

Abstract
The evaluation of the protein binding partner(s) of biologically important proteins is currently an area of intense research, especially since the development of the yeast two-hybrid assay. However, not all protein–protein interactions uncovered by this assay are biologically relevant and another confirmatory assay must be performed. Ideally, this assay should be rapid, versatile and performed under conditions which mimic the ‘normal’ physiological state as closely as possible. Towards this goal, we have constructed two eukaryotic expression vectors that facilitate the purification of a protein of interest, along with any associated proteins, from mammalian cells. These vectors incorporate the following features: (i) a tetracycline-responsive promoter so that the level of protein production can be regulated; (ii) an N-terminal glutathione S-transferase tag or a triple repeat of the HA1 epitope, to facilitate purification of the protein either by glutathione affinity chromatography or immunoprecipitation, respectively, followed by a multiple cloning site; (iii) the gene for the enhanced green fluorescent protein (for detection of the presence of the fusion protein and subcellular localization); (iv) a puromycin marker for the selection of stable transformants; (v) a truncated EBNA protein and oriP sequence for episomal replication of the vector. These latter two features permit expansion of small cultures of transfected cells under puromycin selection, thereby increasing the amount of tagged protein that can be purified. We show that these vectors can be used to direct the doxycycline-inducible expresssion of tagged proteins and to recover tagged CIP1–p21 protein complexes from HeLa cells. Furthermore, we show that these tagged p21-purified complexes contain both cyclin A and Cdk2, which are known to interact with p21, but not β-actin.
INTRODUCTION
Since the development and first description of the yeast two-hybrid assay 10 years ago (1), this simple and powerful assay has been widely employed for the detection of protein–protein interactions. The ability to genetically screen for proteins which interact with a protein of interest in a eukaryotic organism permits the detection of interactions which otherwise would have been difficult to uncover. However, not all interactions detected with this assay are physiologically relevant or meaningful. Accordingly, secondary assays are generally required to confirm that the observed interaction occurs within mammalian cells. An ideal secondary assay would be one that could be employed for a wide variety of different proteins so that new assays or reagents do not have to be generated for every protein screened. This is especially true for those proteins that may be toxic to the cell when overexpressed or are expressed very poorly, since it may be difficult to obtain sufficient quantities of the desired proteins to perform a confirmatory assay. The confirmatory assay should also be rapid and have high specificity and sensitivity so that any false positives from the yeast two-hybrid assay can be quickly confirmed or dismissed. Towards this goal, we have constructed a pair of vectors that employ a tetracycline-responsive promoter which drives the expression of a tripartite protein fusion which can be easily and rapidly purified from cell extracts. The tetracycline-responsive promoter permits generation of stable transfectants even when the gene of interest is toxic to the cell when expressed, as well as a method to control the level of expression of the protein of interest. To circumvent the requirement for high transfection efficiencies or large-scale transfections to obtain a sufficient amount of protein for protein–protein interaction studies, the vectors can replicate as episomes, thereby eliminating the need for chromosomal integration for the generation of stable transformants. We have also included an antibiotic selection marker which is compatible with all commercially available tetracycline-responsive cell lines, greatly facilitating the isolation of stable transformants. We show that one of these vectors can direct the doxycycline-inducible expression of a tripartite protein containing the Cdk inhibitor known as p21/CIP1/WAF1 and that this fusion protein can be recovered from HeLa cell extracts, along with Cdk2 and cyclin A, two proteins with which it is known to associate.
MATERIALS AND METHODS
Construction of vectors
The precise details of the construction of these vectors are available upon request; however, the derivation of the main components of these vectors are: the tetracycline-inducible promoter was recovered from plasmid pTRE-1 (Clontech Laboratories, Palo Alto, CA); the GST gene was excised from plasmid pBC (2). In order to incorporate the PreScission protease site, the GST gene from this plasmid was digested with BstBI–EcoRI and replaced with the corresponding fragment, containing the PreScission protease site (LEVLFQGP) (3), from pGEX6P-1 (Pharmacia, Piscataway, NJ). The triple repeat of the HA1 epitope (YPYDVPDYA) was PCR amplified from pEF2-HA (a gift from Brian Pollack) using primers with convenient restriction sites. The puromycin-selectable marker under the control of the SV40 early promoter was recovered from pPUR (Clontech Laboratories); the enhanced green fluorescent protein (EGFP) gene was PCR amplified from plasmid pEGFP (Clontech Laboratories) using primers containing convenient restriction sites; the truncated EBNA gene and oriP sequence were obtained from a modified pREP10 plasmid (Invitrogen, San Diego, CA) in which the EcoRI site in this region was deleted. Maps of these vectors are presented in Figure 1. The sequence at the multiple cloning site is the same in both plasmids and is: …ATG—GST or HA1x3—GAA TTC ACG TGT GCC GGC TTC GCG AGC TCG AGA TCT GTA CAG GAT CCG TCG—EGFP.
Figure 1.


Maps of pTIP.GEX6P-1.EGFP and pTIP.HA1x3.EGFP. The vectors include a tetracycline-responsive promoter (TIP) controlling expression of the GST gene or a triple repeat of the HA1 epitope. A PreScission protease site is encoded between the GST gene and the multiple cloning site, followed by the gene for EGFP. The plasmids encode a truncated EBNA protein and an EBV oriP sequence, permitting their replication as an episome within mammalian cells. The sequence of the multiple cloning site is: …ATG—HA1x3 or GST–PreScission protease—GAA TTC ACG TGT GCC GGC TTC GCG AGC TCG AGA TCT GTA CAG GAT CCG TCG—EGFP. Pro, promoter; AMP, β lactamase gene; polyA, poly(A) signal sequence.
Transfection of cells
The HeLa Tet-On cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM l-glutamine, 10% fetal bovine serum and 0.2 mg/ml G418.
For transient transfections, HeLa Tet-On cells were seeded into 6-well plates (4 × 105 cells/well). Duplicate wells were transfected with 2 µg of plasmid DNA using the Lipofectamine Plus Reagent kit (Gibco BRL, Gaithersburg, MD) according to the manufacturer’s protocol. One duplicate well contained no doxycycline (uninduced) and the other well contained 1 µg/ml doxycycline (fusion protein induced). The cells were evaluated 12–24 h after transfection for GFP localization and gene expression by observing the cells with a UV inverted microscope and the appropriate GFP filter set.
For the studies involving purification of tagged proteins from cells, a transfection in a 60 mm plate was performed as indicated above. Three days following transfection, the transfected cells were trypsinized and plated into one 15 cm or three 10 cm plates. After incubation for 24 h to allow the cells to attach to the plate, puromycin (final concentration 1 µg/ml) was added to the cells and they were incubated until the plate became ~50% confluent (generally 10–14 days). Doxycycline then was added to the HeLa Tet-On cells to induce expression of the fusion proteins. The cells were generally harvested 36–48 h after induction.
For the development of stable cell lines, 2 days after transfection the transfected cells were 10-fold serially diluted into 10 cm plates and incubated in the absence of selection for an additional 2 days. Ninety-six hours after transfection, puromycin was added to a final concentration of 1 µg/ml to select for cells that had acquired the plasmids. Medium containing puromycin was changed every 3–4 days and when colonies appeared, generally 12–14 days later, individual puromycin-resistant colonies were selected and divided into two wells of a 24-well culture dish. Half of the cells were induced with 1 µg/ml doxycycline and examined for EGFP expression 24 h later; the other half of the cells were maintained in medium lacking doxycycline to prevent induction of transcription from the tetracycline-responsive promoter. Clones were scored as positive when at least 40% of the cells exhibited green fluorescence under UV light in the presence of doxycycline and <1% of the cells were green in the absence of doxycycline.
Preparation of cellular extracts
Forty-eight hours post-induction with doxycycline, the pTIP.GEX6P-1.p21.EGFP transfected cells were washed briefly with ice-cold phosphate-buffered saline (PBS). The cells were then scraped from each of the plates into 1 ml of ice-cold PBS and transferred to a pre-chilled 15 ml polypropylene tube. The cells were pelleted by centrifugation at 1800 r.p.m. for 5 min at 4°C. After removal of excess PBS, the cell pellet was resuspended in 300 µl of cold HKM buffer [20 mM HEPES, pH 7.5, 5 mM KCl, 0.5 mM MgCl2 and 0.5 mM dithiothreitol (DTT)] containing 1× Complete EDTA-free protease inhibitor cocktail prepared according to the manufacturer’s recommendations (Boehringer-Mannheim, Indianapolis, IN). The cells were disrupted by two 15 s pulses of sonication using an Ultrasonics W-22F cell disruptor at setting 7. Following sonication, 5 M NaCl was added to the lysed extracts to yield a final concentration of 100 mM and the extracts were quick frozen on dry ice and stored at –80°C until processed further.
GST purification
The lysed cellular extracts prepared as described above were thawed slowly on ice and cleared of debris by centrifugation at 14 000 r.p.m. for 10 min at 4°C. Approximately 50 µl of a 50% slurry of glutathione immobilized on Sepharose beads (Pharmacia) were first incubated for 30 min at 4°C with 200 µl of a crude Escherichia coli extract to block non-specific protein binding. After three 1 ml washes with ice-cold Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4·H2O, pH 7.0, 10 mM KCl and 1 mM MgSO4·7H2O) to remove the bacterial proteins, the beads were added to the HeLa cell extract prepared as described above. The mixture was brought to a final volume of 500 µl with Z buffer containing protease inhibitors and incubated for 2 h at 4°C, turning end over end. The proteins bound to the beads were pelleted by brief centrifugation in a microfuge, washed four times with 1 ml of ice-cold Z buffer and then eluted with 10 mM glutathione in Z buffer or 8 U of PreScission protease in 50 µl of PreScission protease buffer (50 mM Tris–HCl pH 7.0, 150 mM NaCl, 1 mM EDTA and 1 mM DTT). The eluted samples were applied to a 10% SDS–PAGE gel and electrophoresed.
Immunoblotting
Following electrophoresis, the gels were soaked for 1 h in transfer buffer (25 mM Tris base, pH 8.3, 0.192 M glycine and 20% methanol). The separated proteins were then transferred to Trans-blot nitrocellulose membranes (Bio-Rad, Hercules, CA) using a Mini Protean gel transfer apparatus overnight at 25 V. Following transfer, the filters were stained with Ponceau stain (Sigma, St Louis, MO) to confirm transfer of the proteins. The Ponceau stain was then washed off the filter and the filter was blocked with 5% non-fat dried milk in TTBS (20 mM Tris base, pH 7.5, 500 mM NaCl and 0.1% Tween-20) for 1 h. After three 5 min washes with TTBS, the blots were incubated with mouse monoclonal anti-p21 187 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit polyclonal anti-cyclin A H432 antibody (Santa Cruz Biotechnology) for 1 h at room temperature. The blots were then washed three times for 5 min with TTBS buffer and incubated for 1 h at room temperature with a 1/3000 dilution of goat anti-mouse IgG alkaline phosphatase conjugate (Bio-Rad) or goat anti-rabbit IgG alkaline phosphatase conjugate (Bio-Rad), respectively. The filters were then washed three times for 5 min each with TTBS buffer and incubated with LumiPhos substrate (Gibco BRL) and exposed to X-ray film overnight. In the morning, the film was developed. Following film development, the filters were washed three times with TTBS to remove the Lumiphos substrate and then incubated with either rabbit polyclonal anti-Cdk2 M2 antibody (Santa Cruz) or mouse monoclonal anti-β-actin antibody (Boehringer-Mannheim) for 1 h at room temperature. The filters were washed as before and then incubated with a 1/3000 dilution of goat anti-rabbit IgG HRP conjugate (Chemicon, Temecula, CA) or a 1/300 dilution of sheep anti-mouse IgG HRP conjugate (Amersham, Arlington Heights, IL), respectively, for ~1 h. After washing, the filters were developed with a DAB substrate kit (Vector Laboratories, Burlingame) and photographed.
RESULTS
In order to have regulated levels of expression of the desired protein in mammalian cells so that stable cell lines could be generated even if the protein were toxic to cells, we chose the tetracycline-responsive promoter system (4,5). This system has been well characterized and a number of different cell lines permitting the induction (Tet-On) or repression (Tet-Off) of the tetracycline-responsive promoter are commercially available.
In order to purify proteins from mammalian cell extracts, we used the GST fusion system (6), since it is rapid, widely used and has been shown previously to be effective in retrieving protein complexes from mammalian cell extracts (2). This ‘tag’ does not appear to affect the binding properties of many proteins nor does the presence of C-terminal fusions alter the ability of GST to bind to glutathione, thus making it a useful ‘universal’ tag. A PreScission protease site (from plasmid pGEX6P-1; Pharmacia) was inserted between the end of the GST gene and the multiple cloning site to permit specific protelytic cleavage between the GST tag and the protein of interest while the fusion protein is still bound to the column. This feature can theoretically increase the level of purification since only the protein of interest fused with EGFP (and any associated proteins) should be released from the column with PreScission protease. Downstream of the PreScission protease site are multiple cloning sites for insertion of a gene of interest. The cloning sites in pTIP.GEX6P-1.EGFP include EcoRI (reading frame GAA TTC), unique blunt-ended restriction sites in all three reading frames (PmlI, NaeI and NruI) and XhoI (reading frame XXC TCG AGX).
Under some circumstances, the generation of fusions with GST may be undesirable. We therefore constructed an identical vector containing a triple repeat of the HA1 epitope (7) as the N-terminal tag in place of GST. This tag is much smaller than GST and can be purified by immunoprecipitation with a monoclonal anti-HA1 antibody. Aside from this N-terminal tag, the vector is identical to that of pTIP.GEX6P-1.EGFP and is named pTIP.HA1x3.EGFP. However, the PmlI site is not unique in this vector so only two blunt-ended restriction sites are suitable for cloning in the multiple cloning site.
To follow the efficiency of transfection, the level of protein production and the subcellular location of the protein of interest, the gene for EGFP (8) was inserted downstream of the multiple cloning sites. Production of the tagged proteins can be checked by UV fluoroscopy. If desired, the EGFP gene can be deleted from the vectors by digestion of the plasmid with the restriction enzyme BglII or BsrGI once production of the fusion protein has been confirmed. Thus, when induced, a tripartite fusion protein is produced in mammalian cells, consisting of the GST or HA1 tag at the N-terminal end, the protein of interest, followed by EGFP at the C-terminal end.
To facilitate the production of stable transformants, the puromycin-selectable marker from plasmid pPUR is present in these vectors. This greatly facilitates the generation of stable transformants since co-transfection with another plasmid containing a selectable marker is not required. Thus, all colonies that form in the presence of puromycin have acquired the plasmid containing the tetracycline-responsive promoter; which is not always true when co-transfections are performed. Puromycin selection is compatible with all commercially available tetracycline-responsive cell lines.
Finally, the truncated EBNA protein gene and the oriP region have been inserted in these vectors so that the plasmids can replicate as episomes, thus increasing the number of stably transfected cells compared to vectors requiring plasmid integration. Regulated expression of the fusion proteins may be less variable with these eipsomes since chromosomal integration, which might affect transcription depending upon the site of integration, is not required. In addition, small-scale transfections can be performed and then expanded under puromycin selection to permit the recovery of relatively large quantities of fusion protein.
Maps of the plasmid constructs are shown in Figure 1.
Induction of tagged protein expression by doxycycline
Since these vectors do not require chromosomal integration and are likely to be present within cells at multiple copy numbers, it was possible that control of the tetracycline-responsive promoter would be affected, leading to leaky expression of the downstream fusion proteins. To check whether the induction remained tetracycline responsive in these plasmids, HeLa Tet-On cells (Clontech) were transfected with pTIP.HA1x3.EGFP. Twelve clones were selected and tested for doxycycline inducibility. Approximately one-third of the clones exhibited doxycycline-inducible expression, which was assessed by screening for green fluorescence. The results for a representative, inducible clone are shown in Figure 2A. No green fluorescence is detectable in these transfected cells in the absence of doxycycline, while ~60% of the cells are green in the presence of 1 µg/ml doxycycline (Fig. 2B). We believe that the remaining 40% of cells from this clone which do not appear to be doxycycline-responsive do harbor pTIP.HA1x3.EGFP since the cells are resistant to puromycin. Indeed, when 5 mM sodium butyrate is added to these cells, virtually 100% of the cells turn green, suggesting that the tetracycline-responsive promoter has been silenced in some manner in these cells but that it can be reversed by sodium butyrate. Western blot analysis of extracts from these transfected cells incubated in the absence and presence of doxycycline confirms that little HA1–EGFP fusion protein is produced in the absence of doxycycline (Fig. 3, UN) while it is readily detectable when doxycycline is present (Fig. 3, IN).
Figure 2.

Photomicrograph of HeLa Tet-On cells harboring pTIP.HA1x3.EGFP. (A) Cells grown in the absence of doxycycline visualized under visible (left) and UV light with an EGFP filter (right). (B) HeLa Tet-On cells with pTIP.HA1x3.EGFP induced with 1 µg/ml doxycycline visualized under visible (left) and UV light with an EGFP filter (right). Magnification ×600.
Figure 3.

Cell lysates were prepared from HeLa Tet-On cells containing pTIP.HA1x3.EGFP and electrophoresed by SDS–PAGE. (A) Coomassie blue stained gel containing extracts from uninduced (UN) and induced (IN) cells. (B) Western blot of identical aliquots probed with a polyclonal anti-EGFP antibody. The appropriately sized HA1-tagged EGFP is expressed in the cells exposed to doxycycline (lane IN) but not in those grown without doxycycline.
Purification of GST from crude lysates and PreScission protease elution
We next determined whether the GST–EGFP fusion protein could be recovered in high purity from mammalian cell extracts transfected with pTIP.GEX6P-1.EGFP. About 1 × 105 HeLa Tet-On cells were transfected with pTIP.GEX6P-1.EGFP and selected for puromycin resistance 4 days later. The total transfection mixture was expanded to ~3 × 106 cells in the presence of puromycin and then induced for 48 h with 1 µg/ml doxycycline. About 50% of the cells appeared to produce fusion protein, as determined by UV microscopy. Cell lysates were prepared and the GST–EGFP fusion protein was purified from the cleared cellular extract using glutathione affinity chromatography. Approximately two-thirds of the resin was eluted with 10 mM glutathione while the remaining one-third was eluted with 8 U of PreScission protease. SDS–PAGE confirmed that the GST–EGFP fusion protein was rather highly purified from crude extracts (see Fig. 4A, lane 2) and that the EGFP moiety could be released from the column and GST with PreScission protease (Fig. 4A, lane 3; note that the released protein is ~26 kDa smaller). Western analysis with anti-EGFP antibody (Fig. 4B) confirmed elution of the GST–EGFP with glutathione and EGFP alone with PreScission protease protein.
Figure 4.

Purification of GST fusion proteins from crude extracts. An aliquot of the cleared lysate from HeLa Tet-On cells harboring pTIP.GEXP-EGFP and induced for 48 h with 1 µg/ml doxycycline was incubated with a slurry of glutathione–Sepharose, washed and then eluted with either 10 mM glutathione or 8 U of PreScission protease. (A) Coomassie blue staining; (B) Western blotting with anti-EGFP antibody. Lane 1, total cellular extract; lane 2, glutathione eluted GST–EGFP; lane 3, PreScission protease eluted EGFP.
Purification of protein complexes
In order to confirm that these vectors could be used to recover protein complexes from mammalian cells, the gene for CIP1/p21 was cloned into pTIP.GEX6P-1.EGFP between the unique EcoRI and XhoI sites. This gene was chosen because p21 has been shown to bind to both Cdk2 and cyclin A (9–12) and because overexpression of this protein in cells causes a G1 arrest. Thus, we believed that it would serve as a useful test for ‘toxic’ proteins. After transfecting HeLa Tet-On cells with pTIPGEX6P-1.p21.EGFP and expanding the transfectants for ~2 weeks under puromycin selection, the cells were induced with doxycycline. As shown in Figure 5, ~50% of the cells appeared green under UV light. Furthermore, the green fluorescence was concentrated in the nuclei of the cells, consistent with the fact that p21 is a nuclear protein. Thus, the presence of the GST and EGFP proteins does not prevent nuclear translocation of p21.
Figure 5.
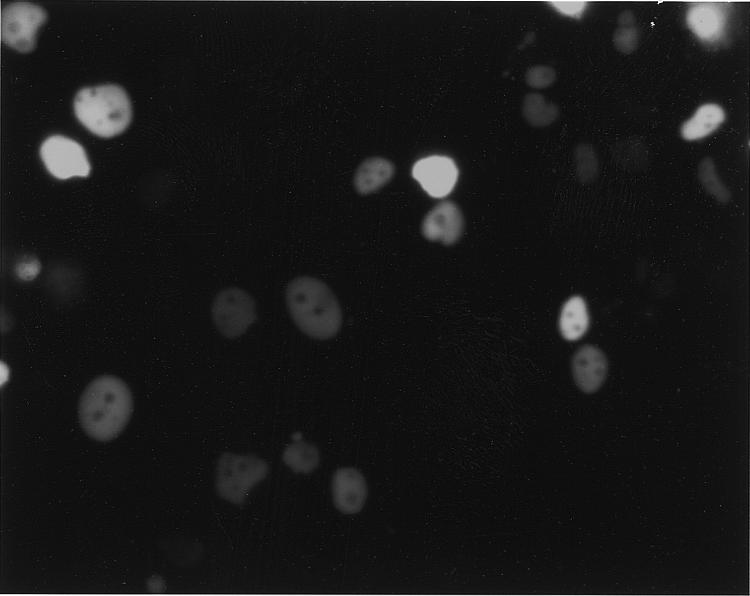
Photomicrograph of HeLa Tet-On cells transfected with pTIP.GEX6P-1.p21.EGFP and induced with 1 µg/ml doxycycline. Approximately half of the cells exhibit green fluorescence of their nuclei, indicating expression and nuclear translocation of the tagged p21 fusion protein. Magnification ×600.
Forty-eight hours after induction, the cells were lysed as indicated in Materials and Methods and glutathione affinity column chromatography was performed to isolate the p21 fusion protein and any associated proteins. The protein which had bound to the glutathione–agarose resin was eluted with 10 mM glutathione and electrophoresed on a 10% SDS–PAGE gel. After electrophoresis, the separated proteins were transferred to a nitrocellulose membrane. After checking for transfer of proteins using Ponceau staining, the filter was probed with anti-p21, anti-cyclin A, anti-Cdk2 and anti-β-actin antibodies.
As shown in Figure 6A, Ponceau staining revealed the presence of an ~73 kDa protein in the glutathione eluate (lane E), the expected molecular weight of the tripartite fusion protein. Anti-p21 antibody staining confirmed that this band was positive for p21 (data not shown). Anti-cyclin A and anti-Cdk2 antibody staining detected significant amounts of these proteins in both the total cell lysate and eluate fractions (Fig. 6B and C, lanes T and E). This binding was specific for p21 since staining with anti-β-actin readily detected the presence of this protein in the total cell extract (Fig. 6D, lane T) but not in the eluted protein fraction (Fig 6D, lane E). Purification of the control GST–EGFP fusion protein from HeLa Tet-On cells failed to recover any detectable cyclin A or Cdk2 [Fig. 7, lanes 2 (αcyclin A) and 4 (αCdk2)]. Thus, glutathione affinity chromatography could specifically capture protein complexes from mammalian cells containing the GST–p21–EGFP fusion protein.
Figure 6.

Purification of p21-containing complexes from HeLa cells. HeLa Tet-On cells transfected with pTIP.GEX6P-1.p21.EGFP were induced and lysed as described in Materials and Methods. The p21 fusion protein was purified from the cleared cell lysates by glutathione–Sepharose affinity chromatography. After extensive washing, the bound proteins were eluted with 10 mM glutathione and subjected to SDS–PAGE and western blotting. (A) Ponceau staining of the total cell lysate (T) and proteins eluted from the glutathione–Sepharose column (E). (B) Filter probed with anti-cyclin A antibody. (C) Filter probed with anti-Cdk2 antibody. (D) Filter probed with anti-actin antibody.
Figure 7.

Purification of GST–EGFP, GST–cyclin A–EGFP and GST–HRS–EGFP from HeLa cells. HeLa Tet-On cells transfected with pTIP.GEX6P-1.EGFP, pTIP.GEX6P-1.cyclinA.EGFP and pTIP.GEX6P-1.HRS.EGFP were induced and lysed as described in Materials and Methods. The EGFP fusion proteins were purified from the cell lysates by glutathione–Sepharose affinity chromatography. After extensive washing, the bound proteins were eluted with PreScission protease digestion and subjected to SDS–PAGE. The lanes containing the control EGFP eluates were probed with anti-cyclin A antibody (lane 2) and anti-Cdk2 antibody (lane 4) while the lane containing the cyclin A–EGFP eluate was probed with anti-Cdk2 antibody (lane 6). The lane containing the HRS–EGFP eluate was probed with anti-STAM antibody (lane 8). The filters were then stripped and probed with anti-EGFP antibody to confirm the presence of the appropriate bait protein in the eluates (lanes 1, 3, 5 and 7).
To verify that other protein complexes could be isolated using these vectors, the cyclin A gene was cloned into pTIP.GEX6P-1.EGFP. After confirming that the proper fusion protein was produced in HeLa Tet-On cells, the cyclin A fusion protein was purified as already described and the eluate was subjected to western blot analysis with anti-Cdk2 antibody. As shown in Figure 7 (lane 6, αCdk2), Cdk2 was easily detected in the eluate, confirming the recovery of cyclin A–Cdk2 complexes from HeLa cells. The control protein PP2A was not detected in the eluate (data not shown). Finally, the STAM (13,14) protein has been reported to interact with HRS (15), based upon immunoprecipitation studies. The HRS gene was PCR amplified from a human cDNA library and cloned into pTIP.GEX6P.EGFP. Although not highly expressed in HeLa Tet-On cells (Fig. 7, lane 7, HRS), purification of this fusion protein from the cells resulted in specific co-purification of STAM (Fig. 7, lane 8, αSTAM). Thus, these vectors should prove useful for a number of different proteins.
DISCUSSION
In this study we report the construction of two mammalian expression vectors that have several features which make them useful for the purification of protein complexes from mammalian cells. When a gene of interest is inserted in the proper reading frames, a tripartite fusion protein can be synthesized in mammalian cells, consisting of GST or HA1, the protein of interest and EGFP. Expression of this fusion protein is driven by a tetracycline-inducible promoter, making it possible to control its level of expression, an especially useful feature for toxic proteins. The GST portion of the fusion protein permits its rapid purification from mammalian cell extracts under mild conditions, allowing the isolation of intact protein complexes from these cells. A PreScission protease site can be used to cleave the protein of interest from the GST moiety directly from the column, theoretically further improving the degree of specificity of the purification method. Alternatively, the smaller HA1 epitope in pTIP.HA1x3.EGFP can be purified from cells by immunoprecipitation with an anti-HA1 monoclonal antibody. In addition, the EGFP portion of the fusion protein facilitates identification of transfected cells, the extent of protein production and the subcellular location(s) of the fusion protein. This feature may be particularly useful for proteins which are difficult to express in cells since the assay for protein production is rapid and non-destructive to the cells. Finally, the EBNA and oriP sequences, along with the puromycin gene, are helpful in generating and selecting stable transformants.
However, the presence of these multiple tags may interfere with binding of some proteins. For example, the EGFP tag may interfere with protein–protein interactions under some conditions, particularly if protein binding is located near the C-terminal domain of the protein and the EGFP fusion site. For this reason, the vectors have been designed so that the EGFP gene can be removed by digestion with BglII or BsrGI once the gene of interest has been cloned into the vector and expressed in the cells to confirm its tetracycline-inducible expression. Similarly, the GST moiety is rather large (~26 kDa) and may cause steric hindrance under some circumstances. If so, use of the pTIP.HA1x3.EGFP vector, with its much smaller tag (<3 kDa), and immunoprecipitation with an anti-HA1 antibody may overcome this difficulty.
Preliminary experiments with a number of different genes inserted into these vectors and transfected into HeLa Tet-On cells indicate that regulation of the tetracycline-inducible promoter may not always be tightly regulated; in some cases, a small percentage of cells appear to express the tagged fusion protein even when the cells are maintained in the ‘uninduced’ state (i.e. in the absence of doxycyline). This appears to be more pronounced within 48 h of transfection and when the cells are trypsinized, so it might be advisable to minimize the number of times the cells are split, especially when dealing with toxic proteins or trying to recover large amounts of fusion protein. In other cases, puromycin-resistant clones are obtained but no EGFP fusion protein is produced even in the presence of high levels of doxycycline. In these cases, high levels of EGFP fusion protein can usually be induced within 24 h by adding 5 mM sodium butyrate to the culture medium, indicating that the cells have acquired the plasmid but have silenced the tetracycline-responsive promoter for some reason. The presence of the EGFP moiety as part of the fusion protein has made it possible to rapidly assess all puromycin-resistant clones for inducibility and production in these difficult cases, which otherwise would be a very time-consuming and expensive procedure if assessed by western blot analysis. In any event, we have been able to select stable cell lines in HeLa Tet-On cells which are tetracycline-inducible with the p21-containing plasmid described in this report and this p21 fusion protein is active since it can induce G1 arrest when expressed at high levels (data not shown).
In conclusion, we believe that these vectors will facilitate the study of protein–protein interactions in mammalian cells and will prove especially helpful for isolating protein complexes containing a protein of interest from mammalian cells, especially if that protein of interest is toxic to the cell when overexpressed.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported in part by grant CA68192 to J.G.
REFERENCES
- 1.Fields S. and Song,O. (1989) Nature, 340, 245–247. [DOI] [PubMed] [Google Scholar]
- 2.Chatton B., Bahr,A., Acker,J. and Kedinger,C. (1995) Biotechniques, 18, 142–145. [PubMed] [Google Scholar]
- 3.Walker P.A., Leong,L.E., Ng,P.W., Tan,S.H., Waller,S., Murphy,D. and Porter,A.G. (1994) Biotechnology, 12, 601–605. [DOI] [PubMed] [Google Scholar]
- 4.Gossen M. and Bujard,H. (1992) Proc. Natl Acad. Sci. USA, 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gossen M., Freundlieb,S., Bender,G., Muller,G., Hillen,W. and Bujard,H. (1995) Science, 268, 1766–1769. [DOI] [PubMed] [Google Scholar]
- 6.Smith D. and Johnson,K. (1988) Gene, 67, 31–40. [DOI] [PubMed] [Google Scholar]
- 7.Wilson I.A., Niman,H.L., Houghten,R.A., Cherenson,A.R., Connolly,M.L. and Lerner,R.A. (1984) Cell, 37, 767–778. [DOI] [PubMed] [Google Scholar]
- 8.Cormack B., Valdivia,R. and Falkow,S. (1996) Gene, 173, 33–38. [DOI] [PubMed] [Google Scholar]
- 9.Lin J., Reichner,C., Wu,X. and Levine,A. (1996) Mol. Cell. Biol., 16, 1786–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harper J., Adami,G., Wei,N., Keyomars,K. and Elledge,S. (1993) Cell, 75, 805–816. [DOI] [PubMed] [Google Scholar]
- 11.Hall M., Bates,S. and Peters,G. (1995) Oncogene, 11, 1581–1588. [PubMed] [Google Scholar]
- 12.Fotedar R., Fitzgerald,P., Rousselle,T., Cannella,D., Doree,M., Messier,H. and Fotedar,A. (1996) Oncogene, 12, 2155–2164. [PubMed] [Google Scholar]
- 13.Takeshita T., Arita,T., Asao,H., Tanaka,N., Higuchi,M., Kuroda,H., Kaneko,K., Munakata,H., Endo,Y., Fujita,T. and Sugamura,K. (1996) Biochem. Biophys. Res. Commun., 225, 1035–1039. [DOI] [PubMed] [Google Scholar]
- 14.Takeshita T., Arita,T., Higuchi,M., Asao,H., Endo,K., Kuroda,H., Tanaka,N., Murata,K., Ishii,N. and Sugamura,K. (1997) Immunity, 6, 449–457. [DOI] [PubMed] [Google Scholar]
- 15.Asao H., Sasaki,Y., Arita,T., Tanaka,N., Endo,K., Kasai,H., Takeshita,T., Endo,Y., Fujita,T. and Sugamura,K. (1997) J. Biol. Chem., 272, 32785–32791. [DOI] [PubMed] [Google Scholar]