. 10.1002/pro.4686
Review Editor: Nir Ben‐Tal
REFERENCES
- Abbasabadi O, Azarakhsh AJ, Nikkhah M, Meratan AA, Ghiasi P, Nemat‐Gorgani M. Disruption of mitochondrial membrane integrity induced by amyloid aggregates arising from variants of SOD1. Int J Biol Macromol. 2013;61:212–7. 10.1016/j.ijbiomac.2013.07.007 [DOI] [PubMed] [Google Scholar]
- Abedini A, Plesner A, Cao P, Ridgway Z, Zhang J, Ling‐Hsien T, et al. Time‐resolved studies define the nature of toxic IAPP intermediates, providing insight for anti‐amyloidosis therapeutics. Elife. 2016;5:e12977. 10.7554/eLife.12977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev Immunol. 1996;14(1):483–510. 10.1146/annurev.immunol.14.1.483 [DOI] [PubMed] [Google Scholar]
- Acín C, Bolea R, Monzón M, Monleón E, Moreno B, Filali H, et al. Classical and atypical scrapie in sheep and goats. Review on the etiology, genetic factors, pathogenesis, diagnosis, and control measures of both diseases. Animals. 2021;11(3):691. 10.3390/ani11030691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adegbuyiro A, Sedighi F, Pilkington AW, Groover S, Legleiter J. Proteins containing expanded polyglutamine tracts and neurodegenerative disease. Biochemistry. 2017;56(9):1199–217. 10.1021/acs.biochem.6b00936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar A, Andleeb A, Waris TS, Bazzar M, Moradi A‐R, Awan NR, et al. Neurodegenerative diseases and effective drug delivery: a review of challenges and novel therapeutics. J Control Release. 2021;330:1152–67. 10.1016/j.jconrel.2020.11.021 [DOI] [PubMed] [Google Scholar]
- Alam P, Bousset L, Melki R, Otzen DE. Α‐synuclein oligomers and fibrils: a spectrum of species, a spectrum of toxicities. J Neurochem. 2019;150(5):522–34. 10.1111/jnc.14808 [DOI] [PubMed] [Google Scholar]
- Alarcón‐Arís D, Recasens A, Galofré M, Carballo‐Carbajal I, Zacchi N, Ruiz‐Bronchal E, et al. Selective α‐synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: potential therapy for Parkinson's disease. Mol Ther. 2018;26(2):550–67. 10.1016/j.ymthe.2017.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MJ, Lacroix JJ, Ramachandran S, Capone R, Whitlock JL, Ghadge GD, et al. Mutant SOD1 forms ion channel: implications for ALS pathophysiology. Neurobiol Dis. 2012;45(3):831–8. 10.1016/j.nbd.2011.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amporndanai K, Rogers M, Watanabe S, Yamanaka K, O'Neill PM, Samar Hasnain S. Novel selenium‐based compounds with therapeutic potential for SOD1‐linked amyotrophic lateral sclerosis. EBioMedicine. 2020;59:102980. 10.1016/j.ebiom.2020.102980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A, Winblad B, et al. Cerebrospinal fluid β‐amyloid (1‐42) in Alzheimer disease: differences between early‐ and late‐onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56(6):673. 10.1001/archneur.56.6.673 [DOI] [PubMed] [Google Scholar]
- Arispe N, Pollard HB, Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta‐protein [a beta P‐(1‐40)] in bilayer membranes. Proc Natl Acad Sci. 1993;90(22):10573–7. 10.1073/pnas.90.22.10573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431(7010):805–10. 10.1038/nature02998 [DOI] [PubMed] [Google Scholar]
- Assayag K, Yakunin E, Loeb V, Selkoe DJ, Sharon R. Polyunsaturated fatty acids induce α‐synuclein‐related pathogenic changes in neuronal cells. Am J Pathol. 2007;171(6):2000–11. 10.2353/ajpath.2007.070373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer S, Dobson CM, Vendruscolo M. Characterization of the nucleation barriers for protein aggregation and amyloid formation. HFSP J. 2007;1(2):137–46. 10.2976/1.2760023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avni A, Swasthi HM, Majumdar A, Mukhopadhyay S. Intrinsically disordered proteins in the formation of functional amyloids from bacteria to humans. Prog Mol Biol Transl Sci. 2019;166:109–43. 10.1016/bs.pmbts.2019.05.005 [DOI] [PubMed] [Google Scholar]
- Bae E‐J, Ho D‐H, Park E, Jung JW, Cho K, Hong JH, et al. Lipid peroxidation product 4‐hydroxy‐2‐nonenal promotes seeding‐capable oligomer formation and cell‐to‐cell transfer of α‐synuclein. Antioxid Redox Signal. 2013;18(7):770–83. 10.1089/ars.2011.4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmik AA, Chinnathambi S. Methylation as a key regulator of tau aggregation and neuronal health in Alzheimer's disease. Cell Commun Signal. 2021;19(1):51. 10.1186/s12964-021-00732-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett PJ, Timothy Greenamyre J. Post‐translational modification of α‐synuclein in Parkinson's disease. Brain Res. 2015;1628:247–53. 10.1016/j.brainres.2015.06.002 [DOI] [PubMed] [Google Scholar]
- Bartels T, Choi JG, Selkoe DJ. α‐Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–10. 10.1038/nature10324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton S, Jacak R, Khare SD, Ding F, Dokholyan NV. The length dependence of the PolyQ‐mediated protein aggregation. J Biol Chem. 2007;282(35):25487–92. 10.1074/jbc.M701600200 [DOI] [PubMed] [Google Scholar]
- Barz B, Liao Q, Strodel B. Pathways of amyloid‐β aggregation depend on oligomer shape. J Am Chem Soc. 2018;140(1):319–27. 10.1021/jacs.7b10343 [DOI] [PubMed] [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Primers. 2015;1(1):15005. 10.1038/nrdp.2015.5 [DOI] [PubMed] [Google Scholar]
- Baumketner A. Amyloid beta‐protein monomer structure: a computational and experimental study. Protein Sci. 2006;15(3):420–8. 10.1110/ps.051762406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke SJS, Paulson HL. Protein aggregation and the ubiquitin proteasome pathway: gaining the UPPer hand on neurodegeneration. Curr Opin Genet Dev. 2003;13(3):253–61. 10.1016/S0959-437X(03)00053-4 [DOI] [PubMed] [Google Scholar]
- Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, et al. Amyloid‐β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat Chem. 2009;1(4):326–31. 10.1038/nchem.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick DC, Kudaibergenova Z, Burnett L, Jeremic AM. Molecular mechanisms of amylin turnover, misfolding and toxicity in the pancreas. Molecules. 2022;27(3):1021. 10.3390/molecules27031021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binolfi A, Theillet F‐X, Selenko P. Bacterial in‐cell NMR of human α‐synuclein: a disordered monomer by nature? Biochem Soc Trans. 2012;40(5):950–4. 10.1042/BST20120096 [DOI] [PubMed] [Google Scholar]
- Blancas‐Mejia LM, Misra P, Dick CJ, Cooper SA, Redhage KR, Bergman MR, et al. Immunoglobulin light chain amyloid aggregation. Chem Commun. 2018;54(76):10664–74. 10.1039/C8CC04396E [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondelle SE, Forood B, Houghten RA, Pérez‐Payá E. Polyalanine‐based peptides as models for self‐associated β‐pleated‐sheet complexes. Biochemistry. 1997;36(27):8393–400. 10.1021/bi963015b [DOI] [PubMed] [Google Scholar]
- Bolognin S, Messori L, Drago D, Gabbiani C, Cendron L, Zatta P. Aluminum, copper, iron and zinc differentially alter amyloid‐Aβ1–42 aggregation and toxicity. Int J Biochem Cell Biol. 2011;43(6):877–85. 10.1016/j.biocel.2011.02.009 [DOI] [PubMed] [Google Scholar]
- Bonfanti S, Lionetti MC, Fumagalli MR, Chirasani VR, Tiana G, Dokholyan NV, et al. Molecular mechanisms of heterogeneous oligomerization of huntingtin proteins. Sci Rep. 2019;9(1):7615. 10.1038/s41598-019-44151-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley D. The evolution of post‐translational modifications. Curr Opin Genet Dev. 2022;76:101956. 10.1016/j.gde.2022.101956 [DOI] [PubMed] [Google Scholar]
- Brady OA, Meng P, Zheng Y, Mao Y, Fenghua H. Regulation of TDP‐43 aggregation by phosphorylation Andp62/SQSTM1: regulation of TDP‐43 aggregation by phosphorylation and P62. J Neurochem. 2011;116(2):248–59. 10.1111/j.1471-4159.2010.07098.x [DOI] [PubMed] [Google Scholar]
- Brais B, Bouchard J‐P, Xie Y‐G, Rochefort DL, Chrétien N, Tomé FMS, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18(2):164–7. 10.1038/ng0298-164 [DOI] [PubMed] [Google Scholar]
- Breydo L, Uversky VN. Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics. 2011;3(11):1163–80. 10.1039/c1mt00106j [DOI] [PubMed] [Google Scholar]
- Britti E, Ros J, Esteras N, Abramov AY. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium‐induced cell death. Cell Calcium. 2020;86:102150. 10.1016/j.ceca.2019.102150 [DOI] [PubMed] [Google Scholar]
- Brodie NI, Popov KI, Petrotchenko EV, Dokholyan NV, Borchers CH. Conformational ensemble of native α‐synuclein in solution as determined by short‐distance crosslinking constraint‐guided discrete molecular dynamics simulations. PLoS Comput Biol. 2019;15(3):e1006859. 10.1371/journal.pcbi.1006859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeck V, Lies PC, Dermaut B. TDP‐43‐mediated neurodegeneration: towards a loss‐of‐function hypothesis? Trends Mol Med. 2014;20(2):66–71. 10.1016/j.molmed.2013.11.003 [DOI] [PubMed] [Google Scholar]
- Budworth H, McMurray CT. A brief history of triplet repeat diseases. In: Kohwi Y, McMurray CT, editors. Trinucleotide Repeat Protocols. Volume 1010. Totowa, NJ: Humana Press; 2013. p. 3–17. 10.1007/978-1-62703-411-1_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buée L, Bussière T, Buée‐Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev. 2000;33(1):95–130. 10.1016/S0165-0173(00)00019-9 [DOI] [PubMed] [Google Scholar]
- Bulic B, Pickhardt M, Schmidt B, Mandelkow E‐M, Waldmann H, Mandelkow E. Development of tau aggregation inhibitors for Alzheimer's disease. Angew Chem Int Ed. 2009;48(10):1740–52. 10.1002/anie.200802621 [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Kanski J. Brain protein oxidation in age‐related neurodegenerative disorders that are associated with aggregated proteins. Mech Ageing Dev. 2001;122(9):945–62. 10.1016/S0047-6374(01)00249-4 [DOI] [PubMed] [Google Scholar]
- Camilleri A, Ghio S, Caruana M, Weckbecker D, Schmidt F, Kamp F, et al. Tau‐induced mitochondrial membrane perturbation is dependent upon cardiolipin. Biochim Biophys Acta – Biomembr. 2020;1862(2):183064. 10.1016/j.bbamem.2019.183064 [DOI] [PubMed] [Google Scholar]
- Capper MJ, Wright GSA, Barbieri L, Luchinat E, Mercatelli E, McAlary L, et al. The cysteine‐reactive small molecule Ebselen facilitates effective SOD1 maturation. Nat Commun. 2018;9(1):1693. 10.1038/s41467-018-04114-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlomagno Y, Zhang Y, Davis M, Lin W‐L, Cook C, Dunmore J, et al. Casein kinase II induced polymerization of soluble TDP‐43 into filaments is inhibited by heat shock proteins. PLoS One. 2014;9(3):e90452. 10.1371/journal.pone.0090452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella R, Bigi A, Cremades N, Cecchi C. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Cell Mol Life Sci. 2022;79(3):174. 10.1007/s00018-022-04166-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle AR, Gill AC. Physiological functions of the cellular prion protein. Front Mol Biosci. 2017;4. 10.3389/fmolb.2017.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Dong X‐P. Epidemiological characteristics of human prion diseases. Infect Dis Poverty. 2016;5(1):47. 10.1186/s40249-016-0143-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G‐f, Ting‐hai X, Yan Y, Zhou Y‐r, Jiang Y, Melcher K, et al. Amyloid beta: structure, biology and structure‐based therapeutic development. Acta Pharmacol Sin. 2017;38(9):1205–35. 10.1038/aps.2017.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zaer S, Drori P, Zamel J, Joron K, Kalisman N, et al. The structural heterogeneity of α‐synuclein is governed by several distinct subpopulations with interconversion times slower than milliseconds. Structure. 2021;29(9):1048–64. 10.1016/j.str.2021.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang Y, Wang Q, Qin Y, Yang X, Xing Z, et al. The function of SUMOylation and its crucial roles in the development of neurological diseases. FASEB J. 2021;35(4):e21510. 10.1096/fj.202002702R [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75(1):333–66. 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a P53 tumor suppressor‐DNA complex: understanding tumorigenic mutations. Science. 1994;265(5170):346–55. 10.1126/science.8023157 [DOI] [PubMed] [Google Scholar]
- Choi ES, Dokholyan NV. SOD1 oligomers in amyotrophic lateral sclerosis. Curr Opin Struct Biol. 2021;66:225–30. 10.1016/j.sbi.2020.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Ernst J, Clubb R, Omichinski JG, Poindexter Kennedy WM, Sakaguchi K, et al. Refined solution structure of the Oligomerization domain of the tumour suppressor P53. Nat Struct Mol Biol. 1995;2(4):321–33. 10.1038/nsb0495-321 [DOI] [PubMed] [Google Scholar]
- Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante‐Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–92. 10.1212/WNL.0b013e3182661eb1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–29. 10.1056/NEJMoa1208760 [DOI] [PubMed] [Google Scholar]
- Cohen SIA, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, et al. Proliferation of amyloid‐Β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci. 2013;110(24):9758–63. 10.1073/pnas.1218402110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Hwang AW, Restrepo CR, Yuan C‐X, Trojanowski JQ, Lee VMY. An acetylation switch controls TDP‐43 function and aggregation propensity. Nat Commun. 2015;6(1):5845. 10.1038/ncomms6845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3(1):a006833. 10.1101/cshperspect.a006833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, et al. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J Am Chem Soc. 2016;138(30):9663–74. 10.1021/jacs.6b05129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon EE, Duff KE. Is tau aggregation toxic or protective? J Alzheimers Dis. 2008;14(4):453–7. [DOI] [PubMed] [Google Scholar]
- Conicella AE, Zerze GH, Mittal J, Fawzi NL. ALS mutations disrupt phase separation mediated by α‐helical structure in the TDP‐43 low‐complexity C‐terminal domain. Structure. 2016;24(9):1537–49. 10.1016/j.str.2016.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet. 2014;23(1):104–16. 10.1093/hmg/ddt402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid‐rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci. 1987;84(23):8628–32. 10.1073/pnas.84.23.8628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina EY, Oliveira Â, Leite JP, Llop J, Gales L, Quintana J, et al. Repurposing benzbromarone for familial amyloid polyneuropathy: a new transthyretin tetramer stabilizer. Int J Mol Sci. 2020;21(19):7166. 10.3390/ijms21197166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzolino M, Carrì MT. Mitochondrial dysfunction in ALS. Prog Neurobiol. 2012;97(2):54–66. 10.1016/j.pneurobio.2011.06.003 [DOI] [PubMed] [Google Scholar]
- Croce KR, Yamamoto A. Dissolving the complex role aggregation plays in neurodegenerative disease. Mov Disord. 2021;36(5):1061–9. 10.1002/mds.28522 [DOI] [PubMed] [Google Scholar]
- Dantuma NP, Bott LC. The ubiquitin‐proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci. 2014;7. 10.3389/fnmol.2014.00070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari AKR, Hughes RM, Wi S, Hung I, Gan Z, Kelly JW, et al. Transthyretin aggregation pathway toward the formation of distinct cytotoxic oligomers. Sci Rep. 2019;9(1):33. 10.1038/s41598-018-37230-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari S, Mallik BS. Conformational dynamics of amyloid‐β (16–22) peptide in aqueous ionic liquids. RSC Adv. 2020;10(55):33248–60. 10.1039/D0RA06609E [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H‐X, Shi Y, Furukawa Y, Zhai H, Ronggen F, Liu E, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci. 2006;103(18):7142–7. 10.1073/pnas.0602046103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Gao K, Jankovic J. The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol. 2014;10(6):337–48. 10.1038/nrneurol.2014.78 [DOI] [PubMed] [Google Scholar]
- Ding F, Dokholyan NV. Dynamical roles of metal ions and the disulfide bond in Cu, Zn superoxide dismutase folding and aggregation. Proc Natl Acad Sci. 2008;105(50):19696–701. 10.1073/pnas.0803266105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Furukawa Y, Nukina N, Dokholyan NV. Local unfolding of Cu, Zn superoxide dismutase monomer determines the morphology of fibrillar aggregates. J Mol Biol. 2012;421(4‐5):548–60. 10.1016/j.jmb.2011.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, LaRocque JJ, Dokholyan NV. Direct observation of protein folding, aggregation, and a prion‐like conformational conversion. J Biol Chem. 2005;280(48):40235–40. 10.1074/jbc.M506372200 [DOI] [PubMed] [Google Scholar]
- Ding F, Tsao D, Nie H, Dokholyan NV. Ab initio folding of proteins with all‐atom discrete molecular dynamics. Structure. 2008;16(7):1010–8. 10.1016/j.str.2008.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig AJ, del Castillo‐Frias MP, Berthoumieu O, Tarus B, Nasica‐Labouze J, Sterpone F, et al. Why is research on amyloid‐β failing to give new drugs for Alzheimer's disease? ACS Chem Nerosci. 2017;8(7):1435–7. 10.1021/acschemneuro.7b00188 [DOI] [PubMed] [Google Scholar]
- Dokholyan NV, Buldyrev SV, Eugene Stanley H, Shakhnovich EI. Discrete molecular dynamics studies of the folding of a protein‐like model. Fold Des. 1998;3(6):577–87. 10.1016/S1359-0278(98)00072-8 [DOI] [PubMed] [Google Scholar]
- Drago D, Bettella M, Bolognin S, Cendron L, Scancar J, Milacic R, et al. Potential pathogenic role of β‐AMYLOID1–42–aluminum complex in Alzheimer's disease. Int J Biochem Cell Biol. 2008;40(4):731–46. 10.1016/j.biocel.2007.10.014 [DOI] [PubMed] [Google Scholar]
- Eisele YS, Monteiro C, Fearns C, Encalada SE, Luke Wiseman R, Powers ET, et al. Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov. 2015;14(11):759–80. 10.1038/nrd4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Agnaf OMA, Brent Irvine G. Review: formation and properties of amyloid‐like fibrils derived from α‐synuclein and related proteins. J Struct Biol. 2000;130(2‐3):300–9. 10.1006/jsbi.2000.4262 [DOI] [PubMed] [Google Scholar]
- Ercan‐Herbst E, Ehrig J, Schöndorf DC, Behrendt A, Klaus B, Ramos BG, et al. A post‐translational modification signature defines changes in soluble tau correlating with oligomerization in early stage Alzheimer's disease brain. Acta Neuropathol Commun. 2019;7(1):192. 10.1186/s40478-019-0823-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller P, Hureau C, La Penna G. Metal ions and intrinsically disordered proteins and peptides: from Cu/Zn amyloid‐β to general principles. Acc Chem Res. 2014;47(8):2252–9. 10.1021/ar400293h [DOI] [PubMed] [Google Scholar]
- Fan H‐C, Ho L‐I, Chi C‐S, Chen S‐J, Peng G‐S, Chan T‐M, et al. Polyglutamine (PolyQ) diseases: genetics to treatments. Cell Transplant. 2014;23(4‐5):441–58. 10.3727/096368914X678454 [DOI] [PubMed] [Google Scholar]
- Fandrich M. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 2002;21(21):5682–90. 10.1093/emboj/cdf573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y‐S, Tsai K‐J, Chang Y‐J, Kao P, Woods R, Kuo P‐H, et al. Full‐length TDP‐43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia‐TDP patients. Nat Commun. 2014;5(1):4824. 10.1038/ncomms5824 [DOI] [PubMed] [Google Scholar]
- Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, et al. TDP‐43 is intercellularly transmitted across axon terminals. J Cell Biol. 2015;211(4):897–911. 10.1083/jcb.201504057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ST, Vieira MNN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB Life. 2007;59(4):332–45. 10.1080/15216540701283882 [DOI] [PubMed] [Google Scholar]
- Ferretti GDS, Quarti J, dos Santos G, Rangel LP, Silva JL. Anticancer therapeutic strategies targeting P53 aggregation. Int J Mol Sci. 2022;23(19):11023. 10.3390/ijms231911023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci. 2004;101(26):9683–8. 10.1073/pnas.0308413101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields CR, Bengoa‐Vergniory N, Wade‐Martins R. Targeting alpha‐synuclein as a therapy for Parkinson's disease. Front Mol Neurosci. 2019;12:299. 10.3389/fnmol.2019.00299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S, Jang SK. Presence of a potent transcription activating sequence in the P53 protein. Science. 1990;249(4972):1046–9. 10.1126/science.2144363 [DOI] [PubMed] [Google Scholar]
- Finder VH, Glockshuber R. Amyloid‐β aggregation. Neurodegener Dis. 2007;4(1):13–27. 10.1159/000100355 [DOI] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo‐EM structures of tau filaments from Alzheimer's disease. Nature. 2017;547(7662):185–90. 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DL. Lipid rafts mediate the synaptic localization of α‐synuclein. J Neurosci. 2004;24(30):6715–23. 10.1523/JNEUROSCI.1594-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α‐Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4(2):160–4. 10.1038/ncb748 [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N. A seeding reaction recapitulates intracellular formation of sarkosyl‐insoluble transactivation response element (TAR) DNA‐binding protein‐43 inclusions. J Biol Chem. 2011;286(21):18664–72. 10.1074/jbc.M111.231209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco G, Chen SW, Williamson PTF, Cascella R, Perni M, Jarvis JA, et al. Structural basis of membrane disruption and cellular toxicity by α‐synuclein oligomers. Science. 2017;358(6369):1440–3. 10.1126/science.aan6160 [DOI] [PubMed] [Google Scholar]
- Galvagnion C. The role of lipids interacting with α‐synuclein in the pathogenesis of Parkinson's disease. J Parkinsons Dis. 2017;7(3):433–50. 10.3233/JPD-171103 [DOI] [PubMed] [Google Scholar]
- Gao X, Carroni M, Nussbaum‐Krammer C, Mogk A, Nillegoda NB, Anna Szlachcic D, et al. Human Hsp70 disaggregase reverses Parkinson's‐linked α‐synuclein amyloid fibrils. Mol Cell. 2015;59(5):781–93. 10.1016/j.molcel.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4(1):13. 10.1186/1750-1326-4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Osswald HL. BACE1 (β‐secretase) inhibitors for the treatment of Alzheimer's disease. Chem Soc Rev. 2014;43(19):6765–813. 10.1039/C3CS60460H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Singh PK, Sahay S, Jha NN, Jacob RS, Sen S, et al. Structure based aggregation studies reveal the presence of helix‐rich intermediate during α‐synuclein aggregation. Sci Rep. 2015;5(1):9228. 10.1038/srep09228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannopoulos PN, Robertson C, Jodoin J, Paudel H, Booth SA, LeBlanc AC. Phosphorylation of prion protein at serine 43 induces prion protein conformational change. J Neurosci. 2009;29(27):8743–51. 10.1523/JNEUROSCI.2294-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Mushynski WE. Aberrant stress‐induced phosphorylation of perikaryal neurofilaments. J Biol Chem. 1996;271(48):30404–9. 10.1074/jbc.271.48.30404 [DOI] [PubMed] [Google Scholar]
- Gitler AD, Tsuiji H. There has been an awakening: emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 2016;1647:19–29. 10.1016/j.brainres.2016.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittings LM, Sattler R. Recent advances in understanding amyotrophic lateral sclerosis and emerging therapies. Fac Rev. 2020;9. 10.12703/b/9-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Tau gene mutations and their effects. Mov Disord. 2005;20(S12):S45–52. 10.1002/mds.20539 [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, et al. Alzheimer's disease‐affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci. 2003;100(18):10417–22. 10.1073/pnas.1834302100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Garcia M, Fusco G, De Simone A. Membrane interactions and toxicity by misfolded protein oligomers. Front Cell Dev Biol. 2021;9:642623. 10.3389/fcell.2021.642623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremer L, Schölzel D, Schenk C, Reinartz E, Labahn J, Ravelli RBG, et al. Fibril structure of amyloid‐β(1‐42) by cryo‐electron microscopy. Science. 2017;358(6359):116–9. 10.1126/science.aao2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey M, Linse S, Nilsson H, Brundin P, Sparr E. Membrane interaction of α‐synuclein in different aggregation states. J Parkinsons Dis. 2011;1(4):359–71. 10.3233/JPD-2011-11067 [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β‐peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–12. 10.1038/nrm2101 [DOI] [PubMed] [Google Scholar]
- Hall D, Hirota N, Dobson CM. A toy model for predicting the rate of amyloid formation from unfolded protein. J Mol Biol. 2005;351(1):195–205. 10.1016/j.jmb.2005.05.013 [DOI] [PubMed] [Google Scholar]
- Hall D, Kardos J, Edskes H, Carver JA, Goto Y. A multi‐pathway perspective on protein aggregation: implications for control of the rate and extent of amyloid formation. FEBS Lett. 2015;589(6):672–9. 10.1016/j.febslet.2015.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham SW, Jeon H‐Y, Jin X, Kim E‐J, Kim J‐K, Shin YJ, et al. TP53 gain‐of‐function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2019;26(3):409–25. 10.1038/s41418-018-0126-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison AF, Shorter J. RNA‐binding proteins with prion‐like domains in health and disease. Biochem J. 2017;474(8):1417–38. 10.1042/BCJ20160499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, et al. Phosphorylated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64(1):60–70. 10.1002/ana.21425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of P53. Nature. 1997;387(6630):296–9. 10.1038/387296a0 [DOI] [PubMed] [Google Scholar]
- Hegde AN, Duke LM, Timm LE, Nobles H. The proteasome and ageing. In: Robin Harris J, Korolchuk VI, editors. Biochemistry and cell biology of ageing: part III biomedical science. Volume 102. Switzerland AG: Springer International Publishing; 2023. p. 99–112. 10.1007/978-3-031-21410-3_5 [DOI] [PubMed] [Google Scholar]
- Hieronymus L, Griffin S. Role of amylin in type 1 and type 2 diabetes. Diabetes Educ. 2015;41:47S–56S. 10.1177/0145721715607642 [DOI] [PubMed] [Google Scholar]
- Hillmer AS, Putcha P, Levin J, Högen T, Hyman BT, Kretzschmar H, et al. Converse modulation of toxic α‐synuclein oligomers in living cells by N′‐benzylidene‐benzohydrazide derivates and ferric iron. Biochem Biophys Res Commun. 2010;391(1):461–6. 10.1016/j.bbrc.2009.11.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota N, Edskes H, Hall D. Unified theoretical description of the kinetics of protein aggregation. Biophys Rev. 2019;11(2):191–208. 10.1007/s12551-019-00506-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnath B, Dokholyan NV. Toxic SOD1 trimers are off‐pathway in the formation of amyloid‐like fibrils in ALS. Biophys J. 2022;121:2084–95. 10.1016/j.bpj.2022.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffner G, Djian P. Protein aggregation in Huntington's disease. Biochimie. 2002;84(4):273–8. 10.1016/S0300-9084(02)01398-6 [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor P53. FEBS Lett. 1997;420(1):25–7. 10.1016/S0014-5793(97)01480-4 [DOI] [PubMed] [Google Scholar]
- Hu W, Zhang X, Tung YC, Xie S, Liu F, Iqbal K. Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement. 2016;12(10):1066–77. 10.1016/j.jalz.2016.01.014 [DOI] [PubMed] [Google Scholar]
- Hu X, Crick SL, Guojun B, Frieden C, Pappu RV, Lee J‐M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid‐beta peptide. Proc Natl Acad Sci. 2009;106(48):20324–9. 10.1073/pnas.0911281106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C‐j, Haataja L, Gurlo T, Butler AE, Xiuju W, Soeller WC, et al. Induction of endoplasmic reticulum stress‐induced β‐cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am J Physiol Endocrinol Metab. 2007;293(6):E1656–62. 10.1152/ajpendo.00318.2007 [DOI] [PubMed] [Google Scholar]
- Huang EP. Metal ions and synaptic transmission: think zinc. Proc Natl Acad Sci. 1997;94(25):13386–7. 10.1073/pnas.94.25.13386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun D‐H, Lee MH, Halliwell B, Jenner P. Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins: proteasomal dysfunction and protein aggregates. J Neurochem. 2004;86(2):363–73. 10.1046/j.1471-4159.2003.01841.x [DOI] [PubMed] [Google Scholar]
- Ilie IM, Caflisch A. Simulation studies of amyloidogenic polypeptides and their aggregates. Chem Rev. 2019;119(12):6956–93. 10.1021/acs.chemrev.8b00731 [DOI] [PubMed] [Google Scholar]
- Inanami O, Hashida S, Iizuka D, Horiuchi M, Hiraoka W, Shimoyama Y, et al. Conformational change in full‐length mouse prion: a site‐directed spin‐labeling study. Biochem Biophys Res Commun. 2005;335(3):785–92. 10.1016/j.bbrc.2005.07.148 [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Investig. 2003;111(2):163–9. 10.1172/JCI200317638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Lisa Flanagan HA, de Silva R, et al. The precursor protein of non‐Aβ component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14(2):467–75. 10.1016/0896-6273(95)90302-X [DOI] [PubMed] [Google Scholar]
- Iyer A, Claessens MMAE. Disruptive membrane interactions of alpha‐synuclein aggregates. Biochim Biophys Acta Proteins Proteom. 2019;1867(5):468–82. 10.1016/j.bbapap.2018.10.006 [DOI] [PubMed] [Google Scholar]
- Jackson GS, Murray I, Hosszu LLP, Gibbs N, Waltho JP, Clarke AR, et al. Location and properties of metal‐binding sites on the human prion protein. Proc Natl Acad Sci. 2001;98(15):8531–5. 10.1073/pnas.151038498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamasbi E, Separovic F, Hossain MA, Ciccotosto GD. Phosphorylation of a full length amyloid‐β peptide modulates its amyloid aggregation, cell binding and neurotoxic properties. Mol Biosyst. 2017;13(8):1545–51. 10.1039/C7MB00249A [DOI] [PubMed] [Google Scholar]
- Joerger AC, Fersht AR. Structural biology of the tumor suppressor P53. Annu Rev Biochem. 2008;77(1):557–82. 10.1146/annurev.biochem.77.060806.091238 [DOI] [PubMed] [Google Scholar]
- Johnson BS, Snead D, Lee JJ, Michael McCaffery J, Shorter J, Gitler AD. TDP‐43 is intrinsically aggregation‐prone, and amyotrophic lateral sclerosis‐linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284(30):20329–39. 10.1074/jbc.M109.010264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GVW, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. 2004;117(24):5721–9. 10.1242/jcs.01558 [DOI] [PubMed] [Google Scholar]
- Johri A, Flint Beal M. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. 2012;342(3):619–30. 10.1124/jpet.112.192138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokar S, Khazaei S, Behnammanesh H, Shamloo A, Erfani M, Beiki D, et al. Recent advances in the design and applications of amyloid‐β peptide aggregation inhibitors for Alzheimer's disease therapy. Biophys Rev. 2019;11(6):901–25. 10.1007/s12551-019-00606-2 [DOI] [PubMed] [Google Scholar]
- Khare SD, Caplow M, Dokholyan NV. The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. Proc Natl Acad Sci. 2004;101(42):15094–9. 10.1073/pnas.0406650101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare SD, Ding F, Dokholyan NV. Folding of Cu, Zn superoxide dismutase and familial amyotrophic lateral sclerosis. J Mol Biol. 2003;334(3):515–25. 10.1016/j.jmb.2003.09.069 [DOI] [PubMed] [Google Scholar]
- Khare SD, Ding F, Gwanmesia KN, Dokholyan NV. Molecular origin of polyglutamine aggregation in neurodegenerative diseases. PLoS Comput Biol. 2005;1(3):230–5. 10.1371/journal.pcbi.0010030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare SD, Dokholyan NV. Common dynamical signatures of familial amyotrophic lateral sclerosis‐associated structurally diverse Cu, Zn superoxide dismutase mutants. Proc Natl Acad Sci. 2006;103(9):3147–52. 10.1073/pnas.0511266103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killinger BA, Melki R, Brundin P, Kordower JH. Endogenous alpha‐synuclein monomers, oligomers and resulting pathology: let's talk about the lipids in the room. NPJ Parkinsons Dis. 2019;5(1):23. 10.1038/s41531-019-0095-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim K‐T. Therapeutic approaches for inhibition of protein aggregation in Huntington's disease. Exp Neurobiol. 2014;23(1):36–44. 10.5607/en.2014.23.1.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TD, Paik SR, Yang C‐H, Kim J. Structural changes in α‐synuclein affect its chaperone‐like activity in vitro. Protein Sci. 2000;9(12):2489–96. 10.1110/ps.9.12.2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimov DK, Thirumalai D. Dissecting the assembly of Aβ16–22 amyloid peptides into antiparallel β sheets. Structure. 2003;11(3):295–307. 10.1016/S0969-2126(03)00031-5 [DOI] [PubMed] [Google Scholar]
- Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 reduces α‐synuclein aggregation and toxicity. J Biol Chem. 2004;279(24):25497–502. 10.1074/jbc.M400255200 [DOI] [PubMed] [Google Scholar]
- Knowles TP, Fitzpatrick AW, Meehan S, Mott HR, Vendruscolo M, Dobson CM, et al. Role of intermolecular forces in defining material properties of protein nanofibrils. Science. 2007;318(5858):1900–3. 10.1126/science.1150057 [DOI] [PubMed] [Google Scholar]
- Knowles TPJ, Buehler MJ. Nanomechanics of functional and pathological amyloid materials. Nat Nanotechnol. 2011;6(8):469–79. 10.1038/nnano.2011.102 [DOI] [PubMed] [Google Scholar]
- Knowles TPJ, Waudby CA, Devlin GL, Cohen SIA, Aguzzi A, Vendruscolo M, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326(5959):1533–7. 10.1126/science.1178250 [DOI] [PubMed] [Google Scholar]
- Korobeynikov VA, Lyashchenko AK, Blanco‐Redondo B, Jafar‐Nejad P, Shneider NA. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. 2022;28(1):104–16. 10.1038/s41591-021-01615-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovač V, Šerbec VČ. Prion protein: the molecule of many forms and faces. Int J Mol Sci. 2022;23(3):1232. 10.3390/ijms23031232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RSG, Budka H. Mutations of the prion protein gene. J Neurol. 2002;249(11):1567–82. 10.1007/s00415-002-0896-9 [DOI] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst C‐A, Klein AM, Li X‐J, Shi‐Hua Li M, Beal F, et al. Huntingtin aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46(6):842–9. [DOI] [PubMed] [Google Scholar]
- Kuijper EC, Bergsma AJ, Pim Pijnappel WWM, Aartsma‐Rus A. Opportunities and challenges for antisense oligonucleotide therapies. J Inherit Metab Dis. 2021;44(1):72–87. 10.1002/jimd.12251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni P, Leite VBP, Roy S, Bhattacharyya S, Mohanty A, Achuthan S, et al. Intrinsically disordered proteins: ensembles at the limits of Anfinsen's dogma. Biophys Rev. 2022;3(1):011306. 10.1063/5.0080512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Walter J. Phosphorylation of amyloid beta (Aβ) peptides – a trigger for formation of toxic aggregates in Alzheimer's disease. Aging. 2011;3(8):803–12. 10.18632/aging.100362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Wirths O, Stüber K, Wunderlich P, Koch P, Theil S, et al. Phosphorylation of the amyloid β‐peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol. 2016;131(4):525–37. 10.1007/s00401-016-1546-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laferrière F, Maniecka Z, Pérez‐Berlanga M, Hruska‐Plochan M, Gilhespy L, Hock E‐M, et al. TDP‐43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci. 2019;22(1):65–77. 10.1038/s41593-018-0294-y [DOI] [PubMed] [Google Scholar]
- Lakhani VV, Ding F, Dokholyan NV. Polyglutamine induced Misfolding of huntingtin Exon1 is modulated by the flanking sequences. PLoS Comput Biol. 2010;6(4):e1000772. 10.1371/journal.pcbi.1000772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AE. Clinical trials of disease‐modifying therapies for neurodegenerative diseases: the challenges and the future. Nat Med. 2010;16(11):1223–6. 10.1038/nm.2220 [DOI] [PubMed] [Google Scholar]
- Lasagna‐Reeves CA, Clos AL, Castillo‐Carranza D, Sengupta U, Guerrero‐Muñoz M, Kelly B, et al. Dual role of P53 amyloid formation in cancer; loss of function and gain of toxicity. Biochem Biophys Res Commun. 2013;430(3):963–8. 10.1016/j.bbrc.2012.11.130 [DOI] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α‐synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14(1):38–48. 10.1038/nrn3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V, Fieńko S, Andres‐Alonso M, Anni D, Ivanova D, Montenegro‐Venegas C, et al. Physiological concentrations of amyloid beta regulate recycling of synaptic vesicles via alpha7 acetylcholine receptor and CDK5/calcineurin signaling. Front Mol Neurosci. 2017;10:221. 10.3389/fnmol.2017.00221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc E, Serban H, Prusiner SB, Burton DR, Williamson RA. Copper induces conformational changes in the N‐terminal part of cell‐surface PrPC. Arch Virol. 2006;151(11):2103–9. 10.1007/s00705-006-0804-1 [DOI] [PubMed] [Google Scholar]
- Lee R v d, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Keith Dunker A, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114(13):6589–631. 10.1021/cr400525m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Harvey TS, Yin Y, Yau P, Litchfield D, Arrowsmith CH. Solution structure of the tetrameric minimum transforming domain of P53. Nat Struct Mol Biol. 1994;1(12):877–90. 10.1038/nsb1294-877 [DOI] [PubMed] [Google Scholar]
- Leitman J, Ulrich Hartl F, Lederkremer GZ. Soluble forms of PolyQ‐expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat Commun. 2013;4(1):2753. 10.1038/ncomms3753 [DOI] [PubMed] [Google Scholar]
- Li H‐Y, Yeh P‐A, Chiu H‐C, Tang C‐Y, Tu B P‐h. Hyperphosphorylation as a defense mechanism to reduce TDP‐43 aggregation. PLoS One. 2011;6(8):e23075. 10.1371/journal.pone.0023075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Guo M, Chen L, Chen Z, Ying F, Chen Y. P53 amyloid aggregation in cancer: function, mechanism, and therapy. Exp Hematol Oncol. 2022;11(1):66. 10.1186/s40164-022-00317-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Michael Babinchak W, Surewicz WK. Cryo‐EM structure of amyloid fibrils formed by the entire low complexity domain of TDP‐43. Nat Commun. 2021;12(1):1620. 10.1038/s41467-021-21912-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X‐L, Chen T, Wong Y‐S, Gang X, Fan R‐R, Zhao H‐L, et al. Involvement of mitochondrial dysfunction in human islet amyloid polypeptide‐induced apoptosis in INS‐1E pancreatic beta cells: an effect attenuated by phycocyanin. Int J Biochem Cell Biol. 2011;43(4):525–34. 10.1016/j.biocel.2010.12.008 [DOI] [PubMed] [Google Scholar]
- Lin MT, Flint Beal M. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95. 10.1038/nature05292 [DOI] [PubMed] [Google Scholar]
- Lippens G, Sillen A, Landrieu I, Amniai L, Sibille N, Barbier P, et al. Tau aggregation in Alzheimer's disease: what role for phosphorylation? Prion. 2007;1(1):21–5. 10.4161/pri.1.1.4055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Moloney A, Meehan S, Morris K, Thomas SE, Serpell LC, et al. Iron promotes the toxicity of amyloid β peptide by impeding its ordered aggregation. J Biol Chem. 2011;286(6):4248–56. 10.1074/jbc.M110.158980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Zaidi T, Iqbal K, Grundke‐Iqbal I, Merkle RK, Gong C‐X. Role of glycosylation in hyperphosphorylation of tau in Alzheimer's disease. FEBS Lett. 2002;512(1‐3):101–6. 10.1016/S0014-5793(02)02228-7 [DOI] [PubMed] [Google Scholar]
- Liu K, Liu Y, Li L, Qin P, Iqbal J, Deng Y, et al. Glycation Alter the process of tau phosphorylation to change tau isoforms aggregation property. Biochim Biophys Acta Mol Basis Dis. 2016;1862(2):192–201. 10.1016/j.bbadis.2015.12.002 [DOI] [PubMed] [Google Scholar]
- López L, Varea O, Navarro S, Carrodeguas J, Sanchez N, de Groot S, et al. Benzbromarone, quercetin, and folic acid inhibit amylin aggregation. Int J Mol Sci. 2016;17(6):964. 10.3390/ijms17060964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Yadong Y, Zhu I, Cheng Y, Sun PD. Structural mechanism of serum amyloid A‐mediated inflammatory amyloidosis. Proc Natl Acad Sci. 2014;111(14):5189–94. 10.1073/pnas.1322357111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J‐X, Qiang W, Yau W‐M, Schwieters CD, Meredith SC, Tycko R. Molecular structure of β‐amyloid fibrils in Alzheimer's disease brain tissue. Cell. 2013;154(6):1257–68. 10.1016/j.cell.2013.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H‐B, Xia Y‐Y, Shu X‐J, Liu Z‐C, Feng Y, Liu X‐H, et al. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc Natl Acad Sci. 2014;111(46):16586–91. 10.1073/pnas.1417548111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Nussinov R. Stabilities and conformations of Alzheimer's β‐amyloid peptide oligomers (Aβ16−22, Aβ16–35, and Aβ10−35): sequence effects. Proc Natl Acad Sci. 2002;99(22):14126–31. 10.1073/pnas.212206899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Nussinov R. Molecular dynamics simulations of alanine rich β‐sheet oligomers: insight into amyloid formation. Protein Sci. 2009;11(10):2335–50. 10.1110/ps.4270102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Shang YC, Wang S. MTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol Med. 2013;19(1):51–60. 10.1016/j.molmed.2012.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Wiedau M. Therapeutic approaches targeting protein aggregation in amyotrophic lateral sclerosis. Front Mol Neurosci. 2020;13:98. 10.3389/fnmol.2020.00098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsool MD, Marsool PP, Reddy YB, Marsool ADM, Lam JR, Nandwana V. Newer modalities in the management of Alzheimer's dementia along with the role of Aducanumab and Lecanemab in the treatment of its refractory cases. Dis Mon. 2023;69(5):101547. 10.1016/j.disamonth.2023.101547 [DOI] [PubMed] [Google Scholar]
- Martinelli A, Lopes F, John E, Carlini C, Ligabue‐Braun R. Modulation of disordered proteins with a focus on neurodegenerative diseases and other pathologies. Int J Mol Sci. 2019;20(6):1322. 10.3390/ijms20061322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maziuk B, Ballance HI, Wolozin B. Dysregulation of RNA binding protein aggregation in neurodegenerative disorders. Front Mol Neurosci. 2017;10. 10.3389/fnmol.2017.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Schumann P, Weydt P, Petri S, Koc Y, Spittel S, et al. Neurofilament light‐chain response during therapy with antisense oligonucleotide Tofersen in sod1‐related als: treatment experience in clinical practice. Muscle Nerve. 2023;67:515–21. 10.1002/mus.27818 [DOI] [PubMed] [Google Scholar]
- Middleton ER, Rhoades E. Effects of curvature and composition on α‐synuclein binding to lipid vesicles. Biophys J. 2010;99(7):2279–88. 10.1016/j.bpj.2010.07.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, et al. Trial of antisense oligonucleotide Tofersen for SOD1 ALS. N Engl J Med. 2022;387(12):1099–110. 10.1056/NEJMoa2204705 [DOI] [PubMed] [Google Scholar]
- Miyake Y, Tanaka K, Fukushima W, Sasaki S, Kiyohara C, Tsuboi Y, et al. Dietary intake of metals and risk of Parkinson's disease: a case‐control study in Japan. J Neurol Sci. 2011;306(1‐2):98–102. 10.1016/j.jns.2011.03.035 [DOI] [PubMed] [Google Scholar]
- Mogk A, Bukau B. Molecular chaperones: structure of a protein disaggregase. Curr Biol. 2004;14(2):R78–80. 10.1016/j.cub.2003.12.051 [DOI] [PubMed] [Google Scholar]
- Mokretar K, Pease D, Taanman J‐W, Soenmez A, Ejaz A, Lashley T, et al. Somatic copy number gains of α‐synuclein (SNCA) in Parkinson's disease and multiple system atrophy brains. Brain. 2018;141(8):2419–31. 10.1093/brain/awy157 [DOI] [PubMed] [Google Scholar]
- Mosselman S, Höppener JWM, Zandberg J, van Mansfeld ADM, Geurts van Kessel AHM, Lips CJM, et al. Islet amyloid polypeptide: identification and chromosomal localization of the human gene. FEBS Lett. 1988;239(2):227–32. 10.1016/0014-5793(88)80922-0 [DOI] [PubMed] [Google Scholar]
- Mummery CJ, Junge C, Kordasiewicz HB, Mignon L, Moore KM, Yun C, et al. Results of the first‐in‐human, randomized, double‐blind, placebo‐controlled phase 1b study of lumbar intrathecal bolus administrations of antisense oligonucleotide (ISIS 814907; BIIB080) targeting tau MRNA in patients with mild Alzheimer's disease. Alzheimers Dement. 2021;17(S9). 10.1002/alz.051871 [DOI] [Google Scholar]
- Munishkina LA, Cooper EM, Uversky VN, Fink AL. The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J Mol Recognit. 2004;17(5):456–64. 10.1002/jmr.699 [DOI] [PubMed] [Google Scholar]
- Musteikytė G, Jayaram AK, Xu CK, Vendruscolo M, Krainer G, Knowles TPJ. Interactions of α‐synuclein oligomers with lipid membranes. Biochim Biophys Acta Biomembr. 2021;1863(4):183536. 10.1016/j.bbamem.2020.183536 [DOI] [PubMed] [Google Scholar]
- Nasica‐Labouze J, Nguyen PH, Sterpone F, Berthoumieu O, Buchete N‐V, Coté S, et al. Amyloid β protein and Alzheimer's disease: when computer simulations complement experimental studies. Chem Rev. 2015;115(9):3518–63. 10.1021/cr500638n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näsström T, Fagerqvist T, Barbu M, Karlsson M, Nikolajeff F, Kasrayan A, et al. The lipid peroxidation products 4‐oxo‐2‐nonenal and 4‐hydroxy‐2‐nonenal promote the formation of α‐synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic Biol Med. 2011;50(3):428–37. 10.1016/j.freeradbiomed.2010.11.027 [DOI] [PubMed] [Google Scholar]
- Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, et al. Structure of the cross‐β spine of amyloid‐like fibrils. Nature. 2005;435(7043):773–8. 10.1038/nature03680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PH, Ramamoorthy A, Sahoo BR, Zheng J, Faller P, Straub JE, et al. Amyloid oligomers: a joint experimental/computational perspective on Alzheimer's disease, Parkinson's disease, type II diabetes, and amyotrophic lateral sclerosis. Chem Rev. 2021;121(4):2545–647. 10.1021/acs.chemrev.0c01122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M. Techniques to study amyloid fibril formation in vitro. Methods. 2004;34(1):151–60. 10.1016/j.ymeth.2004.03.012 [DOI] [PubMed] [Google Scholar]
- Nonaka T, Masuda‐Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, et al. Prion‐like properties of pathological TDP‐43 aggregates from diseased brains. Cell Rep. 2013;4(1):124–34. 10.1016/j.celrep.2013.06.007 [DOI] [PubMed] [Google Scholar]
- Novials A, Jiménez‐Chillarón JC, Franco C, Casamitjana R, Gomis R, Gómez‐Foix AM. Reduction of islet amylin expression and basal secretion by adenovirus‐mediated delivery of amylin antisense CDNA. Pancreas. 1998;17(2):182–6. 10.1097/00006676-199808000-00012 [DOI] [PubMed] [Google Scholar]
- Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti GM, Morgan TE, et al. Clusterin (ApoJ) alters the aggregation of amyloid β‐peptide (Aβ1‐42) and forms slowly sedimenting Aβ complexes that cause oxidative stress. Exp Neurol. 1995;136(1):22–31. 10.1006/exnr.1995.1080 [DOI] [PubMed] [Google Scholar]
- O'Driscoll J, Clare D, Saibil H. Prion aggregate structure in yeast cells is determined by the Hsp104‐Hsp110 disaggregase machinery. J Cell Biol. 2015;211(1):145–58. 10.1083/jcb.201505104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira GAP, Petronilho EC, Pedrote MM, Marques MA, Vieira TCRG, Cino EA, et al. The status of P53 oligomeric and aggregation states in cancer. Biomolecules. 2020;10(4):548. 10.3390/biom10040548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway JM, Tallaksen‐Greene S, Gutekunst C‐A, Bernstein EM, Cearley JA, Wiener HW, et al. Ectopically expressed CAG repeats cause Intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell. 1997;91(6):753–63. 10.1016/S0092-8674(00)80464-X [DOI] [PubMed] [Google Scholar]
- Ouberai MM, Wang J, Swann MJ, Galvagnion C, Guilliams T, Dobson CM, et al. α‐Synuclein senses lipid packing defects and induces lateral expansion of lipids leading to membrane remodeling. J Biol Chem. 2013;288(29):20883–95. 10.1074/jbc.M113.478297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padrick SB, Miranker AD. Islet amyloid: phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry. 2002;41(14):4694–703. 10.1021/bi0160462 [DOI] [PubMed] [Google Scholar]
- Panda D, Samuel JC, Massie M, Feinstein SC, Wilson L. Differential regulation of microtubule dynamics by three‐ and four‐repeat tau: implications for the onset of neurodegenerative disease. Proc Natl Acad Sci. 2003;100(16):9548–53. 10.1073/pnas.1633508100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannuzzo M, Raudino A, Milardi D, La Rosa C, Karttunen M. α‐Helical structures drive early stages of self‐assembly of Amyloidogenic amyloid polypeptide aggregate formation in membranes. Sci Rep. 2013;3(1):2781. 10.1038/srep02781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil SM, Mehta A, Jha S, Alexandrescu AT. Heterogeneous amylin fibril growth mechanisms imaged by total internal reflection fluorescence microscopy. Biochemistry. 2011;50(14):2808–19. 10.1021/bi101908m [DOI] [PubMed] [Google Scholar]
- Pena‐Diaz S, Pujols J, Ventura S. Small molecules to prevent the neurodegeneration caused by α‐synuclein aggregation. Neural Regen Res. 2020;15(12):2260–1. 10.4103/1673-5374.284993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perluigi M, Barone E, Di Domenico F, Butterfield DA. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro‐survival and cell death pathways. Biochim Biophys Acta Mol Basis Dis. 2016;1862(10):1871–82. 10.1016/j.bbadis.2016.07.005 [DOI] [PubMed] [Google Scholar]
- Perneczky R, Jessen F, Grimmer T, Levin J, Flöel A, Peters O, et al. Anti‐amyloid antibody therapies in Alzheimer's disease. Brain. 2023;146:842–9. 10.1093/brain/awad005 [DOI] [PubMed] [Google Scholar]
- Perutz MF. Glutamine repeats and neurodegenerative diseases: molecular aspects. Trends Biochem Sci. 1999;24(2):58–63. 10.1016/S0968-0004(98)01350-4 [DOI] [PubMed] [Google Scholar]
- Petrov D, Mansfield C, Moussy A, Hermine O. ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Front Aging Neurosci. 2017;9. 10.3389/fnagi.2017.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe L, Rué L, Robberecht W, Van Den Bosch L. Translating biological findings into new treatment strategies for amyotrophic lateral sclerosis (ALS). Exp Neurol. 2014;262:138–51. 10.1016/j.expneurol.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Porta S, Yan X, Restrepo CR, Kwong LK, Zhang B, Brown HJ, et al. Patient‐derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP‐43 pathology in vivo. Nat Commun. 2018;9(1):4220. 10.1038/s41467-018-06548-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK. Molecular mechanisms of TDP‐43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci. 2019;12:25. 10.3389/fnmol.2019.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor EA, Ding F, Dokholyan NV. Discrete molecular dynamics. WIREs Comput Mol Sci. 2011;1(1):80–92. 10.1002/wcms.4 [DOI] [Google Scholar]
- Proctor EA, Fee L, Tao Y, Redler RL, Fay JM, Zhang Y, et al. Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis. Proc Natl Acad Sci. 2016;113(3):614–9. 10.1073/pnas.1516725113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Yau W‐M, Jun‐Xia L, Collinge J, Tycko R. Structural variation in amyloid‐β fibrils from Alzheimer's disease clinical subtypes. Nature. 2017;541(7636):217–21. 10.1038/nature20814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Hossen MA, Chowdhury MFI, Bari S, Tamanna N, Sultana SS, et al. Aducanumab for the treatment of Alzheimer's disease: a systematic review. Psychogeriatrics. 2023;23(3):512–22. 10.1111/psyg.12944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Andersen JK. Alpha synuclein aggregation: is it the toxic gain of function responsible for neurodegeneration in Parkinson's disease? Mech Ageing Dev. 2001;122(14):1499–510. 10.1016/S0047-6374(01)00283-4 [DOI] [PubMed] [Google Scholar]
- Rambaran RN, Serpell LC. Amyloid fibrils: abnormal protein assembly. Prion. 2008;2(3):112–7. 10.4161/pri.2.3.7488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasia RM, Bertoncini CW, Marsh D, Hoyer W, Cherny D, Zweckstetter M, et al. Structural characterization of copper(II) binding to α‐synuclein: insights into the bioinorganic chemistry of Parkinson's disease. Proc Natl Acad Sci. 2005;102(12):4294–9. 10.1073/pnas.0407881102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redler RL, Dokholyan NV. The complex molecular biology of amyotrophic lateral sclerosis (ALS). In: Teplow DE, editor. Progress in molecular biology and translational science. Volume 107. London, UK: Elsevier; 2012. p. 215–62. 10.1016/B978-0-12-385883-2.00002-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redler RL, Fee L, Fay JM, Caplow M, Dokholyan NV. Non‐native soluble oligomers of Cu/Zn superoxide dismutase (SOD1) contain a conformational epitope linked to cytotoxicity in amyotrophic lateral sclerosis (ALS). Biochemistry. 2014;53(14):2423–32. 10.1021/bi500158w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redler RL, Wilcox KC, Proctor EA, Fee L, Caplow M, Dokholyan NV. Glutathionylation at Cys‐111 induces dissociation of wild type and FALS mutant SOD1 dimers. Biochemistry. 2011;50(32):7057–66. 10.1021/bi200614y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed‐Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll‐like receptors 2 and 4 are required for fibrillar Aβ‐stimulated microglial activation. J Neurosci. 2009;29(38):11982–92. 10.1523/JNEUROSCI.3158-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss AB, Montufar N, DeLeon J, Pinkhasov A, Gomolin IH, Glass AD, et al. Alzheimer disease clinical trials targeting amyloid: lessons learned from success in mice and failure in humans. Neurologist. 2021;26(2):52–61. 10.1097/NRL.0000000000000320 [DOI] [PubMed] [Google Scholar]
- Reynolds MR, Berry RW, Binder LI. Nitration in neurodegeneration: deciphering the ‘Hows’ ‘NYs’. Biochemistry. 2007;46(25):7325–36. 10.1021/bi700430y [DOI] [PubMed] [Google Scholar]
- Rhoades E, Agarwal J, Gafni A. Aggregation of an Amyloidogenic fragment of human islet amyloid polypeptide. Biochim Biophys Acta Protein Struct Mol Enzymol. 2000;1476(2):230–8. 10.1016/S0167-4838(99)00248-4 [DOI] [PubMed] [Google Scholar]
- Ricchelli F, Fusi P, Tortora P, Valtorta M, Riva M, Tognon G, et al. Destabilization of non‐pathological variants of ataxin‐3 by metal ions results in aggregation/fibrillogenesis. Int J Biochem Cell Biol. 2007;39(5):966–77. 10.1016/j.biocel.2007.01.012 [DOI] [PubMed] [Google Scholar]
- Rivera JF, Gurlo T, Daval M, Huang CJ, Matveyenko AV, Butler PC, et al. Human‐IAPP disrupts the autophagy/lysosomal pathway in pancreatic β‐cells: protective role of P62‐positive cytoplasmic inclusions. Cell Death Differ. 2011;18(3):415–26. 10.1038/cdd.2010.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera NA, Brandt AL, Novakofski JE, Mateus‐Pinilla NE. Chronic wasting disease in cervids: prevalence, impact and management strategies. Vet Med. 2019;10:123–39. 10.2147/VMRR.S197404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo AN, Lin JB, Gates SN, Tse E, Bart SM, Castellano LM, et al. Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase. Nat Commun. 2019;10(1):2393. 10.1038/s41467-019-10150-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooijen V, Bart D, Claessens MMAE, Subramaniam V. Membrane binding of oligomeric α‐synuclein depends on bilayer charge and packing. FEBS Lett. 2008;582(27):3788–92. 10.1016/j.febslet.2008.10.009 [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(S7):S10–7. 10.1038/nm1066 [DOI] [PubMed] [Google Scholar]
- Runwal G, Edwards RH. The membrane interactions of synuclein: physiology and pathology. Annu Rev Pathol. 2021;16(1):465–85. 10.1146/annurev-pathol-031920-092547 [DOI] [PubMed] [Google Scholar]
- Saelices L, Johnson LM, Liang WY, Sawaya MR, Cascio D, Ruchala P, et al. Uncovering the mechanism of aggregation of human transthyretin. J Biol Chem. 2015;290(48):28932–43. 10.1074/jbc.M115.659912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel F, Flavin WP, Iqbal S, Pacelli C, Renganathan SDS, Trudeau L‐E, et al. Effects of serine 129 phosphorylation on α‐synuclein aggregation, membrane association, and internalization. J Biol Chem. 2016;291(9):4374–85. 10.1074/jbc.M115.705095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanches MN, Knapp K, Oliveira AB, Wolynes PG, Onuchic JN, Leite VBP. Examining the ensembles of amyloid‐β monomer variants and their propensities to form fibers using an energy landscape visualization method. J Phys Chem B. 2022;126(1):93–9. 10.1021/acs.jpcb.1c08525 [DOI] [PubMed] [Google Scholar]
- Sarell CJ, Wilkinson SR, Viles JH. Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid‐β from Alzheimer disease. J Biol Chem. 2010;285(53):41533–40. 10.1074/jbc.M110.171355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel MTOR‐independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α‐synuclein. J Biol Chem. 2007;282(8):5641–52. 10.1074/jbc.M609532200 [DOI] [PubMed] [Google Scholar]
- Schaffert L‐N, Carter WG. Do post‐translational modifications influence protein aggregation in neurodegenerative diseases: a systematic review. Brain Sci. 2020;10(4):232. 10.3390/brainsci10040232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab AD, Thurston MJ, Machhi J, Olson KE, Namminga KL, Gendelman HE, et al. Immunotherapy for Parkinson's disease. Neurobiol Dis. 2020;137:104760. 10.1016/j.nbd.2020.104760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seffernick JT, Lindert S. Hybrid methods for combined experimental and computational determination of protein structure. J Chem Phys. 2020;153(24):240901. 10.1063/5.0026025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289(5483):1317–21. 10.1126/science.289.5483.1317 [DOI] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lei L, Nasrabady SE, Elmaleh M, Mendelsohn M, et al. ALS‐associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun. 2016;7(1):10465. 10.1038/ncomms10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirvanyants D, Ding F, Tsao D, Ramachandran S, Dokholyan NV. Discrete molecular dynamics: an efficient and versatile simulation method for fine protein characterization. J Phys Chem B. 2012;116(29):8375–82. 10.1021/jp2114576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Southworth DR. Spiraling in control: structures and mechanisms of the Hsp104 disaggregase. Cold Spring Harb Perspect Biol. 2019;11(8):a034033. 10.1101/cshperspect.a034033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe JD, Cohen AS. Review: history of the amyloid fibril. J Struct Biol. 2000;130(2‐3):88–98. 10.1006/jsbi.2000.4221 [DOI] [PubMed] [Google Scholar]
- Smith AV, Tabrizi SJ. Therapeutic antisense targeting of huntingtin. DNA Cell Biol. 2020;39(2):154–8. 10.1089/dna.2019.5188 [DOI] [PubMed] [Google Scholar]
- Southwell AL, Skotte NH, Frank Bennett C, Hayden MR. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol Med. 2012;18(11):634–43. 10.1016/j.molmed.2012.09.001 [DOI] [PubMed] [Google Scholar]
- Sprunger ML, Jackrel ME. Prion‐like proteins in phase separation and their link to disease. Biomolecules. 2021;11(7):1014. 10.3390/biom11071014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan E, Chandrasekhar G, Chandrasekar P, Anbarasu K, Vickram AS, Rohini Karunakaran R, et al. Alpha‐synuclein aggregation in Parkinson's disease. Front Med. 2021;8:736978. 10.3389/fmed.2021.736978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Ahmad R, Khare SK. Alzheimer's disease and its treatment by different approaches: a review. Eur J Med Chem. 2021;216:113320. 10.1016/j.ejmech.2021.113320 [DOI] [PubMed] [Google Scholar]
- Stawoska I, Wesełucha‐Birczyńska A, Regonesi ME, Riva M, Tortora P, Stochel G. Interaction of selected divalent metal ions with human Ataxin‐3 Q36. J Biol Inorg Chem. 2009;14(8):1175–85. 10.1007/s00775-009-0561-1 [DOI] [PubMed] [Google Scholar]
- Stefani M. Structural polymorphism of amyloid oligomers and fibrils underlies different fibrillization pathways: immunogenicity and cytotoxicity. Curr Protein Pept Sci. 2010;11(5):343–54. 10.2174/138920310791330631 [DOI] [PubMed] [Google Scholar]
- Stommel JM. A leucine‐rich nuclear export signal in the P53 tetramerization domain: regulation of subcellular localization and P53 activity by NES masking. EMBO J. 1999;18(6):1660–72. 10.1093/emboj/18.6.1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneja V, Verma M, Vats A. Toxic species in amyloid disorders: oligomers or mature fibrils. Ann Indian Acad Neurol. 2015;18(2):138–45. 10.4103/0972-2327.144284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray AM, Lam B, McNulty EE, Nalls AV, Mathiason CK, Magadi SS, et al. Clearance of variant Creutzfeldt–Jakob disease prions in vivo by the Hsp70 disaggregase system. Brain. 2022;145(9):3236–49. 10.1093/brain/awac144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumalai D. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr Opin Struct Biol. 2003;13(2):146–59. 10.1016/S0959-440X(03)00032-0 [DOI] [PubMed] [Google Scholar]
- Tiwari MK, Kepp KP. Β‐amyloid pathogenesis: chemical properties versus cellular levels. Alzheimers Dement. 2016;12(2):184–94. 10.1016/j.jalz.2015.06.1895 [DOI] [PubMed] [Google Scholar]
- Tojo K, Sekijima Y, Kelly JW, Ikeda S‐i. Diflunisal stabilizes familial amyloid polyneuropathy‐associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res. 2006;56(4):441–9. 10.1016/j.neures.2006.08.014 [DOI] [PubMed] [Google Scholar]
- Tõugu V, Tiiman A, Palumaa P. Interactions of Zn(ii) and Cu(ii) ions with Alzheimer's amyloid‐Beta peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics. 2011;3(3):250–61. 10.1039/c0mt00073f [DOI] [PubMed] [Google Scholar]
- Tycko R. Amyloid polymorphism: structural basis and neurobiological relevance. Neuron. 2015;86(3):632–45. 10.1016/j.neuron.2015.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umoh ME, Fournier C, Li Y, Polak M, Shaw L, Landers JE, et al. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology. 2016;87(10):1024–30. 10.1212/WNL.0000000000003067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Ding F, Sammond D, Khare S, Buldyrev SV, et al. Molecular dynamics simulation of amyloid β dimer formation. Biophys J. 2004;87(4):2310–21. 10.1529/biophysj.104.040980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. A decade and a half of protein intrinsic disorder: biology still waits for physics: protein intrinsic disorder. Protein Sci. 2013;22(6):693–724. 10.1002/pro.2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. Introduction to intrinsically disordered proteins (IDPs). Chem Rev. 2014;114(13):6557–60. 10.1021/cr500288y [DOI] [PubMed] [Google Scholar]
- Uversky VN, Fink AL. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta Proteins Proteom. 2004;1698(2):131–53. 10.1016/j.bbapap.2003.12.008 [DOI] [PubMed] [Google Scholar]
- Uversky VN, Lee H‐J, Li J, Fink AL, Lee S‐J. Stabilization of partially folded conformation during α‐synuclein oligomerization in both purified and cytosolic preparations. J Biol Chem. 2001;276(47):43495–8. 10.1074/jbc.C100551200 [DOI] [PubMed] [Google Scholar]
- Uversky VN, Oldfield CJ, Keith Dunker A. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37(1):215–46. 10.1146/annurev.biophys.37.032807.125924 [DOI] [PubMed] [Google Scholar]
- Uversky VN, Yamin G, Munishkina LA, Karymov MA, Millett IS, Doniach S, et al. Effects of nitration on the structure and aggregation of α‐synuclein. Mol Brain Res. 2005;134(1):84–102. 10.1016/j.molbrainres.2004.11.014 [DOI] [PubMed] [Google Scholar]
- Vassilakopoulou V, Karachaliou C‐E, Evangelou A, Zikos C, Livaniou E. Peptide‐based vaccines for neurodegenerative diseases: recent endeavors and future perspectives. Vaccine. 2021;9(11):1278. 10.3390/vaccines9111278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Thinakaran G. Membrane rafts in Alzheimer's disease beta‐amyloid production. Biochim Biophys Acta Mol Cell Biol Lipids. 2010;1801(8):860–7. 10.1016/j.bbalip.2010.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villoslada P, Moreno B, Melero I, Pablos JL, Martino G, Uccelli A, et al. Immunotherapy for neurological diseases. Clin Immunol. 2008;128(3):294–305. 10.1016/j.clim.2008.04.003 [DOI] [PubMed] [Google Scholar]
- Vitek GE, Decourt B, Sabbagh MN. Lecanemab (BAN2401): an anti‐beta‐amyloid monoclonal antibody for the treatment of Alzheimer disease. Expert Opin Investig Drugs. 2023;32(2):89–94. 10.1080/13543784.2023.2178414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, et al. Atomic‐resolution structure of a disease‐relevant Aβ(1–42) amyloid fibril. Proc Natl Acad Sci. 2016;113(34):E4976–84. 10.1073/pnas.1600749113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Yin Y‐L, Liu X‐Z, Shen P, Zheng Y‐G, Lan X‐R, et al. Current understanding of metal ions in the pathogenesis of Alzheimer's disease. Transl Neurodegener. 2020;9(1):10. 10.1186/s40035-020-00189-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Nema S, Teagarden D. Protein aggregation—pathways and influencing factors. Int J Pharm. 2010;390(2):89–99. 10.1016/j.ijpharm.2010.02.025 [DOI] [PubMed] [Google Scholar]
- Wang X, Wang W, Li L, Perry G, Lee H‐g, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta Mol Basis Dis. 2014;1842(8):1240–7. 10.1016/j.bbadis.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JM, Darling AL, Uversky VN, Blair LJ. Small heat shock proteins, big impact on protein aggregation in neurodegenerative disease. Front Pharmacol. 2019;10:1047. 10.3389/fphar.2019.01047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells C, Brennan S, Keon M, Ooi L. The role of amyloid oligomers in neurodegenerative pathologies. Int J Biol Macromol. 2021;181:582–604. 10.1016/j.ijbiomac.2021.03.113 [DOI] [PubMed] [Google Scholar]
- Wells GAH, Wilesmith JW, McGill IS. Bovine spongiform encephalopathy: a neuropathological perspective. Brain Pathol. 1991;1(2):69–78. 10.1111/j.1750-3639.1991.tb00642.x [DOI] [PubMed] [Google Scholar]
- Westermark P, Wernstedt C, Wilander E, Hayden DW, O'Brien TD, Johnson KH. Amyloid fibrils in human Insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide‐like protein also present in Normal islet cells. Proc Natl Acad Sci. 1987;84(11):3881–5. 10.1073/pnas.84.11.3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwell‐Roper C, Dai DL, Soukhatcheva G, Potter KJ, van Rooijen N, Ehses JA, et al. IL‐1 blockade attenuates islet amyloid polypeptide‐induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J Immunol. 2011;187(5):2755–65. 10.4049/jimmunol.1002854 [DOI] [PubMed] [Google Scholar]
- Wilcox KC, Zhou L, Jordon JK, Huang Y, Yanbao Y, Redler RL, et al. Modifications of superoxide dismutase (SOD1) in human erythrocytes. J Biol Chem. 2009;284(20):13940–7. 10.1074/jbc.M809687200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that α‐synuclein oligomers are toxic. Proc Natl Acad Sci. 2011;108(10):4194–9. 10.1073/pnas.1100976108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016;351(6269):173–6. 10.1126/science.aad2033 [DOI] [PubMed] [Google Scholar]
- Wright PE, Jane Dyson H. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16(1):18–29. 10.1038/nrm3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, et al. Gain of function of mutant P53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7(5):285–95. 10.1038/nchembio.546 [DOI] [PubMed] [Google Scholar]
- Yamaguchi K‐i, Kuwata K. Formation and properties of amyloid fibrils of prion protein. Biophys Rev. 2018;10(2):517–25. 10.1007/s12551-017-0377-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Shin R‐W, Hasegawa K, Naiki H, Sato H, Yoshimasu F, et al. Iron (III) induces aggregation of hyperphosphorylated τ and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer's disease: τ aggregation by iron (III). J Neurochem. 2004;82(5):1137–47. 10.1046/j.1471-4159.2002.t01-1-01061.x [DOI] [PubMed] [Google Scholar]
- Yamin G, Glaser CB, Uversky VN, Fink AL. Certain metals trigger fibrillation of methionine‐oxidized α‐synuclein. J Biol Chem. 2003;278(30):27630–5. 10.1074/jbc.M303302200 [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Fantini J, Chakrabartty A, Eckert A. A β behavior on neuronal membranes: aggregation and toxicities. Int J Alzheimer's Dis. 2011;2011:1–2. 10.4061/2011/286536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D‐J, Shi S, Zheng L‐F, Yao T‐M, Ji L‐N. Mercury(II) promotes the in vitro aggregation of tau fragment corresponding to the second repeat of microtubule‐binding domain: coordination and conformational transition. Biopolymers. 2010;93(12):1100–7. 10.1002/bip.21527 [DOI] [PubMed] [Google Scholar]
- Yu S, Yin S, Li C, Wong P, Chang B, Xiao F, et al. Aggregation of prion protein with insertion mutations is proportional to the number of inserts. Biochem J. 2007;403(2):343–51. 10.1042/BJ20061592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamel J, Chen J, Zaer S, Harris PD, Drori P, Lebendiker M, et al. Structural and dynamic insights into α‐synuclein dimer conformations. Biophysics. 2019;31:411–423. 10.1101/795997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Stys PK. Role of prions in neuroprotection and neurodegeneration: a mechanism involving glutamate receptors? Prion. 2009;3(4):187–9. 10.4161/pri.3.4.9549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DY, Wang J, Fleeman RM, Kuhn MK, Swulius MT, Proctor EA, et al. Monosialotetrahexosylganglioside promotes early Aβ42 oligomer formation and maintenance. ACS Chem Nerosci. 2022;13(13):1979–91. 10.1021/acschemneuro.2c00221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Xiaoyan H, Khant H, Ludtke SJ, Chiu W, Schmid MF, et al. Interprotofilament interactions between Alzheimer's Aβ 1–42 peptides in amyloid fibrils revealed by CryoEM. Proc Natl Acad Sci. 2009;106(12):4653–8. 10.1073/pnas.0901085106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhu M, Song W‐y, Harmon AC, Chen S. Oxidation and phosphorylation of MAP kinase 4 cause protein aggregation. Biochim Biophys Acta Proteins Proteom. 2015;1854(2):156–65. 10.1016/j.bbapap.2014.11.006 [DOI] [PubMed] [Google Scholar]
- Zhu C, Beck MV, Griffith JD, Deshmukh M, Dokholyan NV. Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc Natl Acad Sci. 2018;115(18):4661–5. 10.1073/pnas.1800187115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zraika S, Hull RL, Udayasankar J, Aston‐Mourney K, Subramanian SL, Kisilevsky R, et al. Oxidative stress is induced by islet amyloid formation and time‐dependently mediates amyloid‐induced beta cell apoptosis. Diabetologia. 2009;52(4):626–35. 10.1007/s00125-008-1255-x [DOI] [PMC free article] [PubMed] [Google Scholar]