

ABSTRACT
Thermoregulation and sleep are tightly coordinated, with evidence that impairments in thermoregulation as well as increases in ambient temperature increase the risk of sleep disturbance. As a period of rest and low demand for metabolic resources, sleep functions to support host responses to prior immunological challenges. In addition by priming the innate immune response, sleep prepares the body for injury or infection which might occur the following day. However when sleep is disrupted, this phasic organization between nocturnal sleep and the immune system becomes misaligned, cellular and genomic markers of inflammation are activated, and increases of proinflammatory cytokines shift from the nighttime to the day. Moreover, when sleep disturbance is perpetuated due to thermal factors such as elevated ambient temperature, the beneficial crosstalk between sleep and immune system becomes further imbalanced. Elevations in proinflammatory cytokines have reciprocal effects and induce sleep fragmentation with decreases in sleep efficiency, decreases in deep sleep, and increases in rapid eye movement sleep, further fomenting inflammation and inflammatory disease risk. Under these conditions, sleep disturbance has additional potent effects to decrease adaptive immune response, impair vaccine responses, and increase vulnerability to infectious disease. Behavioral interventions effectively treat insomnia and reverse systemic and cellular inflammation. Further, insomnia treatment redirects the misaligned inflammatory- and adaptive immune transcriptional profiles with the potential to mitigate risk of inflammation-related cardiovascular, neurodegenerative, and mental health diseases, as well as susceptibility to infectious disease.
KEYWORDS: Thermoregulation, ambient temperature, sleep, sleep disturbance, sleep deprivation, sleep duration, insomnia, immunity, innate immunity, adaptive immunity, inflammation, cytokines, inflammatory disease, vaccine responses, infectious disease, stress, insomnia treatment
Introduction
Thermoregulatory mechanisms are fundamental to sleep, with evidence that thermal factors play a role in the evolution of behavioral strategies for preparation for sleep and the selection of optimal sleeping environments across species [1–3]. As extensively reviewed by others [1], sleep in humans and other mammals coincides with decreases in core body temperature and alterations in thermoregulatory effector activity [1]. Animal studies have found that sleep onset is most likely when core temperature is at its steepest rate of decline, which is accompanied by a decrease in brain temperature [1]. In humans, sleep onset is also coupled with the falling circadian phase of core body temperature rhythm [2]. Further, behavioral quiescence of sleep contributes further to sleep-related decrease in core body temperature [3]. Even during sleep, humans regulate skin temperature microclimates, by unconsciously increasing or decreasing their exposed surface area in response to changes in ambient temperature [3–5]. Finally, experimental disruption of thermoregulation causes persistent insomnia in animals [6–8], and human patients with insomnia are found to have higher rectal and oral temperatures over the sleep period [9,10].
Given the compelling association between thermal regulation and sleep, it is further hypothesized that ambient temperature might contribute to sleep onset and maintenance. Indeed, elevations in ambient temperature and/or deviations in self-regulation of body temperature are linked sleep disturbance [4,5,11]. One study of three geographically distinct pre-industrial societies found sleep onset coincides with a reduction in ambient temperature, and the entire sleep period occurs when ambient temperature was declining [12]. Experimental studies have found that a progressive decrease of ambient temperature within the thermal neutral range (i.e. 27.5°C to 29.5°C) induces increases in amounts of slow wave sleep and improves sleep quality [13]; manipulation of body temperature and more effective reduction in body temperature enhances deep sleep [14]; and mild heat exposure with an increase in ambient temperature (i.e. from 26°C to 32°C) results in increased wakefulness [15,16]. Finally, survey data of 765,000 persons in the United States found that increases in nighttime temperatures amplified self-reported nights of insufficient sleep with the largest effects during the summer and among both lower income and elderly participants [17].
Together these findings demonstrate that impairments in thermoregulation coupled with increases in ambient temperature, for example due to the climate crisis, have the potential to increase the risk of sleep disturbance and represent one of several factors including other environmental (e.g. noise), social (e.g. adversity, socioeconomic stress), and psychological (e.g. anxiety) factors that precipitate and perpetuate insomnia. Indeed, insomnia complaints are highly prevalent and represent one of the most common behavioral complaints, occurring in over a third of the population of the United States [18]. Moreover, among older adults and patients with inflammatory disorders such as cardiovascular disease and some types of depression, who together are likely to show impairment in thermal regulation, the prevalence of insomnia complaints and insomnia disorder is even higher [19]. Such difficulties with sleep has important public health implications, because epidemiologic data show that insomnia is a predictor of chronic disease risk including cardiovascular disease and diabetes, as well as disorders of the brain such as depression and dementia [20–24]. Many of these chronic diseases and mental health disorders are associated with increases in inflammation [25,26], and sleep disturbance is thought to lead to an activation of inflammatory mechanisms, which are hypothesized to contribute to this medical burden [27]. Further, sleep disturbance is prospectively associated with infectious disease risk [27–29], suggesting that sleep disturbance might also have a role of the regulation of antiviral immune responses.
The purpose of the present review is to examine the influence of sleep disturbance on inflammatory mechanisms and immune response to viruses and other pathogens. This review first provides background on the assessment of sleep, and the respective components of the immune system including innate immunity (i.e. inflammatory biology dynamics) and adaptive immunity (i.e. antiviral immune responses). The links between normal sleep patterns as measured by sleep continuity and sleep depth and innate- and adaptive cytokine profiles are first characterized. Then, the impact of experimental sleep loss, and naturalistically occurring sleep disturbances, is considered with assessment of inflammatory biology dynamics including systemic and circulating markers of inflammation, cellular processes, and transcriptional signaling and gene expression profiles. Additionally, the influence of experimental sleep deprivation on vaccine responses is evaluated with examination of the associations between sleep difficulties and infectious disease risk. Whereas infectious disease in turn can have reciprocal effects on sleep, possibly by inducing increases in the inflammatory response and in body temperature. These relationships have been previously reviewed and are beyond the scope of the present review [30–32]. Finally, given that sleep is involved in the homeostatic regulation of a number of physiologic systems such as the hypothalamic pituitary adrenal axis (HPA) and sympathetic nervous system (SNS), the neuroeffector mechanisms linking sleep, sleep disturbance, and the immune are discussed.
Sleep characteristics and assessment of sleep disturbance
Sleep characteristics
Sleep is a behavioral state in which the brain is homeostatically involved in the regulation of the immune system and other physiologic systems [27,29,33]. Characterized by closed eyes and changes in body posture (i.e. typically recumbent position), the state of sleep is associated with a reduced sensitivity to external stimuli and an increased arousal threshold. However, in contrast with coma, this change in consciousness is readily reversible. During sleep, the electroencephalogram (EEG) or polysomnography (PSG) can be used to characterize the activity of the brain which shows an arousal continuum from fully awake to deep sleep. Sleep can also be dimensionally evaluated by physiological, circuit, cellular, and genetic levels of analysis [34].
Two processes are coupled in the regulation of sleep, described as the two-process model [35]. The first process is thought to be driven by homeostatic mechanisms that increase sleep propensity following a prolonged period of time without sleep, and together contribute to sleep onset as well as sleep duration and depth. The second process synchronizes these homeostatic mechanisms with the circadian system and serves to modulate the timing of sleep, as aligned with other behaviors and physiological systems along the 24-h sleep wake cycle [36].
PSG) is the gold standard for assessing sleep behavior, with characterization of sleep continuity, sleep architecture, and rapid eye movement sleep; PSG used standardized scoring methods and criteria to characterize these components of sleep [37]. Sleep continuity is defined by several measures including sleep duration (i.e. total sleep time), duration of time from behaviorally attempting sleep (i.e. turning off the light) until the onset of sleep (i.e. sleep onset latency), and continuous maintenance of sleep with measurement of the amount of time awake after sleep onset (i.e. wake after sleep onset) and the ratio of awake time to the time in bed (i.e. sleep efficiency). Sleep architecture is categorically divided into two major phases, non-rapid eye movement (NREM) and REM sleep, based primarily on the characteristics of the EEG, as shown in Figure 1. NREM sleep is further categorized into three sleep stages: N1, N2, and N3. N1 or Stage 1 sleep is the time between wakefulness and sleep, which often includes slowly rolling eye movements. Whereas this N1 sleep is associated with decreased responsivity to external stimuli, sleep onset does not occur until the emergence of N2 or Stage 2 sleep with slowing of the EEG and the emergence of EEG spindles (i.e. waxing and waning waves of 12–15 Hz) and K-complexes (i.e. single, high negative deflections in the EEG). In healthy young adults, N2 comprises about 50% of the total sleep. With further slowing of the EEG and a preponderance of EEG waves of high amplitude (>75 µV) and low frequency (≤4 Hz), N3 or slow wave sleep is defined. In young adults, around 20% of total sleep time is composed of N3, and the relative amounts of N3 decline to less than 10% in older adults [18]. Finally, REM sleep is characterized by a rapid and low voltage EEG, similar to waking brain activity, which is coupled with rapid and random movement of the eyes and suppression of muscle activity, possibly to mitigate action during dreaming which primarily occurs during REM sleep. REM sleep makes up ~20% of total sleep time. The characteristics of REM sleep, typically reported by PSG, include duration of time from sleep onset to the first REM period (i.e. REM latency) and the frequency of rapid eye movements during REM sleep (i.e. REM density). In adults, total sleep time, sleep efficiency, percentage of slow-wave sleep, percentage of REM sleep, and REM latency all significantly decrease with age; in contrast sleep latency, wake after sleep onset, and percentages of N1 and N2 sleep increase with age [38].
Figure 1.
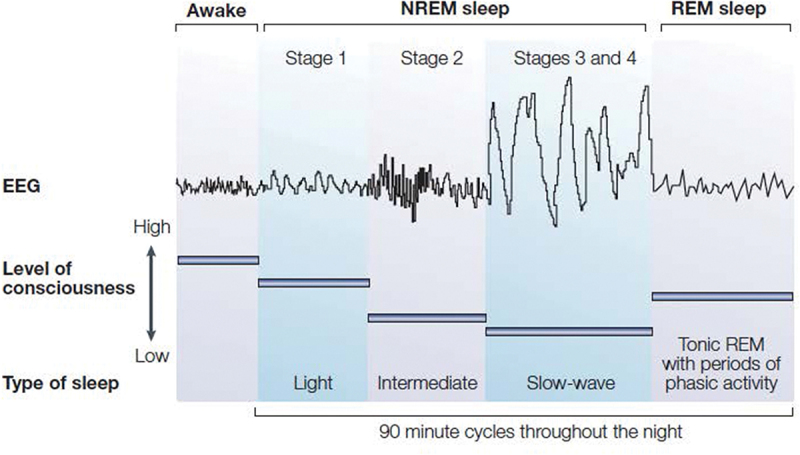
Sleep stages in humans. Electroencephalogram (EEG) patterns during sleep in humans characterize sleep stage which differ in the frequency and amplitude of EEG waves. Stage 1 is denoted by low-amplitude waveforms. Stage 2 shows higher frequency waves or sleep spindles as well as K complexes (not shown). Stages 3 and 4 or slow-wave sleep (SWS) show high-amplitude waves. These three stages also referred to as N1, N2, and N3 sleep comprise non-rapid eye movement (NREM) sleep. Rapid eye movement (REM) sleep is characterized fast, low amplitude, desynchronized brainwaves that resemble the EEG pattern of wakefulness. Figure reprinted from Bryant et al. [28] with permission.
Over the course of the sleep period, humans show a transition from wakefulness to entry into NREM sleep, followed by transition to REM sleep. (Figure 1) At the end of a REM period, a brief arousal or awakening may occur, followed by entry into another NREM sleep period. This cycle from NREM sleep to REM sleep, with each cycle lasting about 80 to 110 minutes, typically repeats four to six times over the course of the night. Additionally, during the early part of the night, sleep architecture reveals that NREM phase is predominated by N3 or SWS, and episodes of REM episodes are short. In contrast, during the latter part of the night, REM episodes are longer and more frequent. Whereas scoring dimensions might suggest that sleep is a quantal behavioral state [37], brain activity shows a continuous progression across the NREM domain from wakefulness to deep sleep. Indeed, spectral analytic methods reveal continuous shifting of mixed EEG frequencies to predominately lower EEG frequencies in the transition from awake to NREM sleep and N3 sleep [39]. Slow-wave activity (SWA), a measure of the density and amplitude of slow waves, is thought to capture the depth of sleep and is markedly increased after prolonged period of wakefulness [40], with potential effects on recovery of immune response following sleep loss [41].
Assessment of sleep and sleep disturbance
As a laboratory-based objective measure of sleep, the PSG is the gold standard for objectively measuring sleep and sleep disturbance with the EEG, electromyogram (EMG), and electrooculogram (EOG) [42]. In addition, the PSG typically includes measurements of heart rate, breathing functions, and blood oxygen saturation, which are important in the screening and identification of sleep apnea and periodic limb movement disorder. Portable PSG devices are now available to allow the recording of sleep in the natural home environment with demonstrated reliability [43].
Despite being the gold standard for sleep assessment, PSG may not adequately characterize sleep occurring at home, or disturbances of sleep that are present over days, weeks, months, or longer. To estimate sleep patterns in a natural sleep setting without the invasiveness of PSG, sleep actigraphy is typically used, which can record sleep-wake activity over multiples days or weeks by employing an activity monitoring device worn on the wrist [42,44]. When coupled with a sleep diary in which the patient reports sleep onset and morning awakening [45], actigraphy estimates sleep patterns including sleep duration, sleep onset latency, and sleep maintenance (i.e. wake after sleep onset, sleep efficiency). As such, actigraphy has a reasonable level of accuracy for estimating sleep continuity indices as compared to PSG [44], although sleep stages and sleep architecture are not reliably differentiated with actigraphy. The long-term recording of sleep-wake activity by the actigraphy captures day-to-day variability in sleep continuity measures, which are increasingly viewed as having impacts on immunity [46].
Beyond these objective measures of sleep including PSG and actigraphy, self-reported sleep quality plays a critical role in the assessment of sleep disturbance and diagnosis of insomnia [42]. Indeed, given that the diagnosis of insomnia disorder relies on patient-reported insomnia complaints and daytime impairment [47], the PSG has not been found to differentiate reliably between those with insomnia disorder vs. those without insomnia [48]. As such, the PSG is not recommended for routine assessment of insomnia [42,49], although it is helpful in the evaluation of other sleep disorders such as sleep apnea which could contribute to patient-reported complaints of poor sleep quality and related daytime impairment [49].
The diagnosis of insomnia disorder as defined by the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-5), relies on subjective complaints of difficulty initiating or maintaining sleep, early awakening, interrupted, or non-restorative sleep and associated impairments in daytime functioning, which must be present at least three nights per week and last for three months or longer [47]. (Table 1) Hence, no objective measure is being used to confirm insomnia diagnosis. Nevertheless, some objective measures of sleep may be helpful in defining more severe phenotypes. For example, insomnia disorder with short sleep duration (less than 5 hours) may have greater adverse consequences for cardiovascular disease risk [50], as well as increases in inflammation, as reviewed below in Section V.
Table 1.
Diagnostic and statistical manual-5th edition: Insomnia Disorder.
| Insomnia Disorder | |
| Diagnostic Criteria | |
| A. | A predominant complaint of dissatisfaction with sleep quantity or quality, associated with one (or more) of the following symptoms: |
|
|
|
|
|
|
| B. | The sleep disturbance causes clinically significant distress or impairment in social, occupational, educational, academic, behavioral, or other important areas of functioning. |
| C. | The sleep difficulty occurs at least 3 nights per week. |
| D. | The sleep difficulty is present for at least 3 months. |
| E. | The sleep difficulty occurs despite adequate opportunity for sleep. |
| F. | The insomnia is not better explained by and does not occur exclusively during the course of another sleep-wake disorder (e.g. narcolepsy, a breathing-related sleep disorder, a circadian rhythm sleep-wake disorder, a parasomnia). |
| G. | The insomnia is not attributable to the physiological effects of a substance (e.g. a drug of abuse, a medication. |
| H. | Coexisting mental disorders and medical conditions do not adequately explain the predominant complaint of insomnia. |
In addition to structured clinical interviews for the diagnosis of insomnia disorder, there is much interest in brief and valid self-report instruments to screen for insomnia complaints and provide an assessment of symptom severity. The Insomnia Severity Index, a 7-item scale, assesses sleep quality, fatigue, psychological symptoms, and quality of life, with high sensitivity and specificity for the detection of insomnia cases [51]. The Pittsburgh Sleep Quality Index, a 19-item self-report questionnaire, evaluates seven clinically derived domains of sleep difficulties (i.e. quality, latency, duration, habitual efficiency, sleep disturbances, use of sleeping medications, and daytime dysfunction), and is used to identify clinically significant sleep impairment [52]. Unfortunately, despite the ease of use of these questionnaires and their validity and reliability in the assessment of insomnia and sleep disturbance, many large-scale epidemiologic studies have relied on single items to assess one or another aspect of sleep or sleep quality, which limits understanding of the influence of sleep on immunity, because there is evidence that various components of sleep such as reported sleep quality, sleep maintenance, and sleep duration have varying and individual contributions to changes in immunity and possibly other physiologic and health outcomes. Furthermore, there appears to be a hierarchical order regarding which sleep items are most associated with daytime consequences, a cardinal component of insomnia disorder; these sleep items follow the following ranking; self-reported dissatisfaction, complaints of nonrestorative sleep, difficulty resuming or maintaining sleep, and difficulty initiating sleep [53]. Additionally, objective EEG data suggest that higher amount of wake after sleep onset (WASO) is associated with greater impairments in daytime functioning [54].
Immune system
The immune system, our body’s defense system, serves to detect and eliminate molecules and cells that display foreign antigens, altered self-antigens, or evidence cellular damage (refer comprehensive textbook) [55]. Distributed throughout the body, immune cells and their function are classified into two interconnected branches, namely, innate immunity and adaptive immunity. Innate immune cells include granulocytes, natural killer (NK) cells, and monocytes with specific types named depending on the tissue, including for example, microglia in the brain and Langerhans cells in the skin. Adaptive immune cells develop in the bone marrow (B lymphocytes or B cells) or in the thymus (T lymphocytes or T cells); these adaptive cells express unique receptors which serve to recognize a specific antigenic peptide.
Innate immune system
As the body’s first line of defense against microbial infection [56], the innate immune system is evolutionally older and is comprised of monocytes, macrophages, and natural killer (NK) cells that traffic among primary (bone marrow, thymus) and secondary (lymph nodes, spleen, mucosa associated lymphatic tissue) lymphoid organs, and circulate via the bloodstream and lymphatic vessels throughout the body. Via surface receptors and cell–to-cell contact, and the release of cytokines, chemokines, and surface molecules, these innate immune cells communicate with other immune and non-immune cells to generate defense responses such as phagocytosis and cytotoxicity.
Innate immune cells including monocytes and macrophages use invariant receptors, which are encoded in the germline and are capable of generating an immunologic response to a wide range of microbial diversity. This recognition strategy, termed pattern recognition [56], facilitates a rapid immune response within minutes to hours which initiates an inflammatory cascade to help contain an infection, and ultimately promote healing and recovery [56]. Among pattern recognition receptors (PRRs), toll-like receptors (TLRs) are found on innate cells such as monocytes that drive inflammation [56] and recognize conserved components of microbes or pathogen-associated molecular patterns (PAMPs), which are expressed by various microbes. An example of a bacterial PAMPs is lipopolysaccharide (LPS), an endotoxin that is the major component of the outer membrane of Gram-negative bacteria [57], which is recognized by TLR4. PRR’s also recognize endogenous danger/damage-associated molecular patterns (DAMPs), which are released by stressed or injured cells; an example is heat-shock proteins (HSPs) [58–60]. Upon recognition of PAMPs or DAMPs, activation of the PRR triggers an acute phase response to drive intracellular processes initiated at the level of the genome to regulate the inflammatory response. For example, PRR binding of PAMPs leads to activation of key intracellular transcription factors such as nuclear factor-κB (NF-κB) and activator protein 1 (AP-1) [61]. In turn, this transcriptional signaling induces transcription of pro-inflammatory immune response genes such as TNF-α and IL-1 and the translation, production, and release of proinflammatory cytokines such as interleukin(IL)-6, tumor necrosis factor (TNF)-α, IL-1, and interferons (IFNs), as well as chemokine such as IL-8. Chemokines signal and recruit other leukocytes to site of challenge whether it is a site of infection or tissue injury.
Low-grade inflammation which is chronic can occur in multiple conditions including cardiovascular disease, diabetes, depression, and aging [19]; the latter instance is denoted as inflammaging [62,63]. Typically, markers of inflammation show only mild elevations above the normal range, with increases in C-reactive protein (CRP), for example, in the moderate (1 to 3 pg/ml) or high (>3 pg/ml) range. Such low-grade inflammation can be further characterized by evaluating circulating levels of IL-6, recognizing that a clinical normative level for IL-6 has not yet been established due in part to the use of different assay methods. In addition to increases in circulating markers of inflammation, there may be changes in circulating number of immune cells with increases in white blood cell counts and neutrophil numbers. Importantly, such inflammatory cytokines does not appear to be driven by typical processes such as infection, but is chronic and persistent and often associated with demographic factors (i.e. older age, nonwhite ethnicity, low socioeconomic status) and health and lifestyle variables (i.e. high body mass index, sedentary activity) [64,65]. As will be reviewed below in Section IV, sleep disturbance and short sleep duration also lead to chronic low-grade inflammation, similar to the effects found with being overweight or sedentary activity.
Adaptive immunity
If the innate immune system does not effectively contain a microbial or viral infection, the adaptive immune response emerges over several days [55]. This adaptive response, initiated in secondary lymphatic tissues, involves the differentiation and proliferation of microbial-specific white blood cells such as T- and B-lymphocytes to provide tailored, specific responses which are based on an immunological memory of having responded to a specific pathogen or antigen in the past. As such, this adaptive response follows a set of sequences and coordinated steps [66].
First, antigen-presenting cells (APCs) such as macrophages cells are recruited to the site of infectious challenge, take up invading antigen, and then migrate to secondary lymphoid organs like the lymph nodes [66]. Within the lymph node, APCs present the antigenic peptide along with the major histocompatibility complex (MHC) II (peptide-MCHII, pMHCII) to naive T-helper (Th) or CD4 T cells. In turn, these antigen-specific CD4 Th cells proliferate and differentiate into effector CD4 T-helper (Th) cells which are specialized to respond with antigen specificity, including such cell types as Th1, Th2, and Th17 cells. In coordination with macrophages, CD8 T cells, and B cells, these specialized Th cell help eliminate the pathogen. For example, CD8 T cells can recognize and kill virus-infected cells by releasing cytotoxins such as perforin, granzyme, and granulosin, whereas activated B cells mature into plasma cells and produce antibodies that bind and neutralize soluble antigens, or signal cells expressing those specific antigens for elimination. Once complete, these antigen-specific Th cells, as well as cytotoxic T cells and B cells, survive, providing immunological memory to respond more rapidly whenever that specific infectious challenge recurs.
This multi-cell response shows a close interplay between the innate and adaptive immune system, and this response is tightly regulated by stimulatory signals (e.g. proinflammatory cytokines), and inhibitory signals (e.g. anti-inflammatory cytokines). Moreover, in the case of an intracellular pathogen such as a virus, transcription factors such as interferon regulatory factors are activated, which induce antiviral immune response genes such as type I interferon leading to the translation of interferon (IFN) and activation of Signal Transducer and Activator of Transcription (STAT)-1 and further production of proinflammatory cytokines [67]. Regulation of this response is critical, thereby averting an inadequate immune response or immune deficiency or one that is too robust resulting in host damage such as autoimmunity or septic shock.
Coordinated engagement of innate immunity along with an adaptive immune response provides a comprehensive, and possibly more robust immunologic response, in which the release, for example, of inflammatory cytokines, IL-1, IL-6, and TNF-α promotes and accelerates the differentiation of T lymphocytes into cytotoxic T cells, leading to greater numbers of these killer lymphocytes. Furthermore, these inflammatory cytokines facilitate redistribution and migration of immune cells from the blood vessels into tissues, via the effects of inflammatory cytokines on vascular permeability, cellular adhesion, and release of chemokines that recruit immune cells to the site of inflammatory activity [66].
Normal noctural sleep and immunity
One function of sleep is to support host defense. Even before the onset of sleep, circadian factors induce an increase in levels of IL-6, which are thought to prepare the immune system for exposure to pathogens during the night, recognizing from an evolutionary perspective that until recently humans slept in groups, typically in confined areas. Indeed, it is speculated that the quiescent period of sleep with its low metabolic demand is teleologically timed to support an effective immune response; responding to immunological challenges has high energy demands. The restorative function of sleep is supported by additional observations as previously reviewed [27] including evidence that sleep helps the body recover from infectious diseases. Moreover, there is a bidirectional relationship between sleep and immune system, and cytokines triggered by an infectious challenge as well as antimicrobial peptides are known to enhance sleep. Additionally, after a night of sleep loss, recovery sleep has a restorative function on the immune system, as many of the alterations in immune cell numbers and functions return to normative levels following recovery sleep.
In addition to the restorative function of sleep, it is also plausible that sleep has a preparatory function. Allostatic theories of physiology have long proposed that prepared physiological systems have substantial survival advantage by anticipating challenges and thereby regulating function to be responsive to challenges [68]. Hence in a recent review on sleep and immune system circuitry, which posited a neurally integrated view of the immune system, the CNS is hypothesized to use the behavioral and metabolic quiescent period of sleep to control both innate and antiviral immune response to be “forward looking” in anticipation of threat, potential injury, and exposure to infectious challenge that will follow wakening [27]. Evidence that nocturnal sleep is homeostatically aligned with preparatory activation of innate immunity and adaptive immune responses is discussed below, as typified by sleep-induced increases in inflammatory activity during the latter part of the night.
In addition to its roles in supporting homeostatic recovery and in initiating a preparatory response of the immune system, sleep is a psychophysiological process, which dynamically regulates two major neuroeffector systems including the HPA and the SNS [28], and both of these effector pathways have potent influences on the innate and adaptive immune system, which modulate and steer balance between inflammatory and anti-viral transcriptional profiles [69], as discussed in Section VII. CNS regulation of sleep is also influenced by circadian oscillators [70–72], and these mechanisms also contribute to the regulation of immunity [73,74]. Hence, to understand the relative influence of sleep as opposed to circadian processes on the immune measures, research has sought to identify aspects of immunity which are regulated primarily by nocturnal sleep, and other immune measures that are driven by circadian mechanism [74,75]. By profiling immune cell numbers and activity with repeated measures across a 24-h sleep-wake cycle vs. a 24-h period of continuous wakefulness, research has provided inferential evidence that divergent profiles between these two conditions identify changes in immune measures that are sleep-dependent; whereas parallel patterns in both conditions suggest that circadian factors are relatively more salient in dynamic variation of immunity [74,75]. The respective influence of sleep and the circadian system on neuroendocrine hormones, HPA axis activity, SNS activity, and circulating profiles of pro- and anti-inflammatory, as reviewed below, is schematically depicted in Figure 2 [74].
Figure 2.
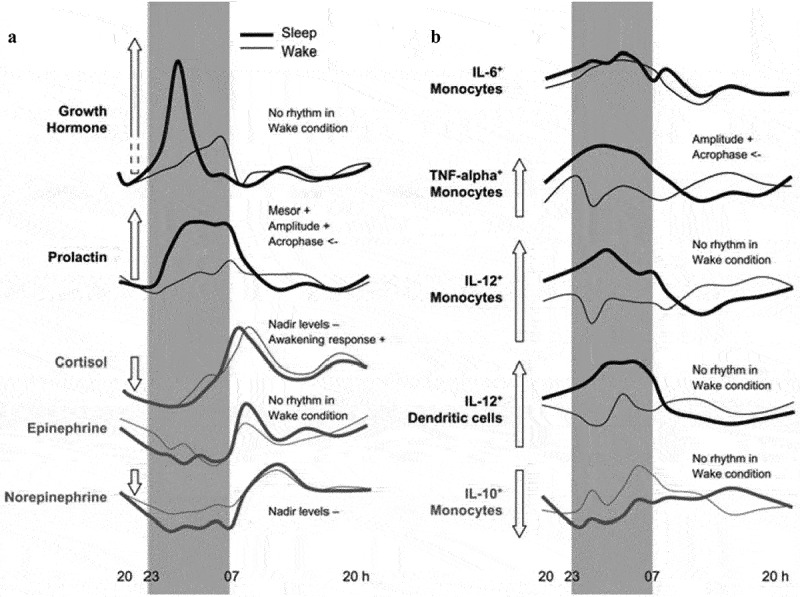
Effects of sleep and the circadian system on (a) neuroendocrine mediators and (b) cytokine production by monocytes cells. Levels of growth hormone (GH), prolactin, cortisol, epinephrine, and norepinephrine, as well as percentages (of total monocytes cells, respectively) of IL-6+, TNF-alpha+,IL-12+, and IL-10+ monocytes and IL-12+ dendritic cells over 24 h during conditions of sleep (thick dark line) and continuous wakefulness (light line). Maximum difference between Sleep and Wake during the first night-half, referenced Mesor concentrations, is schematically depicted in the length of the arrow. Figure reprinted from Lange et al. [74] with permission.
Sleep and the nocturnal profile of innate immunity
Normal sleep is associated with increases in markers of innate immunity such as increases in proinflammatory cytokines. However, as discussed in Section V, there is a seeming paradox; sleep loss also leads to increases in markers of inflammation. To discuss these divergent effects in context, the following section describes how circadian- and sleep-dependent processes coordinate an increase in a number of measures of innate immunity during the night. In short, there a coupling of innate immune response and sleep. In contrast, when sleep is disrupted and there is a loss of sleep, this phasic organization is misaligned; increases of proinflammatory cytokines shift from the nighttime to the day. If sleep disturbance becomes chronic, elevated levels of markers of inflammation such as increases in CRP then become pervasive and are found during both nocturnal and daytime periods. In the following section, the relationships between normal nocturnal sleep and nocturnal immunity are reviewed. Then in Section V, there is consideration of the effects of sleep disturbance and immune responses; such responses have been primarily characterized over the day after acute sleep loss or during chronic sleep disturbance such as insomnia.
It is proposed that sleep induces increases in innate immunity that support host immune response to prior immunological challenges, and also increase proinflammatory cytokine activity in the latter part of the night to prepare the body for pathogen-related infection which might occur during the day following injury or infectious exposure [27,68,76]. Such a preparatory host response primes the immune system, which facilitates a rapid and more robust response to social-environmental or infectious threats. Sleep and its priming of the innate response is characterized by the following sleep-dependent changes: increases in NK activity, increases in circulating levels of the proinflammatory cytokine IL-6 and soluble IL-6 receptor (sIL-6 R), and increases in stimulated production of TNFα. (Figure 2)
One component of the innate immune system regulated by sleep is natural killer (NK) cell activity, which was typically measured by the cytotoxic killing of immortalized myelogenous leukemia cell line, K562, in vitro. NK cells perform cytotoxic clearing of virally infected cells in a natural manner [77]. Over the 24-hour period, counts of NK cells and NK activity show a circadian profile with minimum levels during the early part of the night, and maximal levels during the late morning [78]. However, amount and depth of sleep contribute to the late morning, nocturnal rise of NK cell activity. In persons with normal sleep patterns as indexed by PSG, a robust nocturnal increase in NK activity is found, whereas in persons who show disturbances of sleep with decreases in sleep maintenance, the nocturnal increases in NK cell activity are significantly attenuated [79,80]. Distribution of other innate immune cells, such as monocytes, across of the 24-h period showed a circadian profile that is not linked to sleep.
Inflammation can be measured by circulating levels of inflammatory markers, such as CRP, IL-6, and TNFα; whole blood in vitro stimulated production; or ex vivo intracellular expression of proinflammatory cytokines at rest or following LPS stimulated activation of TLR-4 receptors of monocytes (i.e. CD14 cells) at the single cell level. As noted above, studies have temporally evaluated these markers of inflammation over 24-hour period of normal sleep wake activity as compared to 24 hours of wakefulness. Divergent profiles between these two conditions reveal that sleep, as opposed to circadian influences, have effects on circulating levels of IL-681, as well as effects on the stimulated monocyte production of other inflammatory markers (i.e. TNFα), anti-inflammatory cytokines (i.e. IL-10), and cytokines important in the adaptive immune responses as discussed below (i.e. IL-12) [74].(Figure 2)
One of the most widely used markers of inflammation is CRP, an acute phase protein. Although IL-6 stimulates the production of CRP, concentrations of CRP are relatively stable over the 24-hour period [64], despite within day as well as day-to-day variations in circulating levels of IL-6 [81]. For example, circulating levels of CRP do not show a circadian variation, nor is there an effect of sleep on CRP [82]. Hence, elevated levels of CRP reflect ongoing and sustained chronic or low-grade inflammation.
In regards to IL-6, circulating concentrations show peaks at 7 p.m. and again at 5 a.m [83]. Importantly, the nocturnal increase in circulating IL-6 appears to be sleep-dependent. During a night of early sleep deprivation with wake time between 11 p.m. and 3 a.m., the nocturnal increase of IL-6 is delayed until sleep onset [84]. Moreover, the nocturnal increase of IL-6 is diminished by about 50% during a night of total sleep deprivation [83]. Yet, circadian factors also contribute to nocturnal increases of IL-6; even in the absence of sleep with early night sleep deprivation, there is a transient peak in IL-6 at 1 a.m. [84].. Despite sleep-dependent increases in IL-6 levels, the unstimulated expression of IL-6 in monocytes is not linked to sleep, and increase in the proportion of unstimulated monocytes expressing IL-6 occurs similarly during nocturnal sleep and wake [82]. However, Redwine et al. have found that stimulated production of IL-6 was greater in the late part of the night along with relative increases in REM sleep amounts [84], suggesting that sleep might heighten the preparation of immune cells to respond to challenge.
To activate cells such as neural tissues that do not express the IL-6 receptor, IL-6 must be bound to the trans-signaling molecule, namely, soluble IL-6 receptor (sIL-6 R) [85]. Again, sleep plays a robust role in the regulation of this inflammatory measure, which is signaling and preparing the CNS to respond to infectious challenge. During nocturnal sleep, there is an over 70% increase in levels of sIL-6 R82. Moreover, activation of this signaling molecule is most pronounced during the late part of the night, similar to the preparatory activation aligned with increases in IL-6 levels and stimulated production of IL-6.
Finally, when monocytes are examined at the cellular level, sleep is associated with an increase in whole blood in vitro stimulated production of TNFα [86], a measure of the ability of monocytes to respond to an infectious challenge. Interestingly, sleep does not influence circulating levels of TNFα, soluble receptor (sTNF-R), or resting or unstimulated levels of monocyte expression of TNFα [75], although other studies from the same investigative team have found that sleep reduces circulating levels of TNFα [86] and its production in supernatants of mixed peripheral blood mononuclear cell [87].
Sleep stages, especially REM sleep, appear to regulate dynamic changes in IL-6. Higher levels of IL-6 are found during REM sleep, but not SWS, as compared to awake [84], and IL-6 and IL-6 R increase during the late part of the night, along with increases in REM sleep amounts [82,84]. Finally, amounts of REM sleep correlate with morning levels of whole blood in vitro stimulated production of IL-679. The mechanisms that account for the increase in production of IL-6 associated with REM sleep are not known, but REM sleep co-occurs with increases in SNS activity and release of sympathetic neurotransmitters [88]; such adrenergic activation steers the transcriptional toward inflammatory gene expression [69,89], possibly accounting for the increases in proinflammatory cytokines during the REM sleep and the late part of the night. However, as discussed further below, SNS activity can also have suppressive effects on inflammatory cytokines [90–92].
In sum, consistent with the hypothesis that sleep functions to prepare the host defense response, sleep and especially REM sleep during the late part of the night is linked to increases in NK cell activity, increases in circulating levels of IL-6, and increases in stimulated production of IL-6, and increases monocyte expression of TNFα.
Nocturnal profile of adaptive immunity
As a period of rest, quiescence, and low demand, metabolic resources are available during sleep to support adaptive immune response [27,68,76]. Indeed, sleep is associated with the release of cytokines that facilitate an antiviral immune response, and a shift in immune defenses to coordinate clearance of intracellular challenges, and these responses are synchronized with circadian processes which regulate the distribution of immune cells [74,93–95].
First, in regards to immune cell numbers, circadian factors appear to drive changes in the distribution of immune cells from extravascular spaces into the circulation independent of sleep. During the evening and early night, numbers of leukocytes, granulocytes, monocytes as well as the major lymphocyte subsets, including T-helper cells (CD4+), cytotoxic T cells (CD8+), activated T cells (HLA-DR+) and B cells (CD19+), reach a maximum, which is then followed by a decline throughout the remaining night with a minimum occurring in the hours after awakening [75]. This movement of antigen presenting cells and T cells from the circulation into lymphoid tissue, for example, is thought facilitate APC transfer of antigenic information to T cells [75,96–101]. Cortisol appears to be important in mobilizing this distribution of T cells, as the 24-hour circadian rhythm of cortisol is coupled to changes in the number of T cells in the circulation; in other words, with increasing levels of cortisol, T cell numbers in the blood decrease. Subsequent to the peak in cortisol in the beginning of the wake period, there is a 3-hour lagged decrease in blood T-cell number [74].
The signals that drive nocturnal priming of the inflammatory response, which then support the initiation of the adaptive immune response are not known. However, during the wake period, “danger signals” like reactive oxygen species, nucleotides (e.g. adenosine triphosphate), and heat shock proteins (HSP) are thought to accumulate [60]. These signals can act like classical immunological stimulants leading to activation of the inflammatory response, with priming of the adaptive immune response [93]. Another mechanism focuses on sleep induction of GH and prolactin release during the early night, which act to enhance the proliferation and differentiation of T cells as well as to promote Type 1 cytokine activity [102]. In support of this possibility, and the coordinated interplay between inflammatory and anti-viral immune responses, the release of pro-inflammatory cytokines has been observed to peak during the rest period of sleep, and especially SWS-dominated portion of sleep, in humans as well as in animals, with increases in inflammation detected by mRNA and protein levels in various tissues such as lymph nodes, adipose tissue, and CNS [93].
Sleep disturbance and inflammation
After a night of sleep loss followed by a night of good sleep, common experience tells us that sleep plays a critical role in maintaining mood and cognitive acuity during the day [103,104], with evidence that sleep promotes physiological resilience [76]. We and others have hypothesized that sleep functions to maintain brain and physiological homeostasis [19,76] and, as reviewed above, to support immunity and to prepare the body to respond to possible injury and pathogen exposure during the day. However, when sleep is disrupted, the phasic organization of sleep and immune responses becomes misaligned, and the nocturnal increase of markers of inflammation that occurs during normal sleep shifts leading to morning increase in proinflammatory cytokines. If sleep loss becomes chronic, systemic markers of inflammation such as CRP increase [105], which temporally coincide with impairments in multiple components of cognitive and physiological resilience.
It is not uncommon for sleep to be disturbed with insomnia complaints reported in at least 30% of the population, with even higher rates in older adults [18,106]. As reviewed in the Introduction, thermal factors including elevations in ambient temperature as associated with sleep disturbance and insomnia. Additionally, sleep can be impacted by signal of social adversity. For example, when threat is perceived by the CNS, the normal profile of sleep is likely be disrupted leading to disturbance of sleep continuity such as reduced total sleep time, poor sleep efficiency, and increases in wake after sleep onset. Threats in the current social environment occur following exposure to either acute- or chronic adversity, and disruption of sleep is one behavioral response to such threat [107–110].
Perceived threat also activates physiological responses including activation of the HPA axis, as well as the SNS, which have downstream effects on the immune system leading to increases in markers of inflammation, as well as decreases in antiviral immune responses [30,69,111]. Sleep appears to have a critical role in mediating and transducing the effects of perceived threat on inflammatory responses. Even in the absence of perceived threat, disturbance of sleep (i.e. experimental manipulation of sleep duration), there is an increase in sympathetic outflow [88,112] which is thought to contribute to increases in daytime levels of proinflammatory cytokines including a temporal shift of peak levels of inflammatory cytokines such as IL-6 from the night into the day, as well as overall accumulated increase in levels of inflammatory markers [83].
Experimental sleep loss and inflammation
To test whether sleep disturbance is causing increases in markers inflammation, rather simply being a correlate of inflammation, experimental studies have manipulated sleep duration in humans and assessed daytime levels of systemic, cellular, and genomic markers of inflammation. However, findings are mixed. (Table 2) Indeed, our recent meta-analyses systematically examined whether experimental sleep deprivation altered circulating levels of proinflammatory markers including CRP, IL-6, and TNFα, and found no overall effects of sleep deprivation on any of these markers [105]. Yet, there was heterogeneity in these results related to the subject sample; differences in experimental strategy such as partial night- or total night sleep deprivation; duration of partial (i.e. sleep restriction) or total sleep deprivation over several nights; and various inflammatory outcomes. (Table 2) Hence, the following provides a qualitative evaluation of several studies and highlights differences which might account for various findings.
Table 2.
Studies linking sleep deprivation to daytime levels of markers of inflammation in humans, as modified from [27].
| Type of sleep deprivation |
Findings |
References |
| Systemic markers of inflammation | ||
| Circadian misalignment over 25 days | Increased levels of TNF and CRP, and of the anti-inflammatory cytokine IL-10 | [73] |
| TSD, 88 hours | Increased levels of TNFR1 at 44 and 66 hours Increased levels of IL-6 at 88 hours No change in TNFR2, sIL-2 R or IL-10 levels Progressive increase in CRP level over 88 hours |
[122,125] |
| TSD, 40 hours | Increased levels of IL-1 and IL-2 Increased level of IL-6 Increased levels of E-selectin, ICAM1, IL-1 and IL-1ra Decreased levels of CRP and IL-6 Increased levels of IL-6 and ICAM1 after a night of recovery sleep No change in the level of IL-6 |
[83,113–115,130,131] |
| TSD, 34 hours | Increased level of TNF, but not of TNFR2, IL-6 or CRP | [129] |
| PSD, over 12 nights | Increased levels of IL-6 and CRP, but not of sTNFR1 | [126] |
| PSD, over 10 nights | Increased level of CRP at day 10 | [125] |
| PSD, over 7 nights | Increased level of IL-6 in both sexes, and increased level of TNF in men, but not women | [127] |
| PSD, over 5 nights | Increased level of CRP after sleep deprivation and recovery sleep | [125] |
| PSD, over 2 nights | No change in IL-6 level | [119] |
| PSD, 1 night | Increased levels of IL-6 and TNF in alcoholics with sleep disturbance, but not controlsIncreased level of L-6 No change in level of CRP No change in level of IL-6 |
[41,84,118,121] |
| Sleep fragmentation, over 2 nights | No change in level of CRP or IL-6 | [120] |
| Cellular inflammation | ||
| PSD, 1 night | Increase in both TLR4-stimulated and resting production of IL-6 and TNF by monocytes Prolonged daytime increase in TLR4-stimulated production of IL-6 and TNF by monocytes in women, but not men No change in TLR4-stimulated production of IL-6 and TNF by monocytes in older adults >60 years of age |
[116,117,132] |
| Transcriptional measures of inflammation | ||
| PSD, over 5 nights | Increased mRNA expression levels of IL-1, IL-6 and IL-17 | [128] |
| PSD, 1 night | Increased mRNA expression levels of IL-6 and TNF | [123] |
| Inflammatory signaling and transcriptional profiles | ||
| PSD, 1 night | Increased inflammatory gene expression profiles owing to activation of AP-1 and NF-κB pathways | [123] |
| PSD, 1 night | Increased activation of NF-κB in women, but not in men Increased constitutive monocytic expression of activated STAT1 and STAT5 in night following sleep loss |
[116,133] |
AP-1, activator protein-1; CRP, C-reactive protein; ICAM1, intercellular adhesion molecule 1; IL-1ra, IL-1 receptor antagonist; NF-κB, nuclear factor-κB; PSD, partial sleep deprivation; sIL-2 R, soluble IL-2 receptor; STAT, signal transducer and activator of transcription; TLR4, Toll-like receptor 4; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; TSD, total sleep deprivation.
First, consider modest experimental sleep loss, which mimics the kind of sleep loss that is ubiquitous in society, in which a person experiences short sleep duration (i.e. demands of work schedule) or disturbance in sleep maintenance for one or two nights (i.e. early child care). In studies of normal adults with normal sleep patterns (i.e. sleep period typically from 11 p.m. to 7 a.m.), experimental restriction of the sleep period (i.e. partial night sleep deprivation) or experimental fragmentation of sleep (i.e. forced awakenings for only one or two nights) circulating levels of inflammatory markers did not change [118–120]. Furthermore, if sleep restriction was imposed along with opportunity for intervening daytime naps [121,122], daytime levels of inflammatory cytokines were similar to the control condition. Yet, when there is evidence of an underlying sleep disturbance, sleep deprivation can trigger an increase in circulating levels of cytokines suggesting that sleep disturbance primes the immune system to be more sensitive to challenges. For example, in a population who report chronic sleep disturbance, a single night of sleep loss triggered a greater increase in the circulating levels of IL-6 and TNFα, as compared to responses in comparison controls who reported no sleep problems, consistent with the notion that sleep disturbance primes the inflammatory response to sleep loss [41].
Given that chronic sleep disturbance, as noted below, is associated with increases in IL-6 and possibly TNFα, the reasons for these null findings for the effects of sleep deprivation in on circulating markers of inflammation in healthy volunteers are not known. However, it is possible that the duration of time from exposure to sleep loss to assessment of inflammatory cytokines may be too short for translation of protein to occur; for example, even though a single partial night sleep deprivation does not increase proinflammatory cytokines, partial night sleep deprivation has been found to induce increases in mRNA levels of IL-6 and TNFα [123]. Moreover, other aspects of the innate immune system, namely, NK cell activity as measured in vitro by cytotoxic killing of the target cell K562, which are not dependent on translational expression of cytokines, are highly responsive to sleep deprivation. Indeed, partial night sleep deprivation, either early- or late night, leads to robust reductions in morning levels NK activity [80,124], as well as sensitivity of NK cells to IL-2 activation [80], findings which parallel the suppressive effects of sleep deprivation on adaptive immune responses.
Second, if partial night sleep deprivation is repeated for several nights, there appears to be an increase in circulating levels of inflammation. For example, sleep restriction over 10 nights induces robust increases in circulating levels of CRP [125] and in IL-6 [126]. Moreover, even shorter periods of sleep restriction lasting seven nights have been found to induce increases in plasma concentrations of IL-6 in males and females and increases of TNFα in males only [127]. Likewise, five nights of sleep restriction induce increases in inflammatory transcripts of IL-1β, IL-6, and IL-17 [128] with evidence that such increases persist even after a night of recovery sleep [128].
Finally, total night sleep deprivation has also been found to induce increases in circulating markers of inflammation which are dependent on the duration of repeated exposure. With evaluation of circulating levels of CRP following one to four nights of sleep loss, cumulative increases of CRP occur with progressive increases after each repeated exposure to sleep deprivation [125]. Other studies have found that exposure to only one to two nights of sleep loss is sufficient to induce increases of TNFα [129], although at least four nights of sleep loss is needed to observe elevations in circulating levels of IL-6 [122]. (Table 2) Other markers of inflammation may be more sensitive to the effects of sleep loss although research is limited. Nevertheless, a single night of sleep loss is adequate to produce increases in the expression vascular endothelial markers (i.e. E-selectin, sICAM-1) [130,131].
Increases in circulating levels of inflammatory cytokines such as IL-6 may arise from immune- and nonimmune sources (e.g. adipose tissue). Hence, further research has examined upstream sources for hypothesized inflammatory activation in response to sleep loss. Indeed, changes in stimulated cellular production of inflammatory cytokines and activation of inflammatory transcriptional pathways occur early in the inflammatory cascade, and alteration of these mechanisms appears to be robust and sensitive signals of inflammatory activation following sleep loss [123]. Given that cellular production of IL-6 and TNFα is thought to be due in part to aberrant increases in the sensitivity of Toll-like receptor (TLR) activity following stimulation with LPS, we have examined whether sleep loss alters ex vivo stimulated intracellular monocytes (i.e. CD14 cell) production of IL-6 and TNFα. Using the experimental strategy, partial night sleep deprivation, we have found that TLR-4 stimulated monocyte production of IL-6 and TNFα is markely increased after a single exposure to early night partial sleep deprivation (Figure 3), similar to the effects of this modest sleep loss to increase mRNA levels of IL-6 and TNFα [123]. However, Besedovsky et al. have suggested that this increase in the cellular production of inflammatory cytokines might reflect a rebound effect of sleep recovery [29]; the authors reason that nocturnal sleep is also associated with increases in stimulated monocyte production of TNFα[86]; hence, allowing participants to sleep in the second half of the night prior to blood sampling might lead to a rebound increase in cellular production of cytokines. However, this conclusion is not supported by other observations. The increase in monocyte stimulated production of IL-6 and TNFα following partial sleep deprivation is consistent with observations for circulating inflammatory markers during chronic sleep disturbance (i.e. insomnia) or total night sleep deprivation, namely, that sleep deprivation can also lead to daytime elevations of inflammation, but inducing a temporal shift in circulating levels of IL6 in which the nocturnal peak of IL-6 that normally occurs at 5 a.m. shifts to occur during the day with overall increases in IL-663. It is further unlikely that the morning increase in TLR-4 monocyte production of IL-6 and TNFα is simply a rebound phenomenon. When the daytime profile of stimulated monocyte production of proinflammatory is examined by repeated measures, sleep deprivation leads to activation of the TLR-4 responses which persist throughout the day in females but not males [132]. Finally, we have found that experimentally induced sleep fragmentation (i.e. forced awakening) increases TLR-4 monocyte production of IL-6 and TNFα, similar to partial night sleep deprivation, and this effect is mediated by a selective loss of slow wave sleep (Irwin et al., in review). Together, these data provide converging evidence that modest sleep loss, even during the early part of the night, induces inflammatory activation of cellular monocyte responses.
Figure 3.
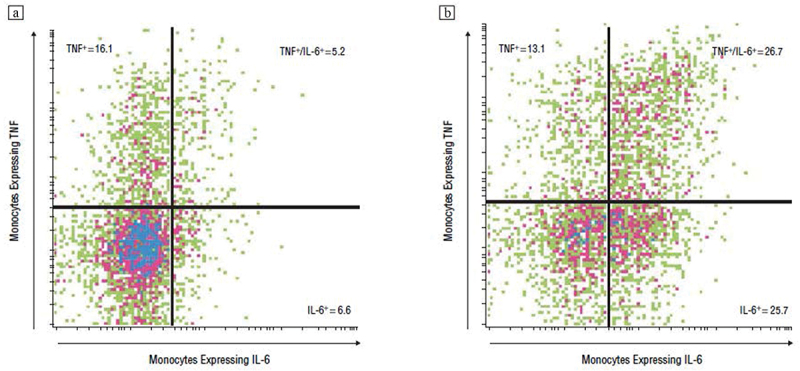
Representative expression of interleukin (IL) 6 and tumor necrosis factor (TNF) α in lipopolysaccharide-stimulated CD14+ cells from a participant (a) at baseline and (b) at partial sleep deprivation (PSD). Numbers indicate the percentages of the fraction of CD14+ cells that are positive for TNF- α alone (upper left), TNF- α and IL-6 (upper right), and IL-6 alone (lower right). In the baseline condition, 72.1% of the CD14+ cells are negative for both IL-6 and TNF-α whereas only 34.5% of the CD14+ cells are negative for both IL-6 and TNF- α in the PSD condition. Reprinted from Irwin et al. [123] with permission.
Examination of transcriptional profiles provides further support for the notion that modest amount of sleep loss activates inflammatory signaling. For example, early night sleep deprivation induces an activation of nuclear factor (NF)-κB, the key transcription control pathway in the inflammatory signaling cascade [133]; nuclear extracts were isolated from peripheral blood mononuclear cells. Similar to the TLR-4-induced activity of monocytes, females show a heightened sensitivity to sleep, and evidence greater increases in NF-κB following sleep deprivation as compared to responses in males [133]. Finally, transcriptome dynamics are substantially influenced by sleep deprivation with evidence of an up-regulation of a gene ensemble that includes the master circadian regulator, several “immediate early genes” marking cellular signal transduction, and multiple inflammatory response genes [123]. (Table 3) For example, among the transcription factor-binding motifs that were over-represented in response to sleep loss were promoters of genes important in metabolism (i.e. cAMP/PKA-induced transcription factors of the CREB/ATF family), as well as inflammation (i.e. PKC-induced AP-1 family, the pro-inflammatory NF-κB/Rel family, and the MAP kinase-inducible ETS transcription factor family typified by ELK1). (Table 3)
Table 3.
Gene transcripts induced by sleep deprivation, as modified from [123].
| Probe Name | Gene symbol | Gene common name | Significant increase(% of participants) |
|---|---|---|---|
| 202644_s_at | TNFAIP3 | tumor necrosis factor,alpha-induced protein 3 | 80 |
| 205067_at | IL1B | interleukin 1, beta | 80 |
| 202014_at | PPP1R15A | protein phosphatase 1, regulatory (inhibitor) subunit 15A | 60 |
| 210042_s_at | CTSZ | cathepsin Z | 60 |
| 204373_s_at | CAP350 | centrosome-associated protein 350 | 60 |
| 217022_s_at | IGHA2 /// MGC27165 | immunoglobulin heavy constant alpha 2 (A2m marker) /// hypothetical protein MGC27165 | 60 |
| 37028_at | PPP1R15A | protein phosphatase 1, regulatory (inhibitor) subunit 15A | 60 |
| 204440_at | CD83 | CD83 antigen (activated B lymphocytes, immunoglobulin superfamily) | 60 |
| 204285_s_at | PMAIP1 | phorbol-12-myristate-13-acetate-induced protein 1 | 60 |
| 201110_s_at | THBS1 | thrombospondin 1 | 60 |
| 205114_s_at | CCL3 /// CCL3L1 /// MGC12815 | chemokine (C-C motif) ligand 3 /// chemokine (C-C motif) ligand 3-like 1 /// chemokine (C-C motif) ligand 3-like, centromeric | 60 |
| 202393_s_at | KLF10 | Kruppel-like factor 10 | 60 |
| 201631_s_at | IER3 | immediate early response 3 | 60 |
| 202859_x_at | IL8 | interleukin 8 | 60 |
| 202887_s_at | DDIT4 | DNA-damage-inducible transcript 4 | 60 |
| 212099_at | RHOB | ras homolog gene family, member B | 60 |
| 202768_at | FOSB | FBJ murine osteosarcoma viral oncogene homolog B | 60 |
| 212830_at | EGFL5 | EGF-like-domain, multiple 5 | 60 |
| 38037_at | HBEGF | heparin-binding EGF-like growth factor | 60 |
| 211506_s_at | IL8 | interleukin 8 | 60 |
| 201044_x_at | DUSP1 | dual specificity phosphatase 1 | 60 |
| 202861_at | PER1 | period homolog 1 (Drosophila) | 60 |
In sum, experimental sleep loss has mixed effects on circulating levels of inflammatory outcomes. Yet, when upstream sources of inflammation are examined, it is evident that even a modest exposure to partial night sleep deprivation is capable of activating the inflammatory cascade, including activation of multiple signal transduction pathways such as NF-κB inflammatory signaling system, increased transcription of IL-6 and TNF mRNA, and increases in the TLR-4 stimulated monocyte production of IL-6 and TNFα. As compared to males, females show heightened risk for inflammatory disorders and display an increased sensitivity to sleep loss, with greater activation of cellular and genomic markers of inflammation.
Naturalistic sleep disturbance and inflammation
The cross-talk between normal nocturnal sleep and the immune system becomes misaligned when sleep disruption is chronically instantiated in a person’s sleep-wake activity pattern [30,31]. Such naturalistic sleep disruption involving loss of sleep or sleep fragmentation can be initially precipitated by acute external events or psychological challenges [134]. However, when nonphysical threats become chronic or disease leads to impaired homeostatic mechanisms, hyperarousal, and loss of physiological flexibility, these disturbances of sleep are perpetuated and become persistent, with nightly reports of poor sleep quality, non-restorative sleep, and insomnia complaints of difficulties initiating sleep, maintaining sleep, and/or waking up too early in the morning [135–138]. Such changes in sleep in response to ecological or physiological inputs also lead to elevations in markers of inflammation, which in turn foment inflammation-related disease, cardiovascular, neurodegenerative, and neoplastic diseases [19,139].
In our recent meta-analytic review that systematically evaluated over 70 studies on sleep and inflammatory outcomes, we assessed evidence that links self-reported sleep disturbance or extremes of sleep duration (i.e. short sleep, long sleep) with circulating markers of inflammation [105]. Here we summarize the major conclusions from these naturalistic, case-control studies which have used a diverse variety of methods to assess sleep disturbance including single survey item, multiple symptoms reporting, validated questionnaire, or diagnosis. Similarly, these studies have evaluated sleep duration using various measures including single survey items, validated questionnaire, sleep diary, actigraphy, or PSG. The vast majority of this research examined only three inflammatory markers, namely, CRP, IL-6, and TNF-α [105].
Normal nocturnal sleep and inflammatory dynamics are homeostatically aligned, as reviewed above, and normal sleep leads to increases in nocturnal levels of inflammation. Given these observations under normal conditions, it is important to ask how it is that sleep disturbance or loss of normal leads to increases in daytime levels of inflammatory cytokines. First, similar to the effects of sleep deprivation which shifts peak levels of IL-6 from the night to the day [83], patients with chronic sleep disturbance or insomnia disorder also show a shift in peak IL-6 from the night to the day, with greater 24-hour cumulative levels of IL-6 as compared to comparison controls [140]. Together, these data suggest that there is a misalignment of sleep and immunity with chronic sleep disturbance. In other words, the homeostatic mechanisms that normally serve to dampen daytime markers of inflammation are impaired in those with sleep disturbance. Second, sleep disturbance can be viewed to have physiologic effects similar to other conditions of chronic social adversities [76], and such loss of sleep leads to chronic HPA activation [141]. In turn, there is reduced glucocorticoid sensitivity which steers the immune system toward a pro-inflammatory transcriptional profiles and away from antiviral profile, as previously reviewed [69]. Third, sleep disturbance leads to increases in SNS outflow as well as SNS overactivity [76,142] which activate NFκB, related inflammatory transcriptional pathways, and cellular inflammation [69]. Finally during the subsequent night, higher levels of inflammatory cytokines can lead to decreases in sleep amounts, sleep fragmentation, and increases REM sleep at the expense of SWS sleep [31,143,144]. In turn, such disruptions in sleep continuity and sleep architecture in turn lead to further increases in inflammatory cytokines over the following day [143].
Across various methods of assessment of sleep, meta-analytic findings indicate that sleep disturbance is associated with higher levels of CRP (Figure 4) and with higher levels of IL-6 [105]. (Figure 5) However, the number of studies was too small to detect an effect of sleep disturbance on TNFα. It is of interest that the associations between sleep disturbance and increases in CRP and IL-6 were consistently found, even though the assessment of sleep disturbance varied from single questions about sleep quality to validated questionnaires and diagnostic interviews, which systematically assessed sleep disturbance. Further, similar results were found whether sleep duration was evaluated by subjective reports or objective means such as actigraphy. Nevertheless, the assessment methods used to evaluate sleep disturbance may indeed account to heterogeneity in the findings. For example, Patel et al found that sleep duration assessed by self-report correlated with levels of CRP and IL-6, whereas sleep duration assessed objectively by PSG correlated inversely with levels of TNFα[145].
Figure 4.
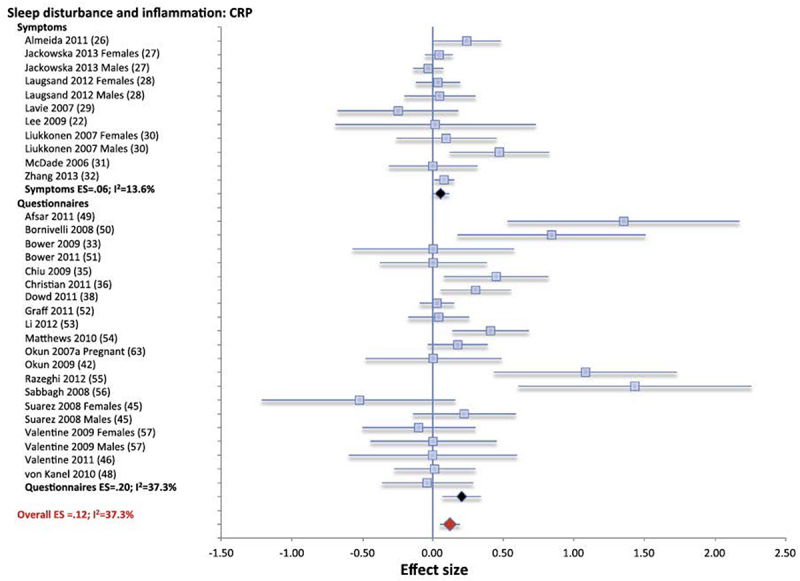
Forest plot of sleep disturbance associated with inflammation as indexed by C-reactive protein (CRP). Sleep disturbance is assessed by self-reported symptoms and questionnaires. Results are expressed as effect sizes (ES) and 95% confidence intervals. Reprinted from Irwin et al. [105] with permission.
Figure 5.
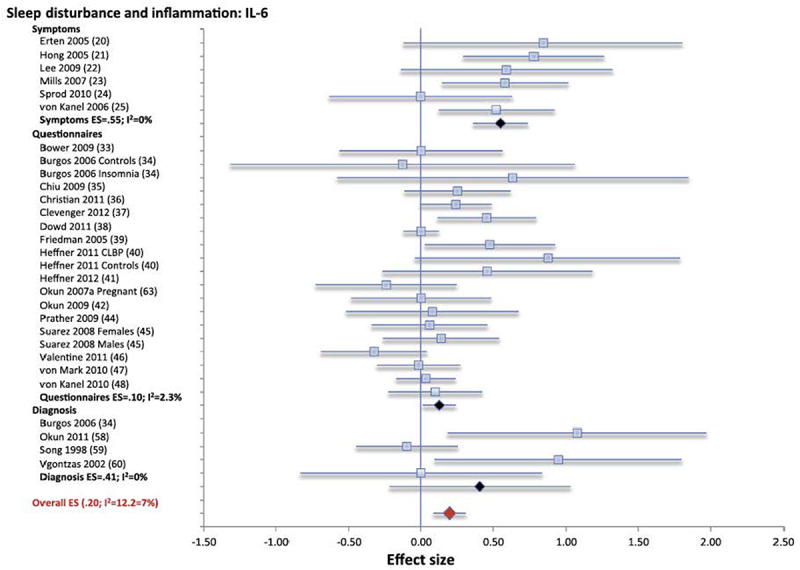
Forest plot of sleep disturbance associated with inflammation as indexed by circulating levels of interleukin-6 (IL-6). Sleep disturbance is assessed by self-reported symptoms and questionnaires. Results are expressed as effect sizes (ES) and 95% confidence intervals. Reprinted from Irwin et al. [105] with permission.
Consistent with experimental sleep deprivation findings, as described above, shorter sleep duration showed null or small effects on inflammatory markers [105]. For example, when sleep duration was evaluated continuously using self-report or objective methods, a small effect was noted for CRP but not IL-6; shorter sleep duration was associated with higher CRP. Interestingly, meta-analytic findings that focused on extremes of sleep duration, long sleep duration, as referenced to 7 to 8 hours of sleep but not short sleep duration, was associated with increases in CRP and in IL-6. However, persons who report long sleep duration often have a variety of medical comorbidities such as diabetes, cancer, and cardiovascular disease which are associated with increases in markers of inflammation. Because the quality of these meta-analytic data cannot go beyond the quality of the individual studies included, it is important to note that many studies did not comprehensively evaluate medical morbidities, or control for the confounding influence on morbidity on increases in inflammation in long sleepers. Some evidence suggests that nightly variability in sleep duration is associated with increases in morbidity, and notably one study has found that variability in sleep duration is also associated with increases in CRP [146].
Overall, results from this meta-analysis suggest that larger effect sizes were found in studies where the sample population was younger aged, and with a greater proportion of female subjects [105]. However, the effect of age and sex in these cross-sectional studies was small and statistically significant for only two subsamples; sleep disturbance predicting IL-6, with greater likelihood in females; and sleep duration continuously predicting CRP, with greater likelihood in younger samples. Other studies have reported ethnic differences, in which increases of circulating markers inflammation at the extremes of short sleep duration are more likely in African Americans as compared to other ethnic groups [147]. Finally, it is possible that quality of social ties may buffer the adverse effects of sleep disturbance on circulating markers of inflammation, although data are limited. Nevertheless, poor sleep efficiency has been found to be associated higher levels of CRP [148] and IL-6 [149], and these relationship are more robust in women with poor social relationships.
This cross-sectional evidence provides support for an effect of short sleep duration on circulating markers of inflammation, which are further supported by prospective data that sleep disturbance may be causally linked to increases in these markers of inflammation. For example, both self-reported sleep disturbance and short sleep duration predict subsequent increases in CRP in a sample of over 3000 persons [150]. Moreover, when short sleep duration (<5 hours) and sleep fragmentation (nocturnal wakening for >90 minutes) are objectively ascertained by in home PSG and actigraphy, these objective measures of short sleep duration were found to be prospectively associated with increases in inflammatory burden as indexed by a composite measure of levels of CRP, IL-6, TNFα, sTNF-RII, and IFN-γ, which further mediate mortality risk [151]. Other prospective findings demonstrate that sleep quality predicts increases in inflammatory cytokines, but only in females [152], consistent with experimental data that show that females show a heightened activation of inflammatory transcriptional pathways in response to sleep loss as compared to males [132,133]. Likewise, there are sex difference for the prospective effects of long sleep duration on markers of inflammation, in which long sleep duration predicts higher levels of CRP and IL-6 in females more than males [153], although other studies have found that long sleep duration predicts greater increases in males than females [154].
As compared to multiple other demographic (e.g, age, race) and biobehavioral (e.g. body mass index, physical activity) factors [105,155], sleep disturbance has effects on markers of inflammation that are equivalent, or in some cases greater. Additionally, as noted below, the reduction in CRP following insomnia treatment is comparable to the anti-inflammatory effects of a healthy dietary pattern and exercise [156,157].
The clinical implications of sleep disturbance of adverse health outcomes, as well as mortality risk, have been previously reviewed and indicate that subjectively reported sleep disturbance, short sleep duration (<7 h), and possibly long sleep duration (>8 h) are associated with increased risk of a number of disorders related to inflammation including diabetes, cardiovascular disease, and dementia [19,29]. In regards to mortality risk, a meta-analysis of 40 prospective cohort studies involving over two million participants found that extremes of sleep duration (<7 h) increased all- cause mortality risk [158]. To the extent that inflammation mediates the association between sleep disturbances and mortality has begun to be examined. In about 3,000 older adults followed prospectively followed over a 9-year period, self-reported short sleep duration predicted all-cause mortality in models adjusting for demographic, lifestyle, and health factors. When a composite of inflammatory markers (levels of IL-6, TNF, and CRP) was included in the model, the mortality risk profile was significantly attenuated suggesting that the association between sleep duration and mortality was partially mediated by inflammation [159]. One other study assessed sleep duration using actigraphy and found that short sleep duration (<5 h) was related to all-cause mortality. Moreover, when a composite of circulating markers of inflammation, including CRP, IL-6, TNFα, sTNF-RII, and IFN-γ, was included in the risk model, there was again a substantial attenuation of the sleep duration to mortality risk association [151].
Treatment of insomnia and reversal of inflammatory risk
As further evidence that sleep disturbance is causally linked to inflammation, clinical trial results have shown that treatment of insomnia leads to reversal of inflammatory activation profiles. Cognitive behavioral therapy for insomnia (CBT-I) is recommended as the first-line treatment for insomnia [160]. CBT-I combines cognitive therapy, stimulus control, sleep restriction, sleep hygiene, and relaxation, and has a robust efficacy profile as compared to other behavioral approaches, with more durable effects than pharmacological treatment [161]. Recent evidence has also found that mindfulness-based treatments including mindfulness meditation and tai-chi are non-inferior in the treatment of insomnia [162,163] with additional effects on neuroeffector mechanisms such as decreases in stress response pathways that are activated in association with insomnia [164–166]. Moreover, CBT-I, tai chi, and mindfulness all have been found to have durable effects in the treatment of insomnia, with improvement in insomnia symptoms sustained over the long term one-year follow-up [162,163]. With administration of any one of the treatment approaches for insomnia, improvements in insomnia are coupled with decreases in sympathetic activity, decreases in levels of systemic inflammation including CRP, decreases in cellular inflammation such as stimulated monocyte production of proinflammatory cytokines, and a reversal of inflammatory transcriptional profile [167–169]. Insomnia treatment effects on systemic inflammation especially robust in those who achieve full clinical remission of insomnia [170]. For example in older adults with insomnia disorder, CBT-I induced decreases of CRP over the 16-month follow-up period as compared to a universal behavior therapy, sleep education [170]. The clinical significance of these changes is notable, as decreases in the proportion with high CRP in response to insomnia treatment was comparable to the benefits reported with vigorous physical activity [171] or weight loss [172]. Furthermore, in those who received tai chi treatment, there were robust decreases in the percentages of monocytes producing TNFα, IL-6, or combined TNFα and IL-6 expression, with similar findings in CBT-I, although effects of CBT-I were not sustained over the 16-month follow-up as compared to tai chi [169]. Finally, both CBT-I and tai chi, as compared to sleep education therapy, showed a down-regulation of transcriptional profiles related to inflammation and an upregulation of genes related to IFN and the antiviral response [169]. In older adults with sleep disturbance, but not insomnia, further research has found that tai chi can reduce severity of insomnia complaints [173], and also boost anti-viral memory T-cell responses to varicella zoster virus [174] and response to a varicella zoster vaccine [174]. Separately, we have found that CBT has been found to improve depressive symptoms including insomnia complaints in rheumatoid arthritis patients, along with reduced the stimulated production of the proinflammatory cytokine, IL-6 [175]; that mindfulness meditation reduces proinflammatory response gene profiles [176] in lonely adults; and that a yogic meditation decreases NF-κB-related transcription of pro-inflammatory cytokines and increases IRF1-related transcription of innate anti-viral response genes [177]. These data from clinical trials show that treatment of insomnia, or insomnia complaints associated with medical or psychosocial co-morbidity, leads to a reduction in inflammatory and an increase in anti-viral profiles.
Sleep disturbance, adaptive immunity, and infectious disease risk
Normal nocturnal sleep, as described in Section IV, contributes to regulation of adaptive immunity and the Th1/Th2 cytokine balance, with a shift toward Th1 cytokine profile (i.e. ratio of IFN-γ to IL-4) in the early part of the night [178]. Similarly, IL-12 and IL-2, which are essential for the formation and maintenance of adaptive immunity, appear to be regulated by sleep with evidence of increases in the stimulated production of IL-2 during sleep in humans [75,80], although these effect were not found within CD4 T cells [179,180]. Nevertheless, there is a striking increase of IL-12 producing cells during sleep compared with nocturnal wakefulness [178] and conversely, there is a sleep-dependent decrease in monocyte production of IL-10 [95] and CD4 T cell production of IL-4 production by CD4 T cells [180] at the specific cell-subset level in whole blood [179]. Together these findings linking normal sleep to cytokines involved in the regulation of the adaptive immunity suggest that loss of sleep or sleep disturbance might alter adaptive immune response, leading to decreases in vaccination responses and increases in infectious disease risk.
Experimental and naturalistic disturbance of sleep and adaptive immunity
Experimental disruption of sleep leads to an opposing profile, as compared to normal sleep, on the adaptive immune response. Sleep deprivation, for example, decreases T cell production of IL-2 [75,181], induces increases in Th2 cytokine activity [95,180], decreases the IFN-γ/IL-4 ratio [102], and alters monocyte production of cytokines important in coordinating the adaptive immune response included decreases in production of IL-12 and increases in production of IL-10 [95]. However, in contrast to the effects of a single night of sleep deprivation, the occurrence of sleep deprivation over several nights leads to only minimal changes in T cell derived cytokines such as IL-2, IL-4 and IFN-γ[182].
Findings regarding the association between naturalistically occurring sleep disturbance and cytokine measures related to adaptive immunity are limited. However, consistent with alterations of Th1/Th2 cytokine balance following sleep deprivation, men who complain of insomnia symptoms were found to have lower stimulated production of IFN-γ and a lower IFN-γ/IL-4 ratio as compared to those without insomnia complaints [183]. In addition, in abstinent alcohol-dependent persons who have persistent sleep disturbance, noted especially for a loss of slow wave sleep, a relative shift toward a Type 2 response (i.e. lower ratio of stimulated production of IFN-γ/IL-10) was found as compared to responses in comparison controls without sleep disturbance [79]. In regards to immune cell numbers possibly associated with adaptive immunity, patients with chronic insomnia also show lower numbers of CD3+, CD4+, and CD8 + T cells as compared to those without insomnia [184], and females with short sleep (<6 h per night) have lower numbers of naïve T cells as compared to those with normal sleep duration [185]. Lower number of naïve T cells may lead to impairments in ability to respond to novel antigens [185], which has been further interrogated by examining the association between short sleep duration and antibody response after vaccination as reviewed in the following section.
Experimental and naturalistic disturbance of sleep and vaccine responses
Vaccination mimics how adaptive immunity responds to an infectious challenge and has been used as an experimental model to provide a translational perspective on the impact of sleep disturbance on clinically relevant infectious endpoints. One early study in animals found that sleep deprivation was associated with slowed clearing of influenza following infectious challenge [186], although this observation was not subsequently replicated [187–189]. In humans, data are limited to a small number of studies (Table 4), all of which have examined healthy adults except for one study of older adults [190]. Hence, it is not yet known whether sleep loss might alter vaccination responses in those who are known to evidence an attenuated vaccine response under basal conditions and who are at risk for infectious disease, such as older adults [63].
Table 4.
Studies linking sleep deprivation to vaccination responses.
| Type of sleep deprivation | Findings | References |
|---|---|---|
| Sleep deprivation before vaccination | ||
| PSD over 4 nights before vaccination; |
Decrease in antibody titers to influenza virus vaccine, Trivalent types A and B, at 10 days after vaccination, but no difference at 3 or 4 weeks after vaccination | [191] |
| TSD over 1 night before vaccination | Decrease in antibody titers to influenza virus vaccine, Type A, at 5 days after vaccination in males, but not females. No difference at 10, 17, or 52 days after vaccination in either sex | [192] |
| Sleep deprivation after vaccination | ||
| TSD over 1 night after vaccination | Decrease in antibody titers to hepatitis A virus vaccine at 4 weeks after vaccination | [193] |
| TSD over 1 night after vaccination | Decrease in hepatitis A specific Th cell response over 52 weeks follow-up, and decrease hepatitis A IgG1, but IgG2, IgG3, or IgG4, over 20 weeks follow-up | [194] |
| TSD over 1 night after vaccination | Decrease in hepatitis B specific Th cell response over 20 weeks, but not 52 weeks, follow-up, and decrease in hepatitis B IgG1, over 52 weeks follow-up | [194] |
Two strategies have been used to examine experimentally whether sleep loss alters vaccine responses in humans: one approach experimentally manipulates sleep before vaccination, whereas the other approach manipulates sleep after vaccination, typically in the first 24 hours following challenge with a vaccine. Experimentally induced sleep deprivation prior to vaccination has been found to attenuate response to the influenza vaccine. The first study used partial sleep deprivation, mimicking the most ubiquitous type of sleep disturbance found in those who are older or are experiencing social adversity and are most vulnerable to infectious diseases. After four nights of partial night sleep deprivation with sleep time restricted to 4 hours, subjects were exposed to the trivalent influenza vaccine, types A and B. In those who underwent sleep deprivation, antibody titers were reduced by over 50% as compared to immune responses in those who maintained a regular sleep schedule [191]. Another study examined the effects of a single night of total sleep deprivation before vaccine exposure to influenza A. In contrast to repeated nights of partial sleep loss, a single night of total sleep deprivation had mixed effects on antibody response, and a reduction in antibody response to influenza A was found in males, but not females [192]. Furthermore, total sleep deprivation induced a decrease in antibody response only during the first five days after vaccination; antibody responses did not differ between the two conditions at later timepoints including 10 to 52 days after vaccination [192]. The early response may be due to initial effects of sleep deprivation on the IgM response, which are not found with the later IgG response, although differences in IgM vs. IgG levels were not evaluate as the assessment method relied only on hemagglutination inhibition antibody titers against the H1N1 virus [192].
The second strategy has involved administration of the vaccine followed by experimental manipulation of sleep. In healthy adults who were administered the Hepatitis A vaccine and then exposed to total sleep deprivation, a reduction in antibody titers to Hepatitis A was found in the sleep deprivation condition as compared to normal sleep schedule, with effects lasting 28 days after vaccination challenge [193]. Similarly, in another study of healthy adults, subjects were challenged with either hepatitis A vaccine or with hepatitis B vaccine followed by exposure to a single night of total sleep deprivation [194]. Rather than simply evaluating antibody titers, this study examined specific antibody subtypes (i., IgG1, IgG2, IgG3, and IgG4) as well as Ag-specific Th cell response (i.e. IFN) to either Hepatitis A- and Hepatitis B challenge [194]. Further, the durability of immune responses was evaluated over one year follow-up. In those who were exposed to sleep deprivation, Ag-specific IgG1 levels to Hepatitis A, but not levels to other IgG subtypes, were significantly reduced as compared to the control condition, with similar effects for Hepatitis B [194]. Furthermore, effects of sleep loss were markedly durable, with reduction in IgG1 levels persisting over one year follow-up [194]. Not only were there reductions in IgG1 levels, but sleep deprivation also reduced the number of antigen-specific CD4 T cells, and the production of the Th1 effector cytokine IFN-γ [194]. Interestingly, durability of response was associated with the amount of slow wave sleep during the night after vaccination, suggesting that EEG slow oscillatory activity in the period after vaccination may predict long-term increases in antigen-specific persisting T cells HAV [194].
Given that exposure to sleep deprivation occurred within 24 hours after vaccination, these findings suggest that sleep loss may be altering immunologic processes occurring early in response to infectious challenge. For example, it is possible that sleep deprivation is altering APC–T cell interactions before the differentiation of effector T cells, which then disrupts the coordinated accumulation of APCs and T cells in lymphoid tissue and strength of the Th1 cytokine signal and maintenance of the response. Indeed, reductions in immunologic memory to HAV persisted for at least one year, which further emphasizes the clinical significance of sleep loss on vaccine efficacy [194]. Finally, in regard to the clinical implications, anecdotal observations found that three of the fourteen subjects assigned to sleep deprivation failed to achieve levels above the hepatitis A threshold defined as necessary for clinical protection; all subjects assigned to normal sleep condition achieved antibody levels consistent with protective clinical response [193].
Prospective cohort studies yield similar results suggesting that naturalistically occurring sleep disturbance characterized by short sleep duration, low sleep efficiency, and/or insomnia diagnosis are associated with lower antibody responses to vaccination for influenza [195–197] and hepatitis B [198], but not meningitis C [199]. First, in regards to short sleep duration, Prather et al. found that sleep duration less than 6 hours per night as confirmed by sleep diary or by actigraphy was prospectively related to decreased likelihood of protection to a hepatitis B vaccination in 125 midlife adults [198]; clinical protection status was defined as anti-hepatitis B surface antigen immunoglobulin G ≥ 10 mIU/ml.
These findings were recently extended by examining response to influenza A vaccination in 83 healthy adults [200]. Again, self-reported shorter sleep duration before vaccination, but not after vaccination, was associated with lower levels of antibodies to influenza over 4 months follow-up. No associations were found between sleep efficiency and reported sleep quality and vaccination responses [200]. In contrast in older adults, no relationship was found between self-reported sleep duration, sleep latency or sleep efficiency and IgG response to influenza vaccination, possibly because responses in this age cohort are already attenuated due to their age [190]. Finally, no association between insomnia diagnosis and response to influenza vaccination was demonstrated in another study of healthy mid-life adults [197].
Together, these observations from experimental and prospective cohort studies suggest that short sleep duration, at least in healthy adults, is likely associated with an attenuated adaptive immunologic response, and possibly clinical protection, following influenza- or hepatitis vaccine administration, although null findings are also reported. Nevertheless, the findings further suggest the possibility that sleep disturbance, and especially short sleep duration might be associated with reduced response to other vaccines, and possibly related to increased infectious disease risk, another area of investigation, as review below.
Naturalistic disturbance of sleep and infectious disease risk
The most striking feature linking sleep disturbance to adaptive immune response is the compelling association between short sleep duration, whether experimentally manipulated or naturalistically occurring, on Th1 cytokine regulation and vaccine responses. Hence, consistent with this evidence, sleep duration but not other aspects of sleep (i.e. sleep quality, sleep efficiency) are associated with infectious disease risk outcomes in both basic research, and clinical translational studies. Briefly, basic research has found that prolonging sleep duration leads to decreases in bacterial load and improved survival in a variety of infectious disease models, as previously reviewed [27,30,31]. For example, using a genetic approach in Drosophila, a prolonged sleep duration was associated with increased resistance to bacterial infection as indexed by bacterial load as compared to Drosophila with normal sleep. Additionally, Drosphophla with prolonged sleep duration show improved survival [201]. However, in other studies in Drosophla, sleep deprivation led to increased, not decreased, resistance to bacterial infection [202]. In mice exposed to experimentally induced sepsis, sleep deprivation was associated with increased mortality as compared to mice with uninterrupted sleep who were also exposed to sepsis[203]. Other data indicate the sleep deprivation can reduce resistance to other pathogens such as parasites. In mice exposed to malaria, sleep deprivation was found to increase the number of infected cells and decrease survival as compared to mice who had uninterrupted sleep; interestingly, these adverse effects were reversed following a night of recovery sleep [204]. In contrast, sleep deprivation did not alter severity of infection with Trichinella spiralis in rats [205].
In humans, short sleep duration (≤5 h) is prospectively associated with a greater than 70% increase in the risk of pneumonia in 56,953 females who were followed over 4 years, with similar associations between perceived inadequate sleep and pneumonia [206]. In addition, this study also found that long sleep duration (≥9 h) was associated with pneumonia, similar to the association between long sleep duration and mortality risk [20], although it is not fully understood whether underlying medical morbidity is contributing to the association between long sleep duration and morbid and mortality outcomes. In another nationally representative sample of 22,726 Americans enrolled in the National Health and Nutrition Examination Surveys (NHANES), self-reported sleep duration and physician reported diagnosis of a sleep disorder was characterized in association with reported history of a head or chest cold or a respiratory infection, including influenza or pneumonia, in the past 30 days [207]. Again, short sleepers (≤5 hours) showed about a 30% increase in the risk of a head or chest infection, and about an 82% increases in the risk of respiratory infection as compared to those sleeping 7 to 8 hours per night. In addition, those with a history of physician reported sleep disorder showed similar increases in the risk of infection [207].
Similarly in adolescents, short sleep duration as objectively characterized by actigraphy was associated with self-report increases in the number of illness events, including symptoms related to respiratory infections [208], although no such association between self-reported sleep duration and self-reported upper- respiratory tract infections was found in a Swedish cohort [209]. Finally, whereas there is an abundance of evidence that the prevalence of sleep disturbance and depressive symptoms has markedly increased in the midst of the Covid-19 pandemic [210–214], little is known about the potential influence of sleep disturbance on risk of infection, hospitalization, and mortality related to Covid-19. One recent study of 46,535 participants obtained information on sleep behaviors including sleep duration, daytime sleepiness, insomnia complaints with assignment of a composite sleep score based on severity (i.e. sleep duration <6) and frequency of symptoms [215]. Of the sample, 8422 participants tested positive (COVID+), and 38,111 participants were tested but never positive (COVID-). Mortality over 30-days, primary outcome, was examined in COVID + and COVID – groups. In COVID+ participants, high scores on the sleep composite indicating significant sleep disturbance was associated with an over 2-fold increase in the risk of mortality in COVID + participants; no such association was found in the COVID – participants. In addition, secondary outcome analyses examined rates of hospitalization, and found that sleep disturbance as defined by the composite scores predicted an over 50% increases in the rate of hospitalization in the COVID + group [215].
Experimental models of infectious disease risk yield similar findings with evidence that both short sleep duration and poor sleep efficiency are associated with reduced resistance to infectious illness. In a study of 153 healthy adults, self-reported sleep duration and sleep efficiency were obtained for 14 days, followed by experimental inoculation with the rhinovirus (i.e. common cold) and objective evaluation of signs of illness and its severity. Among those sleeping less 7 hours of sleep, there was nearly a 3-fold greater risk of developing the common cold infection as compared to those who reported sleeping 8 or more hours [216]. Furthermore, among those who reported sleep efficiency of 92% or less, there was a 5 times greater likelihood of developing a common cold infection as compared to those with a sleep efficiency of 98% or more [216]. These findings are striking given the rather modest disturbance of sleep efficiency. For example, a sleep efficiency of 85% or less typically characterizes those with sleep disturbance or insomnia; hence, a sleep efficiency of 92% might be conceptualized as in the normative range. Given that treatment with selective serotonin reuptake inhibitor improves sleep complaints and normalizes vaccine responses to the zoster vaccine in depressed patients [217], further research is need to evaluate whether targeting sleep problems might mitigate infectious disease risk profiles in vulnerable older adults.
The evolutionary advantage of sleep and its contribution to dynamic variations in adaptive immune responses, vaccine response, and infectious disease risk are not known. Possibly, because sleep is quiescent period, it is speculated that energy resources can be reallocated from daytime functions to support immune responses to infectious challenge, which are known to be metabolically demanding [93]. Consonant with this notion, circadian processes such as change in the 24 hours rhythm of cortisol drive the redistribution of immune cells (i.e. naïve T cell, B cells) from the circulation to the lymph nodes, where they are first exposed to foreign antigens with initiation of an adaptive immune response. It is speculated that available metabolic resources during sleep support the demands on immune-cell division and differentiation, so that an effective adaptive immune response can be generated.
Neuroeffector mechanisms linking sleep and immunity
The central nervous system (CNS) is linked to the immune system by two principal effector pathways, namely the HPA axis and the autonomic nervous system. (Figure 6) As previously reviewed, HPA axis activation leads to the release of cortisol, and this glucocorticoid hormone is anti-inflammatory, acting to suppress transcriptional expression of antiviral immune response genes, as well as inflammatory immune response genes. Three mechanisms are thought to contribute to the immune suppressive effects of cortisol [27]. First, glucocorticoids can bind to receptor on gene promoter sequences, and thereby interrupt expression of antiviral and proinflammatory gene transcription. Second, when glucocorticoids bind to the glucocorticoid receptors, there is a transcriptional activation of anti-inflammatory genes, which inhibit activation of inflammatory transcriptional signaling pathways induced by the transcription factor NF-κB. Given that NF-κB is key in initiating the inflammatory cascade, inflammatory activation is effectively abrogated. Finally, in order for the proinflammatory transcription factors, NF-κB and AP-1, to initiate transcription, these factors must bind to gene promoters; glucocorticoids lead to protein-protein interactions that interfere with bind of proinflammatory transcription factors, thereby antagonizing inflammatory gene transcription [69].
Figure 6.
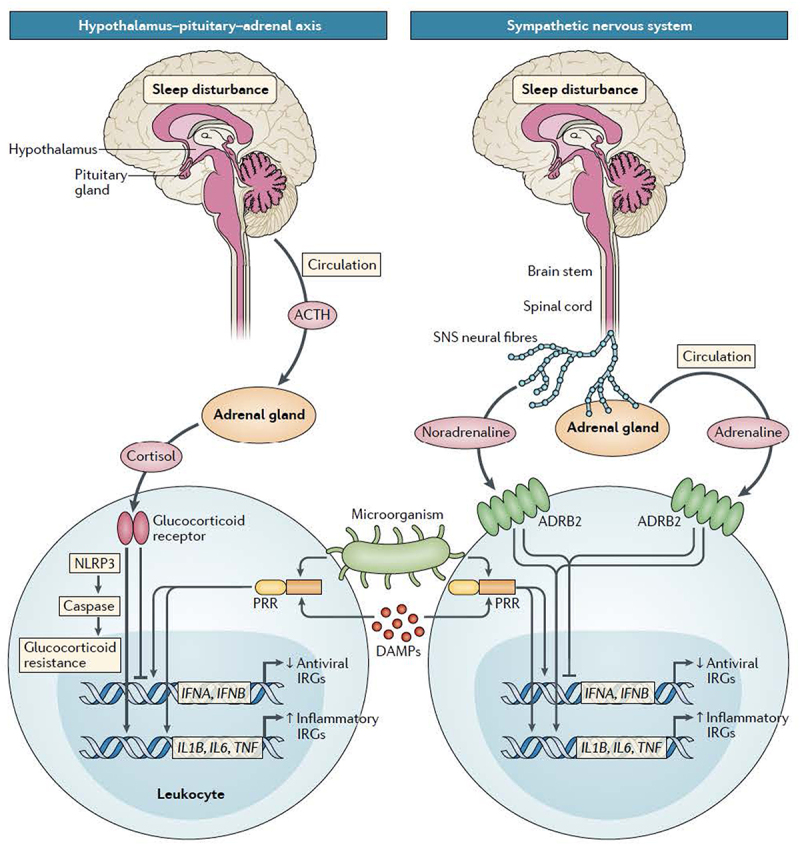
Model of effects of sleep disturbance on the regulation of immune response gene profiles a | In response to activation of the hypothalamic–pituitary–adrenal (HPA) axis, the circulating neuroendocrine glucocorticoid hormone, cortisol, is produced, which regulates by inhibits inflammatory and antiviral immune response genes (IRGs). With sustained engagement of the HPA axis, as might occur with insomnia, glucocorticoid resistance develops, and typical suppression of pro- inflammatory gene networks (such as those encoding IL-1β.
IL-6 and tumor necrosis factor (TNF)) does not occur. However, cortisol continues suppress antiviral gene profiles (such as those encoding IFNα and IFNβ). Glucocorticoid resistance is thought to develop when damage- associated molecular patterns (DAMPs) trigger triggering of pattern recognition receptors (PRRs), leading to activation of inflammatory signaling pathways involving nuclear factor- κB (NF- κB) and the NOD-, LRR- and pyrin domain- containing protein 3 (NLRP3) inflammasome. NLRP3 stimulation in turn activates caspase 1, which cleaves the glucocorticoid receptor, leading to glucocorticoid resistance.b | During sleep disturbance, activation of the SNS leads to release of the neurotransmitter norepinephrine from SNS nerve fibers, as well as release of epinephrine from the adrenal glands into the blood. Both norepinephrine and epinephrine stimulate β2-adrenergic receptors (ADRB2) on leukocytes, which suppresses transcription of antiviral IRGs, and enhances transcription of inflammatory IRGs. ACTH, adrenocorticotropic hormone. Reprinted from Irwin [27] with permission.
SNS activation is the second neuroeffector pathway linking the CNS to regulation of immunity. The SNS is thought to have a more nuanced role in the regulation of immunity and acts to steer or balance adaptive and pro-inflammatory immune responses [218]. Throughout the many lymphoid tissues, SNS neural fibers innervate these microenvironments, making synaptic like contact with immune cells to modulate immune response gene transcription [219–221]. Indeed, SNS nerve fibers innervate all the following tissues: primary and secondary lymphoid organs, vasculature and peri-vascular tissues, and most visceral organs and musculoskeletal structures. When SNS fibers are activated, release of norepinephrine occurs and this sympathetic neurotransmitter crosses the synaptic-like cleft and binds to β-adrenergic receptors. In turn, β-adrenergic receptor binding activates the Gs protein–adenylyl cyclase–cAMP–PKA signaling cascade and regulation of leukocyte gene expression. SNS activation has divergent effects on adaptive and inflammatory gene expression. Adrenergic signaling suppresses Th1-type gene expression (such as IFNG and IL12B), and also activates T helper 2 (Th2)-type cytokine genes including IL4 and IL5 which together suppress the adaptive cytokine immune response profiles [69]. In addition, SNS activation suppresses Type I IFN-mediated immune responses. In contrast with the suppressive effects on the adaptive immune, the SNS has been found to induce an activation of NF-κB [222] along with upregulation of pro-inflammatory cytokine gene expression, inducing increases in the transcription of IL1B, TNF and IL6 genes [218]. Whereas these data suggest that SNS activity can activate the inflammatory response, it would be simplistic to suggest that sympathetic neurotransmitters are pro-inflammatory, as substantial evidence has also found that. the sympathetic activity can have anti-inflammatory effects, including inhibition of the release of TNFα [90–92]. Indeed, as reviewed by Pongratz and Straub [223], it might be more accurate to state that sympathetic neurotransmitters (i.e. norepinephrine) modulate inflammatory cytokine production and their release depending on a number of contextual factors including the state of activation state of the cell [224], how proximal the cell is to the source of the neurotransmitters and/or neurotransmitter concentration [225], and the co-expression of other factors capable of modulating adrenergic activation and/or density adrenergic receptor expression [226] including activation of vagal activity [227]. Finally, the timing of sympathetic activity in relation to the inflammatory response requires consideration, with evidence at the systemic level that the SNS signal can be proinflammatory at the initial phase of inflammation (e.g. sleep deprivation), but can become anti-inflammatory when inflammation becomes chronic (e.g. chronic social adversity) [223]. In addition to changes in adaptive and inflammatory gene profiles and related immune response, the distribution of immune cell types showing a high density of β-adrenergic receptors are preferentially influenced, leading to a selective distribution of these immune cells as characterized by a mobilization of hematopoietic stem cells, natural killer cells and splenic neutrophils and monocytes into the circulation [69].
The sleep circadian profile is synchronized with HPA activity, which indicates a substantial influence of both sleep and circadian processes on innate and adaptive immune responses. During the early part of the night, which is characterized by an enrichment of slow wave sleep, the 24 hour profile of hypothalamic corticotropin-releasing hormone (CRH), pituitary corticotropin (ACTH) and adrenal corticosteroids shows an absolute minimum [30,74]. During the late part of the night, which is characterized by a REM sleep dominance, HPA activity is increases typically reaching its maximum shortly after wakening. Although experimental evidence is limited, increases of markers of the adaptive immune system during the early part of the night might be mechanistically related to low levels of cortisol which occur during this period along with a predominance of slow wave sleep [96]. One study collected blood samples during a period of slow wave sleep, and then added to cortisol ex vivo to these samples; a decrease in percentage of CD4+ and CD8 + T cells producing IFN-γ, IL-2 and TNF was found [74]. Conversely, if blood samples were collected during the late part of the night in which there is a maximum in circadian oscillation of cortisol, selective blockade of cortisol binding to the glucocorticoid receptor leads to an increase of CD4+ and CD8 + T cells producing IL-2, IFN-γ, and TNFα [74,228]. Other studies indicate that sleep, as compared to circadian processes, has a more salient role, in the regulation of the inflammatory cytokine IL-6. Early night sleep deprivation interrupts early night increases in IL-6, which indicates that sleep is needed to support activation of inflammatory or innate immune responses during the night [74,84].
Sleep disturbance that becomes chronic induces repeated and recurring activation of the HPA axis. In turn, chronic elevation of cortisol leads to glucocorticoid resistance. In this instance, there is a decreased sensitivity to cortisol and glucocorticoids show a reduced potency or ability to constrain an inflammatory response [229]. For example in patients with chronic insomnia and short sleep duration, there is evidence of both increases in cortisol and increases in inflammation as evidenced by elevated levels of CRP and IL-6; elevated levels of cortisol do not effectively reduce inflammation because immune cells no longer “hear” this message [230]. The mechanisms that lead to glucocorticoid resistance with chronic sleep disturbance are not known. However, chronic stressors have been found to induce increases in endogenous damage-associated molecular patterns (DAMPs), along with activation of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome. When the NLRP3 inflammasome is upregulated, leading to caspase- mediated cleavage of the glucocorticoid receptor, glucocorticoid resistance emerges [231]. A second mechanism pertains to the polymorphic variation in the gene encoding the glucocorticoid receptor; such variation correlates with anti-inflammatory signaling of glucocorticoids at the cellular level. This latter observations might explain heterogeneity in risk of inflammatory disorders among short sleepers at the population level; namely that chronic short sleep duration is associated with variance in glucocorticoid sensitivity [232].
Sleep also regulates sympathetic outflow, and during normal nocturnal sleep, SNS activity decreases along with an increase in parasympathetic outflow. Additionally, depth of sleep and stage of sleep are related to changes in SNS activity [233]. During NREM and slow wave sleep, which predominantly occur in the first half of the night, sympathetic outflow decreases, as indexed by microneurographic recording of sympathetic nerve cell activity [234]. In contrast during REM sleep, which occurs mainly during the latter part of the night, there is a marked increase in SNS outflow reaching levels similar to waking states during the day [234]. The measurement of plasma levels of norepinephrine by repeated blood sampling every 30 minutes during the night has also shown that circulating levels of norepinephrine, but not epinephrine, decrease during the nocturnal period as compared to wakefulness. In addition, lower levels of norepinephrine are found during slow wave sleep as compared to awake, whereas levels of norepinephrine during REM sleep are similar to awake [88]. In sum, SNS activity measured by either sympathetic neural activity or release of norepinephrine show similar changes during sleep, with decreases during the nocturnal period due to predominant decreases during slow wave sleep, but not REM sleep.
Experimental manipulation of sleep shows that that sleep is causally linked to nocturnal regulation of sympathetic activity. In recumbent persons who are deprived of sleep, the nocturnal decrease of sympathetic activity is not found as compared to sleep persons who are also recumbent. In addition to increasing sympathetic activity, sleep deprivation leads to increases in nocturnal sympathovagal balance, as assessed by heart rate variability. Interestingly, increases in sympathovagal balance that occur with sleep deprivation, persist into the following day leading to daytime increases in sympathetic tone as compared to those who are not exposed to sleep deprivation [112,235]. Naturalistic observations are in agreement with these experimental findings. In patients with insomnia, for example, there are increases in levels of norepinephrine, epinephrine, and other markers of sympathetic outflow [50] as compared to controls, and persons with insomnia who have co-morbid short sleep duration, show even greater increases in measures of sympathetic activity (i.e. urinary catecholamines and their metabolites) as compared to those with insomnia without short sleep duration. Separately, persons with insomnia who are co-morbid for short sleep duration also show greater increases in inflammatory biomarkers as compared to those who do not express this more severe phenotype of insomnia [50].
The mediating path linking sleep disturbance, increases in sympathetic activity, and subsequent increases of inflammatory outcomes is not yet fully established, although correlative findings suggest that nocturnal elevations in sympathetic activity are temporally associated with increases in circulating markers of inflammation during the day. For example, when sympathetic activity is estimated by measures of heart rate variability during NREM and REM sleep, higher sympathetic tone correlates with higher levels of IL-6 in the morning [236]. Moreover, experimentally induced sleep deprivation leads to increases in sympathetic activity, which corresponds to decreases in natural killer cell activity. Similar relationships between increases in sympathetic tone during the night and decreases in NK activity are found in patients with insomnia. Because the effects of sleep deprivation to reduce NK activity was abrogated by pretreatment with a β-adrenergic antagonist, propranolol [237], it appears that SNS activation is causally mediating these effects.
Multiple other effector mechanisms are associated with sleep-wake activity, including the regulation of neuroendocrine hormones such as prolactin, melatonin, and growth hormone. Nevertheless, despite robust evidence suggesting that melatonin, for example, can alter immunity, little is known whether sleep drives changes in these hormones which alter the sleep-wake profiles of innate or- adaptive immune responses, as reviewed by [93].
In summary, chronic sleep disturbance activates HPA and SNS pathways [50] and these neuroeffector mechanisms contribute to decreases in antiviral immunity, and increases in inflammatory responses. (Figure 6) This change in balance of these adaptive and inflammatory transcriptional profiles, coined a conserved transcriptional response to adversity (CTRA) occurs with chronic sleep disturbance, as well as in association with multiple psychosocial adversities such as depression, which are typically co-morbid with sleep disturbance [69,238].
Conclusions
Chronic sleep disturbance, as occurs with insomnia disorder, leads to activation of the inflammatory response, and when such inflammation is sustained, it can be damaging to the person. Indeed, such inflammation is known to contribute to major disease risk including depression, dementia, cardiovascular disease, and inflammatory disorders. (Figure 7) The reasons for sleep disturbance are varied as sleep patterns are influenced in part by environmental factors and perceived threats. Such social adversities can also increase inflammation, which also disrupts sleep as extensively discussed in other reviews10−12. In the presence of chronic adversities and/or intrinsic low grade inflammation, the beneficial crosstalk between sleep and the immune system becomes imbalanced. In turn, this dysregulation in sleep further leads to increases in inflammation and decreases in antiviral immune responses. Moreover, further elevations in inflammation induce sleep fragmentation with decreases in sleep efficiency, decreases in slow wave sleep, and increases in REM sleep, further fomenting inflammation, inflammatory disease risk, and vulnerability to infectious disease. Fortunately, there is evidence that this spiral can be interrupted; behavioral interventions and as well pharmacologic treatments can treat insomnia and redirect a misaligned inflammatory transcriptional profile with the potential to mitigate risk of inflammation-related cardiovascular, neurodegenerative, and mental health diseases, and possibly alter susceptibility to infectious disease. Knowing that sleep is impacted by multiple factors such as physical activity, the circadian rhythm of sleep wake cycles, and the homeostatic drive to sleep, there is substantial opportunity to refine interventions that consider the separate and overlapping influences of these extrinsic and intrinsic factors on sleep continuity and sleep depth. Finally, research that advances understanding of sleep with assessment of sleep architecture and spectral analyses to evaluate sleep depth will inform development of specific interventions that target components of sleep to modify precise pathways between sleep, inflammatory and antiviral dynamics, and morbidity risk.
Figure 7.
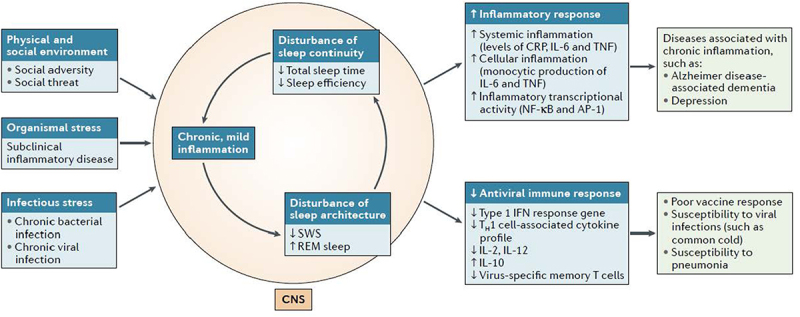
Model showing proposed links chronic perceived stress, organismal stress, or infectious stress and the induction of chronic, mild inflammation, which is associated with decreases in total sleep time, sleep efficiency and SWS, with increases in rapid eye movement (REM) sleep. Together these changes in sleep continuity and sleep architecture characterize sleep disturbance. Sleep disturbance, in turn, shifts the transcriptional profile toward increased inflammatory activity and decreased antiviral responses. The increase in the inflammatory transcriptional state is thought to contribute clinical manifestations of inflammatory disorders, whereas the decrease in antiviral responses is believed to result in poor vaccine responses and increased susceptibility to infectious disease.
AP-1, activator protein 1; CNS, central nervous system; CRP, C- reactive protein; NF- κB, nuclear factor- κB; TH1 cell, T helper 1 cell; TNF, tumor necrosis factor.As reprinted from Irwin [27] with permission.
Funding Statement
This work was supported by the National Cancer Institute [R01CA207130]; National Cancer Institute [R01 CA160245]; National Institute on Aging [R01 AG026364]; National Institute on Aging [R01AG051944]; National Institute on Aging [R01 AG056424].
Abbreviations
ACTH, adrenocorticophic hormone; AP-1, activator protein 1; APC, antigen presenting cell; cAMP, cyclic adenosine monophosphate; ATF, activating transcription factor; CB T-I, cognitive behavorial therapy for insomnia; CNS, central nervous system; CREB, cAMP response element-binding protein; CRH, corticotropin releasing hormone; CRP, C- reactive protein; CTRA, conserved transcriptional response to adversity; DAMP, danger/damage-associated molecular patterns; DSM-5, Diagnostic and Statistical Manual-5; EEG, electroencephalogram; EMG, electromyogram; EOG, electrooculogram; ETS. Erythroblast Transformation Specific;
HPA, hypothalamic pituitary adrenal axis; HSP, heat shock protein; Hz, herz; IgG, immunoglobulin G; IFN, interferon; IL, interleukin; LPS, lipopolysaccharide; MHC, major histocompatibility; NF- κB, nuclear factor- κB; NHANES, National Health and Nutrition Examination Surveys; NLRP3, NLR family pyrin domain containing 3; NK, natural killer; NREM, non rapid eye movement sleep; PAMP, pathogen associated molecular pattern; PKA, protein kinase A; PKC, protein kinase C; PRR, pattern recognition receptor; PSG, polysomnography; REM, rapid eye movement sleep; sIL6R, soluble interleukin-6 receptor; sICAM, soluble intercellular adhesion molecule; SNS, sympathetic nervous system; STAT, Signal Transducer and Activator of Transcription; SWA, slow wave activity; TH1 cell, T helper 1 cell; TLR-4, toll-like receptor 4; TNF, tumor necrosis factor-α; WASO, wake after sleep onset.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Szymusiak R. Body temperature and sleep. Handb Clin Neurol. 2018;156:341–351. [DOI] [PubMed] [Google Scholar]
- [2].Lack LC, Gradisar M, Van Someren EJ, et al. The relationship between insomnia and body temperatures. Sleep Med Rev. 2008;12(4):307–317. [DOI] [PubMed] [Google Scholar]
- [3].Harding EC, Franks NP, Wisden W. The temperature dependence of sleep. Front Neurosci. 2019;13:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Muzet A, Libert JP, Candas V. Ambient temperature and human sleep. Experientia. 1984;40(5):425–429. [DOI] [PubMed] [Google Scholar]
- [5].Okamoto-Mizuno K, Nigai Y, Iizuka S. The effect of ambient temperature change on the covered area of the body during sleep. J Home Econ Jpn. 2003;54:1025–1030. [Google Scholar]
- [6].Szymusiak R, Satinoff E. Maximal REM sleep time defines a narrower thermoneutral zone than does minimal metabolic rate. Physiol Behav. 1981;26(4):687–690. [DOI] [PubMed] [Google Scholar]
- [7].Szymusiak R, Danowski J, McGinty D. Exposure to heat restores sleep in cats with preoptic/anterior hypothalamic cell loss. Brain Res. 1991;541(1):134–138. [DOI] [PubMed] [Google Scholar]
- [8].McGinty D, Alam MN, Szymusiak R, et al. Hypothalamic sleep-promoting mechanisms: coupling to thermoregulation. Arch Ital Biol. 2001;139(1–2):63–75. [PubMed] [Google Scholar]
- [9].Monroe LJ. Psychological and physiological differences between good and poor sleepers. J Abnorm Psychol. 1967;72(3):255–264. [DOI] [PubMed] [Google Scholar]
- [10].Vgontzas AN, Tsigos C, Bixler EO, et al. Chronic insomnia and activity of the stress system: a preliminary study. J Psychosom Res. 1998;45(1 Spec No):21–31. [DOI] [PubMed] [Google Scholar]
- [11].Raymann RJ, Swaab DF, Van Someren EJ. Cutaneous warming promotes sleep onset. Am J Physiol Regul Integr Comp Physiol. 2005;288(6):R1589–1597. [DOI] [PubMed] [Google Scholar]
- [12].Yetish G, Kaplan H, Gurven M, et al. Natural sleep and its seasonal variations in three pre-industrial societies. Curr Biol. 2015;25(21):2862–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Togo F, Aizawa S, Arai J, et al. Influence on human sleep patterns of lowering and delaying the minimum core body temperature by slow changes in the thermal environment. Sleep. 2007;30(6):797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Reid KJ, Krauchi K, Grimaldi D, et al. Effects of manipulating body temperature on sleep in postmenopausal women. Sleep Med. 2021;81:109–115. [DOI] [PubMed] [Google Scholar]
- [15].Okamoto-Mizuno K, Tsuzuki K, Mizuno K. Effects of mild heat exposure on sleep stages and body temperature in older men. Int J Biometeorol. 2004;49(1):32–36. [DOI] [PubMed] [Google Scholar]
- [16].Okamoto-Mizuno K, Tsuzuki K, Mizuno K. Effects of humid heat exposure in later sleep segments on sleep stages and body temperature in humans. Int J Biometeorol. 2005;49(4):232–237. [DOI] [PubMed] [Google Scholar]
- [17].Obradovich N, Migliorini R, Mednick SC, et al. Nighttime temperature and human sleep loss in a changing climate. Sci Adv. 2017;3(5):e1601555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ohayon MM. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev. 2002;6(2):97–111. [DOI] [PubMed] [Google Scholar]
- [19].Irwin MR. Why sleep is important for health: a psychoneuroimmunology perspective. Annu Rev Psychol. 2015;66(1):143–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kripke DF, Garfinkel L, Wingard DL, et al. Mortality associated with sleep duration and insomnia. Arch Gen Psychiatry. 2002;59(2):131–136. [DOI] [PubMed] [Google Scholar]
- [21].Mallon L, Broman JE, Hetta J. Sleep complaints predict coronary artery disease mortality in males: a 12-year follow-up study of a middle-aged Swedish population. J Intern Med. 2002;251(3):207–216. [DOI] [PubMed] [Google Scholar]
- [22].Mallon L, Broman JE, Hetta J. Relationship between insomnia, depression, and mortality: a 12-year follow-up of older adults in the community. Int Psychogeriatr. 2000;12(3):295–306. [DOI] [PubMed] [Google Scholar]
- [23].Dew MA, Hoch CC, Buysse DJ, et al. Healthy older adults’ sleep predicts all-cause mortality at 4 to 19 years of follow-up. Psychosom Med. 2003;65(1):63–73. [DOI] [PubMed] [Google Scholar]
- [24].Phillips B, Mannino DM. Does insomnia kill? Sleep. 2005;28(8):965–971. [DOI] [PubMed] [Google Scholar]
- [25].Ferrucci L, Harris TB, Guralnik JM, et al. Serum IL-6 level and the development of disability in older persons. J Am Geriatr Soc. 1999;47(6):639–646. [DOI] [PubMed] [Google Scholar]
- [26].Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25(12):1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Irwin MR. Sleep and inflammation: partners in sickness and in health. Nat Rev Immunol. 2019;19(11):702–715. [DOI] [PubMed] [Google Scholar]
- [28].Bryant PA, Trinder J, Curtis N. Sick and tired: does sleep have a vital role in the immune system. Nat Immunol. 2004;4(6):457–467. [DOI] [PubMed] [Google Scholar]
- [29].Besedovsky L, Lange T, Haack M. The Sleep-Immune Crosstalk in Health and Disease. Physiol Rev. 2019;99(3):1325–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Irwin MR, Opp MR. Sleep health: reciprocal regulation of sleep and innate immunity. Neuropsychopharmacology. 2017;42(1):129–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci. 2009;10(3):199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Krueger JM, Majde JA. Microbial products and cytokines in sleep and fever regulation. Crit Rev Immunol. 1994;14(3–4):355–379. [DOI] [PubMed] [Google Scholar]
- [33].Horne J. Why we sleep: the function of sleep in humans and other mammals. Oxford: Oxford University Press; 1988. [Google Scholar]
- [34].Buysse DJ. Sleep health: can we define it? Does it matter? Sleep. 2014;37(1):9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Borbely AA, Daan S, Wirz-Justice A, et al. The two-process model of sleep regulation: a reappraisal. J Sleep Res. 2016;25(2):131–143. [DOI] [PubMed] [Google Scholar]
- [36].Brown RE, Basheer R, McKenna JT, et al. Control of sleep and wakefulness. Physiol Rev. 2012;92(3):1087–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rechtschaffen A, Kales A. A manual of standardized terminology, techniques and scoring system for sleep stages of human subjects. Bethesda: N Inst Neurol Dis & Blindness; 1968. [DOI] [PubMed] [Google Scholar]
- [38].Ohayon MM, Carskadon MA, Guilleminault C, et al. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep. 2004;27(7):1255–1273. [DOI] [PubMed] [Google Scholar]
- [39].Ehlers CL, Havstad JW, Garfinkel A, et al. Nonlinear analysis of EEG sleep states. Neuropsychopharmacology. 1991;5(3):167–176. [PubMed] [Google Scholar]
- [40].Irwin M, Gillin JC, Dang J, et al. Sleep deprivation as a probe of homeostatic sleep regulation in primary alcoholics. Biol Psychiatry. 2002;51(8):632–641. [DOI] [PubMed] [Google Scholar]
- [41].Irwin M, Rinetti G, Redwine L, et al. Nocturnal proinflammatory cytokine-associated sleep disturbances in abstinent African American alcoholics. Brain Behav Immun. 2004;18(4):349–360. [DOI] [PubMed] [Google Scholar]
- [42].Buysse DJ, Ancoli-Israel S, Edinger JD, et al. Recommendations for a standard research assessment of insomnia. Sleep. 2006;29(9):1155–1173. [DOI] [PubMed] [Google Scholar]
- [43].Finan PH, Richards JM, Gamaldo CE, et al. Validation of a wireless, self-application, ambulatory electroencephalographic sleep monitoring device in healthy volunteers. J Clin Sleep Med. 2016;12(11):1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ancoli-Israel S, Cole R, Alessi C, et al. The role of actigraphy in the study of sleep and circadian rhythms. Sleep. 2003;26(3):342–392. [DOI] [PubMed] [Google Scholar]
- [45].Monk TH, Reynolds CF, Kupfer DJ, et al. The pittsburgh sleep diary. J Sleep Res. 1994;3(2):111–120. jsr003002111. pii. [PubMed] [Google Scholar]
- [46].Okun ML, Reynolds CF 3rd, Buysse DJ, et al. Sleep variability, health-related practices, and inflammatory markers in a community dwelling sample of older adults. Psychosom Med. 2011;73(2):142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].American Psychiatric Association., American Psychiatric Association . DSM-5 task force. In: Diagnostic and statistical manual of mental disorders: DSM-5. 5th ed. Washington D.C: American Psychiatric Association; 2013. pp. 362–368. [Google Scholar]
- [48].Vgontzas AN, Kales A, Bixler EO, et al. Usefulness of polysomnographic studies in the differential diagnosis of insomnia. Int J Neurosci. 1995;82(1–2):47–60. [DOI] [PubMed] [Google Scholar]
- [49].Littner M, Hirshkowitz M, Kramer M, et al. Practice parameters for using polysomnography to evaluate insomnia: an update. Sleep. 2003;26(6):754–760. [DOI] [PubMed] [Google Scholar]
- [50].Vgontzas AN, Fernandez-Mendoza J, Liao D, et al. Insomnia with objective short sleep duration: the most biologically severe phenotype of the disorder. Sleep Med Rev. 2013;17(4):241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Morin CM, Belleville G, Belanger L, et al. The insomnia severity index: psychometric indicators to detect insomnia cases and evaluate treatment response. Sleep. 2011;34(5):601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cole JC, Motivala SJ, Buysse DJ, et al. Validation of a 3-factor scoring model for the pittsburgh sleep quality index in older adults. Sleep. 2006;29(1):112–116. [DOI] [PubMed] [Google Scholar]
- [53].Ohayon MM, Riemann D, Morin C, et al. 3rd. Hierarchy of insomnia criteria based on daytime consequences. Sleep Med. 2012;13(1):52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kierlin L, Olmstead R, Yokomizo M, et al. Diagnostic and Statistical Manual criteria for insomnia related impairment in daytime functioning: polysomnographic correlates in older adults. Sleep Med. 2012;13(7):958–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Paul WE. Fundamental Immunology. Philadelphia: Wolters Kluwer; 2013. [Google Scholar]
- [56].Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. [DOI] [PubMed] [Google Scholar]
- [57].Barton GM. A calculated response: control of inflammation by the innate immune system. J Clin Invest. 2008;118(2):413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5. [DOI] [PubMed] [Google Scholar]
- [59].Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol. 2013;14(7):668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat Immunol. 2007;8(1):11–13. [DOI] [PubMed] [Google Scholar]
- [61].Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–436. nature04870 [pii. [DOI] [PubMed] [Google Scholar]
- [62].Piber D, Olmstead R, Cho JH, et al. Inflammaging: age and systemic, cellular, and nuclear jnflammatory biology in older adults. J Gerontol A Biol Sci Med Sci. 2019;74(11):1716–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(1):S4–9. [DOI] [PubMed] [Google Scholar]
- [64].Ishii S, Karlamangla AS, Bote M, et al. Gender, obesity and repeated elevation of C-reactive protein: data from the CARDIA cohort. PLoS One. 2012;7(4):e36062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].O’Connor MF, Bower JE, Cho HJ, et al. To assess, to control, to exclude: effects of biobehavioral factors on circulating inflammatory markers. Brain Behav Immun. 2009;23(7):887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Murphy K. Janeway’s Immunobiology. New York NY: Garland Science; 2011. [Google Scholar]
- [67].Gao B, Wang H, Lafdil F, et al. STAT proteins - key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J Hepatol. 2012;57(2):430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87(3):873–904. [DOI] [PubMed] [Google Scholar]
- [69].Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol. 2011;11(9):625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Fung CH, Vitiello MV, Alessi CA, et al. Report and research agenda of the american geriatrics society and national institute on aging bedside-to-bench conference on sleep, circadian rhythms, and aging: new avenues for improving brain health, physical health, and functioning. J Am Geriatr Soc. 2016;64(12):e238–e247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sack RL, Auckley D, Auger RR, et al. Circadian rhythm sleep disorders: part I, basic principles, shift work and jet lag disorders. An American Academy of sleep medicine review. Sleep. 2007;30(11):1460–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sack RL, Auckley D, Auger RR, et al. Circadian rhythm sleep disorders: part II, advanced sleep phase disorder, delayed sleep phase disorder, free-running disorder, and irregular sleep-wake rhythm. An American academy of sleep medicine review. Sleep. 2007;30(11):1484–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wright KP Jr., Drake AL, Frey DJ, et al. Influence of sleep deprivation and circadian misalignment on cortisol, inflammatory markers, and cytokine balance. Brain Behav Immun. 2015;47:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lange T, Dimitrov S, Born J. Effects of sleep and circadian rhythm on the human immune system. Ann N Y Acad Sci. 2010;1193(1):48–59. [DOI] [PubMed] [Google Scholar]
- [75].Born J, Lange T, Hansen K, et al. Effects of sleep and circadian rhythm on human circulating immune cells. J Immunol. 1997;158(9):4454–4464. [PubMed] [Google Scholar]
- [76].McEwen BS. Sleep deprivation as a neurobiologic and physiologic stressor: allostasis and allostatic load. Metabolism. 2006;55(10 Suppl 2):S20–23. [DOI] [PubMed] [Google Scholar]
- [77].Whiteside TL, Herberman RB. The role of natural killer cells in human disease. Clin Immunol Immunopathol. 1989;53(1):1–23. [DOI] [PubMed] [Google Scholar]
- [78].Kronfol Z, Nair M, Zhang Q, et al. Circadian immune measures in healthy volunteers: relationship to hypothalamic-pituitary-adrenal axis hormones and sympathetic neurotransmitters. Psychosom Med. 1997;59(1):42–50. [DOI] [PubMed] [Google Scholar]
- [79].Redwine L, Dang J, Hall M, et al. Disordered sleep, nocturnal cytokines, and immunity in alcoholics. Psychosom Med. 2003;65(1):75–85. [DOI] [PubMed] [Google Scholar]
- [80].Irwin M, McClintick J, Costlow C, et al. Partial night sleep deprivation reduces natural killer and cellular immune responses in humans. FASEB J. 1996;10(5):643–653. [DOI] [PubMed] [Google Scholar]
- [81].Vgontzas AN, Bixler EO, Lin HM, et al. IL-6 and its circadian secretion in humans. Neuroimmuno modulation. 2005;12(3):131–140. [DOI] [PubMed] [Google Scholar]
- [82].Dimitrov S, Lange T, Benedict C, et al. Sleep enhances IL-6 trans-signaling in humans. FASEB J. 2006;20(12):2174–2176. [DOI] [PubMed] [Google Scholar]
- [83].Vgontzas AN, Papanicolaou DA, Bixler EO, et al. Circadian interleukin-6 secretion and quantity and depth of sleep. J Clin Endocrinol Metab. 1999;84(8):2603–2607. [DOI] [PubMed] [Google Scholar]
- [84].Redwine L, Hauger R, Gillin J, et al. Effects of sleep and sleep deprivation on interleukin-6, growth hormone, cortisol, and melatonin levels in humans. J ClinEndocrin Metab. 2000;85(10):3597–3603. [DOI] [PubMed] [Google Scholar]
- [85].Muller-Newen G, Kuster A, Hemmann U, et al. Soluble IL-6 receptor potentiates the antagonistic activity of soluble gp130 on IL-6 responses. J Immunol. 1998;161(11):6347–6355. [PubMed] [Google Scholar]
- [86].Dimitrov S, Besedovsky L, Born J, et al. Differential acute effects of sleep on spontaneous and stimulated production of tumor necrosis factor in men. Brain Behav Immun. 2015;47:201–210. [DOI] [PubMed] [Google Scholar]
- [87].Uthgenannt D, Schoolmann D, Pietrowsky R, et al. Effects of sleep on the production of cytokines in humans. Psychosom Med. 1995;57(2):97–104. [DOI] [PubMed] [Google Scholar]
- [88].Irwin M, Thompson J, Miller C, et al. Effects of sleep and sleep deprivation on catecholamine and interleukin-2 levels in humans: clinical implications. J Clin Endocrin Metab. 1999;84(6):1979–1985. [DOI] [PubMed] [Google Scholar]
- [89].Powell ND, Sloan EK, Bailey MT, et al. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A. 2013;110(41):16574–16579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Martelli D, Farmer DGS, McKinley MJ, et al. Anti-inflammatory reflex action of splanchnic sympathetic nerves is distributed across abdominal organs. Am J Physiol Regul Integr Comp Physiol. 2019;316(3):R235–R242. [DOI] [PubMed] [Google Scholar]
- [91].Martelli D, Yao ST, McKinley MJ, et al. Neural control of inflammation by the greater splanchnic nerves. Temperature. 2014;1(1):14–15. doi: 10.4161/temp.29135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Martelli D, Yao ST, McKinley MJ, et al. Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol. 2014;592(7):1677–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Besedovsky L, Lange T, Born J. Sleep and immune function. Pflugers Arch. 2012;463(1):121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Opp MR, Born J, Irwin MR, et al. Sleep and the Immune System. In: Psychoneuroimmunology, vols I and II. 4th ed. 2007. p. 579–618 .New York:Academic Press. [Google Scholar]
- [95].Lange T, Dimitrov S, Fehm HL, et al. Shift of monocyte function toward cellular immunity during sleep. Arch Inten Med. 2006;166(16):1695–1700. [DOI] [PubMed] [Google Scholar]
- [96].Westermann J, Lange T, Textor J, et al. System consolidation during sleep - a common principle underlying psychological and immunological memory formation. Trends Neurosci. 2015;38(10):585–597. [DOI] [PubMed] [Google Scholar]
- [97].Lissoni P, Rovelli F, Brivio F, et al. Circadian secretions of IL-2, IL-12, IL-6 and IL-10 in relation to the light/dark rhythm of the pineal hormone melatonin in healthy humans. Nat Immun. 1998;16(1):1–5. [DOI] [PubMed] [Google Scholar]
- [98].Petrovsky N, Harrison LC. The chronobiology of human cytokine production. Int Rev Immunol. 1998;16(5–6):635–649. [DOI] [PubMed] [Google Scholar]
- [99].Hunter CA. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat Rev Immunol. 2005;5(7):521–531. [DOI] [PubMed] [Google Scholar]
- [100].Brombacher F, Kastelein RA, Alber G. Novel IL-12 family members shed light on the orchestration of Th1 responses. Trends Immunol. 2003;24(4):207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Petrovsky N, Harrison LC. Diurnal rhythmicity of human cytokine production: a dynamic disequilibrium in T helper cell type 1/T helper cell type 2 balance? J Immunol. 1997;158:5163–5168. [PubMed] [Google Scholar]
- [102].Lange T, Dimitrov S, Fehm HL, et al. Sleep-like concentrations of growth hormone and cortisol modulate type1 and type2 in-vitro cytokine production in human T cells. Int Immunopharmacol. 2006;6(2):216–225. [DOI] [PubMed] [Google Scholar]
- [103].Lo JC, Groeger JA, Cheng GH, et al. Self-reported sleep duration and cognitive performance in older adults: a systematic review and meta-analysis. Sleep Med. 2016;17:87–98. [DOI] [PubMed] [Google Scholar]
- [104].Pace-Schott EF, Spencer RM. Sleep-dependent memory consolidation in healthy aging and mild cognitive impairment. Curr Top Behav Neurosci. 2015;25:307–330. [DOI] [PubMed] [Google Scholar]
- [105].Irwin MR, Olmstead R, Carroll JE. Sleep disturbance, sleep duration, and inflammation: a systematic review and meta-analysis of cohort studies and experimental sleep deprivation. Biol Psychiatry. 2016;80(1):40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ohayon MM. Prevalence and correlates of nonrestorative sleep complaints. Arch Internal Med. 2005;165(1):35–41. [DOI] [PubMed] [Google Scholar]
- [107].Kuhlman KR, Chiang JJ, Bower JE, et al. Persistent low positive affect and sleep disturbance across adolescence moderate link between stress and depressive symptoms in early adulthood. J Abnorm Child Psychol. 2020;48(1):109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Hall SJ, Ferguson SA, Turner AI, et al. The effect of working on-call on stress physiology and sleep: a systematic review. Sleep Med Rev. 2017;33:79–87. [DOI] [PubMed] [Google Scholar]
- [109].Kim EJ, Dimsdale JE. The effect of psychosocial stress on sleep: a review of polysomnographic evidence. Behav Sleep Med. 2007;5(4):256–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Song Y, Carlson GC, McGowan SK, et al. Sleep disruption due to stress in women veterans: a comparison between caregivers and noncaregivers. Behav Sleep Med. 2021;19(2):243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Pike JL, Smith TL, Hauger RL, et al. Chronic life stress alters sympathetic, neuroendocrine, and immune responsivity to an acute psychological stressor in humans. Psychosom Med. 1997;59(4):447–457. [DOI] [PubMed] [Google Scholar]
- [112].Irwin MR, Ziegler M. Sleep deprivation potentiates activation of cardiovascular and catecholamine responses in abstinent alcoholics. Hypertension. 2005;45(2):252–257. [DOI] [PubMed] [Google Scholar]
- [113].Moldofsky H, Lue FA, Davidson JR, et al. Effects of sleep deprivation on human immune functions. Faseb J. 1989;3(8):1972–1977. [DOI] [PubMed] [Google Scholar]
- [114].Vgontzas AN, Pejovic S, Zoumakis E, et al. Daytime napping after a night of sleep loss decreases sleepiness, improves performance, and causes beneficial changes in cortisol and interleukin-6 secretion. Am J Physiol Endocrinol Metab. 2007;292(1):E253–261. [DOI] [PubMed] [Google Scholar]
- [115].Haack M, Kraus T, Schuld A, et al. Diurnal variations of interleukin-6 plasma levels are confounded by blood drawing procedures. Psychoneuroendocrinology. 2002;27(8):921–931. [DOI] [PubMed] [Google Scholar]
- [116].Irwin MR, Witarama T, Caudill M, et al. Sleep loss activates cellular inflammation and signal transducer and activator of transcription (STAT) family proteins in humans. Brain Behav Immun. 2015;47:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Carroll JE, Carrillo C, Olmstead R, et al. Sleep deprivation and divergent toll-like receptor-4 activation of cellular inflammation in aging. Sleep. 2015;38(2):205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Abedelmalek S, Chtourou H, Aloui A, et al. Effect of time of day and partial sleep deprivation on plasma concentrations of IL-6 during a short-term maximal performance. Eur J Appl Physiol. 2013;113(1):241–248. [DOI] [PubMed] [Google Scholar]
- [119].Schmid SM, Hallschmid M, Jauch-Chara K, et al. Disturbed glucoregulatory response to food intake after moderate sleep restriction. Sleep. 2011;34(3):371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Stamatakis KA, Punjabi NM. Effects of sleep fragmentation on glucose metabolism in normal subjects. Chest. 2010;137(1):95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Faraut B, Boudjeltia KZ, Dyzma M, et al. Benefits of napping and an extended duration of recovery sleep on alertness and immune cells after acute sleep restriction. Brain Behav Immun. 2011;25(1):16–24. [DOI] [PubMed] [Google Scholar]
- [122].Shearer WT, Reuben JM, Mullington JM, et al. Soluble TNF-alpha receptor 1 and IL-6 plasma levels in humans subjected to the sleep deprivation model of spaceflight. J Allergy Clin Immunol. 2001;107(1):165–170. [DOI] [PubMed] [Google Scholar]
- [123].Irwin MR, Wang M, Campomayor CO, et al. Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med. 2006;166(16):1756–1762. [DOI] [PubMed] [Google Scholar]
- [124].Irwin M, Mascovich A, Gillin JC, et al. Partial sleep deprivation reduces natural killer cell activity in humans. Psychosom Med. 1994;56(6):493–498. [DOI] [PubMed] [Google Scholar]
- [125].Meier-Ewert HK, Ridker PM, Rifai N, et al. Effect of sleep loss on C-reactive protein, an inflammatory marker of cardiovascular risk. J Am Coll Cardiol. 2004;43(4):678–683. [DOI] [PubMed] [Google Scholar]
- [126].Haack M, Sanchez E, Mullington JM. Elevated inflammatory markers in response to prolonged sleep restriction are associated with increased pain experience in healthy volunteers. Sleep. 2007;30(9):1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Vgontzas AN, Zoumakis E, Bixler EO, et al. Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J Clin Endocrinol Metab. 2004;89(5):2119–2126. [DOI] [PubMed] [Google Scholar]
- [128].van Leeuwen WM, Lehto M, Karisola P, et al. Sleep restriction increases the risk of developing cardiovascular diseases by augmenting proinflammatory responses through IL-17 and CRP. PLoS One. 2009;4(2):e4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Chennaoui M, Sauvet F, Drogou C, et al. Effect of one night of sleep loss on changes in tumor necrosis factor alpha (TNF-α) levels in healthy men. Cytokine. 2011;56(2):318–324. [DOI] [PubMed] [Google Scholar]
- [130].Sauvet F, Leftheriotis G, Gomez-Merino D, et al. Effect of acute sleep deprivation on vascular function in healthy subjects. J Appl Physiol. 2010;108(1):68–75. [DOI] [PubMed] [Google Scholar]
- [131].Frey DJ, Fleshner M, Wright KP Jr. The effects of 40 hours of total sleep deprivation on inflammatory markers in healthy young adults. Brain Behav Immun. 2007;21(8):1050–1057. [DOI] [PubMed] [Google Scholar]
- [132].Irwin MR, Carrillo C, Olmstead R. Sleep loss activates cellular markers of inflammation: sex differences. Brain Behav Immun. 2010;24(1):54–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Irwin MR, Wang M, Ribeiro D, et al. Sleep loss activates cellular inflammatory signaling. Biol Psychiatry. 2008;64(6):538–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Mai E, Buysse DJ. Insomnia: prevalence, impact, pathogenesis, differential diagnosis, and evaluation. Sleep Med Clin. 2008;3(2):167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].LeBlanc M, Merette C, Savard J, et al. Incidence and risk factors of insomnia in a population-based sample. Sleep. 2009;32(8):1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Savard J, Villa J, Ivers H, et al. Prevalence, natural course, and risk factors of insomnia comorbid with cancer over a 2-month period. J Clin Oncol. 2009;27(31):5233–5239. [DOI] [PubMed] [Google Scholar]
- [137].Bonnet MH, Arand DL. Hyperarousal and insomnia: state of the science. Sleep Med Rev. 2010;14(1):9–15. [DOI] [PubMed] [Google Scholar]
- [138].Riemann D, Spiegelhalder K, Feige B, et al. The hyperarousal model of insomnia: a review of the concept and its evidence. Sleep Med Rev. 2010;14(1):19–31. [DOI] [PubMed] [Google Scholar]
- [139].Irwin MR, Vitiello MV. Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 2019;18(3):296–306. [DOI] [PubMed] [Google Scholar]
- [140].Vgontzas AN, Zoumakis M, Papanicolaou DA, et al. Chronic insomnia is associated with a shift of interleukin-6 and tumor necrosis factor secretion from nighttime to daytime. Metabolism. 2002;51(7):887–892. [DOI] [PubMed] [Google Scholar]
- [141].Vgontzas AN, Chrousos GP. Sleep, the hypothalamic-pituitary-adrenal axis, and cytokines: multiple interactions and disturbances in sleep disorders. Endocrinol Metab Clin North Am. 2002;31(1):15–36. [DOI] [PubMed] [Google Scholar]
- [142].Irwin M, Clark C, Kennedy B, et al. Nocturnal catehcolamines and immune function in insomniacs, depressed patients, and control subjects. Brain Beh Immun. 2003;17(5):365–372. [DOI] [PubMed] [Google Scholar]
- [143].Bjurstrom MF, Olmstead R, Irwin MR. Reciprocal relationship between sleep macrostructure and evening and morning cellular inflammation in rheumatoid arthritis. Psychosom Med. 2016. DOI: 10.1097/PSY.0000000000000363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Irwin MR, Olmstead R, Valladares EM, et al. Tumor necrosis factor antagonism normalizes rapid eye movement sleep in alcohol dependence. Biol Psychiatry. 2009;66(2):191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Patel SR, Zhu X, Storfer-Isser A, et al. Sleep duration and biomarkers of inflammation. Sleep. 2009;32(2):200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Park H, Tsai KM, Dahl RE, et al. Sleep and inflammation during adolescence. Psychosom Med. 2016;78(6):677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Grandner MA, Buxton OM, Jackson N, et al. Extreme sleep durations and increased C-reactive protein: effects of sex and ethnoracial group. Sleep. 2013;36(5):769–779E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Okun ML, Coussons-Read ME. Sleep disruption during pregnancy: how does it influence serum cytokines? J Reprod Immunol. 2007;73(2):158–165. [DOI] [PubMed] [Google Scholar]
- [149].Friedman EM, Hayney MS, Love GD. Social relationships, sleep quality, and interleukin-6 in aging women. Proc Nat Acad Sci. 2005;102(51):18757–18762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Cho HJ, Seeman TE, Kiefe CI, et al. Sleep disturbance and longitudinal risk of inflammation: moderating influences of social integration and social isolation in the coronary artery risk development in young adults (CARDIA) study. Brain Behav Immun. 2015;46:319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Smagula SF, Stone KL, Redline S, et al. Actigraphy- and polysomnography-measured sleep disturbances, inflammation, and mortality among older men. Psychosom Med. 2016;78(6):686–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Prather AA, Epel ES, Cohen BE, et al. Gender differences in the prospective associations of self-reported sleep quality with biomarkers of systemic inflammation and coagulation: findings from the heart and soul study. J Psychiatr Res. 2013;47(9):1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Prather AA, Vogelzangs N, Penninx BW. Sleep duration, insomnia, and markers of systemic inflammation: results from the Netherlands Study of Depression and Anxiety (NESDA). J Psychiatr Res. 2015;60:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Bakour C, Schwartz S, O’Rourke K, et al. Sleep duration trajectories and systemic inflammation in young adults: results from the national longitudinal study of adolescent to adult health (Add health). Sleep. 2017;40(11):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].O’Connor MF, Irwin MR. Links between behavioral factors and inflammation. Clin Pharmacol Ther. 2010;87(4):479–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Neale EP, Batterham MJ, Tapsell LC. Consumption of a healthy dietary pattern results in significant reductions in C-reactive protein levels in adults: a meta-analysis. Nutr Res. 2016;36(5):391–401. [DOI] [PubMed] [Google Scholar]
- [157].Hayashino Y, Jackson JL, Hirata T, et al. Effects of exercise on C-reactive protein, inflammatory cytokine and adipokine in patients with type 2 diabetes: a meta-analysis of randomized controlled trials. Metabolism. 2014;63(3):431–440. [DOI] [PubMed] [Google Scholar]
- [158].Liu TZ, Xu C, Rota M, et al. Sleep duration and risk of all-cause mortality: a flexible, non-linear, meta-regression of 40 prospective cohort studies. Sleep Med Rev. 2017;32:28–36. [DOI] [PubMed] [Google Scholar]
- [159].Hall MH, Smagula SF, Boudreau RM, et al. Association between sleep duration and mortality is mediated by markers of inflammation and health in older adults: the health, aging and body composition study. Sleep. 2015;38(2):189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Morgenthaler T, Kramer M, Alessi C, et al. Practice parameters for the psychological and behavioral treatment of insomnia: an update. An American academy of sleep medicine report. Sleep. 2006;29(11):1415–1419. [PubMed] [Google Scholar]
- [161].Morin CM, Bootzin RR, Buysse DJ, et al. Psychological and behavioral treatment of insomnia:update of the recent evidence (1998-2004). Sleep. 2006;29(11):1398–1414. [DOI] [PubMed] [Google Scholar]
- [162].Irwin MR, Olmstead R, Carrillo C, et al. Tai Chi Chih compared with cognitive behavioral therapy for the treatment of insomnia in survivors of breast cancer: a randomized, partially blinded, noninferiority trial. J Clin Oncol. 2017;35(23):2656–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Garland SN, Carlson LE, Stephens AJ, et al. Mindfulness-based stress reduction compared with cognitive behavioral therapy for the treatment of insomnia comorbid with cancer: a randomized, partially blinded, noninferiority trial. J Clin Oncol. 2014;32(5):449–457. [DOI] [PubMed] [Google Scholar]
- [164].Motivala SJ, Sollers J, Thayer J, et al. Tai chi chih acutely decreases sympathetic nervous system activity in older adults. J Gerontol A Biol Sci Med Sci. 2006;61(11):1177–1180. [DOI] [PubMed] [Google Scholar]
- [165].Park J, Lyles RH, Bauer-Wu S. Mindfulness meditation lowers muscle sympathetic nerve activity and blood pressure in African-American males with chronic kidney disease. Am J Physiol Regul Integr Comp Physiol. 2014;307(1):R93–R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Ardi Z, Golland Y, Shafir R, et al. The effects of mindfulness-based stress reduction on the association between autonomic interoceptive signals and emotion regulation selection. Psychosom Med. 2021;83(8):852–862. [DOI] [PubMed] [Google Scholar]
- [167].Black DS, Irwin MR, Olmstead R, et al. Tai chi meditation effects on nuclear factor-kappaB signaling in lonely older adults: a randomized controlled trial. Psychother Psychosom. 2014;83(5):315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Irwin M, Olmstead R, Breen E, et al. Tai Chi, cellular inflammation, and transcriptome dynamics in breast cancer survivors with insomnia: a randomized controlled trial. J Natl Cancer Inst. 2014;50(50):295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Irwin MR, Olmstead R, Breen EC, et al. Cognitive behavioral therapy and tai chi reverse cellular and genomic markers of inflammation in late-life insomnia: a randomized controlled trial. Biol Psychiatry. 2015;78(10):721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Irwin MR, Olmstead R, Carrillo C, et al. Cognitive behavioral therapy vs. Tai Chi for late life insomnia and inflammatory risk: a randomized controlled comparative efficacy trial. Sleep. 2014;37(9):1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Ford ES. Does exercise reduce inflammation? Physical activity and C-reactive protein among U.S. adults. Epidemiology. 2002;13(5):561–568. [DOI] [PubMed] [Google Scholar]
- [172].Esposito K, Pontillo A, Di Palo C, et al. Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA. 2003;289(14):1799–1804. [DOI] [PubMed] [Google Scholar]
- [173].Irwin MR, Olmstead R, Motivala SJ. Improving sleep quality in older adults with moderate sleep complaints: a randomized controlled trial of Tai Chi Chih. Sleep. 2008;31(7):1001–1008. [PMC free article] [PubMed] [Google Scholar]
- [174].Irwin MR, Olmstead R, Oxman MN. Augmenting immune responses to varicella zoster virus in older adults: a randomized, controlled trial of Tai Chi. J Am Geriatr Soc. 2007;55(4):511–517. [DOI] [PubMed] [Google Scholar]
- [175].Zautra AJ, Davis MC, Reich JW, et al. Comparison of cognitive behavioral and mindfulness meditation interventions on adaptation to rheumatoid arthritis for patients with and without history of recurrent depression. J Consult Clin Psychol. 2008;76(3):408–421. [DOI] [PubMed] [Google Scholar]
- [176].Creswell JD, Irwin MR, Burklund LJ, et al. Mindfulness-based stress reduction training reduces loneliness and pro-inflammatory gene expression in older adults: a small randomized controlled trial. Brain Behav Immun. 2012;26(7):1095–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Black DS, Cole SW, Irwin MR, et al. Yogic meditation reverses NF-kappa B and IRF-related transcriptome dynamics in leukocytes of family dementia caregivers in a randomized controlled trial. Psychoneuroendocrinology. 2013;38(3):348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Dimitrov S, Lange T, Nohroudi K, et al. Number and function of circulating human antigen presenting cells regulated by sleep. Sleep. 2007;30(4):401–411. [DOI] [PubMed] [Google Scholar]
- [179].Bollinger T, Bollinger A, Skrum L, et al. Sleep-dependent activity of T cells and regulatory T cells. Clin Exp Immunol. 2009;155(2):231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Dimitrov S, Lange T, Tieken S, et al. Sleep associated regulation of T helper 1/T helper 2 cytokine balance in humans. Brain Behav Immun. 2004;18(4):341–348. [DOI] [PubMed] [Google Scholar]
- [181].Irwin M, Thompson J, Miller C, et al. Effects of sleep and sleep deprivation on catecholamine and interleukin-2 levels in humans: clinical implications. J Clin Endocrin Metab. 1999;84(6):1979–1985. [DOI] [PubMed] [Google Scholar]
- [182].Dinges DF, Douglas SD, Hamarman S, et al. Sleep deprivation and human immune function. Adv Neuroimmunol. 1995;5(2):97–110. [DOI] [PubMed] [Google Scholar]
- [183].Sakami S, Ishikawa T, Kawakami N, et al. Coemergence of insomnia and a shift in the Th1/Th2 balance toward Th2 dominance. Neuroimmunomodulation. 2002;10(6):337–343. 71474. [DOI] [PubMed] [Google Scholar]
- [184].Savard J, Laroche L, Simard S, et al. Chronic insomnia and immune functioning. Psychosom Med. 2003;65(2):211–221. [DOI] [PubMed] [Google Scholar]
- [185].Carroll JE, Irwin MR, Levine M, et al. Epigenetic aging and immune senescence in women with insomnia symptoms: findings from the Women’s health initiative study. Biol Psychiatry. 2017;81(2):136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Brown R, Pang G, Husband AJ, et al. Suppression of immunity to influenza virus infection in the respiratory tract following sleep disturbance. Regional Immunology. 1989;2(5):321–325. [PubMed] [Google Scholar]
- [187].Renegar KB, Floyd RA, Krueger JM. Effects of short-term sleep deprivation on murine immunity to influenza virus in young adult and senescent mice. Sleep. 1998;21(3):241–248. [PubMed] [Google Scholar]
- [188].Renegar KB, Floyd R, Krueger JM. Effect of sleep deprivation on serum influenza-specific IgG. Sleep. 1998;21(1):19–24. [PubMed] [Google Scholar]
- [189].Renegar KB, Crouse D, Floyd RA, et al. Progression of influenza viral infection through the murine respiratory tract: the protective role of sleep deprivation. Sleep. 2000;23(7):859–863. [PubMed] [Google Scholar]
- [190].Ayling K, Fairclough L, Tighe P, et al. Positive mood on the day of influenza vaccination predicts vaccine effectiveness: a prospective observational cohort study. Brain Behav Immun. 2018;67:314–323. [DOI] [PubMed] [Google Scholar]
- [191].Spiegel K, Sheridan JF, Van Cauter E. Effect of sleep deprivation on response to immunization. JAMA. 2002;288(12):1471–1472. [DOI] [PubMed] [Google Scholar]
- [192].Benedict C, Brytting M, Markstrom A, et al. Acute sleep deprivation has no lasting effects on the human antibody titer response following a novel influenza A H1N1 virus vaccination. BMC Immunol. 2012;13(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Lange T, Perras B, Fehm HL, et al. Sleep enhances the human antibody response to hepatitis A vaccination. Psychosom Med. 2003;65(5):831–835. [DOI] [PubMed] [Google Scholar]
- [194].Lange T, Dimitrov S, Bollinger T, et al. Sleep after vaccination boosts immunological memory. J Immunol. 2011;187(1):283–290. [DOI] [PubMed] [Google Scholar]
- [195].Miller GE, Cohen S, Pressman S, et al. Psychological stress and antibody response to influenza vaccination: when is the critical period for stress, and how does it get inside the body? Psychosom Med. 2004;66(2):215–223. [DOI] [PubMed] [Google Scholar]
- [196].Pressman SD, Cohen S, Miller GE, et al. Loneliness, social network size, and immune response to influenza vaccination in college freshmen. Health Psychol. 2005;24(3):297–306. [DOI] [PubMed] [Google Scholar]
- [197].Taylor DJ, Kelly K, Kohut ML, et al. Is insomnia a risk factor for decreased influenza vaccine response? Behav Sleep Med. 2017;15(4):270–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Prather AA, Hall M, Fury JM, et al. Sleep and antibody response to hepatitis B vaccination. Sleep. 2012;35(8):1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Burns VE, Drayson M, Ring C, et al. Perceived stress and psychological well-being are associated with antibody status after meningitis C conjugate vaccination. Psychosom Med. 2002;64(6):963–970. [DOI] [PubMed] [Google Scholar]
- [200].Prather AA, Pressman SD, Miller GE, et al. Temporal links between self-reported sleep and antibody responses to the influenza vaccine. Int J Behav Med. 2021;28(1):151–158. [DOI] [PubMed] [Google Scholar]
- [201].Kuo TH, Williams JA. Increased sleep promotes survival during a bacterial infection in Drosophila. Sleep. 2014;37(6):1077–1086, 1086A–1086D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Williams JA, Sathyanarayanan S, Hendricks JC, et al. Interaction between sleep and the immune response in Drosophila: a role for the NFkappaB relish. Sleep. 2007;30(4):389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Friese RS, Bruns B, Sinton CM. Sleep deprivation after septic insult increases mortality independent of age. J Trauma. 2009;66(1):50–54. [DOI] [PubMed] [Google Scholar]
- [204].Lungato L, Gazarini ML, Paredes-Gamero EJ, et al. Paradoxical sleep deprivation impairs mouse survival after infection with malaria parasites. Malar J. 2015;14(1):183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Ibarra-Coronado EG, Velazquez-Moctezuma J, Diaz D, et al. Sleep deprivation induces changes in immunity in Trichinella spiralis-infected rats. Int J Biol Sci. 2015;11(8):901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Patel SR, Malhotra A, Gao X, et al. A prospective study of sleep duration and pneumonia risk in women. Sleep. 2012;35(1):97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Prather AA, Leung CW. Association of unsufficient sleep with respiratory infection among adults in the United States. JAMA Intern Med. 2016;176(6):850–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Orzech KM, Acebo C, Seifer R, et al. Sleep patterns are associated with common illness in adolescents. J Sleep Res. 2014;23(2):133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Ghilotti F, Pesonen AS, Raposo SE, et al. Physical activity, sleep and risk of respiratory infections: a Swedish cohort study. PLoS One. 2018;13(1):e0190270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Cheshmehzangi A, Chen H, Su Z, et al. How does the COVID-19 fuel insomnia? Brain Behav Immun Health. 2022;21:100426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Li X, Yu H, Bian G, et al. Prevalence, risk factors, and clinical correlates of insomnia in volunteer and at home medical staff during the COVID-19. Brain Behav Immun. 2020;87:140–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Li Y, Chen B, Hong Z, et al. Insomnia symptoms during the early and late stages of the COVID-19 pandemic in China: a systematic review and meta-analysis. Sleep Med. 2021. DOI: 10.1016/j.sleep.2021.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Morin CM, Vezina-Im LA, Ivers H, et al. Prevalent, incident, and persistent insomnia in a population-based cohort tested before (2018) and during the first-wave of COVID-19 pandemic (2020). Sleep. 2022;45(1). DOI: 10.1093/sleep/zsab258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Xu F, Wang X, Yang Y, et al. Depression and insomnia in COVID-19 survivors: a cross-sectional survey from Chinese rehabilitation centers in Anhui province. Sleep Med. 2021. DOI: 10.1016/j.sleep.2021.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Li P, Zheng X, Ulsa MC, et al. Poor sleep behavior burden and risk of COVID-19 mortality and hospitalization. Sleep. 2021;44(8). DOI: 10.1093/sleep/zsab138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Cohen S, Doyle WJ, Alper CM, et al.Sleep habits and susceptibility to the common cold.Arch Intern Med.2009;169(1):62–67;169/1/62 [pii [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Irwin MR, Levin MJ, Laudenslager ML, et al. Varicella zoster virus-specific immune responses to a herpes zoster vaccine in elderly recipients with major depression and the impact of antidepressant medications. Clin Infect Dis. 2013;56(8):1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Cole SW, Arevalo JM, Takahashi R, et al. Computational identification of gene-social environment interaction at the human IL6 locus. Proc Natl Acad Sci U S A. 2010;107(12):5681–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Felten SY, Madden KS, Bellinger DL, et al. The role of the sympathetic nervous system in the modulation of immune responses. Adv Pharmacol. 1998;42(1):583–587. [DOI] [PubMed] [Google Scholar]
- [220].Felten DL. Neurotransmitter signaling of cells of the immune system: important progress, major gaps. Brain Behav Immun. 1991;5(1):2–8. [DOI] [PubMed] [Google Scholar]
- [221].Felten DL, Ackerman KD, Wiegand SJ, et al. Noradrenergic sympathetic innervation of the spleen: i. Nerve fibers associate with lymphocytes and macrophages in specific compartments of the splenic white pulp. J Neurosci Res. 1987;18(1):28–36. [DOI] [PubMed] [Google Scholar]
- [222].Bierhaus A, Wolf J, Andrassy M, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A. 2003;100(4):1920–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Pongratz G, Straub RH. The sympathetic nervous response in inflammation. Arthritis Res Ther. 2014;16(6):504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Harle P, Mobius D, Carr DJ, et al. An opposing time-dependent immune-modulating effect of the sympathetic nervous system conferred by altering the cytokine profile in the local lymph nodes and spleen of mice with type II collagen-induced arthritis. Arthritis Rheum. 2005;52(4):1305–1313. [DOI] [PubMed] [Google Scholar]
- [225].Li W, Knowlton D, Woodward WR, et al. Regulation of noradrenergic function by inflammatory cytokines and depolarization. J Neurochem. 2003;86(3):774–783. [DOI] [PubMed] [Google Scholar]
- [226].Lorton D, Lubahn C, Bellinger DL. Potential use of drugs that target neural-immune pathways in the treatment of rheumatoid arthritis and other autoimmune diseases. Curr Drug Targets Inflamm Allergy. 2003;2(1):1–30. [DOI] [PubMed] [Google Scholar]
- [227].Olofsson PS, Rosas-Ballina M, Levine YA, et al. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev. 2012;248(1):188–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Lange T, Born J. The immune recovery function of sleep - tracked by neutrophil counts. Brain Behav Immun. 2011;25(1):14–15. [DOI] [PubMed] [Google Scholar]
- [229].Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16(1):22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Floam S, Simpson N, Nemeth E, et al. Sleep characteristics as predictor variables of stress systems markers in insomnia disorder. J Sleep Res. 2015;24(3):296–304. [DOI] [PubMed] [Google Scholar]
- [231].Paugh SW, Bonten EJ, Evans WE. Inflammasome-mediated glucocorticoid resistance: the receptor rheostat. Mol Cell Oncol. 2016;3(1):e1065947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Walsh CP, Lim A, Marsland AL, et al. Circulating Interleukin-6 concentration covaries inversely with self-reported sleep duration as a function of polymorphic variation in the glucocorticoid receptor. Brain Behav Immun. 2019;78:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Boudreau P, Yeh WH, Dumont GA, et al. Circadian variation of heart rate variability across sleep stages. Sleep. 2013;36(12):1919–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Somers VK, Dyken ME, Mark AL, et al. Sympathetic-nerve activity during sleep in normal subjects. N Engl J Med. 1993;328(5):303–307. [DOI] [PubMed] [Google Scholar]
- [235].Tobaldini E, Cogliati C, Fiorelli EM, et al. One night on-call: sleep deprivation affects cardiac autonomic control and inflammation in physicians. Eur J Intern Med. 2013;24(7):664–670. [DOI] [PubMed] [Google Scholar]
- [236].Bell KA, Kobayashi I, Chen Y, et al. Nocturnal autonomic nervous system activity and morning proinflammatory cytokines in young adult African Americans. J Sleep Res. 2017;26(4):510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].De Lorenzo BH, de Oliveira Marchioro L, Greco CR, et al. Sleep-deprivation reduces NK cell number and function mediated by beta-adrenergic signalling. Psychoneuroendocrinology. 2015;57:134–143. [DOI] [PubMed] [Google Scholar]
- [238].Slavich GM, Irwin MR. From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull. 2014;140(3):774–815. [DOI] [PMC free article] [PubMed] [Google Scholar]