

Abstract
Circularizing oligonucleotide probes, so-called padlock probes, have properties that should prove valuable in a wide range of genetic investigations, including in situ analyses, genotyping and measurement of gene expression. However, padlock probes can be difficult to obtain by standard oligonucleotide synthesis because they are relatively long and require intact 5′- and 3′-end sequences to function. We describe a PCR-based protocol for flexible small-scale enzymatic synthesis of such probes. The protocol also offers the advantage over chemical synthesis that longer probes can be made that are densely labeled with detectable functions, resulting in an increased detection signal. The utility of probes synthesized according to this protocol is demonstrated for the analysis of single nucleotide variations in human genomic DNA both in situ and in solution.
INTRODUCTION
DNA and RNA analyses, whether they are aimed at monitoring nucleotide sequence variations or measuring copy numbers of specific sequences, must be sufficiently precise to detect small numbers of target sequences against a large background of irrelevant nucleic acids and to distinguish target sequence variants that may differ in only single nucleotide positions. So-called padlock probes exhibit very high specificity and are promising for simultaneous detection of numerous target sequences and for localized detection reactions, two properties where PCR exhibits important limitations.
Padlock probes are oligonucleotides that become circularized by DNA ligation in the presence of an appropriate DNA (1) or RNA target sequence (Nilsson et al., submitted for publication) (Fig. 1). The reaction is highly specific as it requires that two target-complementary segments, one at each end of the probe, hybridize to a target sequence for circularization to occur and because DNA ligation is strongly inhibited by any mismatches at the ligation junction (2,3). Furthermore, since only intramolecular probe ligation is scored, cross-reactions are unlikely to arise between many simultaneously added probes. This is in contrast to intermolecular probe reactions such as PCR, where the simultaneous presence of many primer pairs dramatically increases the risk of false products. Finally, highly sensitive detection of circularized probes is possible, e.g. by amplifying reacted probes via a rolling circle replication mechanism (4–6).
Figure 1.
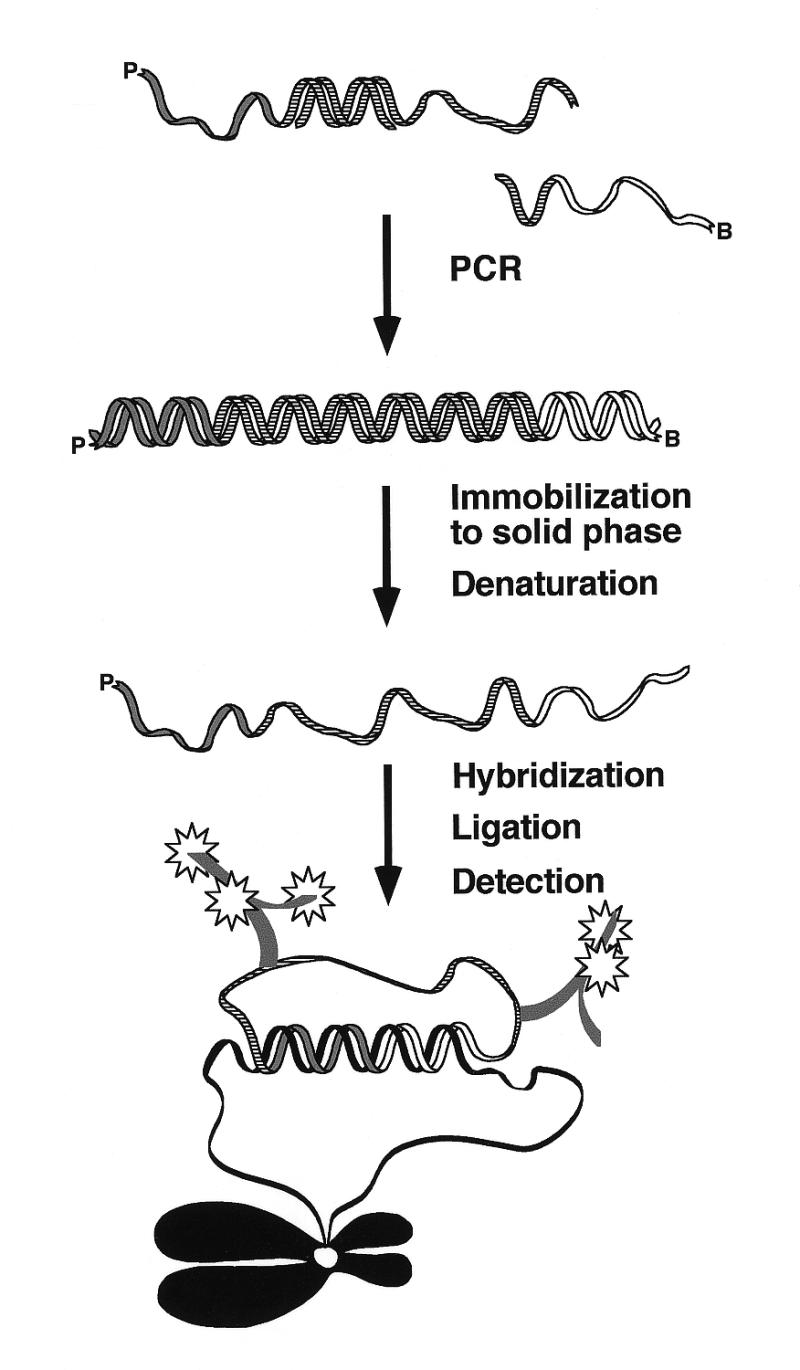
A schematic overview of enzymatic synthesis of padlock probes. Two primers are used to amplify an oligonucleotide template by PCR. The 5′-phosphorylated primer, marked P, will define the 5′-end of the padlock. The complement of the 5′-biotinylated primer, marked B, will form the 3′-end of the padlock. The double-stranded PCR product is immobilized on paramagnetic streptavidin-coated beads via the 5′-biotin and the complementary strand is released under denaturing conditions. The single-stranded padlock probe is hybridized to the target sequence, ligated and detected using fluorescent-labeled antibodies against labeled nucleotides incorporated during PCR.
Padlock probes are typically 70–100 nt in length. They are therefore relatively expensive to synthesize, especially if detectable functions such as haptens or fluorophores are incorporated during or after synthesis. Moreover, many mechanisms contribute to the accumulation of imperfect products during chemical synthesis. The proportion of truncated sequences increases with length, as does the problem of separating full-length oligonucleotides from truncated ones using either HPLC or gel purification. Oligonucleotides missing bases at either end cannot contribute to probe circularization reactions, due to the strict requirement for correctly matched ends for ligation to occur. We have previously addressed this problem by developing a novel support for chemical synthesis that facilitates isolation of full-length oligonucleotides (7). Here we present an alternative, PCR-based method which is suitable for synthesizing padlock probes on a small scale in a cost-effective and flexible manner or for construction of probes longer than 100 or so nucleotides. We demonstrate the utility of probes synthesized in this manner for sensitive detection of single nucleotide differences in a repeated sequence in metaphase chromosomes and in a single copy sequence in total genomic DNA in solution.
MATERIALS AND METHODS
Oligonucleotides
The oligonucleotides used to synthesize the padlock probes are presented in Table 1. All oligonucleotides were purchased from Interactiva (Ulm, Germany), except αchr12 [5′-P-AAATCTCCAACTGGAAACTG-(HEG)2-CBIO)7-(HEG)2-ATTTGGTCTCAAAGTGATTG-3′, where P is the 5′-phosphate, HEG is hexaethylene glycol and CBIO is biotin attached to a C residue modified with a primary amine, synthesized according to Nilsson et al. (1).
Table 1. Oligonucleotide and linker segment templates used to synthesize padlock probes.

Target-complementary sequences are shown in bold and the complements to the target complementary sequences are in bold italics.
Construction of padlock probes
Probes were generated by PCR with a 5′-phosphorylated forward primer at 0.1 µM and a 5′-biotinylated reverse primer at 0.05 µM in Pfu buffer [20 mM Tris–HCl pH 8.8, 2 mM MgSO4, 10 mM KCl, 10 mM (NH4)2SO4, 0.1% Triton X-100, 0.1 mg/ml nuclease-free BSA], 50 µM each dATP, dCTP, dGTP and dTTP (Amersham Pharmacia Biotech, Piscataway, NJ) and 0.05 U/µl cloned Pfu polymerase (Stratagene, La Jolla, CA) and using 5 fmol oligonucleotide as template. After 30 cycles of 94, 55 and 72°C with 1 min at each temperature, dNTPs were added to a final concentration of 200 µM before the concluding extension at 72°C for 7 min.
Probes of variable lengths but with identical 5′- and 3′-ends were synthesized in two amplification reactions. In the first PCR one biotinylated forward primer was applied with one of six alternative reverse primers at 0.1 µM each (for primers see Table 1) in Taq buffer (50 mM KCl, 10 mM Tris–HCl pH 8.3, 1.5 mM MgCl2, 12.5 µg/ml BSA) with 200 µM each dNTP and 0.02 U/µl Taq polymerase (PE Biosystems, Foster City, CA) using 0.1 amol λ DNA as template. No extra nucleotides were added before the last extension step. The PCR products were diluted 5000 times and used as templates in a second PCR with the same biotinylated forward primer as in the first PCR and a 5′-phosphorylated reverse primer, using cloned Pfu polymerase in Pfu buffer, but with one-third of the dTTPs replaced by digoxigenin-11-dUTP (Boehringer-Mannheim GmbH, Mannheim, Germany) to label the probes. Pfu polymerase was inhibited by dinitrophenyl-11-dUTP (Molecular Probes, Leiden, The Netherlands), so for labeling with this hapten Taq polymerase was used in Taq buffer, with 50 µM each dNTP and one-third of the dTTP exchanged for dinitrophenyl-11-dUTP.
DNA was precipitated with 0.1 vol of 3 M Na acetate pH 5.0, and 2 vol of ice-cold 95% ethanol. The concentration of resuspended DNA was determined by spectrophotometry (GeneQuant; Amersham Pharmacia Biotech). An aliquot of 4 µg DNA in 1 M NaCl was bound to 100 µl of paramagnetic beads (Dynabeads M-280; Dynal, Oslo, Norway), pre-washed with 100 µl of washing buffer (50 mM NaCl, 10 mM Tris–HCl, pH 7.0), for 30 min on a rotator at room temperature. The beads were washed three times with washing buffer before adding 50 µl of 0.15 M LiOH and 0.15 M NaCl. After 5 min the solution was removed and neutralized with 50 µl of 0.15 M NH4Cl. The released strands were precipitated with 2 µg of glycogen (Boehringer-Mannheim) added as carrier, resuspended in 25 µl of water and quantitated by spectrophotometry.
Ligation
Ligation reactions were carried out in Tth buffer (20 mM Tris–HCl pH 7.9, 100 mM KCl, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.1% Triton X-100, 1 mM NAD) and 0.25 U/µl Tth ligase, using the temperature profile 92°C for 2 min and 55°C for 15 min.
In situ analysis
Metaphase chromosome preparations, spread on microscope slides, were obtained from lymphocyte cultures using standard techniques of colcemid treatment, hypotonic shock, methanol/acetic acid fixation and RNase A treatment. DNA was denatured by placing the slides on a temperature controlled block (GenE thermal cycler; Techne, Cambridge, UK), adding 70% formamide in 2× SSC (1× SSC = 150 mM NaCl, 15 mM sodium citrate, pH 7.0), sealing with a coverslip and heating the slides at 68°C for 2 min. The slides were washed in ice-cold 2× SSC for 2 min and dehydrated in a series of 70, 85 and 100% ethanol for 2 min each. Hybridization was performed with a probe concentration of 1 fmol/µl for each labeled probe in 20% formamide, 10% dextran sulfate, 2× SSC and denatured sonicated salmon sperm at 0.5 µg/µl. A 24 × 50 mm coverslip was placed over 30 µl of hybridization solution spotted onto each slide and sealed with rubber glue. The slides were incubated in a moist chamber at 37°C overnight. The coverslip was removed and excess probe washed away with 2× SSC at 37°C for 5 min, followed by dehydration in an ethanol series. Ligation was carried out in Tth buffer supplemented with 8.7% glycerol, 0.1 µg/µl sonicated salmon sperm DNA and 0.1 µg/µl BSA, using a Tth ligase concentration of 0.25 U/µl. The slides were placed on a temperature controlled block, 60 µl of ligation solution was spotted onto each slide and a 24 × 50 mm coverslip was added before incubation at 55°C for 20 min. The reactions were stopped by immersing the slides in 3.3× SSC and 50 mM EDTA at 55°C for 2 min. Unreacted padlock probes were removed by washing in 30% formamide in 2× SSC at 42°C and 2× SSC at 55°C for 10 min, before cooling the slides to room temperature in 2× SSC and 0.05% Tween-20. Reacted probes were visualized by adding fluorophore-conjugated Fab fragments directed against digoxigenin and dinitrophenyl at a concentration of 4 ng/µl or 2 ng/µl fluorescein-labeled avidin (Vector Laboratories Inc., Burlingame, CA) in 2× SSC and 0.05% Tween-20 (70 µl of solution for each slide covered with a 60 × 24 mm coverslip). The slides were incubated at 37°C for 30 min and then washed with gentle agitation twice in 2× SSC and 0.05% Tween-20 for 7 min each time. The slides were dehydrated in an ethanol series and then counterstained with 100 ng/µl DAPI (Sigma, St Louis, MO) in Vectashield (Vector Laboratories Inc.) under a 50 × 24 mm coverslip. The slides were viewed under a microscope (Axioskop; Zeiss, Oberkochen, Germany) and images were captured with a CCD camera (CE 200A; Photometrics GmbH, Munich, Germany).
PCR analysis of padlock probes
Two padlock probes, WD14wt and WD14mut, were generated by PCR. Ten micrograms of genomic DNA were digested using 50 U MnlI restriction enzyme in 100 µl Tth buffer at 37°C for 19 h. For each DNA sample two 20 µl ligation reactions were performed, containing 500 ng genomic DNA and 50 fmol of either WD14wt or WD14mut probes and 0.25 U Tth ligase. After an initial 5 min denaturation at 94°C, the ligation reactions were thermally cycled 10 times at 94 (30 s) and 55°C (15 min). To stop ligation before return to ambient temperature (hot stop), 0.5 U of calf intestinal phosphatase was added in 1 µl of Tth buffer before a concluding 20 min incubation at 55°C. The phosphatase was inactivated for 10 min at 94°C. Ligated probes from each ligation reaction were amplified by PCR across the ligation junction using 0.1 µM primers 5′-ATTGTAATAGCGTTGACCAC-3′ and 5′-AATTCACTGGCCGTCGTTCG-3′ and 0.02 U/µl AmpliTaq Gold (PE Biosystems) in Taq polymerase buffer. After 7 min at 94°C, the samples were cycled 35 times at 94, 50 and 72°C for 30 s at each temperature, before a concluding extension step at 72°C for 7 min. Amplification products were analyzed by agarose gel electrophoresis in an ethidium bromide stained 2% agarose gel and the image was captured using a digital camera.
RESULTS
Construction of padlock probes
We synthesized padlock probes by PCR, using two amplification primers and a support for immobilizing the amplification products. In this procedure one of the primers is equipped with a 5′-phosphate, and this primer will form the 5′-ends of the padlock probes. The other primer has a 5′-biotin for immobilization of amplification products, allowing release of the complementary strand under denaturing conditions. The complement of this second primer defines the 3′-ends of the padlock probes (Fig. 1). The 3′-ends of the two amplification primers are designed to recognize a piece of DNA suitable as the target-non-complementary part of the circularizable probes, herein referred to as the linking segment.
DNA polymerases lacking proof-reading activity, such as Taq DNA polymerase, are known to leave extra nucleotides at the 3′-ends of PCR products, while polymerases with strong 3′→5′ exonuclease activity can produce PCR products that lack one or more nucleotides at the 3′-end (8). Both extra and missing nucleotides inhibit circularization of padlock probes, due to the requirement for perfectly matched ends. The addition or removal of nucleotides at the 3′-terminus is influenced by the nucleotide concentration in the PCR. Lower nucleotide concentrations tend to decrease the non-templated addition of nucleotides by polymerases that lack 3′→5′ exonucleolytic activity, but lower nucleotide concentrations also reduce the yield of amplification products. We found that amplification using Pfu polymerase yielded 60–70% products of the correct size and with a corresponding ligation efficiency (Fig. 2), while Taq polymerase, with nucleotide concentrations that resulted in similar ligation efficiencies, resulted in ~50% lower probe yield.
Figure 2.
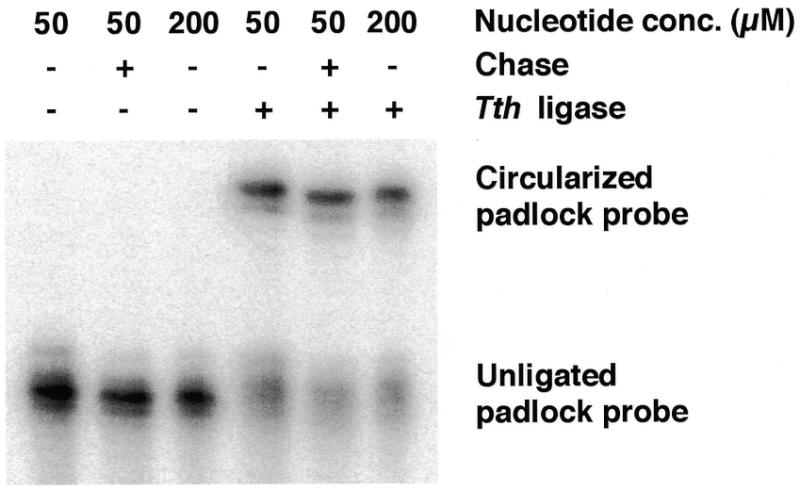
Ligation of PCR-generated padlock probes analyzed by denaturing polyacrylamide gel electrophoresis. Padlock probes were generated using chrAB and chrA as primers and link oligonucleotide as template. DNA was labeled during PCR by addition of 20 µCi [32P]dCTP (NEN, Boston, MA) to the reactions. No extra dNTP was added to the non-chase sample and to a control that was run with 200 µM each dNTP. The padlock probe was circularized using a 2-fold excess (0.5 µM) of the oligonucleotide 5′-AATAGAAATGTTCAACTCCTTTAGCTGGGTACACACATCA-3′ as template in a 10 µl reaction, The reaction was stopped by adding 10 µl of loading buffer containing 10 mM EDTA. The samples were run on a 6% denaturing polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA) and quantified using a PhosphorImager system (Molecular Dynamics, Sunnyvale, CA).
The probes can be labeled during amplification through incorporation of modified nucleotide triphosphates. In order to save on these reagents, we performed the PCR in the presence of 50 µM dNTP (including 17 µM labeled dUTP) and then added 200 µM unlabeled dNTP at the concluding extension step. This chase with unlabeled dNTP gives probes of the same quality on gel inspection as when, as judged by ligation efficiency, the PCR is performed with a dNTP concentration of 200 µM during the entire reaction. In contrast, probes generated with low nucleotide concentrations and without this chase step exhibited 8% lower ligation efficiency, due to the appearance of shorter products (Fig. 2).
Comparison between padlock probes derived by PCR or by chemical synthesis
We have previously applied padlock probes to distinguish two α-satellite sequences in human chromosomes 13 and 21 in situ, on the basis of a single nucleotide difference (3). We now used enzymatic synthesis to construct two padlock probes aimed at the same target sequences as in the previous study. Probes specific for the two sequence variants were labeled by including dUTPs modified with either dinitrophenyl or digoxigenin. The dinitrophenyl and digoxigenin residues were visualized using fluorescein- and rhodamine-labeled antibody fragments specific for the two respective haptens (Fig. 3).
Figure 3.
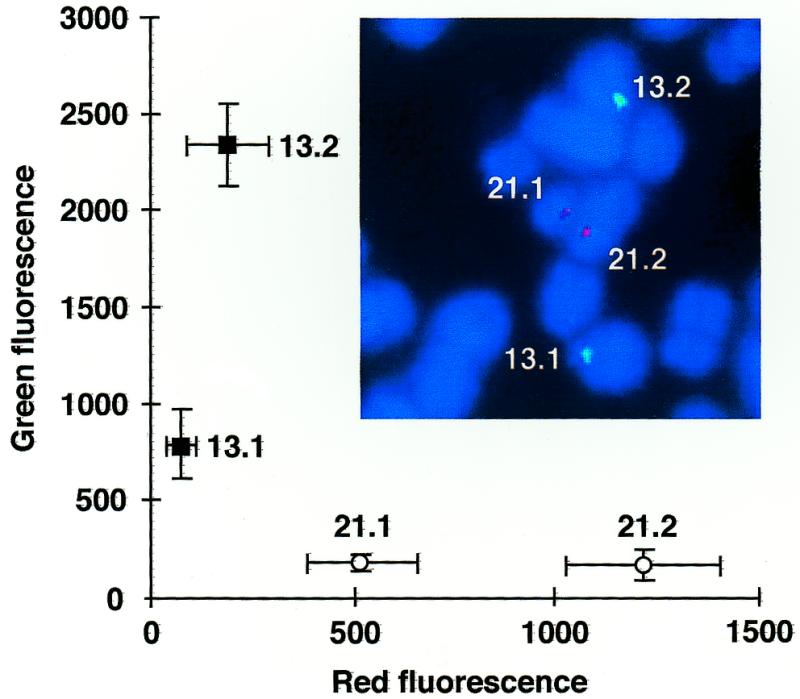
PCR-generated padlock probes identify individual chromosomes 13 and 21. In situ hybridization was performed with cenT and cenC padlock probes labeled with dinitrophenyl-11-dUTP and digoxigenin-11-dUTP, respectively. Probes were visualized with fluorescein-labeled anti-dinitrophenyl Fab fragment (Molecular Probes) and rhodamine-labeled anti-digoxigenin Fab fragment (Boehringer-Mannheim GmbH). The green and red signals were quantitated from 10 metaphase images, where the background was subtracted and the strongest signal was set to 100%. The mean signal intensities are shown with the standard errors of the means.
In order to compare fluorescence intensities from different probes, a chemically synthesized padlock probe specific for an α-satellite sequence present in chromosome 12 (1) was used as an internal fluorescence standard. The fluorescence at the centromeres of chromosomes 12 and 21 was measured in at least 15 metaphase plates from one individual. A ratio was calculated of the mean intensities for that homolog in each pair which yielded the strongest signal. By comparing these normalized fluorescence ratios, the enzymatically synthesized probes were estimated to fluoresce three times brighter than the corresponding chemically synthesized probe (data not shown).
Optimal length of padlock probes
We further investigated how the length of the linking segment of padlock probes influenced the in situ detection of sequences in metaphase chromosomes. Probes with the same target-complementary sequence as one of the probes in the previous experiment, but with linking segments of 50, 100, 150, 200, 400 and 800 nt, were synthesized using one forward primer and varying the position of the reverse primer on the template used in the PCR. The strongest signal was observed using probes with a linking segment of 100 nt and a total length of 140 nt (Fig. 4), while probes with linking segments longer than 200 nt gave no signal. Longer ligation probes resulted in greater background fluorescence.
Figure 4.

Effect of probe length on signal intensity. In situ hybridization was performed with probes of the indicated lengths synthesized by PCR and labeled with digoxigenin-11-dUTP. A chemically synthesized 90mer probe, αchr12, was used as an internal fluorescence standard. The variable length probes and the αchr12 probe were visualized with fluorescein-labeled anti-digoxigenin Fab fragments and with fluorescein-labeled avidin (Vector Laboratories Inc.), respectively. A ratio was calculated between the signal intensities from the most strongly labeled chromosome 21 and 12 homologs in each metaphase. The mean ratio from at least 17 metaphases with the standard error of the mean is shown. The number of T residues indicates the number of positions where a labeled nucleotide can be incorporated during PCR.
Detection of single nucleotide variants in single copy genes
We used probes synthesized according to the present protocol to analyze single copy genes in total human genomic DNA. Ligation reactions were performed in solution in the presence of genomic DNA from individuals homozygous for either the wild-type sequence of the ATP7B gene or for a mutant variant of this gene, differing in a single nucleotide position (C3207A). In homozygous form this mutation causes the copper transport deficiency Wilson’s disease (9). Ligation of probes specific for the mutant or the wild-type sequence was monitored by PCR amplification of the probes across the ligation junction, a method previously used by Zhang et al. for RNA detection (10). Abundant PCR product was only generated in the presence of probes specific for that sequence variant present in the genomic DNA sample. A faint band arising from wild-type genomic DNA probed with a mutant-specific padlock probe is probably due to mismatch ligation (Fig. 5).
Figure 5.
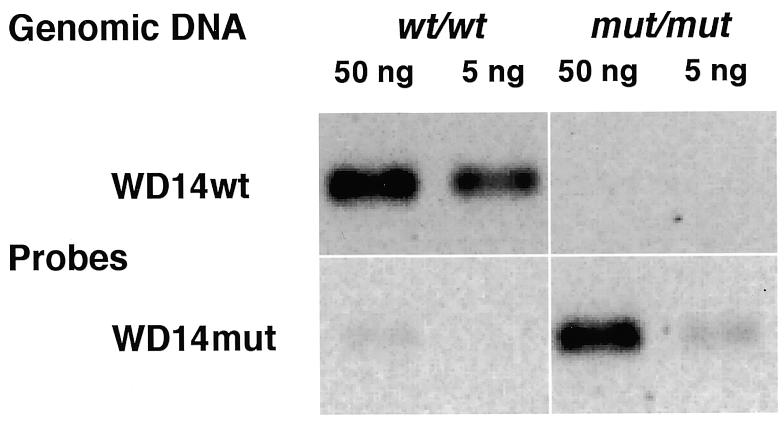
Padlock probes detect single nucleotide variations in genomic DNA. Two padlock probes generated by PCR, WD14wt and WD14mut, were ligated in the presence of genomic DNA from individuals homozygous for the wild-type sequence of the ATP7B gene or for the mutant variant (C3207A) and the amplification products were examined by agarose gel electrophoresis.
DISCUSSION
The padlock probe design places stringent requirements on probe oligonucleotide quality; it is crucial that both ends of the padlock probes are intact for ligation to occur. Among the DNA polymerases tested for probe synthesis, we found that the Pfu DNA polymerase produced probes with high ligation efficiency in the greatest yield. Sixty to seventy percent of PCR-synthesized probes can be circularized (Fig. 2), compared to ~90% of chemically synthesized probes of high quality (for comparison see fig. 3 in Kwiatkowski et al., 7). The appearence of a ligated n – 1 band in Figure 2 is most likely due to internal single nucleotide deletions in the primers used in PCR, whereas the unligated n – 1 band is probably missing a nucleotide at the 5′- or 3′-end of the padlock probe (7). Still, the PCR approach is a valuable complement to chemical synthesis because of the convenience of changing labels as required and the ability to synthesize longer probes than is possible chemically. Moreover, small quantities of probes can be synthesized at a lower cost compared to chemical synthesis.
Compared to chemically synthesized probes with the same target-complementary sequences and of an equal length, enzymatically synthesized probes resulted in a 3-fold greater signal in in situ analysis. This probably reflects denser labeling of the probes produced by PCR. We further found that the staining pattern of different chromosomes was better resolved using enzymatically synthesized probes specific for single nucleotide sequence variants (see Fig. 3) compared to ones generated by chemical synthesis, probably due to the increased signal strength (unpublished results). Moreover, we demonstrate that the possibility of generating longer probes can permit even stronger detection signals, resulting in an increased sensitivity of detection (Fig. 4). The lack of signal from probes with linking segments longer than 200 nt may be due to poor access of longer probes to target sequences in metaphase chromosomes, as well as increased non-specific binding.
Probes generated by PCR were shown herein to permit detection of a single nucleotide variation in both repeated and single copy sequences in total genomic DNA. For enhanced sensitivity of detection, ligated probes were amplified across the ligation junction by PCR, using linking segment-specific primers. We and others have previously reported similar results using chemically synthesized probes (6,11). Since in this procedure a standard pair of primers can be used to amplify ligation products from many different padlock probes, some of the problems of multiplex PCR could be avoided in this manner. Alternatively, increased sensitivity of detection of circularized probes can also be achieved by employing a rolling circle replication mechanism (4,5).
Enzymatic construction of padlock probes should allow production of ligation probes for multiplex detection of large numbers of target sequences. The present protocol is a useful complement to chemical synthesis for small-scale pilot studies, for probe optimization and for sensitive in situ detection.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Francis Barany for kindly providing the Tth DNA ligase used in this study. This work was supported by grants from the Beijer Foundation, the Swedish Medical and Technological Research Councils and the Swedish Cancer Foundation. A.I. and M.N. were supported by EMBO long-term fellowships.
REFERENCES
- 1.Nilsson M., Malmgren,H., Samiotaki,M., Kwiatkowski,M., Chowdhary,B.P. and Landegren,U. (1994) Science, 265, 2085–2088. [DOI] [PubMed] [Google Scholar]
- 2.Landegren U., Kaiser,R., Sanders,J. and Hood,L. (1988) Science, 241, 1077–1080. [DOI] [PubMed] [Google Scholar]
- 3.Nilsson M., Krejci,K., Koch,J., Kwiatkowski,M., Gustavsson,P. and Landegren,U. (1997) Nature Genet., 16, 252–255. [DOI] [PubMed] [Google Scholar]
- 4.Lizardi P.M., Huang,X., Zhu,Z., Bray-Ward,P., Thomas,D.C. and Ward,D.C. (1998) Nature Genet., 19, 225–232. [DOI] [PubMed] [Google Scholar]
- 5.Banér J., Nilsson,M., Mendel-Hartvig,M. and Landegren,U. (1998) Nucleic Acids Res., 22, 5073–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas D.C., Nardone,G.A. and Randall,S.K. (1999) Arch. Pathol. Lab. Med., 123, 1170–1176. [DOI] [PubMed] [Google Scholar]
- 7.Kwiatkowski M., Nilsson,M. and Landegren,U. (1996) Nucleic Acids Res., 24, 4632–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu G. (1993) DNA Cell Biol., 12, 763–770. [DOI] [PubMed] [Google Scholar]
- 9.Tanzi R.E., Petrukhin,K., Chernov,I., Pellequer,J.L., Wasco,W., Ross,B., Romano,D.M., Parano,E., Pavone,L., Brzustowicz,L.M. et al. (1993) Nature Genet., 5, 344–350. [DOI] [PubMed] [Google Scholar]
- 10.Zhang D.Y., Brandwein,M., Hsuih,T.C.H. and Li,H. (1998) Gene, 211, 277–285. [DOI] [PubMed] [Google Scholar]
- 11.Nilsson M. (1998) Acta Universitatis Uppsaliensis, Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine, no. 759. Uppsala University, Uppsala, Sweden.