

Abstract
Ribosome biogenesis requires, in addition to rRNA molecules and ribosomal proteins, a multitude of trans-acting factors. Recently it has become clear that in the yeast Saccharomyces cerevisiae many RNA helicases of the DEAD-box and related families are involved in ribosome biogenesis. Here we show that the previously uncharacterised open reading frame YDL031w (renamed DBP10 for DEAD-box protein 10) encodes an essential putative RNA helicase that is required for accurate ribosome biogenesis. Genetic depletion of Dbp10p results in a deficit in 60S ribosomal subunits and an accumulation of half-mer polysomes. Furthermore, pulse–chase analyses of pre-rRNA processing reveal a strong delay in the maturation of 27SB pre-rRNA intermediates into 25S rRNA and 7S pre-rRNA. Northern blot analyses indicate that this delay leads to higher steady-state levels of 27SB species and reduced steady-state levels of 7S pre-rRNA and 25S/5.8S mature rRNAs, thus explaining the final deficit in 60S subunit and the formation of half-mer polysomes. Consistent with a direct role in ribosome biogenesis, Dbp10p was found to be located predominantly in the nucleolus.
INTRODUCTION
Ribosome biogenesis constitutes a major cellular activity in actively growing cells. Indeed, in an exponentially growing yeast ~2 × 105 new ribosomes have to be produced during each cell cycle (1,2). To meet the large demand of mature rRNAs, eukaryotic organisms encode the pre-rRNAs in clusters of rDNA repeats. In Saccharomyces cerevisiae, the small ribosomal subunit is composed of 32 ribosomal proteins (r-proteins) and the 18S rRNA, whereas the large subunit contains 46 r-proteins and three rRNA species (5.8S, 25S and 5S) (3). Three of the four rRNAs (18S, 25S and 5.8S) are co-transcribed as a single large precursor by RNA polymerase I, while the 5S rRNA is independently transcribed as a pre-rRNA by RNA polymerase III.
The 35S pre-rRNA, which is the longest detectable precursor, contains the sequences of the mature 18S, 5.8S and 25S rRNAs separated by two internal transcribed spacers (ITS1 and ITS2) and flanked by two external transcribed spacers (5′ ETS and 3′ ETS). During ribosome biogenesis, which mainly takes place in the nucleolus, a specialised compartment of the nucleus (4,5), the 35S precursor is processed through a series of sequential endo- and exo-nucleolytic cleavages to give rise to mature rRNAs 18S, 25S and 5.8S (Fig. 1). Furthermore, shortly after transcription, it undergoes approximately 108 covalent modifications of nucleotides (55 2′-O-ribose methylations, 43 pseudo-uridinylations, approximately 10 base methylations) that are mostly guided by small nucleolar RNAs (snoRNAs) (6). All these maturation steps occur in large pre-ribosomal particles that contain, in addition to r-proteins, numerous trans-acting proteins and snoRNAs that associate transiently with pre-rRNA sequences (7–9). Current analyses indicate the presence of approximately 75–100 different snoRNAs per yeast genome (10) and to date more than 60 protein trans-acting factors have been reported to act in ribosome biogenesis (11,12). Thus, it is generally believed that the pre-ribosomal particle is a highly dynamic structure in which many structural rearrangements are required for maturation of ribosomal subunits to occur. We and others have shown that there are many putative RNA helicases of the DEAD-box and related families amongst the trans-acting factors (13). These proteins are characterised by eight conserved motifs. Based on the in vitro RNA-dependent ATPase and helicase activities of some of these proteins (14–19), it has been suggested that they are involved in the local unwinding of structured RNAs. Nevertheless it cannot be excluded that these proteins also possess RNA–protein dissociating activities. Indeed, unwinding of RNA–RNA duplexes (rRNA–rRNA or rRNA–snoRNA) or dissociation of RNA–protein complexes are expected to occur during the dynamic and complex process of ribosome biogenesis.
Figure 1.

Pre-rRNA processing in S.cerevisiae. (A) Structure and processing sites of the 35S pre-rRNA. This precursor contains the sequences for the mature 18S, 5.8S and 25S rRNAs, which are separated by the two internal transcribed spacers ITS1 and ITS2 and flanked by two external transcribed spacers 5′ ETS and 3′ ETS. The location of various probes (labelled from B to H) used in this study are indicated. Bars represent mature rRNA species and lines the transcribed spacers. (B) Pre-rRNA processing pathway. The 35S pre-rRNA is cleaved at site A0 by the endonuclease Rnt1p, generating the 33S pre-rRNA. This molecule is subsequently processed at sites A1 and A2, resulting in the separation of the pre-rRNAs destined for the small and large ribosomal subunits. It is proposed that the early pre-rRNA cleavages A0–A2 require a large snoRNP complex, which may be assisted by the putative ATP-dependent RNA helicases Dbp4p, Dbp8p, Fal1p, Rok1p and Rrp3p. Final maturation of the 20S precursor takes place in the cytoplasm, where endonucleolytic cleavage at site D yields the mature 18S rRNA. The 27SA2 precursor is processed by two alternative pathways that both lead to the formation of mature 5.8S and 25S rRNAs. In the major pathway, the 27SA2 precursor is cleaved at site A3 by the RNase MRP complex. The putative ATP-dependent RNA helicase Dpb3p assists in this processing step. The 27SA3 precursor is exonucleolytically digested 5′→3′ up to site B1S to yield the 27SBS precursor, a reaction requiring the exonucleases Xrn1p and Rat1p. A minor pathway processes the 27SA2 molecule at site B1L, producing the 27SBL pre-rRNA. While processing at site B1 is completed, the 3′ end of mature 25S rRNA is generated by processing at site B2. The subsequent processing at ITS2 appears to be identical for both 27SB species. Cleavage at sites C1 and C2 releases the mature 25S rRNA and the 7S pre-rRNA. The latter undergoes exosome-dependent 3′ to 5′ exonuclease digestion to the 3′ end of the mature 5.8S rRNA. It has been proposed that Dob1p/Mtr4p, a putative ATP-dependent RNA helicase, assists the exosome activity. The data presented in this study suggest that Dbp10p is required for a late step in the assembly of 60S ribosomal subunits, a process that may also involve six other putative ATP-dependent RNA helicases: Dbp6p, Dbp7p, Dbp9p, Spb4p, Drs1p and Mak5p. The pre-rRNA processing phenotype of the Drs1p-depleted and Mak5p-depleted strains has not been described. Data concerning Dbp8p and Dbp9p are unpublished results from our laboratory.
Depending on the molecular aspect of the terminal molecular phenotype observed upon helicase inactivation or depletion, it has been suggested that RNA helicases may be involved in ribosome assembly or pre-rRNA processing, although these two processes are intrinsically linked. The absence of a functional RNA helicase may result in an unstable pre-ribosomal particle and rapid degradation of pre-rRNA intermediates (20,21). Alternatively, the depletion of an RNA helicase may result in the accumulation of pre-rRNA intermediates that are no longer processed into mature rRNAs (22).
So far, 12 putative RNA helicases have been described that are involved in ribosome biogenesis (20–32) (Fig. 1). The activity of these proteins seems to be highly specific for distinct steps since they are mostly essential and they generally cannot replace each other. Here, we describe the analysis of the DEAD-box protein Dbp10p, which is encoded by the open reading frame (ORF) YDL031w. Depletion of Dbp10p results in an inhibition of the processing of the 27SB pre-rRNA intermediates, which in turn results in a deficit in 60S ribosomal subunits.
MATERIALS AND METHODS
Strains, media and microbiological methods
Saccharomyces cerevisiae strains described in this study were all derivatives of W303 (MATa/MATα ura3-1/ura3-1 ade2-1/ade2-1 his3-11,15/his3-11,15 leu2-3,112/leu2-3,112 trp1-1/trp1-1) (33). YBF1 (MATa/MATα DBP10/dbp10::kanMX4) was obtained by transforming W303 with the SFH-PCR product dbp10::kanMX4. YBF1-1A (MATa DBP10) and YBF1-1B (MATa dbp10::kanMX4) are both meiotic segregants of YBF1. YBF1-1B requires a plasmid-borne copy of DBP10 for cell viability. Genetic manipulations and preparation of media were according to established procedures (34,35). Yeast cells were transformed using the lithium acetate method (36). Escherichia coli DH10B was used for all recombinant DNA techniques (37). Tetrad dissections were performed using a Singer MS manual micromanipulator.
Deletion of the DBP10 gene
Deletion-disruption of the DBP10 ORF was obtained by transformation of a PCR-synthesised kanMX4 marker cassette with DBP10 short flanking homology regions (SFH–PCR) into W303 (38). The kanMX4 module was generated by PCR using Pfu polymerase (Stratagene), the plasmid pFA6a-kanMX4 as the DNA template and the following oligonucleotides: DBP10-P1 5′-C ATT TAA GTT ACC ACC ACA TAT ATC TCT TTA ACC TAG AAT ATG CGT ACG CTG CAG GTC GAC-3′ (the DBP10 5′ upstream sequence with the underlined start codon is in bold type, the kanMX4 5′ upstream sequence is in plain type) and DBP10-P2 5′-AAA CTA TAT ATC TGT AGA TCT AGA TGG TTT TAA ACT ATC TAA TCG ATG AAT TCG AGC TCG-3′ (the DBP10 3′ downstream sequence with the underlined stop codon is in bold type, the kanMX4 3′ upstream sequence is in plain type). After concentration by ethanol precipitation, the SFH–PCR dbp10::kanMX4 product was used to transform W303. Transformants were selected on YPD plates containing 200 mg/l G418 (Gibco BRL). The correct integration of the kanMX4 module at the DBP10 genomic locus was verified by Southern analysis and resulted in the heterozygous diploid strain YBF1 (DBP10/dbp10::kanMX4).
Cloning of the DBP10 gene and recovery of the dbp10-1 allele
The DBP10 gene was amplified by PCR using Pfu polymerase, genomic DNA from W303 as a template and the following oligonucleotides: DBP10-P3 5′-GGC GGG GGT ACC AGG GTC GTC CAA AGA TTG-3′ (the DBP10 5′ upstream sequence starting at –516 with respect to the ATG codon is in bold type with the introduced KpnI site underlined) and DBP10-P4 5′-CCC GCC CGC ATG CGA TGC CCT CCT ACT CCT ATT-3′ (the DBP10 3′ downstream sequence starting at +327 with respect to the stop codon is in bold type with the introduced SphI site underlined). After digestion with KpnI and SphI restriction enzymes, the PCR product was gel-purified (Gene Clean, BIO101, Inc.) and cloned as a 3.8 kb fragment into the KpnI–SphI-restricted plasmid YCplac33. The resulting pool of plasmids was used to transform the heterozygous YBF1 strain. Several independent transformants were selected and subjected to sporulation and tetrad analysis. All the transformants analysed, except one, gave tetrads of four wild-type growing spores, and all the G418R spores were Ura+, indicating that they contained a fully functional plasmid-borne DBP10 allele. Interestingly, one transformant gave tetrads with a 2:2 segregation of wild-type to slow growing spores, with the latter all being G418R and Ura+. Plasmid rescue, back-transformation and additional subcloning of the insert confirmed the isolation of a mutant allele of DBP10 that we named dbp10-1.
A wild-type DBP10 allele was also cloned from a genomic YEplac181 library (39). Briefly, the haploid strain YBF1-1B [YCplac33-dbp10-1] was transformed with the library and screened for 5-FOA-resistant clones by replica-plating on 5-FOA-containing plates. Several clones were isolated, and one was used for subcloning a 4.4 kb EcoRV fragment containing DBP10 into the SmaI-restricted plasmid YCplac22, yielding YCplac22-DBP10. A 4 kb EcoRI insert from YCplac22-DBP10 was further subcloned into the EcoRI-restricted plasmid YCplac111 yielding YCplac111-DBP10.
Construction of the GAL::DBP10 allele and in vivo depletion of Dbp10p
DBP10 was amplified by PCR using Pfu polymerase, the plasmid YCplac22-DBP10 as a template and the following oligonucleotides: DBP10-P4 (described above) and DBP10-P5 5′-GGG CGG CCG CAT GCG CAG GCG TGC AGA AAA GAA AA-3′ (the DBP10 ORF sequence starting with the second codon is in bold type with the introduced SphI site underlined). The PCR product was digested with SphI, gel purified and subcloned as a 3.3 kb fragment into the SphI-restricted plasmid pAS24 (40) to yield the plasmid pAS24-DBP10 in which the expression of HA-DBP10 is under the control of the GAL1-10 promoter.
The strain YBF1-1B [pAS24-DBP10] was obtained after transformation of YBF1-1B [YCplac33-dbp10-1] with the plasmid pAS24-DBP10 followed by plasmid shuffling on SGal plates containing 5-FOA. Growth behaviour on galactose and glucose media was further studied to test the faithful complementation and the shut off under non-permissive conditions.
For in vivo depletion, strain YBF1-1B [pAS24-DBP10] was grown in YPGal medium until mid-exponential phase (OD600 = 1) and then inoculated in YPD medium. Cell growth was monitored over a period of 36 h, during which the culture was regularly diluted into fresh YPD medium to maintain exponential growth. Samples for western blot analysis, polysome analyses and RNA extraction were taken at different times.
Sucrose gradient analyses
Polysome analysis and ribosome subunit quantification were performed as previously described (24).
Construction of a plasmid-borne HA-DBP10 allele and indirect immunofluorescence
A plasmid-borne, HA-epitope-tagged DBP10 allele was constructed by fusion PCR (41). Briefly, two PCR series were performed using Pfu polymerase, the plasmid YCplac111-DBP10 as a template and the following oligonucleotide couples: M13/pUC sequencing primer (–20) (New England Biolabs) and DBP10-B 5′-GTC ATA GGG ATA GCC CGC ATA GTC AGG AAC ATC GTA TGG GTA TGC CAT ATT CTA GGT TAA AGA GAT (the sequence of the HA epitope is in plain type with the overlapping part underlined and the DBP10 ORF sequence is in bold with the start codon underlined); DBP10-A 5′-AGC TTT GGG GGT TGA AAT-3′ (internal sequence of DBP10) and DBP10-C 5′-GCG GGC TAT CCC TAT GAC GTC CCG GAC TAT GCA CTC GAG GCA GGC GTG CAG AAA AGA-3′ (the sequence of the HA epitope is in plain type with the overlapping part underlined, the DBP10 ORF sequence starting with the second codon is in bold). The fusion PCR was performed using the products of the two PCR series as DNA templates and the two oligonucleotides DBP10-A and M13/pUC. The final PCR product was digested with SalI, gel purified and subcloned as a 2.4 kb fragment into the SalI-restricted plasmid YCplac111-DBP10 to yield YCplac111-HA-DBP10. The strain YBF1-1B [YCplac111-HA-DBP10] was obtained after transformation of the strain YBF1-1B [YCplac33-dbp10-1] followed by plasmid shuffling on 5-FOA containing plates. The plasmid-borne HA-DBP10 allele complemented the dbp10::kanMX4 null allele to the wild-type extent at all the tested temperatures (18, 30 and 37°C). In addition, western blot analysis with monoclonal mouse anti-HA antibodies detected a single protein exclusively in whole cell lysates of strains harbouring the plasmid-borne HA-DBP10 allele (data not shown). This protein, corresponding to the HA-Dbp10p, had the expected molecular mass of ~115 kDa. Indirect immunofluorescence analyses were performed as previously described (22).
RNA analysis
Pulse–chase labelling of pre-rRNAs and RNA preparations were performed as described previously (20). Steady-state levels of pre-rRNAs were assessed by northern and primer extension analyses according to Venema et al. (42). The oligonucleotides used for northern analysis and primer extension are the following: B (5′ ETS, 5′ to A0) 5′-GGT CTC TCT GCT GCC GG-3′; C (ITS1, D/A2) 5′-GAA ACG GTT TTA ATT GTC CTA TAA C-3′; D (ITS1, A2/A3) 5′-TGT TAC CTG GGC CC-3′; E (ITS1, A3/B1) 5′-AAT TTC CAG TTA CGA AAA TTC TTG-3′; F (ITS2, E/C2) 5′-GGC CAG CAA TTT CAA GTT A-3′; G (ITS2, C1/C2) 5′-GAA CAT TGT TCG CCT AGA-3′; H (5.8S) 5′-TTT CGC TGC GTT CTT CAT C-3′; 5S 5′-GGT CAC CCA CTA CAC TAC TCG G-3′. For localisation of the oligonucleotides see Figure 1.
RESULTS
DBP10 is essential for cell growth
The genome of the yeast S.cerevisiae encodes 26 proteins of the DEAD-box family of putative RNA helicases (13). Most of them have been characterised by genetic analysis or biochemical methods. Towards the characterisation of all DEAD-box proteins of a eukaryotic organism, we functionally analysed the so far uncharacterised ORF YDL031w, which we renamed DBP10 (DEAD-box protein 10). Dbp10p is a 995 amino acid protein with a predicted molecular mass of 113 kDa and a predicted pI of 9.3. The protein sequence contains, in addition to the conserved helicase core domain, lysine-rich and highly charged N- and C-terminal extensions of 180 and 468 amino acids, respectively. The N-terminal extension contains a bipartite nuclear localisation signal (KKKAENKDIKKKKNSKK) (43). Sequence comparison revealed a highly conserved protein in Schizosaccharomyces pombe (Swissprot: Q09719; 43% identical, 59% similar), a well conserved human EST sequence (AA459771) and a more distantly conserved protein in Caenorhabditis elegans [Sanger Centre (Cambridge, UK) CE00337, 33% identical, 57% similar].
As a first step in the functional characterisation of Dbp10p, we assessed the impact of a chromosomal deletion of the DBP10 ORF. To do this, one genomic copy of the entire DBP10 ORF was replaced by the heterologous kanMX4 marker in the diploid W303 strain yielding the YBF1 strain (DBP10/dbp10::kanMX4). Upon sporulation and tetrad dissection, a 2:2 segregation of viable to non-viable spores was observed, where all viable spores were G418s, suggesting an essential role for Dbp10p. To distinguish between a defect in germination and vegetative growth, analysis of the meiotic progeny of YBF1 [YCplac33-DBP10] was carried out. Tetrads with four viable spores were obtained and further analysed for growth on 5-FOA medium. All the G418R spore clones were 5-FOAs indicating that Dbp10p is essential for vegetative growth.
Two conditional systems for the phenotypic analysis of Dbp10p
Two different conditional systems were generated to further characterise the cellular function of Dbp10p. First, a plasmid-borne, cold-sensitive and recessive dbp10-1 mutant allele was isolated during the cloning of the DBP10 gene by PCR amplification (Materials and Methods). Compared to the wild-type strain (YBF1-1A), the dbp10-1 mutant strain (YBF1-1B [YCplac33-dbp10-1]) showed a slight growth defect at low temperatures (Fig. 2A). Secondly, a transcriptionally repressible GAL::DBP10 fusion gene was constructed by placing the DBP10 ORF under the control of the inducible GAL1-10 promoter, which allows the expression of the gene in presence of galactose but its shut off in presence of glucose (Materials and Methods). The resulting plasmid pAS24-DBP10, which drives the expression of an N-terminally HA-tagged Dbp10-fusion protein, fully complemented the dbp10-disrupted strain YBF1-1B on YPGal, but it permitted only residual growth on YPD plates (Fig. 2B). In YPGal liquid medium, the GAL::DBP10 strain (YBF1-1B [pAS24-DBP10]) exhibited the same growth rate as the wild-type strain (YBF1-1A). Six hours after a shift from YPGal to YPD liquid medium, the growth rate of the GAL::DBP10 strain progressively slowed to a doubling time of >8 h after 36 h in glucose medium, whereas the wild-type strain growth rate remained the same (Fig. 2C). Accordingly, western blot analysis revealed that the levels of HA-Dbp10p began to decrease rapidly after the shift and it became undetectable after 18 h in glucose medium (Fig. 2D).
Figure 2.
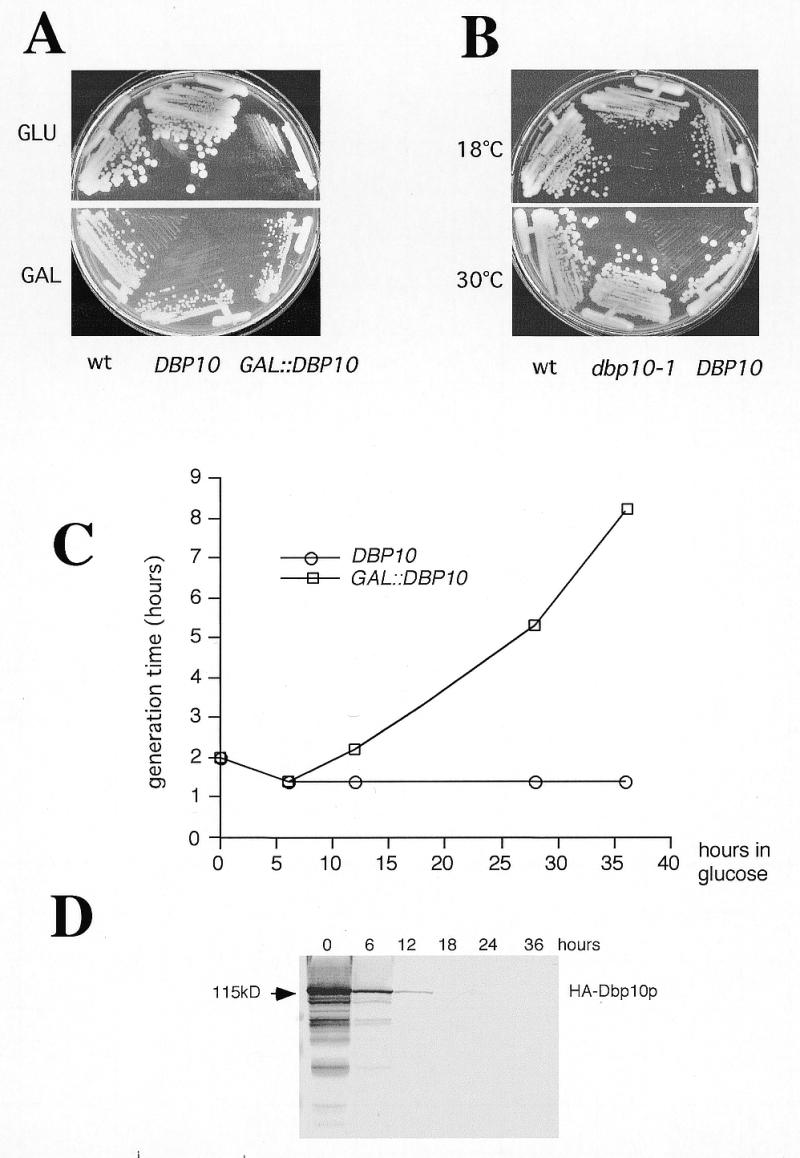
Mutations in DBP10 or depletion of Dbp10p results in growth inhibition. (A) Growth of YBF1-1A (wt), YBF1-1B [YCplac33-DBP10] (DBP10) and YBF1-1B [pAS24-DBP10] (GAL::DBP10) on YPD (glucose) or YPGal (galactose) plates. The plates were incubated at 30°C for 3 and 2 days, respectively. (B) Growth of YBF1-1A (wt), YBF1-1B [YCplac33-DBP10] (DBP10) and YBF1-1B [YCplac33-dbp10-1] (dbp10-1) strains on YPD plates at 16 and 30°C. The plates were incubated for 6 and 2 days, respectively. (C) Growth curves of YBF1-1B [YCplac33-DBP10] (DBP10, circles) and YBF1-1B [pAS24-DBP10] (GAL::DBP10, squares) strains at 30°C after shifting logarithmic cultures from YPGal to YPD for up to 36 h. Data are given as doubling times at different times in YPD medium. (D) Depletion of Dbp10p. The GAL::DBP10 strain was grown in YPGal and shifted to YPD for up to 36 h and cell extracts were prepared from samples harvested at the indicated times and assayed by western blotting. Equal amounts of proteins were loaded in each lane (~70 µg) as judged by red ponceau staining of the blot (data not shown). Pre-stained markers (Bio-Rad) were used as standards for molecular mass estimation. Monoclonal mouse anti-HA 16B12 antibodies followed by alkaline phosphatase coupled goat anti-rabbit IgG were used to detect HA-Dbp10p. The HA-Dbp10p signal is indicated by an arrow.
The GAL::DBP10 and dbp10-1 strains exhibit altered polysome profiles due to a deficit in 60S ribosomal subunits
Many DEAD box proteins have been found to be involved in translation initiation or ribosome biogenesis (13). To test whether Dbp10p was involved in one of these processes, we performed polysome analyses on the conditional dbp10 strains described above.
Polysome extracts prepared from the GAL::DBP10 and wild-type strains grown in YPGal and then shifted to YPD were analysed on 7–50% (w/v) sucrose gradients. As shown in Figure 3C and D, a comparison of the profiles revealed that depletion of Dbp10p is accompanied by an overall decrease of polysomes, a deficit in free 60S ribosomal subunit versus an excess of free 40S subunits and the appearance of half-mer polysomes. Notably, a double peak at the ‘usual’ position of the free 60S ribosomal subunit peak appeared reproducibly at the 12 h depletion time point (Fig. 3C). At later time points, the free 60S ribosomal subunit peak almost disappeared and the double peak was no longer detectable (Fig. 3D). To confirm a deficit in 60S ribosomal subunits relative to 40S subunits, the total ribosomal subunits were quantified in a run-off experiment on sucrose gradients. In accordance with a defect in 60S subunit biogenesis, the 60S/40S ratio dropped from 1.77 to 1.67 and 1.6 after 12 and 18 h of depletion, respectively (data not shown).
Figure 3.

The dbp10-1 mutation and the depletion of Dbp10p result in a deficit in free 60S ribosomal subunits and in the accumulation of half-mer polysomes. The wild-type strain YFB1-1A (A) and the dbp10-1 mutant strain YFB1-1B [YCplac33-dbp10-1] (B) were grown at 30°C and room temperature, respectively. The GAL::DBP10 strain YFB1-1B [pAS24-DBP10] was grown at 30°C in YPGal and shifted to YPD for 12 h (C) and 18 h (D). Cells were harvested at an OD600 of 0.8, and cell extracts were resolved in 7–50% (w/v) sucrose gradients. The A254 was measured continuously. Sedimentation is from left to right. The peaks of free 40S and 60S ribosomal subunits, 80S free couples/monosomes and polysomes are indicated. Half-mers are labelled with asterisks.
Polysome extracts prepared from the dbp10-1 strain grown in YPD medium at 30°C and at room temperature revealed that the dbp10-1 mutation also led to a deficit in free 60S ribosomal subunits and the appearance of half-mer polysomes (Fig. 3B and data not shown). The presence of a double peak at the position of the free 60S subunit peak was observed when the mutant strain was grown at room temperature or at 18°C. Altogether these results strongly suggest a role for Dbp10p in 60S ribosomal subunit biogenesis.
The formation of 25S and 5.8S mature rRNAs is impaired upon Dbp10p depletion
Since polysome analysis indicated a defect in 60S ribosomal subunit synthesis when Dbp10p function was impaired, we analysed the kinetics of rRNA maturation in the GAL::DBP10 strain by pulse–chase labelling experiments. To do so, the YBF1-1B [pAS24-DBP10] [YCplac33] and YBF1-1A [YCplac33] strains were grown in SGal–Ura and then shifted to SD–Ura medium for 18 h. At this time point, the doubling time of the GAL::DBP10 strain was 4 h, versus 2 h for the wild-type strain. Cells were pulse-labelled for 2 min with [5,6-3H]uracil prior to a chase with cold uracil. Total RNA was extracted at given time points during the chase, separated on agarose–formaldehyde gels, blotted onto Nylon membrane and analysed by fluorography. In the wild-type strain, the 35S precursor was rapidly converted into 32S and then into 27S and 20S pre-rRNAs, which were further processed into 25S and 18S mature rRNAs, respectively (Fig. 4A, lanes 1–5). After 5 min of chase, most of the labelled species were mature rRNAs. In the GAL::DBP10 strain, the processing of the 35S precursor was slowed down, and part of it being still detectable after 60 min of chase. The synthesis of the 18S rRNA was only mildly delayed and was not significantly reduced (Fig. 4A, compare 18S rRNA levels in lanes 2–5 with 7–10). Traces of an aberrant 23S species appeared at early chase time points. In contrast, formation of the 25S mature rRNA was clearly slowed down and above all strongly reduced (Fig. 4A, compare ratios 25S/18S lanes 5 and 10). At the same time, its immediate precursor, the 27SB pre-rRNA, persisted until the end of the chase (Fig. 4A, compare lanes 4 and 5 with 9 and 10), thus indicating that the processing step corresponding to the maturation of the 27SB precursor into 25S and 5.8S rRNA species was likely impaired. In agreement with this, analysis of the low-molecular weight RNAs confirmed that formation of 5.8S rRNAs was delayed and strongly reduced in the GAL::DBP10 strain, whereas synthesis of the 5S rRNA and tRNAs were not significantly affected (Fig. 4B).
Figure 4.
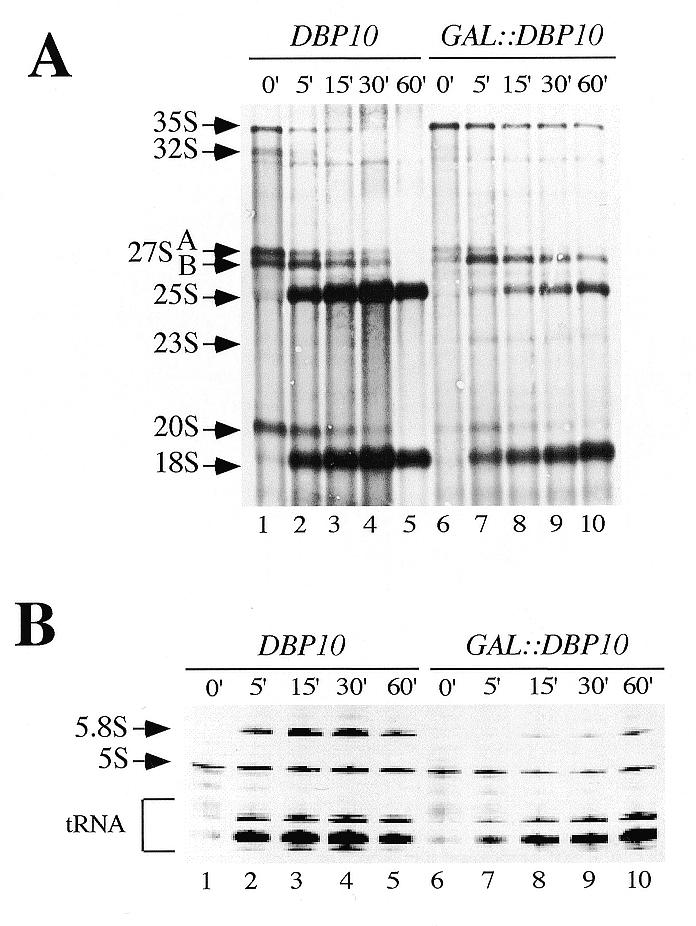
Dbp10p depletion leads to reduced synthesis of the mature 25S and 5.8S rRNAs. Strains YBF1-1A [YCplac33] (DBP10) and YBF1-1B [pAS24-DBP10, YCplac33] (GAL::DBP10) were grown at 30°C in Sgal–Ura and shifted to SD–Ura for 18 h. Cells were pulse-labelled with [5,6-3H]uracil for 2 min, and then chased with an excess of cold uracil for 5, 15, 30 and 60 min. Total RNA was extracted and separated on 1.2% agarose–6% formaldehyde (A) or 7% polyacrylamide–50% urea gels (B), transferred to nylon membrane and visualised by fluorography. Approximately 25 000 c.p.m. were loaded in each lane. The positions of the different pre-rRNAs and mature rRNAs and tRNAs are indicated.
Similar experiments performed with [methyl-3H]methionine revealed that the overall methylation of the pre-rRNAs was not affected upon Dbp10p depletion (data not shown). Finally, consistent with the results observed with the GAL::DBP10 strain, a defect in 25S and 5.8S rRNAs synthesis associated with a delay in the maturation of the 27SB pre-rRNA was also observed in the dbp10-1 mutant strain grown at room temperature (data not shown).
Altogether these results indicated that the deficit in 60S ribosomal subunits observed upon Dbp10p depletion and in the dbp10-1 mutant strain was linked to a defect in the maturation of the 27SB pre-rRNA into mature 25S and 5.8S rRNAs.
Depletion of Dbp10p results in accumulation of the late 27S pre-rRNA precursors
To study the pre-rRNA processing phenotype observed upon Dbp10p depletion in more detail, steady-state levels of the mature rRNAs and pre-rRNA intermediates were assessed by northern blot analyses. Total RNA was extracted from the GAL::DBP10 strain at various time points during the depletion, separated on agarose–formaldehyde gels and blotted onto a nylon membrane. The same blot was repeatedly hybridised with different probes specific for the different pre-rRNA intermediates (for probes see Fig. 1A). As seen in Figure 5A, the methylene blue staining of the mature rRNA species showed a clear deficit in 25S rRNA and a very minor decrease of 18S rRNA steady-state levels upon Dbp10p depletion. Consistent with the results of the pulse–chase experiments, hybridisation with probe F revealed that the 27SB pre-rRNAs accumulated with ongoing depletion, especially at the 12 and 18 h time points (Fig. 5F, compare lanes 1 and 2 with lanes 3 and 4). On the other hand, the steady-state levels of the 27SA pre-rRNAs started to decrease at the 18 h time point as shown with probes D and E (Fig. 5D and E, lanes 4–6). Concomitantly with the diminution of the 27SA pre-rRNAs pools, the 35S precursor accumulated as detected by all the probes used (Fig. 5B–F), the 20S pre-rRNA steady-state levels decreased (Fig. 5C, probe C) and a 23S aberrant precursor, detected by probes B, C and D, arose (Fig. 5B–D). The latter stems from the direct cleavage of the 35S pre-rRNA at site A3 and was indicative of a mild inhibition of early cleavages at sites A0, A1 and A2 (44). This is consistent with the slight reduction in the 18S rRNA steady-state levels observed upon Dbp10p depletion. As shown in Figure 5H, analyses of low molecular RNAs revealed a clear decrease of 7S pre-rRNA steady-state levels and a diminution in mature 5.8S rRNAs upon depletion. The 5S rRNA steady-state levels remained relatively constant during the depletion time course with a decrease at the later time points. Primer extension analysis performed with probe G indicated that during the time course of Dbp10p depletion, 27SBS/27SBL ratios remained unchanged and processing at sites A2, A3, B1L and B1S was accurate at the nucleotide level (data not shown).
Figure 5.
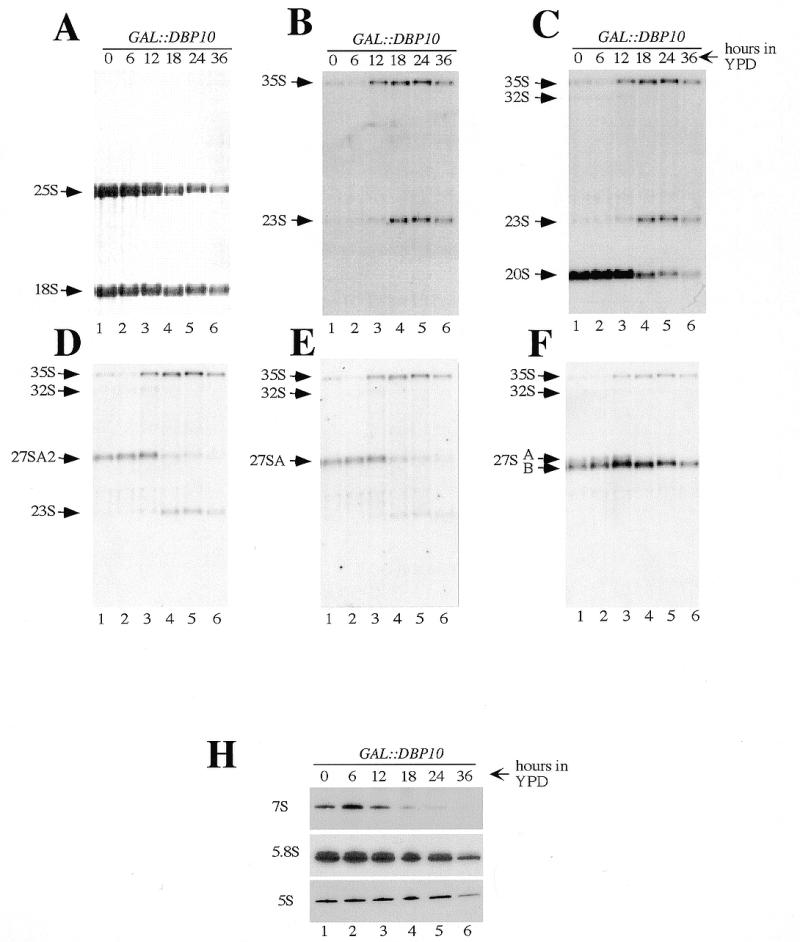
Depletion of Dbp10p affects the steady-state levels of pre-rRNA and mature rRNA species. Strain YBF1-1B [pAS24-DBP10] (GAL::DBP10) was grown in YPGal and shifted for up to 36 h in YPD. Cells were harvested at the indicated times and total RNA was extracted. Equal amounts (5 µg) of total RNA were resolved on 1.2% agarose–6% formaldehyde and transferred to a nylon membrane, which was stained with methylene blue (A), and then consecutively hybridised with the different probes indicated in Figure 1A [(B–F) probes B–F respectively]. Equal amounts (2.5 µg) of the same RNA samples were also resolved on 7% polyacrylamide–50% urea gel, transferred to a nylon membrane and hybridised consecutively with probe E, 5.8S and 5S (H). The positions of the different pre-rRNAs and mature rRNAs are indicated.
A comparison between pre-rRNA and rRNA steady-state levels from the dbp10-1 mutant and the wild-type strains grown at three different temperatures (30°C, room temperature and 18°C) showed that the major difference between the two strains resided in a reduced steady-state level of mature 25S rRNA and increased 27SB pre-rRNA steady-state levels in the mutant strain, especially at the more restrictive temperatures (Fig. 6A–F, compare lane 2 to 5 and lane 3 to 6). Steady-state levels of other precursors were not affected, except at 18°C where the 35S pre-rRNA steady-state level is clearly increased and the 23S aberrant precursor appeared.
Figure 6.
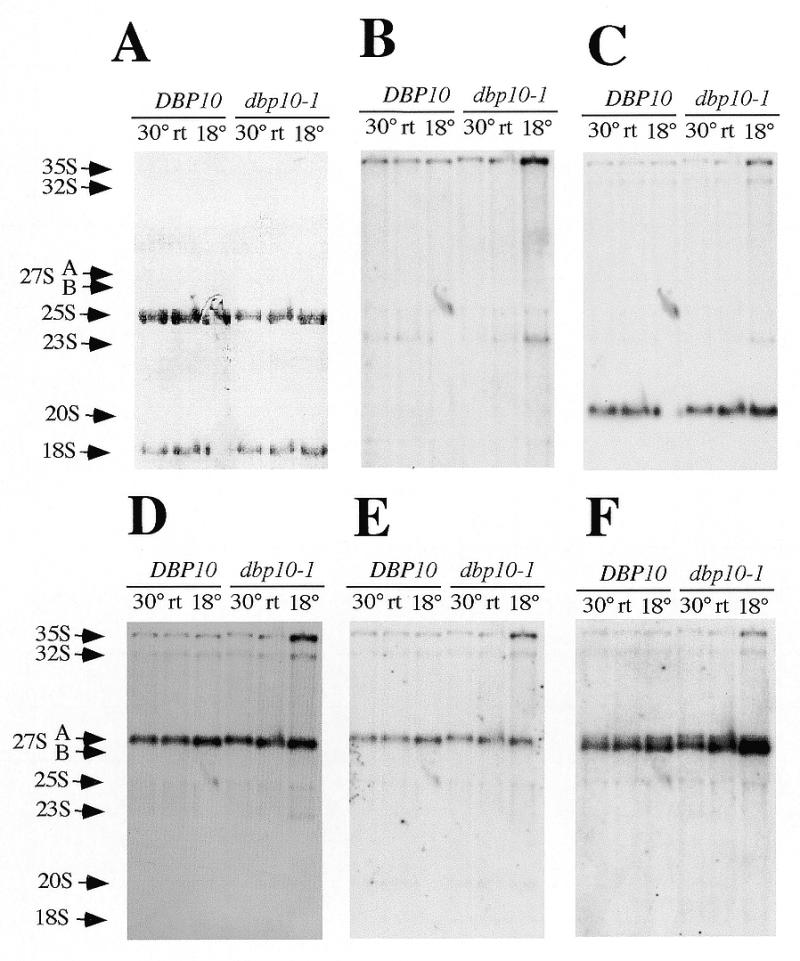
Steady-state levels of pre-rRNAs and mature rRNAs are altered in the dbp10-1mutant strain. Strains YBF1-1A (DBP10) and YBF1-1B [YCplac33-dbp10-1] (dbp10-1) were grown in YPD at 30°C, room temperature (rt) and 18°C as indicated on the top of the lanes. When the cultures reached the exponential phase, cells were harvested and total RNA was extracted. Equal amounts (5 µg) of total RNA were resolved on 1.2% agarose–6% formaldehyde and transferred to a nylon membrane, which was stained with methylene blue (A), and then consecutively hybridised with the different probes indicated in Figure 1A [(B–F) probes B–F respectively]. The positions of the different pre-rRNAs and mature rRNAs are indicated. The absence of 18S rRNA and 20S signals in the lane corresponding to DBP10 strain grown at 18°C is due to a technical artefact during migration of the gel.
Altogether these results confirmed that the primary and major defect occurring in ribosome biogenesis when Dbp10p function was impaired (either by depletion or due to mutation) was an inhibition of the maturation of the 27SB intermediates into 25S mature rRNA and 7S pre-rRNAs, which corresponded to processing events at sites C1 and C2.
Dbp10p localises to the nucleus with enrichment in the nucleolus
Most of the trans-acting factors involved in ribosome biogenesis are located in the nucleolus, whereas components of the ribosome are mainly found in the cytoplasm. In order to distinguish between a trans-acting factor and a component of the ribosome, the subcellular localisation of Dbp10p was determined by indirect immunofluorescence. For this purpose, a centromeric plasmid-borne HA-DBP10 allele under the control of its cognate promoter and expressing a fully functional N-terminally HA-epitope-tagged Dbp10p was constructed (YCplac111-HA-DBP10, see Materials and Methods). Indirect immunofluorescence was performed with the YBF1-1B [YCplac111-HA-DBP10] strain following growth in YPD medium to an OD600 of 0.6. The HA-Dbp10p was detected with primary monoclonal mouse anti-HA antibodies and secondary goat anti-mouse rhodamine-conjugated antibodies (Fig. 7A), the nucleus was visualised by staining DNA with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Fig. 7C) and the nucleolus was visualised by the detection of the Nop1p with primary polyclonal rabbit anti-Nop1 and secondary goat anti-rabbit fluorescein-conjugated antibodies (Fig. 7B). The HA-Dbp10p was restricted to the nucleus and highly enriched in a crescent-shaped area (Fig. 7A) corresponding to the nucleolus as shown by its colocalisation with Nop1p (Fig. 7F, overlap in yellow). A small part of HA-Dbp10p was also detected throughout the nucleoplasm as indicated by the colocalisation with the DAPI staining (Fig. 7E, overlap in magenta). This may represent its normal distribution within the nucleus or may be due to the presence of the HA-epitope, or the fact that the gene is located on a plasmid, despite its cognate promoter. No signal was detected with the anti-HA antibody in control cells not harbouring the HA-DBP10 allele (data not shown). The predominant nucleolar localisation of the HA-Dbp10 protein and its absence in the cytoplasm were consistent with the role of Dbp10p as a trans-acting factor in 60S ribosomal subunit biogenesis.
Figure 7.
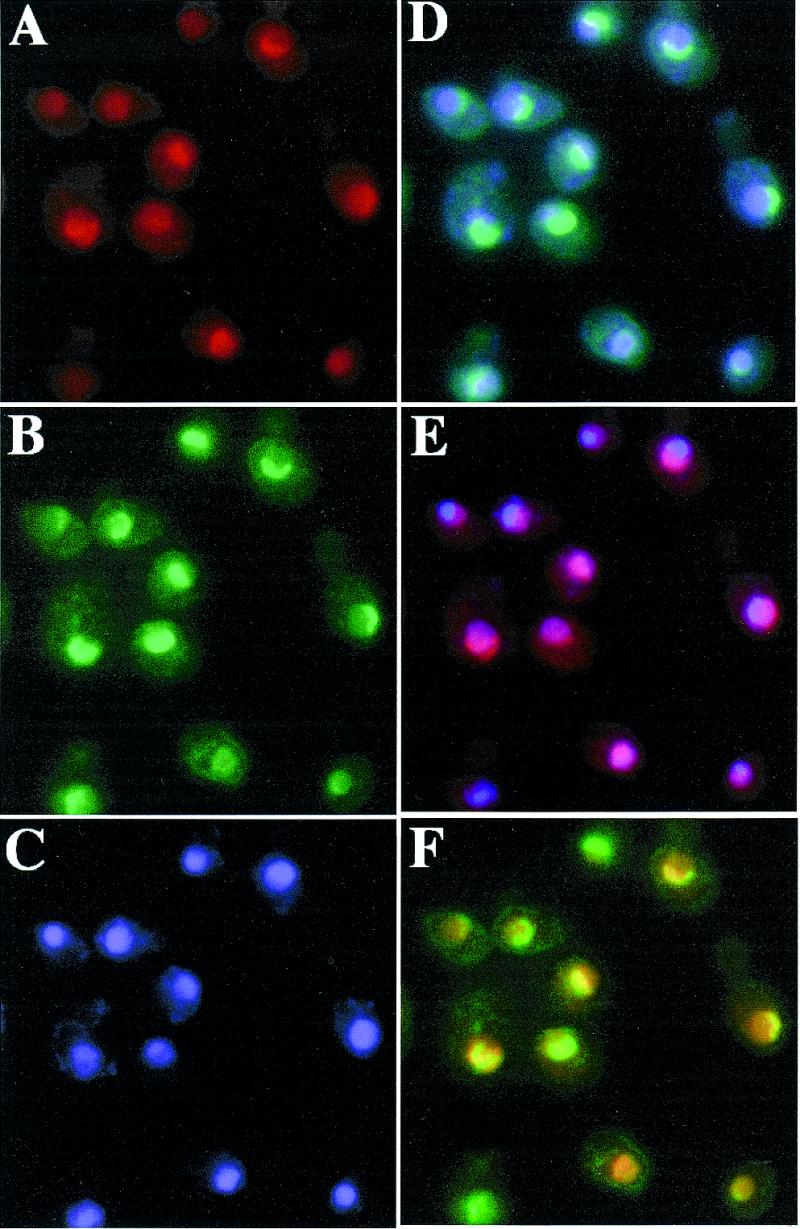
HA-Dbp10p localises to the nucleolus. Indirect immunofluorescence was performed with cells expressing HA-Dbp10p from the cognate promoter (strain YBF1-1B [YCplac111-HA-DBP10]) (A). HA-Dbp10p was detected by the monoclonal mouse anti-HA 16B12 antibody, followed by decoration with a goat anti-mouse rhodamine-conjugated antibody. (B) Nop1p was detected by polyclonal rabbit anti-Nop1p antibodies, followed by decoration with a goat anti-rabbit fluorescein-conjugated antibody. (C) Chromatin DNA was stained using DAPI. Pseudo-colours were assigned to the digitised micrographs (A–C) and images were merged. The overlapping distributions are revealed in: (D) cyan for Nop1p and chromatin DNA colocalisation; (E) magenta for HA-Dbp10p and chromatin DNA colocalisation; and (F) yellow for Nop1p and HA-Dbp10p colocalisation.
DISCUSSION
In this paper, we describe the functional analysis of the ORF YDL031w. YDL031w contains all eight conserved motifs characteristic of the proteins of the DEAD-box family of putative RNA helicases, and therefore we renamed it DBP10. Like most genes encoding putative RNA helicases involved in ribosome biogenesis, DBP10 is essential for vegetative growth. Thus, to pursue the phenotypic analysis, we constructed two different conditional systems: the cold sensitive dbp10-1 mutant strain and the conditional lethal GAL::DBP10 strain.
Polysome analyses and quantification of total ribosomal subunits in these strains revealed a deficit in 60S ribosomal subunits leading to the formation of half-mer polysomes. These phenotypic features are characteristic of mutants affecting either structural components of the 60S ribosomal subunit (45–47) or trans-acting factors involved in its biogenesis (11,20,32,48). Rather than being a structural component of the 60S ribosomal subunit, Dbp10p is more likely a trans-acting factor involved in its biogenesis since a HA-epitope tagged Dbp10p was mostly localised in the nucleolus and was not detected in the cytoplasm.
Analyses of rRNA synthesis by pulse–chase and northern blots confirmed that the deficit in 60S ribosomal subunits observed in the dbp10-1 and GAL::DBP10 strains is due to an inhibition of the pre-rRNA processing, which causes a deficit of the 25S and 5.8S mature rRNAs. The most striking pre-rRNA processing defect in these strains is the accumulation of the normal 27SB intermediates to the detriment of the mature 25S rRNA and of the 7S pre-rRNAs precursor to the 5.8S rRNAs. The 7S pre-rRNAs and the 25S rRNA are generated from the 27SB intermediates through processing reactions at sites C1 at the 5′ end of the 25S rRNA and C2 in ITS2. Little is known about these processing steps that are likely to be coupled. The sequences surrounding the sites are important but the nature of the cleavage reactions (endonucleolytic and/or exonucleolytic) is still under question and so far no trans-acting factors have been specifically implicated in these cleavage steps. It is not clear whether Dbp10p is directly involved in the cleavage reactions or whether it plays a role in the assembly process of a pre-ribosomal particle. In the latter case, an assembly defect would result in stable 27SB pre-rRNA intermediates that cannot be processed due to an incorrect structure or composition of the pre-ribosomal particle.
Because the specific substrate and the interacting partners of Dbp10p are not identified, it is not possible to precisely define its role in 60S ribosomal subunit biogenesis. However, and interestingly, one intriguing peculiarity that we observed during the phenotypic analysis of Dbp10p is the presence of two close peaks in the polysome profiles at the position where a single peak corresponding to free 60S ribosomal subunit is normally observed. The additional ‘60S-like’ peak arose in the GAL::DBP10 strain after 12 h of depletion and in the dbp10-1 mutant strain grown at restrictive temperatures (room temperature and 18°C). Remarkably, at the same time, these strains harboured the highest 27SB pre-rRNA steady-state levels. It is likely that the ‘60S-like’ additional peak corresponds to a sort of pre-ribosomal particle that accumulates when Dbp10p function is impaired. The ‘60S-like’ particle has a sedimentation coefficient very close to that of the mature 60S subunit, which leads to some overlap of the two peaks in polysomes gradients. Furthermore, compared to the overall mature 60S subunits the ‘60S-like’ particles represent a very minor population. Thus, we were not able to determine the protein composition of this peculiar particle. Indeed, no satisfying purification of pre-ribosomal particles has ever been reported. Overcoming these technical limitations will greatly help our understanding of the ribosome assembly process, which is still very poor, and isolation of mutants (such as DBP10) leading to accumulation of stable pre-particles will certainly be of great help.
In addition to Dbp10p, four components (Nop2p, Spb1p, Nip7p, Spb4p) have been described that show a delayed processing at sites C1 and C2. Mutations in the corresponding genes lead to the accumulation of the 27SB pre-rRNAs and delayed synthesis of the 5.8S and 25S mature rRNAs. Nop2p and Spb1p are two putative RNA methyltransferases. Whereas in the case of Nop2p the delay in 27SB processing is accompanied by a delay in methylation at the site UmGmΨUC2922 (48), no specific methylation defect has been described for Spb1p (49,50). Nip7p is both a nucleolar and cytoplasmic protein that interacts with a component of the exosome and with Nop8p (51,52). Spb4p is a putative RNA helicase of the DEAD-box protein family (22). As for Dbp10p, there is no strong experimental evidence that these factors are directly involved in the pre-rRNA processing reactions per se and the favoured hypothesis is that they rather play a role in the assembly process of the pre-ribosomal particles. We believe that the absence of the corresponding functional proteins lead to particles in which the C1 and C2 cleavage sites are no longer accessible for cleavage and further processing.
In addition to the defect in the maturation of the 27SB intermediates, at later time points of Dbp10p depletion, an inhibition of the early cleavages at sites A0, A1 and A2 was apparent, leading to a diminution of the 18S rRNA pools. This was revealed by the accumulation of the 35S pre-rRNA and the 23S aberrant intermediate, and the decrease of the 27SA2 and 20S intermediates. Inhibition of the early processing steps has been reported for numerous mutations that specifically affect 60S ribosomal subunit biogenesis (52–55). Indeed, while many mutations inhibit the formation of the 18S rRNA without affecting that of the 25S and 5.8S rRNAs, very few affect the latter without at least delaying the A0-A2 cleavages. It has been proposed that the early processing machinery has a mechanism for checking that processing factors needed for the subsequent steps are assembling correctly (12). The role of Dbp10p in 25S/5.8S rRNAs appears to be specific since neither the synthesis of the 5S rRNAs and the tRNAs nor rRNA methylation were significantly affected. There is only a very minor effect on the overall synthesis of the 18S rRNA and the steady-state levels of the 40S ribosomal subunit.
Dbp10p is the seventh putative RNA helicase described that is involved in 60S ribosomal subunit biosynthesis (Fig. 1). Two have been assigned a function in the pre-rRNA processing itself, Dbp3p in the cleavage at site A3 performed by RNase MRP and Dob1p in assisting the exosome in trimming the 3′ end of the 7S pre-rRNA (23,31). Two proteins still lack precise descriptions of their pre-rRNA processing defects, their role in 60S synthesis being solely deduced from aberrant polysome profiles (32,56). The remaining three have been assigned a function in the assembly process with an effect on pre-rRNA processing as a consequence (Dbp6p, Dbp7p and Spb4p) (20–22). Regarding the consequences on pre-rRNA processing, two classes of trans-acting factors have been evoked (discussed in 20–22,50). One class of factors has mutations that lead to the depletion of the 27S intermediates, which is thought to be due to a rapid degradation of the pre-rRNA due to the instability of the pre-particles formed in the absence of these factors. This is the case for the Dbp6 and Dbp7 proteins. The second class of factors has mutations that lead to the accumulation of the normal 27S intermediates, which suggests that a stable pre-particle is formed but that it is no longer adequate for processing to occur. This is the case for the Spb4p and Dbp10p proteins. It has also been suggested that factors belonging to the first class act in the early steps of the 60S ribosomal assembly pathway, whereas factors belonging to the second class act in later steps (discussed in 57). To date, our knowledge on the assembly process is too scarce to confirm or reject either hypothesis.
The 27SB intermediates that accumulated in the dbp10 and spb4 conditional strains migrate in run-off sucrose gradients in a manner similar to particles with a sedimentation coefficient of approximately 66S, as do the 27SB intermediates present in a wild-type strain (data not shown; 22). However, no additional ‘60S-like’ peak was detected in polysome gradients of the conditional spb4 strains. Furthermore, each of these two helicases is essential and functionally not redundant. Indeed, over-expression of Dbp10p does not suppress the growth defect of the sbp4-1 strain and does not bypass the lethal phenotype of the spb4 null strain. Reciprocally, over-expression of Spb4p does not suppress the growth defect of the dbp10-1 strain and does not bypass the lethal phenotype of the dbp10 null strain. This suggests that various and distinct assembly steps (still undefined) are required for generating functional 27SB-containing pre-particles, and it becomes more and more clear that numerous structural rearrangements have to occur, and that these are most likely driven by RNA helicases.
It is obvious that characterisation of all the factors required for ribosome biogenesis is an essential step in the complete understanding of this process. One next important step concerning the RNA helicases as ribosome biogenesis trans-acting factors will be the demonstration of their enzymatic activities, the determination of their substrate specificity and the discovery of their interacting partners.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dieter Kressler and Jesús de la Cruz for SPB4 strains and plasmids, and stimulating discussions. We are grateful to D. Kressler, J. de la Cruz and N. K. Tanner for comments on the manuscript. We acknowledge Manuel Rojo for help with the immunofluorescence and Ed Hurt (Heidelberg) for anti-Nop1p antibodies. This work was supported by a grant from the Swiss National Science Foundation (to P.L.).
REFERENCES
- 1.Warner J.R. (1999) Trends Biol. Sci., 24, 437–440. [DOI] [PubMed] [Google Scholar]
- 2.Li B., Nierras,C.R. and Warner,J.R. (1999) Mol. Cell. Biol., 19, 5393–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mager W.H., Planta,R.J., Ballesta,J.-P.G., Lee,J.C., Mizuta,K., Suzuki,K., Warner,J.R. and Woolford,J. (1997) Nucleic Acids Res., 25, 4872–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mélèse T. and Xue,Z. (1995) Curr. Opin. Cell Biol., 7, 319–324. [DOI] [PubMed] [Google Scholar]
- 5.Scheer U. and Hock,R. (1999) Curr. Opin. Cell Biol., 11, 385–390. [DOI] [PubMed] [Google Scholar]
- 6.Lafontaine D.L.J. and Tollervey,D. (1998) Trends Biochem. Sci., 23, 383–388. [DOI] [PubMed] [Google Scholar]
- 7.Bachellerie J.-P. and Cavaillé,J. (1997) Trends Biochem. Sci., 22, 257–261. [DOI] [PubMed] [Google Scholar]
- 8.Trapman J., Planta,R.J. and Raué,H.A. (1976) Biochim. Biophys. Acta, 442, 275–284. [DOI] [PubMed] [Google Scholar]
- 9.Kruiswijk T., Planta,R.J. and Krop,J.M. (1978) Biochim. Biophys. Acta, 517, 378–389. [DOI] [PubMed] [Google Scholar]
- 10.Samarsky D.A. and Fournier,M.J. (1999) Nucleic Acids Res., 27, 161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kressler D., Linder,P. and de la Cruz,J. (1999) Mol. Cell. Biol., 19, 7897–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venema J. and Tollervey,D. (1999) Annu. Rev. Genet., 33, 261–311. [DOI] [PubMed] [Google Scholar]
- 13.de la Cruz J., Kressler,D. and Linder,P. (1999) Trends Biochem. Sci., 24, 192–198. [DOI] [PubMed] [Google Scholar]
- 14.Hirling H., Scheffner,M., Restle,T. and Stahl,H. (1989) Nature, 339, 562–564. [DOI] [PubMed] [Google Scholar]
- 15.Iost I., Dreyfus,M. and Linder,P. (1999) J. Biol. Chem., 274, 17677–17683. [DOI] [PubMed] [Google Scholar]
- 16.Liang L., Diehl-Jones,W. and Lasko,P. (1994) Development, 120, 1201–1211. [DOI] [PubMed] [Google Scholar]
- 17.Schwer B. and Gross,C.H. (1998) EMBO J., 17, 2086–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y., Wagner,J.D.O. and Guthrie,C. (1998) Curr. Biol., 8, 441–451. [DOI] [PubMed] [Google Scholar]
- 19.Tseng S.S.-I., Weaver,P.L., Liu,Y., Hitomi,M., Tartakoff,A.M. and Chang,T.-H. (1998) EMBO J., 17, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kressler D., de la Cruz,J., Rojo,M. and Linder,P. (1998) Mol. Cell. Biol., 18, 1855–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daugeron M.C. and Linder,P. (1998) RNA, 4, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de la Cruz J., Kressler,D., Rojo,M., Tollervey,D. and Linder,P. (1998) RNA, 4, 1268–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de la Cruz J., Kressler,D., Tollervey,D. and Linder,P. (1998) EMBO J., 17, 1128–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kressler D., de la Cruz,J., Rojo,M. and Linder,P. (1997) Mol. Cell. Biol., 17, 7283–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang W.-Q., Clark,J.A. and Fournier,M.J. (1997) Mol. Cell. Biol., 17, 4124–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Day C.L., Chavanikamannil,F. and Abelson,J. (1996) Nucleic Acids Res., 24, 3201–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wickner R.B. (1996) Microbiol. Rev., 60, 250–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zagulski M., Becam,A.-M., Grzybowska,E., Lacroute,F., Migdalski,A., Slonimski,P.P., Sokolowska,B. and Herbert,C.J. (1994) Yeast, 10, 1227–1234. [DOI] [PubMed] [Google Scholar]
- 29.Rasmussen T.P. and Culbertson,M.R. (1998) Mol. Cell. Biol., 18, 6885–6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venema J., Bousquet-Antonelli,C., Gelugne,J.-P., Caizergues-Ferrer,M. and Tollervey,D. (1997) Mol. Cell. Biol., 17, 3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weaver P.L., Sun,C. and Chang,T.-H. (1997) Mol. Cell. Biol., 17, 1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ripmaster T.L., Vaughn,G.P. and Woolford,J.L.,Jr (1992) Proc. Natl Acad. Sci. USA, 89, 11131–11135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas B.J. and Rothstein,R. (1989) Cell, 56, 619–630. [DOI] [PubMed] [Google Scholar]
- 34.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (eds) (1994) In Current Protocols in Molecular Biology, Chapter 13. John Wiley & Sons, Inc.
- 35.Kaiser C., Michaelis,S. and Mitchell,A. (1994) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 36.Gietz D., St. Jean,A., Woods,R.A. and Schiestl,R.H. (1992) Nucleic Acids Res., 20, 1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 38.Wach A., Brachat,A., Pöhlmann,R. and Philippsen,P. (1994) Yeast, 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- 39.de la Cruz J., Iost,I., Kressler,D. and Linder,P. (1997) Proc. Natl Acad. Sci. USA, 94, 5201–5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt A., Bickle,M., Beck,T. and Hall,M.N. (1997) Cell, 88, 531–542. [DOI] [PubMed] [Google Scholar]
- 41.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Gene, 77, 51–59. [DOI] [PubMed] [Google Scholar]
- 42.Venema J., Planta,R.J. and Raué,H.A. (1998) In Martin,R. (ed.), Protein Synthesis: Methods and Protocols. Humana Press, Totowa, NJ, pp. 257–270.
- 43.Robbins J., Dilsworth,S.M., Laskey,R.A. and Dingwall,C. (1991) Cell, 64, 615–623. [DOI] [PubMed] [Google Scholar]
- 44.Venema J. and Tollervey,D. (1995) Yeast, 11, 1629–1650. [DOI] [PubMed] [Google Scholar]
- 45.Deshmukh M., Tsay,Y.-F., Paulovich,A.G. and Woolford,J.L.,Jr (1993) Mol. Cell. Biol., 13, 2835–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moritz M., Paulovich,A.G., Tsay,Y.F. and Woolford,J.L.,Jr (1990) J. Cell Biol., 111, 2261–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vilardell J. and Warner,J.R. (1997) Mol. Cell. Biol., 17, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong B., Brockenbrough,J.S., Wu,P. and Aris,J.P. (1997) Mol. Cell. Biol., 17, 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pintard L., Kressler,D. and Lapeyre,B. (2000) Mol. Cell. Biol., 20, 1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kressler D., Rojo,M., Linder,P. and de la Cruz,J. (1999) Nucleic Acids Res., 27, 4598–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zanchin N.I.T. and Goldfarb,D.S. (1999) Mol. Cell. Biol., 19, 1518–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zanchin N.I.T., Roberts,P., DeSilva,A., Sherman,F. and Goldfarb,D.S. (1997) Mol. Cell. Biol., 17, 5001–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Nues R.W., Rientjes,J.M., Morre,S.A., Mollee,E., Planta,R.J., Venema,J. and Raue,H.A. (1995) J. Mol. Biol., 250, 24–36. [DOI] [PubMed] [Google Scholar]
- 54.Allmang C. and Tollervey,D. (1998) J. Mol. Biol., 278, 67–78. [DOI] [PubMed] [Google Scholar]
- 55.Berges T., Petfalski,E., Tollervey,D. and Hurt,E.C. (1994) EMBO J., 13, 3136–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohtake Y. and Wickner,R.B. (1995) Mol. Cell. Biol., 15, 2772–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kressler D., Doere,M., Rojo,M. and Linder,P. (1999) Mol. Cell. Biol., 19, 8633–8645. [DOI] [PMC free article] [PubMed] [Google Scholar]