

Abstract
We have developed a novel method, termed loop-mediated isothermal amplification (LAMP), that amplifies DNA with high specificity, efficiency and rapidity under isothermal conditions. This method employs a DNA polymerase and a set of four specially designed primers that recognize a total of six distinct sequences on the target DNA. An inner primer containing sequences of the sense and antisense strands of the target DNA initiates LAMP. The following strand displacement DNA synthesis primed by an outer primer releases a single-stranded DNA. This serves as template for DNA synthesis primed by the second inner and outer primers that hybridize to the other end of the target, which produces a stem–loop DNA structure. In subsequent LAMP cycling one inner primer hybridizes to the loop on the product and initiates displacement DNA synthesis, yielding the original stem–loop DNA and a new stem–loop DNA with a stem twice as long. The cycling reaction continues with accumulation of 109 copies of target in less than an hour. The final products are stem–loop DNAs with several inverted repeats of the target and cauliflower-like structures with multiple loops formed by annealing between alternately inverted repeats of the target in the same strand. Because LAMP recognizes the target by six distinct sequences initially and by four distinct sequences afterwards, it is expected to amplify the target sequence with high selectivity.
INTRODUCTION
Nucleic acid amplification is one of the most valuable tools in virtually all life science fields, including application-oriented fields such as clinical medicine, in which diagnosis of infectious diseases, genetic disorders and genetic traits is particularly benefited by this new technique. In addition to the widely used PCR-based detection (1,2), several amplification methods have been invented. They include nucleic acid sequence-based amplification (NASBA) (3), self-sustained sequence replication (3SR) (4) and strand displacement amplification (SDA) (5,6). Each of these amplification methods has its own innovation to re-initiate new rounds of DNA synthesis. For example, PCR uses heat denaturation of double-stranded DNA products to promote the next round of DNA synthesis. 3SR and NASBA eliminate heat denaturation by using a set of transcription and reverse transcription reactions to amplify the target sequence. Similarly, SDA eliminates the heat denaturation step in cycling DNA synthesis by employing a set of restriction enzyme digestions and strand displacement DNA synthesis with modified nucleotides as substrate.
These methods can amplify target nucleic acids to a similar magnitude, all with a detection limit of less than 10 copies and within an hour or so, but still have shortcomings to overcome (7,8). They require either a precision instrument for amplification or an elaborate method for detection of the amplified products due to poor specificity of target sequence selection. Despite the simplicity and the obtainable magnitude of amplification, the requirement for a high precision thermal cycler in PCR prevents this powerful method from being widely used, such as in private clinics as a routine diagnostic tool. On the other hand, NASBA and 3SR, which do not use thermal cycling, are compromised in specificity, resulting mainly from the necessity to use a relatively low temperature of 40°C for amplification. SDA largely overcomes these shortcomings by using four primers and isothermal conditions for amplification, but still has weak points: increased backgrounds due to digestion of irrelevant DNA contained in the sample and the necessity to use costly modified nucleotides as substrate. Although the use of multiple primers, such as in nested PCR and SDA, has improved amplification specificity for the target sequence, residual co-amplification of irrelevant sequences still causes a general setback in nucleic acid amplification, particularly for diagnostic use.
We have recently developed a novel method that can amplify a few copies of DNA to 109 in less than an hour under isothermal conditions and with greater specificity. We describe the mechanism, sensitivity and specificity of this amplification method, termed loop-mediated isothermal amplification (LAMP).
MATERIALS AND METHODS
DNA oligonucleotides
Primer BIP for M13mp18 (M13BIP) consisted of the complementary sequence (24 nt) of B1, a TTTT linker and B2 (24 nt): 5′-CGACTCTAGAGGATCCCCGGGTAC-TTTT-TGTTGTGTGGAATTGTGAGCGGAT-3′. Primer FIP for M13mp18 (M13FIP) consisted of F1c (25 nt), a TTTT linker and the complementary sequence of F2c (22 nt): 5′-ACAACGTCGTGACTGGGAAAACCCT-TTTT-GTGCGGGCCTCTTCGCTATTAC-3′. Primers B3 and F3 for M13mp18 (M13B3 and M13F3) were 5′-ACTTTATGCTTCCGGCTCGTA-3′ and 5′-GTTGGGAAGGGCGATCG-3′, respectively. The probes used for Southern blot hybridization were 5′-AAGCTTGGCACTGGCCGTCGT-3′ (M13-281) and 5′-GTTACCCAACTTAATCGCCTTGCAGCACAT-3′ (M13-333). The primers used for amplification of the HBs region of hepatitis virus B (HBV) DNA were 5′-GATAAAACGCCGCAGACACATCCTTCCAACCTCTTGTCCTCCAA-3′ (HBVBIP), 5′-CCTGCTGCTATGCCTCATCTTCTTTGACAAACGGGCAACATACCTT-3′ (HBVFIP), 5′-CAAAATTCGCAGTCCCCAAC-3′ (HBVB3) and 5′-GGTGGTTGATGTTCCTGGA-3′ (HBVF3). The primers used for prostate-specific antigen mRNA amplification were 5′-TGTTCCTGATGCAGTGGGCAGCTTTAGTCTGCGGCGGTGTTCTG-3′ (PSABIP), 5′-TGCTGGGTCGGCACAGCCTGAAGCTGACCTGAAATACCTGGCCTG-3′ (PSAFIP), 5′-TGCTTGTGGCCTCTCGTG-3′ (PSAB3) and 5′-GGGTGTGGGAAGCTGTG-3′ (PSAF3).
Reaction mixture for LAMP
LAMP was carried out in a total 25 µl reaction mixture containing 0.8 µM each FIP and BIP, 0.2 µM each F3 and B3, 400 µM each dNTP, 1 M betaine (Sigma), 20 mM Tris–HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 4 mM MgSO4, 0.1% Triton X-100 and the specified amounts of double-stranded target DNA. The mixture was heated at 95°C for 5 min, then chilled on ice, 8 U Bst DNA polymerase large fragment (New England Biolabs) were added, followed by incubation at 65°C for 1 h and heating at 80°C for 10 min to terminate the reaction.
Analysis of product
Aliquots of 5 µl of LAMP products and 1 µl of the products digested with restriction enzymes were electrophoresed in 2% agarose gels (0.5× TBE) followed by staining with SYBR Green I (Molecular Probes Inc.). Southern blot analyses were performed by transfer to Hybond N+ nylon membrane (Amersham-Pharmacia). Oligonucleotide probes end-labeled with a DIG Oligonucleotide Tailing Kit (Roche Diagnostics) were used for detection according to the manufacturer’s protocol. An aliquot of 5 µl of LAMP products was also run for 14 h in a 0.7% alkaline agarose gel containing 50 mM NaOH and 1 mM EDTA, followed by neutralization with 1 M Tris–HCl (pH 8.0), and staining with SYBR Green I.
Amplification of HBV DNA cloned in pBR322
HBV viral DNA (type adr) was cut with BamHI, inserted into the BamHI site of pBR322, digested with EcoRV and then used as template. The LAMP reaction mixture was the same as for M13mp18 DNA except for the use of 1.6 µM each HBVFIP and HBVBIP primers and 0.2 µM each HBVF3 and HBVB3 primers. The LAMP reaction was carried out at 60°C for 45 min. Aliquots of 2 µl of the amplification products were mixed with 300 µl of 1/10 000 diluted original SYBR Green I in 10 mM Tris–HCl (pH 8.0), and 1 mM EDTA, incubated at room temperature for 30 min and quantified for fluorescent intensity with a Shimadzu RF-5000 spectrophotometer.
RESULTS AND DISCUSSION
The LAMP method
This method relies on auto-cycling strand displacement DNA synthesis that is performed by a DNA polymerase with high strand displacement activity and a set of two specially designed inner and two outer primers. In the initial steps of the LAMP reaction, all four primers are used, but later during the cycling reaction only the inner primers are used for strand displacement DNA synthesis. The inner primers are called the forward inner primer (FIP) and the backward inner primer (BIP), respectively, and each contains two distinct sequences corresponding to the sense and antisense sequences of the target DNA, one for priming in the first stage and the other for self-priming in later stages. For ease of explanation, the sequences (typically 23–24 nt) inside both ends of the target region for amplification in a DNA are designated F2c and B2, respectively (Fig. 1). Two inner sequences (typically 23–24 nt) 40 nt from the ends of F2c and B2 are designated F1c and B1 and two sequences (17–21 nt) outside the ends of F2c and B2 are designated F3c and B3. Given this structure, the sequences of FIP and BIP were designed as follows. FIP contains F1c, a TTTT spacer and the sequence (F2) complementary to F2c. BIP contains the sequence (B1c) complementary to B1, a TTTT spacer and B2. The two outer primers consist of B3 and the sequence (F3) complementary to F3c, respectively. A DNA sample containing the target sequence and the four primers is heat denatured and rapidly cooled on ice. The LAMP reaction is then initiated by addition of the Bst DNA polymerase large fragment and carried out at 65°C for 1 h.
Figure 1.

Schematic representation of the mechanism of LAMP. (A) Steps in the LAMP reaction. This figure shows the process that starts from primer FIP. However, it should be remembered that DNA synthesis can also begin from primer BIP. (B) Schematic presentation of the structure of LAMP products in a linearized DNA form. B+, B–, F+ and F– stand for the DNA structures shown in the boxes on the left. +, the target sequence flanked by B1 and F1c; –, the complementary sequence.
The mechanism and expected reaction steps of LAMP are illustrated in Figure 1. Inner primer FIP hybridizes to F2c in the target DNA and initiates complementary strand synthesis (Fig. 1A). Outer primer F3, which is a few bases shorter and lower in concentration than FIP, slowly hybridizes to F3c in the target DNA and initiates strand displacement DNA synthesis, releasing a FIP-linked complementary strand, which can form a looped out structure at one end (structure 4). This single-stranded DNA serves as template for BIP-initiated DNA synthesis and subsequent B3-primed strand displacement DNA synthesis, leading to the production of a dumb-bell form DNA (structure 6), which is quickly converted to a stem–loop DNA by self-primed DNA synthesis (structure 7). This stem–loop DNA then serves as the starting material for LAMP cycling, the second stage of the LAMP reaction.
To initiate LAMP cycling, FIP hybridizes to the loop in the stem–loop DNA (structure 7) and primes strand displacement DNA synthesis, generating as an intermediate one gapped stem–loop DNA with an additional inverted copy of the target sequence in the stem and a loop formed at the opposite end via the BIP sequence (structure 8). Subsequent self-primed strand displacement DNA synthesis yields one complementary structure of the original stem–loop DNA (structure 10) and one gap repaired stem–loop DNA with a stem elongated to twice as long (double copies of the target sequence) and a loop at the opposite end (structure 9). Both these products then serve as template for a BIP-primed strand displacement reaction in the subsequent cycles, a part of which is designated the elongation and recycling step, illustrated in the right half of Figure 1A. Thus, in LAMP the target sequence is amplified 3-fold every half cycle.
The final products are a mixture of stem–loop DNAs with various stem lengths and cauliflower-like structures with multiple loops formed by annealing between alternately inverted repeats of the target sequence in the same strand (Fig. 1, structures 16–18). The structures of the cycling intermediate and final products are schematically illustrated in Figure 1B in linearized DNA form.
The use of four primers (recognition of six distinct sequences) in the initial steps of LAMP and two primers (recognition of four distinct sequences) during the subsequent steps ensures high specificity for target amplification. Moreover, in LAMP four primers (six distinct recognition sequences) are simultaneously used to initiate DNA synthesis from the original unamplified DNA to generate a stem–loop DNA for subsequent LAMP cycling, during which the target is recognized by four sequences. Therefore, target selectivity is expected to be higher than those obtained in PCR and SDA.
LAMP amplification of M13 DNA as a model
In order to demonstrate the mechanism, the efficiency, the specificity and the ease of use of LAMP, we chose M13mp18 DNA as a model target DNA and prepared four primers that met the LAMP requirements (Fig. 2A). The reaction was carried out at 65°C for 1 h and the products were separated by agarose gel electrophoresis and identified by restriction enzyme digestion and Southern blot hybridization with appropriate probes (Fig. 2A and B). The LAMP reaction produced many bands of different sizes from ~300 bp to the loading well (Fig. 3A, lane 4). Production of the bands depended on the presence of the inner primers, the template and DNA polymerase. When the products were analyzed by alkaline agarose gel electrophoresis, smeared DNA between bands and at the well (shown in Fig. 3A, lane 4) was shifted to bands of <10 kb (Fig. 3E). Thus, we attributed the slow migrating DNA and the DNA in the loading well to replicating intermediates containing single-stranded loops, as shown in Figure 1A.
Figure 2.

(A) Nucleotide sequence of M13mp18 used for designing the inner and outer primers. The nucleotide sequence of the sense strand of M13mp18 DNA is shown. DNA sequences used for primer design are shown by heavy lines. Probe sequences used for Southern blot hybridization are indicated by dotted lines and the restriction sites for BamHI, PstI and PvuII are indicated by boxes. Numbers at the left end correspond to the positions in M13mp18 (GenBank accession no. X02513). (B) Schematic representation of the anticipated structure of the amplified product. B+, B–, F+ and F– in the first row are as in Figure 1B. The second row indicates the probes used for Southern blot hybridization. The restriction sites for PstI, PvuII and BamHI are shown as lines and the sizes of the restriction fragments are in the boxes.
Figure 3.

Restriction analysis and Southern blot hybridization of the amplified M13mp18 DNA. (A) Electrophoretic analysis of the LAMP amplified M13mp18 product. Six hundred copies of M13mp18 DNA were amplified by LAMP with the specific primers designed on the sequences shown in Figure 2 and run on a 2% agarose gel followed by SYBR Green I staining. Lane M, 100 bp ladder used as size marker (New England Biolabs); lane 1, M13mpl8 DNA digested with PvuII; lane 2, LAMP without Bst DNA polymerase; lane 3, LAMP without target M13 DNA; lane 4, complete LAMP; lanes 5–7, complete LAMP products after digestion with BamHI, PstI and PvuII, respectively (one fifth of the digests were loaded). (B–D) Southern blot analysis of the LAMP products. The 2% agarose gel shown in (A) was used for Southern blot hybridization with M13-281 DNA (B), M13-333 DNA (C) and M13BIP (D) as probes. (E) Alkaline agarose gel electrophoresis of the LAMP products. Lane m, λ DNA HindIII digests; lane 4, the same sample as in (A).
To confirm the structure, the amplified products were digested with several restriction endonucleases and their sizes analyzed by electrophoresis. BamHI cuts B1, PstI cuts between F1c and B1, and PvuII cuts between F1c and F2c (Fig. 2A). Consequently, if the amplification products had exactly the structures shown in Figure 1, the products would be fragmented to 101 and 230 bp fragments by BamHI digestion, 137 and 194 bp fragments by PstI digestion, and 237, 315 and 347 bp fragments by PvuII digestion. As shown in Figure 3A, the sizes of the fragments generated were approximately 100 and 230 bp for BamHI, 140 and 200 bp for PstI, and 240, 320 and 350 bp for PvuII digestion (Fig. 3A, lanes 5–7), in good agreement with the predicted sizes (Fig. 2B). To further confirm their structure, the restriction digests were analyzed by Southern blot hybridization using the sequence complementary to the inner region between F1c and B1 (M13-281), the sequence complementary to the inner region between F1c and F2c (M13-333), and the BIP primer itself as hybridization probes. M13-281 and M13-333 hybridized to the 194 bp PstI product, but not to the 137 bp fragment (Fig. 3B and C, lane 6). In contrast, BIP hybridized to the 137 bp, but not the 194 bp fragment (Fig. 3D, lane 6). Similarly, Southern blot results of the PvuII and BamHI digests perfectly agreed with the conclusion that the amplified DNA originated from target M13 DNA (Fig. 3B–D, lanes 5 and 7). The structures of the amplified products were also confirmed by cloning and sequencing. Several bands shown in Figure 3A, lane 4, were isolated and cloned after digestion with mung bean nuclease. The sequences of the cloned DNAs perfectly agreed with the expected nucleotide sequences (data not shown).
Optimized conditions for LAMP
Since hybridization of the four primers to the target DNA in the initial step was critical for the efficiency of LAMP, the sequences and sizes of the primers were chosen so that their melting temperatures (Tm) fell within certain ranges. The F2 and B2 sequences in FIP and BIP were chosen such that their Tm values fell between 60 and 65°C, the optimal temperature for Bst polymerase. The Tm values of F1c and B1c were set slightly higher than those of F2 and B2 in order that a looped out structure formed immediately after release of the single-stranded DNA from the template. Furthermore, the Tm values of the outer primers (F3 and B3) were set lower than those of F2 and B2 in order to ensure that synthesis occurred earlier from the inner primers than from the outer primers. In addition, the outer primers were used at 1/4–1/10 the concentration of the inner primers.
The formation of a stem–loop DNA (sees Fig. 1, structure 7) from a dumb-bell structure (structure 6) is critical for LAMP cycling. We examined the effect of various sizes of loop between F2c (B2c) and F1c (B1c) on amplification efficiencies and found that a loop of 40 bases or longer gave the best results (data not shown).
The efficiency of LAMP depends on the size of the target DNA because one rate limiting step for amplification in this method is strand displacement DNA synthesis. We tested various sizes of target DNA and found that the best results could be obtained with 130 to 200 bp DNAs. DNA of more than 500 bp amplified, but very poorly. Therefore, the size of target DNA should be set to less than 300 bp, including F2 and B2.
DNA polymerase is another critical factor for efficient amplification. The best amplification was obtained with Bst polymerase or BcaBEST DNA polymerase (TaKaRa) for less than 10–23 mole target DNA. Z-Taq DNA polymerase (TaKaRa) was less efficient under the current conditions, but might be useful when polymerase has to be added before heat denaturation of the target DNA, because it is thermostable.
Chemicals destabilizing the DNA helix were found to markedly elevate amplification efficiencies in LAMP. The presence of 0.5–1.5 M betaine (N,N,N-trimethylglycine) or l-proline, which reduce base stacking (9–11), stimulated not only the overall rate of the reaction, but also increased target selectivity with a significant reduction in amplification of irrelevant sequences.
Sensitivity of LAMP
LAMP is highly sensitive and able to detect DNA at as few as six copies in the reaction mixture. As shown in Figure 4A, in a 45 min LAMP reaction six copies of the HBV target were amplified to a detectable level. As expected, use of inner primers that do not form a looped out structure led to no amplification (Fig. 4B, lanes 5–8). In the absence of one of the outer primers no significant amplification occurred with 60 copies of the HBV target (Fig. 4B, lanes 2–4), indicating a strict requirement for recognition of six distinct sequences in the target DNA in LAMP.
Figure 4.

Sensitivity of LAMP. (A) Time course of the LAMP reaction with various amounts of HBV DNA. Various numbers of copies of HBV DNA were amplified by LAMP. At various times, the reaction was terminated and the amounts of products quantified by measuring fluorescence intensity of SYBR Green I. (B) Requirements for primers in the LAMP reaction. Sixty copies of HBV DNA were amplified by LAMP with omission of one or two of the primers. The products were electrophoresed in 2% agarose gels and stained. –, the corresponding primer was omitted from the reaction; B2 and F2, BIP and FIP were replaced by B2 and F2, respectively, which do not contain B1c and F1c and, therefore, are unable to form the looped out structure. (C) The effect of the presence of genomic DNA on sensitivity. Various numbers of copies of HBV DNA were amplified at 60°C for 60 min in the absence or presence of 100 ng of human genomic DNA and the products separated by gel electrophoresis. Lane M, 100 bp ladder size markers (TaKaRa); lanes 1–4, LAMP carried out in the absence of human genomic DNA; lanes 5–8, LAMP carried out in the presence of 100 ng genomic DNA; lanes 1 and 5, LAMP without HBV DNA; lanes 2 and 6, with six copies; lanes 3 and 7, with 60 copies; lanes 4 and 8, with 600 copies of HBV DNA. Lanes 9 and 10 are EarI digests (1/5 vol) of the same amplified DNAs as in lanes 2 and 6, respectively. (D) Nested PCR of HBV DNA under similar conditions. Single and nested PCR reactions were performed in a 50 µl reaction mixture containing 2.5 U AmpliTaq Gold (PE Biosystems), 0.2 µM each primer (first PCR, HBVB2/HBVF2 or HBVB1/HBVF1; second PCR, HBVB1/HBVF1), 1× GeneAmp PCR buffer (10 mM Tris–HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.01% gelatin) and 0.2 mM dNTPs. The sequences of the primers used were 5′-CCAACCTCTTGTCCTCCAA-3′ for HBVB2, 5′-GACAAACGGGCAACATACCTT-3′ for HBVF2, 5′-GGATGTGTCTGCGGCGTTTTATC-3′ for HBVB1 and 5′-AGAAGATGAGGCATAGCAGCAGG-3′ for HBVF1. Both the first and second round nested PCRs were carried out as follows: preincubation at 95°C for 10 min; 40 cycles each of 30 s at 95°C, 30 s at 60°C and 1 min at 72°C. One microliter of the first PCR products (HBVB2/HBVF2) was subjected to second PCR. An aliquot of 10 µl of the reaction products was analyzed by 4% agarose gel (0.5× TBE) electrophoresis followed by staining with SYBR Green I.
LAMP seems less prone to the presence of irrelevant DNA than PCR. The presence of 100 ng of human genomic DNA in the LAMP reaction for six copies of HBV target neither significantly adversely affected the amplification efficiency nor generated significant background (Fig. 4C). Single PCR, which was performed under the same conditions as for LAMP, failed to amplify six copies of HBV in the absence and 60 copies in the presence of 10–100 ng of the genomic DNA, though nested PCR overcame this problem (Fig. 4D).
LAMP for a RNA target
LAMP is also applicable to RNA upon use of reverse transcriptase (RTase) together with DNA polymerase (12). This method (reverse transcription-coupled LAMP) easily detected prostate-specific antigen (PSA) mRNA in one PSA-expressing LNCaP cell mixed with 1 000 000 PSA-negative K562 cells (13–15; Fig. 5, lane 6). This amplification depended on both RTase and Bst polymerase (Fig. 5, lanes 2 and 4) and the product was authenticated by Sau3AI digestion (Fig. 5, lanes 8 and 9).
Figure 5.
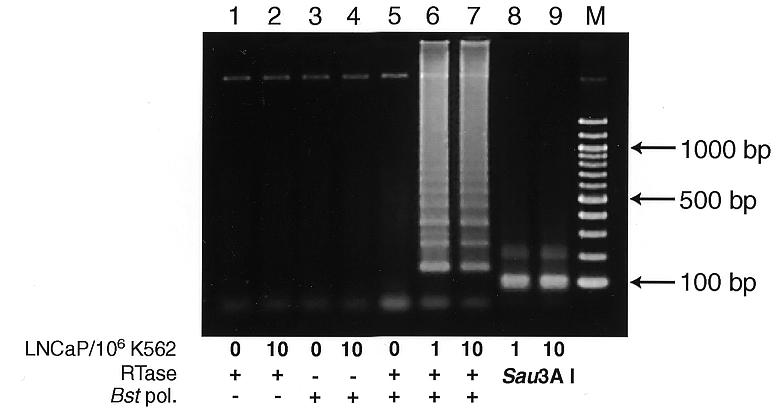
Detection of PSA mRNA by reverse transcription-coupled LAMP (RT-LAMP). Various numbers of LNCaP cells were mixed with 106 PSA-non-producing K562 cells and total RNA was extracted. RT-LAMP was carried out in the same reaction mixture as for M13mp18 DNA amplification except that 1.6 µM each PSAFIP and PSABIP, 0.2 µM each PSAF3 and PSAB3, 0.8 M betaine, 5 mM DTT, 16 U Bst polymerase, 100 U ReverTra Ace (Toyobo) and 5 µg of extracted RNA were used. All the above components were mixed at once on ice and were incubated at 65°C for 45 min. The products were electrophoresed in 2% agarose gel followed by SYBR Green I staining. + and –, RT-LAMP carried out in the presence and absence of Bst DNA polymerase or ReverTra Ace, respectively. Lanes 8 and 9, the same products (1/5 vol) as in lanes 6 and 7, respectively, but digested with Sau3AI; lane M, 100 bp ladder (New England Biolabs).
Advantages of LAMP
(i) LAMP amplifies DNA with high efficiency under isothermal conditions without a significant influence of the co-presence of non-target DNA. Its detection limit is a few copies, being comparable to that of PCR. (ii) The products are a mixture of stem–loop DNAs with various sizes of stem and cauliflower-like structures with multiple loops induced by annealing between alternately inverted repeats of the target sequence in the same strand, the latter of which would enable their simple, easy and selective detection, such as via mechanisms similar to multivalent antigen–antibody interactions. (iii) LAMP is highly specific for the target sequence. This is attributable to recognition of the target sequence by six independent sequences in the initial stage and by four independent sequences during the later stages of the LAMP reaction. This partly alleviates the general problem of backgrounds associated with all nucleic acid amplification methods. (iv) LAMP is simple and easy to perform once the appropriate primers are prepared, requiring only four primers, a DNA polymerase and a regular laboratory water bath or heat block for reaction. (v) By combination with reverse transcription, LAMP can amplify RNA sequences with high efficiency.
Acknowledgments
ACKNOWLEDGEMENT
We thank Dr K. Tatsumi for helpful comments on this manuscript.
REFERENCES
- 1.Saiki R.K., Scharf,S., Faloona,F., Mullis,K.B., Horn,G.T., Erlich,H.A. and Arnheim,N. (1985) Science, 230, 1350–1354. [DOI] [PubMed] [Google Scholar]
- 2.Saiki R.K., Gelfand,D.H., Stoffel,S., Scharf,S.J., Higuchi,R., Horn,G.T., Mullis,K.B. and Erlich,H.A. (1988) Science, 239, 487–491. [DOI] [PubMed] [Google Scholar]
- 3.Compton J. (1991) Nature, 350, 91–92. [DOI] [PubMed] [Google Scholar]
- 4.Guatelli J.C., Whitfield,K.M., Kwoh,D.Y., Barringer,K.J., Richman,D.D. and Gingeras,T.R. (1990) Proc. Natl Acad. Sci. USA, 87, 1874–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker G.T., Fraiser,M.S., Schram,J.L., Little,M.C., Nadeau,J.G. and Malinowski,D.P. (1992) Nucleic Acids Res., 20, 1691–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker G.T., Little,M.C., Nadeau,J.G. and Shank,D.D. (1992) Proc. Natl Acad. Sci. USA, 89, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abramowitz S. (1996) Trends Biotechnol., 14, 397–401. [DOI] [PubMed] [Google Scholar]
- 8.Vrana K.E. (1996) Trends Biotechnol., 14, 413–415. [DOI] [PubMed] [Google Scholar]
- 9.Rees W.A., Yager,T.D., Korte,J. and von Hippel,P.H. (1993) Biochemistry, 32, 137–144. [DOI] [PubMed] [Google Scholar]
- 10.Baskaran N., Kandpal,R.P., Bhargava,A.K., Glynn,M.W., Bale,A. and Weissman,S.M. (1996) Genome Res., 6, 633–638. [DOI] [PubMed] [Google Scholar]
- 11.Rajendrakumar C.S., Suryanarayana,T. and Reddy,A.R. (1997) FEBS Lett., 410, 201–205. [DOI] [PubMed] [Google Scholar]
- 12.Whiting S.H. and Champoux,J.J. (1998) J. Mol. Biol., 278, 559–577. [DOI] [PubMed] [Google Scholar]
- 13.Katz A.E., Olsson,C.A., Raffo,A.J., Cama,C., Perlman,H., Seaman,E., O’Toole,K.M., McMahon,D., Benson,M.C. and Buttyan,R. (1994) Urology, 43, 765–775. [DOI] [PubMed] [Google Scholar]
- 14.Katz A.E., de Vries,G.M., Begg,M.D., Raffo,A.J., Cama,C., O’Toole,K., Buttyan,R., Benson,M.C. and Olsson,C.A. (1995) Cancer, 75, 1642–1648. [DOI] [PubMed] [Google Scholar]
- 15.Gala J.L., Heusterspreute,M., Loric,S., Hanon,F., Tombal,B., Van Cangh,P., De Nayer,P. and Philippe,M. (1998) Clin. Chem., 44, 472–481. [PubMed] [Google Scholar]