

Abstract
The murine Ung gene encodes both mitochondrial (Ung1) and nuclear (Ung2) forms of uracil-DNA glycosylase. The gene contains seven exons organised like the human counterpart. While the putative Ung1 promoter (PB) and the human PB contain essentially the same, although differently organised, transcription factor binding elements, the Ung2 promoter (PA) shows limited homology to the human counterpart. Transient transfection of chimaeric promoter–luciferase constructs demonstrated that both promoters are functional and that PB drives transcription more efficiently than PA. mRNAs for Ung1 and Ung2 are found in all adult tissues analysed, but they are differentially expressed. Furthermore, transcription of both mRNA forms, particularly Ung2, is induced in mid-gestation embryos. Except for a strong conservation of the 26 N-terminal residues in Ung2, the subcellular targeting sequences in the encoded proteins have limited homology. Ung2 is transported exclusively to the nucleus in NIH 3T3 cells as expected. In contrast, Ung1 was sorted both to nuclei and mitochondria. These results demonstrate that although the catalytic domain of uracil-DNA glycosylase is highly conserved in mouse and man, regulatory elements in the gene and subcellular sorting sequences in the proteins differ both structurally and functionally, resulting in altered contribution of the isoforms to total uracil-DNA glycosylase activity.
INTRODUCTION
Uracil in DNA may result from deamination of cytosine (resulting in a U:G mispair) or misincorporation of dUMP during DNA replication (resulting in a U:A pair) (1). In bacteria and yeast, uracil-DNA glycosylase is required for prevention of GC to AT transition mutations (2,3). The highly homologous human enzyme is encoded by the UNG gene, and is thought to have a similar function, but this has not been demonstrated experimentally (reviewed in 4). Recently, it was demonstrated that human nuclear UNG2 encoded by UNG is present in replication foci where it interacts with replication factor A (RPA) and proliferating cell nuclear antigen (PCNA) and is apparently required for removal of misincorporated U residues (5). Other enzymes with uracil-excising activity include T(U)DG-mismatch glycosylase (6), SMUG1 (7), cyclin-like UDG2 (8) and MBD4 (9). In vitro these contribute to a very minor fraction of the total uracil-DNA glycosylase activity, but nothing is known about their possible contributions in preventing mutations.
The gene for human uracil-DNA glycosylase (UNG) consists of seven exons and encodes nuclear (UNG2) and mitochondrial (UNG1) isoforms (10,11). mRNAs for UNG2 and UNG1 arise from transcription from alternative promoters, promoter A (PA) and promoter B (PB) respectively, and the subsequent use of alternative splicing (11). The enzymes have unique N-terminal amino acid sequences and a common catalytic domain of known structure (12,13). The human UNG gene is regulated mainly at the level of transcription initiation in synchronised cells in culture (14). Furthermore, the two resulting mRNAs are differentially expressed (15). Mouse cDNAs corresponding to the human UNG2 and UNG1 proteins have been identified (11,16) but expression of the corresponding mRNAs or proteins has not been investigated. The putative mouse proteins Ung2 and Ung1 consist of 306 and 295 amino acids, respectively, and the identical catalytic domain shares 91% identity with the human catalytic domain. The N-terminal amino acid sequence of the nuclear form of the enzyme is highly conserved, with the first 13 amino acids being identical in the human and mouse proteins. Moreover, the N-terminal amino acids interacting with RPA and PCNA are identical in the mouse and human nuclear proteins which implies that the mouse homologue also interacts with these proteins (5,17). In contrast, limited sequence similarity can be found in the N-terminal sequence of the mitochondrial forms of the proteins.
A previous report on the structure of the mouse Ung gene (16) is largely consistent with our sequencing data presented in this study, including the nucleotide sequence of the mouse promoter B (16). Here we present the complete sequence of the murine Ung gene, including introns and both promoters, which are demonstrated to be functional. In addition to giving the complete sequence and a somewhat modified structure of exons and introns in the Ung gene, we also present the first report of the functionality of the murine promoters. Furthermore, we have analysed the expression of mRNA for Ung1 and Ung2 in adult and embryonic tissues as well as the subcellular distribution of Ung activity in NIH 3T3 cells in relation to replicative status. Analysis of subcellular sorting of Ung1 and Ung2 in fusion with EGFP as reporter protein in NIH 3T3 cells demonstrates that Ung2 is sorted exclusively to the nucleus, while Ung1 sorts both to mitochondria and nuclei. This is different from human UNG1 that contains a dominant mitochondrial sorting signal (17), but resembles the behaviour of another mammalian DNA glycosylase that displays a dual sorting pattern (18).
MATERIALS AND METHODS
Primers and probes
The following primers were used in this study (all sequences are given in 5′ to 3′ direction). Up-Pa: GTA GGT AGG TAA GCA GCA GG; Lp-1B: GGA GTC AGA GTC GGG GTT G; ProA+: AGA ATC CTA AGC CAG CAT TGA AG; ProB+: GTG GTG AGA CGA GCG GGG T; ProB5′–: TTT AAG CTT GCT GCC TGG TGC TGC CCC; ProB–: TTT AAG CTT GGT CCA GCC GAG CAG GGG; mE2F-c: GGG AGC TGT TTT GCA TCG AAA AGC CCA CGT G; mE2F-p: GCA GCA CCA GGC AGA TCG AGA CTG CGG TGC; mE2F-r: GGG AGG TGT TTT GCA TCG AAA AGC CTG CGT. All oligonucleotides were from Eurogentech, Belgium. Nucleotides to be mutated are shown in bold.
mUng1 cDNA was excised from its cloning vector by digestion with EcoRI–XhoI. The 1.95 kb mUng1 insert was isolated and specific probes to detect mUng1 or mUng2 mRNA were generated by amplifying a fragment spanning exon1A down to the splice acceptor site in exon1B (11) by PCR using the primers Up-PA and Lp-1B and mouse genomic DNA (WEHI-Cl13) as template. The fragment was digested with RsaI giving exon1A (357 bp) and exon1B (655 bp) specific probes. All probes were labelled in random primer extension reactions (Rediprime‘ labelling kit, Amersham Pharmacia Biotech, Buckinghamshire, UK) with [α-32P]dCTP from Amersham Pharmacia Biotech.
Isolation of mouse Ung genomic clone
A λ2001 library derived from mouse strain 129SV ES cells (19) was screened with the mUng1 cDNA as a probe. One million phages were screened from which one positive clone was purified through tertiary screening.
Purification of Phage λ DNA
Plate lysate stocks of purified phage, prepared by standard technology (20), were used to infect 50 ml of Escherichia coli NM539 (OD600 = 0.3–0.5). The culture was incubated at 37°C until complete lysis occurred and then centrifuged at 8250 r.p.m. for 15 min. The supernatant was collected and treated with 2 µg/ml RNase A and 1 µg/ml DNaseI (Sigma Chemical Co., St Louis, MO) overnight at 4°C. Solid NaCl was added to a final concentration of 1 M and incubated for 1 h at 4°C. Debris was removed by centrifugation at 11 000 g for 10 min and DNA was precipitated by the addition of PEG8000 to a final concentration of 10% w/v. After incubation on ice for 1 h the DNA was collected by centrifugation at 11 000 g for 10 min, resuspended in 1× TE buffer and extracted with phenol/chloroform and ether. After precipitation in isopropanol the DNA was resuspended in TE.
Analysis of λ clones
A 6.5 kb SacI fragment from the positive λ clone was subcloned into pGEM3 (Promega, Madison, WI). The clone was sequenced by primer walking on an Applied Biosystems Model 373A Sequencing System using AmpliTaq FS cycle sequencing kit from ABI (PE BioSystems, Foster City, CA). The sequencing revealed that the purified clone was truncated at a BamHI site in intron 2. We therefore bought a P1 clone from Genome Systems (St Louis, MO) containing the Ung gene from 129 SV ES cells, from which we isolated a fragment containing the promoter regions and intron 2. This fragment was subcloned into the KpnI site of the pBluescript SK+ polylinker (Stratagene, La Jolla, CA) giving mUngKpnI. The sequence of the upstream region was determined as described above. The sequences were analysed and assembled using the AutoAssembler software (PE BioSystems).
Northern blot analysis
For analysis of Ung1 and Ung2 transcripts in different mouse tissues we used commercial Multiple Tissue Northern Blots (MTN Blots) from Clontech Laboratories (Palo Alto, CA). These were hybridised to radiolabelled probes specific for Ung1 and Ung2 as well as β-actin mRNA. The hybridisations were carried out overnight in ExpressHyb‘ solution (Clontech) supplemented with 100 µg/ml sonicated salmon sperm DNA at 68°C. The blots were washed with 2× SSC/0.5% SDS for 40 min at room temperature with three changes of wash solution, followed by washes with 0.1× SSC/0.5% SDS twice for 20 min at 65°C. The membranes were visualised on a Molecular Dynamics PhosphorImager SF after 48 h. The membranes were stripped by boiling in 0.5% SDS for 10 min.
Constructs for promoter studies
All chimaeric promoter–luciferase constructs were made in the pGL2-Basic vector (Promega) that carries the coding region of the firefly luciferase as a reporter gene. A 1.74 kb fragment comprising promoter A, exon1A and promoter B (ProA+B) was amplified by PCR using primers ProA+ and ProB– and mUngKpnI as template. The 1.44 kb fragment lacking the duplicated area of PB (ProA+B5′) was amplified using the primers ProA+ and ProB5′–. All PCR reactions were performed using the Expand‘High Fidelity PCR system from Roche Diagnostics (Mannheim, Germany) as recommended by the manufacturer. All the primers had HindIII linkers at their 5′ termini, the PCR fragments were therefore purified and digested with HindIII and ligated into HindIII digested pGL2-Basic. To generate a construct containing only promoter A, the 1.74 kb ProA+B insert was digested with HindIII and PstI. This gives a 972 bp fragment containing promoter A down to 31 nucleotides upstream of translation start of mUng2. The fragment was blunted with T4 DNA polymerase (New England Biolabs, Beverly, MA) and ligated into SmaI digested pGL2-Basic giving the construct ProA. Two constructs were made from PB by PCR using primers ProB+ as forward primer with ProB5′– or ProB– as lower primers giving rise to a 314 and a 610 bp fragment, respectively. These fragments were ligated to the pGEM“-T Easy vector (Promega) which subsequently was digested with SacII and SpeI to release the inserts. The inserts were ligated into SmaI–NheI digested pGL2-Basic giving rise to ProB5′ and ProB, respectively. All transformations were performed into heat-shock competent E.coli JM109. All constructs were verified by restriction enzyme digestion and sequencing using vector-specific primers GL1 and GL2 (Promega). The constructs were purified by Qiagen Maxi prep (Qiagen, Valencia, CA) prior to transfection and extracted with phenol/chloroform, precipitated with absolute ethanol and resuspended in 1× TE buffer.
Site-directed mutagenesis
Mutagenesis was performed by QuickChange‘ Site-Directed mutagenesis kit as recommended by the manufacturer (Stratagene). Two base pair changes were made in the consensus sequences for binding of transcription factors using the mutagenesis primers mE2F-c, mE2F-p and mE2F-r. All mutations were confirmed by sequencing as described above.
Cell culture
Mouse NIH 3T3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; high glucose from Gibco BRL, Paisley, UK) supplemented with 10% foetal calf serum (FCS), 2 mM l-glutamine and 4.5 mg/ml gentamicine (Gibco BRL). For transfection, cells were grown to 70% confluence in 175 cm2 tissue culture flasks in a humidified atmosphere containing 5% CO2. The cells were split at 4 × 105 cells into 50 mm tissue culture plates the day before transfection. To monitor DNA synthesis in freely cycling cells, 5 × 105 cells in 50 mm dishes were cultured for 48 h. For density inhibition, the cells were grown for 6 days with two changes of medium. The cells were pulse-labelled with 0.5 µCi/ml [3H]thymidine (Amersham Pharmacia). After incubation the incorporation was stopped on ice, the cells washed three times with ice-cold phosphate-buffered saline (PBS), harvested by trypsination and resuspended in 500 µl PBS. Samples were precipitated by adding 1 ml 1% trichloroacetic acid and collected on GFC filters (Whatman, Kent, UK). Incorporated activity was quantified by scintillation counting.
Transfection and luciferase assays
NIH 3T3 cells plated at 8 × 105 and 60% confluency were transiently transfected with 3 µg of test reporter plasmid DNA and 60 ng internal control pRL-TK (Promega) using FuGENE‘6 transfection reagent (Roche Diagnostics) as recommended by the manufacturer. For cAMP stimulation, 8-bromo-cAMP (Sigma) or forskolin were added to final concentrations of 1 mM or 25 µM respectively, 20 h prior to harvest. After washing twice in PBS the cells were lysed and luciferase activity was measured as luminescence according to the Dual-Luciferase‘ Reporter Assay System manual (Promega) in a Turner Designs Model TD-29/20 Luminometer (Turner Designs, Sunnyvale, CA).
Subcellular fractionation
NIH 3T3 cells were plated at 3 × 106 cells into 15 cm culture dishes. For density inhibition cells were left for 6 days as described above. Freely cycling cells were grown to 70% confluency. The cells were washed twice with PBS and harvested by trypsination. Cells from three dishes were pooled in 30 ml 10% FCS in PBS and centrifuged at 180 g for 10 min at 4°C, resuspended in 25 ml PBS and centrifuged as before. The cells were resuspended in 5 ml lysis buffer [10 mM Tris–HCl, pH 7.5, 10 mM NaCl, 1 mM DTT, 0.5 M EDTA, two tablets of Complete protease inhibitor (Roche Diagnostics)] and allowed to swell on ice for 15 min (cycling) or 30 min (density inhibited). The cell suspensions were transferred to a 7 ml Dounce Homogenizer, added 25 µl 1 M MgCl2 and immediately homogenized by 30 strokes with pestle B. Following centrifugation at 650 g for 2 min, the post-nuclear supernatant (PNS) was removed and the nuclei resuspended in 5 ml lysis buffer. After repeating the previous step, the two PNS were combined. The nuclei were resuspended in 1 ml lysis buffer and homogenized for 3 min in a Branson Model 250 sonicator (output 3, duty cycle 30%) and snap-frozen in liquid nitrogen. Mitochondria were pelleted from the combined PNS by centrifugation at 17 000 g in a swing-out rotor at 4°C for 15 min. The cytosol fraction was taken off, its volume measured and aliquots were snap-frozen. The mitochondria were resuspended in 1 ml lysis buffer and aliquots were snap-frozen. Uracil-DNA glycosylase assays were performed as described previously (21) but with incubation at 37°C for 30 min.
Studies of intracellular transport
Ung1 and Ung2 in pBluescript (11) were mutagenised at bp 983 (GAG to GAC) and bp 985 (CTG to CGG) in order to make an AgeI site prior to the stop codon. The AgeI blunted-EcoRI fragments of Ung1 and Ung2 from pBluescript and the AgeI blunted-EcoRI fragment of pEGFP*N1 (Clontech; mutated in it start codon ATG to CTG) were ligated to make pUng1EGFP and pUng2EGFP. Oligonucleotides encoding amino acids 1–38 of Ung1 were ligated in front of pEGFP*NI. pUNG11–28EGFP and pUNG1EGFP were made as described before (11). pUng21–48EGFP was made by ligating the XmaI–EcoRI fragment of mUNG2 in pBluescript with XmaI–EcoRI fragment of pEGFP*N1. The different pUng1-EGFP constructs were verified by DNA sequencing.
Human HeLa and mouse NIH 3T3 cells on coverslips were transfected with the different constructs using FuGENETM. The cells were analysed on a Zeiss LSM 510 laser scanning microscope equipped with a 40×/NA = 1.4 oil immersion objective. We used the 488 nm laser line for excitation of EGFP (detected at 505 nm < λEGFP < 530 nm). The pinhole diameter was kept at 106 µm. Images were exported to Adobe Photoshop (Adobe Systems Inc., San Jose, CA).
RESULTS
Structure of the murine Ung gene and promoter analysis
Sequencing of the isolated and purified λ-clone and the P1-clone and comparison with cDNA sequence information (11) revealed the gene structure presented in Figure 1. The gene spans 10 kb and comprises seven exons, six introns (1a, 1b, 2–5) and two promoters (GenBank accession no. AF174485). One promoter (PB) is located in intron 1a and the other putative promoter (PA) is located upstream of exon 1A. The introns vary in size from 385 bp (intron 3) to 3589 bp (intron 2) and contain many mouse-specific repetitive sequence elements, including a 54 bp TG-repeat in the untranslated part of exon 6. The exon–intron boundaries are conserved and one intron, intron 3, is very similar to the corresponding intron in the human gene, but otherwise the introns are poorly conserved. The gene encodes two putative proteins with identical catalytic domains that share 91% identity with the human catalytic domains (11).
Figure 1.

Structure of the murine uracil-DNA glycosylase (Ung) gene. Exons are depicted as boxes with their start and stop positions indicated above. Arrows indicate the positions of the promoter regions. Blocks of mouse repetitive sequences are schematically illustrated as grey boxes under the line. Details can be found in GenBank under the accession number AF174485.
In this report, the previously reported gene structure (16) is extended by the identification of a new upstream exon and its promoter. There are also minor discrepancies in the two studies regarding the sizes of some of the introns. We have sequenced a region spanning 1500 bp upstream of translation initiation start in exon 1A which contains the putative promoter A (PA), and identified putative transcription factor binding elements in this region.
Transfection of NIH 3T3 cells using various constructs containing each of the promoters alone, or both putative promoters (PA, exon 1A and PB), linked to the luciferase gene as reporter, demonstrated that both promoters are functional (Fig. 2). The entire promoter area (ProA+B) expresses the firefly luciferase 200-fold over background (pGL2 Basic), a level set as 100% expression. Interestingly, the construct containing PB alone (ProB) drives expression almost as efficient as the entire promoter area (89%). The 603 bp PB contains two direct repeats of ~280 bp. The two repeats are very similar, with 80% identity, leading to duplication of many putative transcription-factor binding sites. Both repeats resemble the human PB in that they contain many of the same transcription factor binding elements in a similar organisation. However, the repeat closer to exon 1A is here called the conserved part of the promoter, whereas the repeat closer to the exon 1B is marginally less related to the human counterpart and thus designated the duplicated part of the promoter. When the duplicated area of PB is removed from ProA+B (in ProA+B5′), the activity is reduced 5.2-fold. A 1.7-fold reduction in activity is seen when the duplicated part is removed from PB alone (in ProB5′). Hence, the duplication has apparently created a more active promoter. In agreement with this, the murine PB (ProB) is significantly more active than PA (3.6-fold), while human PB is some 2-fold less active than human PA (15).
Figure 2.

Activity of promoter–luciferase constructs. The chimaeric promoter constructs are illustrated schematically with the luciferase coding regions depicted as hatched boxes and the exons 1A as grey boxes. Putative transcription factor binding elements are shown as triangles and the positions in the Ung gene are indicated. The pGL2-Basic vector is omitted for simplicity. Activities of promoter constructs after transient transfection in NIH 3T3 cells were measured as the ratio of firefly and renilla luciferase activities and are given as percent of the activity expressed from ProA+B (the pGL2-Basic negative control had only 0.45% activity). The data shown are the mean ± SD of three independent experiments each performed in triplicate. Due to the presence of stop codons in intron 1a/PB, translation from exon 1A would not give a functional luciferase protein. Thus, luciferase activity resulting from transfection of ProA+B and ProA+B5′ are due to transcription from PB.
The first potential transcription factor binding-site in PA is located 7 bp upstream of transcription start in exon 1A (as determined from the Ung2 cDNA; GenBank accession no. X08975). There are no consensus sequence elements for binding of transcription factors in the successive 75 bp down to translation start at position 1414 (Fig. 3). Like the human PA, the murine promoter is TATA-less, GC rich and contains binding elements for Sp1, v-Myb, c-Myc and USF transcription factors, although not in similar organisation. The 45 bp region containing the c-Myc and USF elements is in fact the only highly conserved region of the promoter. Unlike the human gene, the mouse promoter does not contain a CCAAT-element or any obvious initiator elements in close proximity to transcription initiation. Furthermore, the mouse PA has a binding site for c-AMP responsive element binding protein 1 (CREB-1). Enhancement of effective cAMP levels by 8-Br-cAMP or forskolin had a weak stimulatory effect on transcription from PA but no effect on PB (data not shown). An E2F site with a different recognition sequence from the conserved E2F sites is generated in the mid-area of PB and one consensus E2F-binding element is present in each 280 bp repeat. Mutagenesis of the non-consensus E2F-binding site in PB reduced transcription 1.6-fold. Mutagenesis of the E2F sites in the conserved repeat reduced transcription 1.3-fold in ProB and ProB5′. No effect was observed by mutating the E2F-element in the duplicated part of ProB.
Figure 3.

Schematic presentation of mouse promoter A. Exon 1A is shown as a black box with an arrow indicating translation start. Putative binding sites for transcription factors are identified by search for highly correlated sequence fragments versus the TFMATRIX database (35). The nucleotide sequence of a highly conserved region (1270–1314) is shown as well as the sequence and localisation of some putative cis-acting elements for binding of transcription factors.
Differential expression of Ung mRNA and protein
To examine expression of the two types of Ung mRNA in vivo, we hybridised northern blots with probes specific for the nuclear and mitochondrial mRNA forms of mouse uracil-DNA glycosylase, and with a probe detecting both forms simultaneously. Figure 4A shows the mRNA levels in different tissues from adult mouse. The gene is expressed in all tissues examined, but the levels of expression, as well as the relative amounts of Ung1 and Ung2 mRNA vary. The highest levels of Ung1 mRNA are found in heart and skeletal muscle that are rich in mitochondria. High mRNA levels are also found in liver followed by spleen and testis. Low Ung1 mRNA levels are found in brain, lung and kidney. The highest levels of Ung2 mRNA are found in testis and spleen, which are tissues that contain a good proportion of proliferating cells. Intermediate Ung2 mRNA levels are found in liver and lung, while low levels are found in heart, brain, skeletal muscle and kidney. Figure 4B shows the mRNA levels in mouse embryos at different stages of development. Ung1 and Ung2 are both expressed in all stages of development. mRNA for Ung2 dominates quantitatively at days 11 and 15 of gestation. Transcription of both forms is increased from day 7 to 11 with mRNA for Ung2 showing the highest increase. The mRNA levels of both forms are gradually reduced from day 11 resulting in only slightly elevated mRNA levels at day 17 compared to day 7.
Figure 4.
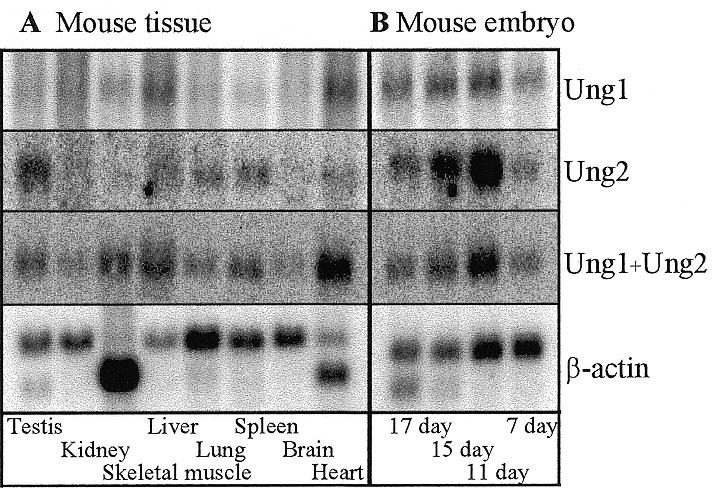
Expression of Ung1, Ung2 and β-actin mRNA in different tissues of adult mice (A) and from different stages of embryonic development (B). The blots were successively hybridised with Ung1, Ung2, Ung1 + Ung2 and β-actin probes. The blots were stripped prior to each new hybridisation. The images were quantified on a Molecular Dynamics PhosphorImager.
Since the identification of cDNA for Ung2 no attempt has been made to differentially measure the activities of the two forms in cells. Since the northern analysis indicated replication-associated expression of Ung2, we measured uracil-DNA glycosylase activity in different cellular compartments in freely cycling and in density inhibited cells. Figure 5A shows that the DNA synthesis is abolished in NIH 3T3 cells after 6 days of culture, thus confirming growth inhibition. In cycling cells, nuclei contained more activity per cell than mitochondria (Fig. 5B). In density inhibited cells in contrast, most of the activity was found in the mitochondria (Fig. 5B). Whereas the activity in mitochondria increased 1.3-fold in growth inhibited compared to cycling cells, levels in nuclei were reduced 3.7-fold. Thus, the activity measurements indicate that Ung1 is the dominating species in resting cells and Ung2 dominates in freely cycling cells.
Figure 5.

Distribution of uracil-DNA glycosylase activity between different cell compartments in mouse cells. Pulse labelling with [3H]thymidine of density inhibited (triangles) and freely cycling (diamonds) NIH 3T3 cells (A). The data are means ± SEM for three independent experiments. Freely cycling and density inhibited NIH 3T3 cells were fractionated and uracil-DNA glycosylase activity was measured in nuclei (black bar) and mitochondria (white bar) (B).
Intracellular transport of mouse Ung proteins
In order to investigate the subcellular localisation of murine Ung1 and Ung2, we prepared several fusion constructs with EGFP (an enhanced form of green fluorescent protein) as reporter. The sorting of these fusion proteins was compared with the corresponding human fusion proteins. Mitochondrial localisation signals frequently seem to have the potential to form amphiphilic helices (22). Amino acids 4–20 of the Ung1 N-terminal sequence (Fig. 6A) may form such an amphiphilic helix (Fig. 6C), although not as perfect as the corresponding helix generated by amino acids 11–28 in human UNG1 (Fig. 6A and B). As expected, much of the Ung11–20EGFP fusion protein was sorted to the mitochondria in NIH 3T3 cells but surprisingly, a significant fraction was also transported to the nucleus (Fig. 7A), in contrast to the exclusive mitochondrial sorting of the corresponding human product (Fig. 7C). Even the full-length Ung1 in front of EGFP did not completely abolish nuclear sorting (Fig. 7B). Although some cells with very low expression of Ung1EGFP displayed mitochondrial sorting exclusively (Fig. 7B, lower, right panel), nuclear sorting of Ung1EGFP is evident in most cells, whereas this is not observed for human UNG11–28EGFP (Fig. 7C) or UNG1EGFP (Fig. 7D), irrespective of expression level. To elucidate whether the differences in sorting pattern between murine Ung1 and human UNG1 was due to lower capacity for import to the mitochondria in NIH 3T3 cells than in HeLa cells, we also transfected the constructs into HeLa cells. Again, the murine Ung1 constructs did not sort exclusively to mitochondria (Fig. 7E and F) whereas the human UNG1 constructs sorted exclusively to mitochondria, irrespective of expression levels (Fig. 7G and H). Hence, it appears that sequence differences between the murine and human proteins account for the different sorting patterns.
Figure 6.

The N-terminal sequences of the murine Ung1 and human UNG1 proteins may form amphiphilic helices. Alignment of the N-terminal sequences of human UNG1 and mouse Ung1 (A). Potential amphiphilic helix-forming residues are underlined. When amino acids 11–28 of UNG1 (B) and 4–21 of Ung1 (C) are plotted in an α-helical wheel with 3.6 amino acids/turn their intrinsic potential to form amphiphilic helices emerges. Charged and polar residues are shaded and positively charged residues are indicated by +.
Figure 7.
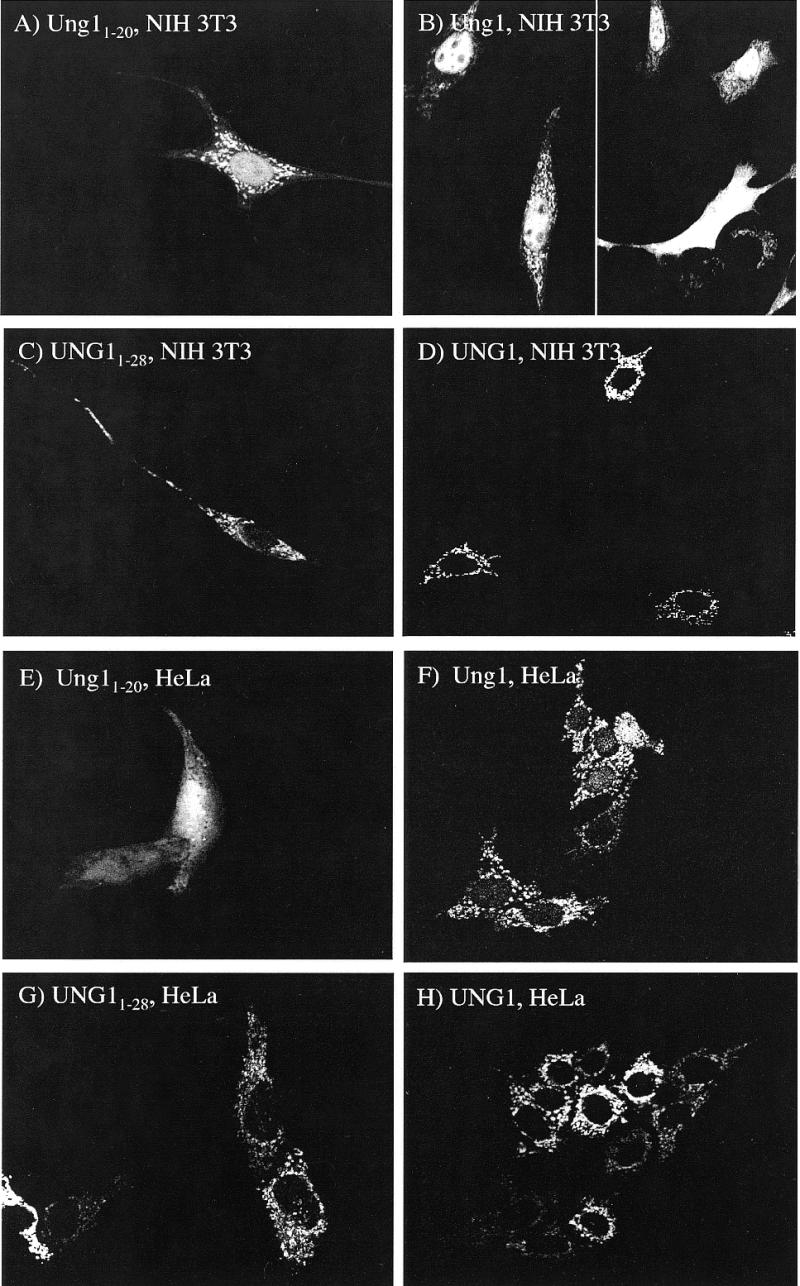
Subcellular localisation of murine and human mitochondrial forms in fusion with EGFP in NIH 3T3 and HeLa cells. Cells were transient transfected with constructs expressing Ung11–20EGFP (A), Ung1EGFP (B), UNG11–28EGFP (C) and UNG1EGFP (D) in NIH 3T3. Ung11–20EGFP (E) and Ung1EGFP (F), UNG11–28EGFP (G) and UNG1EGFP (H) in NIH 3T3.
A construct containing the highly conserved full-length Ung2 showed complete nuclear localisation into both NIH 3T3 and HeLa cells (Fig. 8A and B). However, the N-terminal part of Ung2 (residues 1–48) clearly resulted in incomplete sorting to the nucleus in both cell types (Fig. 8C and D). Hence, the unique N-terminal sequence of Ung2 is not sufficient for nuclear localisation of the protein, analogous to the sorting of human UNG2 in HeLa cells (17). Transfection of a construct containing EGFP only (Fig. 8E) is distributed throughout the cell.
Figure 8.
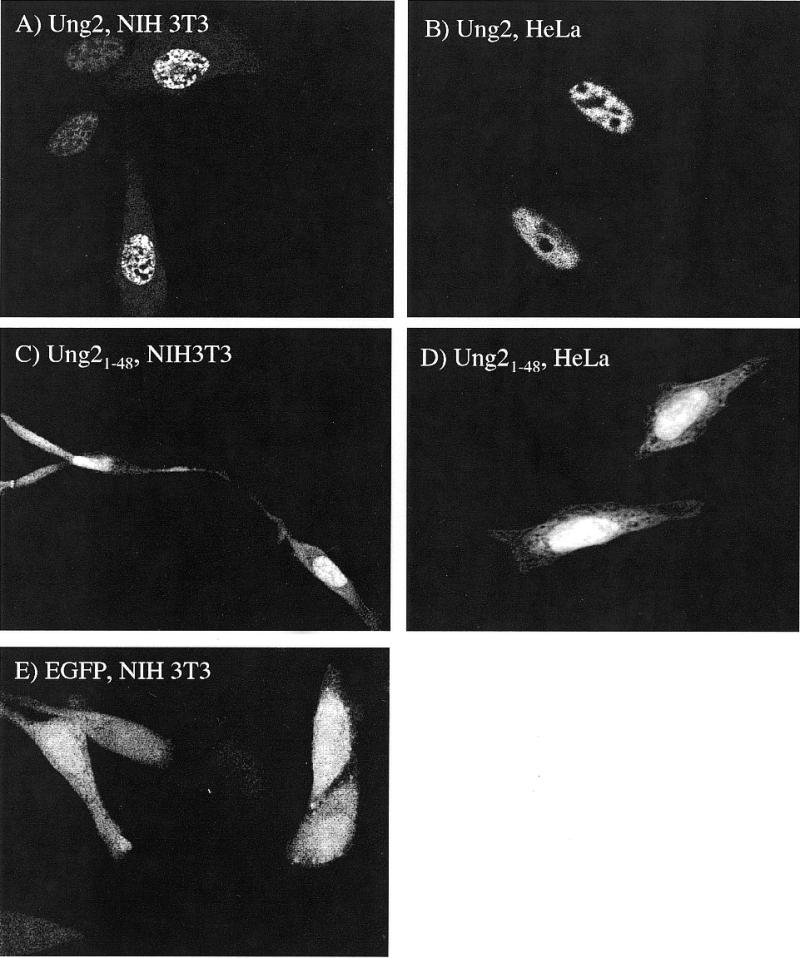
Subcellular localisation of murine and human nuclear forms in fusion with EGFP in NIH 3T3 and HeLa cells. Ung2EGFP in NIH 3T3 (A) and HeLa (B) and Ung21–48EGFP in NIH 3T3 (C) and HeLa (D). EGFP control is shown in (E).
DISCUSSION
The murine Ung gene and the human counterpart UNG are largely similarly organised and both genes encode the nuclear as well as the mitochondrial forms of uracil-DNA glycosylase having highly conserved catalytic domains. However, there are significant differences in promoter structure and function, and apparently also in the subcellular sorting signals in the proteins encoded. We have demonstrated that the relative strengths of the two mouse promoters are inversed compared to the human promoters in that the mouse mitochondrial PB (as evaluated from transient transfection of promoter–luciferase reporter constructs) is significantly stronger than the nuclear PA. The relatively increased strength of mouse PB compared to the human PB may be ascribed to the duplication of a 280 bp segment in the promoter.
In general the UDG activity correlates well with the proliferative status of the cell (23–25). However, these early studies do not differentiate between the nuclear and mitochondrial forms and the nuclear form has generally been assumed to be the major contributor to the activity. We find that Ung2 mRNA expression is correlated with the replicative status of tissues whereas Ung1 is highly expressed in tissues rich in mitochondria. This is in agreement with similar studies in human cells (15). At the level of protein expression, nuclear Ung 2 dominates in cycling NIH 3T3 cells, but not to the same extent as human nuclear UDG-activity in HeLa cells where 70% was nuclear using similar conditions for subcellular fractionation (26). Apparently, the relatively strong murine PB results in a higher contribution from Ung1 to the total activity in cycling mouse cells as compared with human cells. In growth inhibited cells, the mitochondria contain 3.5-fold more activity than nuclei. From this we conclude that Ung1 apparently constitutes the major UNG protein in resting cells, but not in freely cycling cells, thus corroborating the mRNA expression data. Some UDG activity was also measured in the cytosol fraction in cycling cells, most probably due to leakage from the nuclei, since very low cytosolic UDG activity was observed in density-inhibited cells (data not shown). The poorly conserved mitochondrial sorting sequence in murine Ung1 is apparently not as dominant as the sorting signal in human UNG1 since the Ung1EGFP fusion protein is sorted both to nuclei and mitochondria in mouse cells, as well as in human cells. Sorting of human UNG1 was exclusively to mitochondria in both cell types irrespective of expression levels. The human UNG1 mitochondrial localisation signal directs transport to the mitochondria even when placed in front of UNG2 in HeLa cells (17). Comparison between sorting of human UNG1 and murine Ung1 strongly suggests that the different patterns are caused by different strengths of the sorting signals. There is no good reason to assume that the translocation machinery should be more saturated by murine Ung1 than by human UNG1. Thus, sorting of Ung1 to the nuclei as an artefact created by over-expressing the protein does not seem to be a likely explanation for the observed sorting pattern. We propose, therefore, that Ung1 is sorted to both compartments as a consequence of a weaker mitochondrial sorting signal. Indeed, dual sorting patterns is not without precedence as hNTH1 was shown to be sorted to both mitochondria and the nucleus in COS-7 cells (18). The expression and sorting patterns tempt us to speculate that Ung1 might ensure sufficient nuclear Ung activity, and that Ung1 may function as a back-up for the Ung2-activity. It should be pointed out, however, that in human UNG2, the N-terminal sequence functions both as a sorting signal, and as a motif for interaction with PCNA and RPA in replication foci (5). The very N-terminal sequence of Ung2 is highly conserved and is likely to serve a similar function, which cannot be served by the unrelated N-terminal sequence in Ung1. However, Ung1 may be involved in the repair of deaminated cytosine outside S-phase, a function not dependent on interaction with PCNA or RPA (4). We must await development of antibodies specific for Ung1 and Ung2 to further test this hypothesis.
Although PA in the mouse, like the human counterpart, contains many features of a housekeeping gene, the organisation and number of different motifs are dissimilar. The only well-conserved region (spanning positions 1270–1314) contains a USF/c-Myc site, indicating an important role in regulation of transcription. Indeed, mutations in this region in the human PA significantly reduced transcription (15). Interestingly, PA contains a CRE element known to bind c-AMP responsive element binding protein (CREB-1). Its location 355 bp upstream of the transcription start site makes it a candidate enhancer element that might participate in promoter clearance prior to stimulation of transcription in late G1, as observed in the human DNA polymerase β promoter (27,28). Using the dual-luciferase-assay we see a weak stimulatory effect of 8-Br-cAMP or forskolin on the transcription from PA (data not shown). However, our data indicate that trans effects due to the presence of a CRE element in the HSV-tk promoter in pRL-TK (GenBank accession no. AF025846), might interfere with the assay as has been reported previously (29). Consequently, we cannot exclude a possible effect of the CREB element and the findings must be verified using a different reporter system.
In the embryo, we see a dramatic increase in mRNA level from day 7 to 11. The induction is stronger for Ung2 than for Ung1 and it may be mediated through MZF-1, a putative enhancer element in PA which is highly expressed in early stages of development predominantly in cells of hematopoietic origin (30,31). The induction is initiated in the transition from the phases of gastrulation to organogenesis in mouse development where retinoic acid is thought to initiate distinct developmental events through activation of a series of transcription factors, including MZF-1 (30, reviewed in 32). Vitamin A is metabolically activated endogenously to retinoic acid from day 7.5 in the primitive streak in developing mouse embryos and in many organs later in organogenesis (33,34), which coincides with the induction of Ung2 mRNA seen from day 7 to 11. The ability to activate vitamin A is apparently reduced later in development and it is lost in some organs between days 12.5 and 14.5 of gestation (34), which corresponds with the drop in Ung mRNA after day 11 of gestation. We speculate that retinoic acid might be a regulator of Ung expression at least in some cell types under embryonic development. Further investigation is needed to address this question.
In conclusion, there seems to be species-related differences in both regulation of transcription and intracellular localisation of the nuclear and mitochondrial forms of the Ung proteins resulting in different relative contribution from the isoforms to total uracil-DNA glycosylase activity in mouse compared to man. This might be important in the interpretations of phenotypes of Ung knockout mice.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Camilla Skjelbred and Mansour Akbari for technical assistance and Dr Frank Skorpen for helpful discussion. This work was supported by The Norwegian Cancer Society, The Research Council of Norway, The Cancer Fund at the Regional Hospital, Trondheim and the Svanhild and Arne Must Fund for Medical Research.
DDBJ/EMBL/GenBank accession no. AF174485
REFERENCES
- 1.Lindahl T. (1993) Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 2.Duncan B.K. and Weiss,B. (1982) J. Bacterol., 151, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Impellizzeri K.J., Anderson,B. and Burgers,P.M. (1991) J. Bacteriol., 173, 6807–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krokan H.E., Standal,R. and Slupphaug,G. (1997) Biochem. J., 325, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otterlei M., Warbrick,E., Nagelhus,T.A., Haug,T., Slupphaug,G., Akbari,M., Aas,P.A., Steinsbekk,K., Bakke,O. and Krokan,H.E. (1999) EMBO J., 18, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neddermann P., Gallinari,P., Lettieri,T., Schmid,D., Truong,O., Hsuan,J.J., Wiebauer,K. and Jiricny,J. (1996) J. Biol. Chem., 271, 12767–12774. [DOI] [PubMed] [Google Scholar]
- 7.Haushalter K.A., Stukenberg,P.T., Kirschner,M.W. and Verdine,G.L. (1999) Curr. Biol., 9, 174–185. [DOI] [PubMed] [Google Scholar]
- 8.Muller S.J. and Caradonna,S. (1991) Biochim. Biophys. Acta, 1088, 197–207. [DOI] [PubMed] [Google Scholar]
- 9.Hendrich B., Hardeland,U., Ng,H.-H., Jiricny,J. and Bird,A. (1999) Nature, 401, 301–304. [DOI] [PubMed] [Google Scholar]
- 10.Haug T., Skorpen,F., Kvaløy,K., Eftedal,I., Lund,H. and Krokan,H.E. (1996) Genomics, 36, 408–416. [DOI] [PubMed] [Google Scholar]
- 11.Nilsen H., Otterlei,M., Haug,T., Solum,K., Nagelhus,T.A., Skorpen,F. and Krokan,H.E. (1997) Nucleic Acids Res., 25, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mol C.D., Arvai,A.S., Sanderson,R.J., Slupphaug,G., Kavli,B., Krokan,H.E., Mosbaugh,D.W. and Tainer,J.A. (1995) Cell, 82, 701–708. [DOI] [PubMed] [Google Scholar]
- 13.Slupphaug G., Mol,C.D., Kavli,B., Arvai,A.S., Krokan,H.E. and Tainer,J.A. (1996) Nature, 384, 87–92. [DOI] [PubMed] [Google Scholar]
- 14.Slupphaug G., Olsen,L.C., Helland,D., Aasland,R. and Krokan,H.E. (1991) Nucleic Acids Res., 19, 5131–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haug T., Skorpen,F., Aas,P.A., Malm,V., Skjelbred,C. and Krokan,H.E. (1998) Nucleic Acids Res., 26, 1449–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svendsen P.C., Yee,H.A., Winkfein,R.J. and van de Sande,J.H. (1997) Gene, 189, 175–181. [DOI] [PubMed] [Google Scholar]
- 17.Otterlei M., Haug,T., Nagelhus,T.A., Slupphaug,G., Lindmo,T. and Krokan,H.E. (1998) Nucleic Acids Res., 26, 4611–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takao M., Aburatani,H., Kobayashi,K. and Yasui,A. (1998) Nucleic Acids Res., 26, 2917–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warren A.J., Colledge,W.H., Carlton,M.B., Evans,M.J., Smith,A.J. and Rabbitts,T.H. (1994) Cell, 78, 45–57. [DOI] [PubMed] [Google Scholar]
- 20.Maniatis T., Fritsch,E.F. and Sambrook,J. (1982) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 21.Krokan H. and Wittwer,C.U. (1981) Nucleic Acids Res., 9, 2599–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neupert W. (1997) Annu. Rev. Biochem., 66, 863–917. [DOI] [PubMed] [Google Scholar]
- 23.Weng Y. and Sirover,M.A. (1993) Mutat. Res. DNA Repair, 293, 133–141. [DOI] [PubMed] [Google Scholar]
- 24.Aprelikova O.N. and Tomilin,N.V. (1982) FEBS Lett., 137, 193–195. [DOI] [PubMed] [Google Scholar]
- 25.Vilpo J.A. (1988) Mutat. Res., 193, 207–217. [DOI] [PubMed] [Google Scholar]
- 26.Wittwer C.U. and Krokan,H. (1985) Biochim. Biophys. Acta, 832, 308–318. [DOI] [PubMed] [Google Scholar]
- 27.Narayan S., Beard,W.A. and Wilson,S.H. (1995) Biochemistry, 34, 73–80. [DOI] [PubMed] [Google Scholar]
- 28.Narayan S., He,F. and Wilson,S.H. (1996) J. Biol. Chem., 271, 18508–18513. [DOI] [PubMed] [Google Scholar]
- 29.Benzakour O., Kanthou,C., Dennehy,U., Al Haq,A., Berg,L.-P., Kakkar,V.V. and Cooper,D.N. (1995) Biochem. J., 309, 385–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hui P., Guo,X. and Bradford,P.G. (1995) Biochemistry, 34, 16493–16502. [DOI] [PubMed] [Google Scholar]
- 31.Robertson K.A., Hill,D.P., Kelley,M.R., Tritt,R., Crum,B., van Epps,S., Srour,E., Rice,S. and Hromas,R. (1998) Leukemia, 12, 690–698. [DOI] [PubMed] [Google Scholar]
- 32.Zile M.H. (1998) J. Nutr., 128, 455S–458S. [DOI] [PubMed] [Google Scholar]
- 33.Duester G. (1998) J. Nutr., 128, 459S–462S. [DOI] [PubMed] [Google Scholar]
- 34.Rubin W.W. and LaMantia,A. (1999) Dev. Neurosci., 21, 113–125. [DOI] [PubMed] [Google Scholar]
- 35.Heinemeyer T., Wingender,E., Reuter,I., Hermjakob,H., Kel,A.E., Kel,O.V., Ignatieva,E.V., Ananko,E.A., Podkolodnaya,O.A., Kolpakov,F.A., Podkolodny,N.L. and Kolchanov,N.A. (1998) Nucleic Acids Res., 26, 364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]