

Abstract
Aim
The purpose of this study is to evaluate the safety and pharmacokinetics of the novel morpholino oligomer NS‐089/NCNP‐02 which can induce exon 44 skipping, in patients with DMD. Additionally, we aimed to identify markers predictive of therapeutic efficacy and determine the optimal dosing for future studies.
Methods
This is an open‐label, dose‐escalation, two‐center phase I/II trial in ambulant patients with DMD, presence of an out‐of‐frame deletion, and a mutation amenable to exon 44 skipping. Part 1 is a stepwise dose‐finding stage (4 weeks) during which NS‐089/NCNP‐02 will be administered intravenously at four dose levels once weekly (1.62, 10, 40, and 80 mg/kg); Part 2 is a 24‐week evaluation period based on the dosages determined during Part 1. The primary (safety) endpoints are the results of physical examinations, vital signs, 12‐lead electrocardiogram and echocardiography tests, and adverse event reporting. Secondary endpoints include expression of dystrophin protein, motor function assessment, exon 44 skipping efficiency, plasma and urinary NS‐089/NCNP‐02 concentrations, and changes in blood creatine kinase levels.
Discussion
Exon‐skipping therapy using ASOs shows promise in selected patients, and this first‐in‐human study is expected to provide critical information for subsequent clinical development of NS‐089/NCNP‐02.
Keywords: antisense oligonucleotide, DMD, Duchenne muscular dystrophy, exon 44, exon‐skipping, first‐in‐human study, NS‐089/NCNP‐02, pharmacokinetics, safety, study protocol
The purpose of this study is to evaluate the safety and pharmacokinetics of the novel morpholino oligomer NS‐089/NCNP‐02 which can induce exon 44 skipping, in patients with DMD. Additionally, we aimed to identify markers predictive of therapeutic efficacy and determine the optimal dosing for future studies. Exon‐skipping therapy using ASOs shows promise in selected patients, and this first‐in‐human study is expected to provide critical information for subsequent clinical development of NS‐089/NCNP‐02.
1. BACKGROUND
Globally, the X‐linked recessive disorder Duchenne muscular dystrophy (DMD) is reported to occur with a birth prevalence of 19.8 per 100 000 males. 1 DMD is the most common form of childhood‐onset muscular dystrophy, caused by mutations in the DMD gene that result in absent or insufficient levels of the functional cytoskeletal protein dystrophin. 2 This underlying pathophysiology results in progressive muscular degeneration and damage and, ultimately, early mortality. 3
Owing to its chromosomal localization, DMD predominantly affects male children, while females are generally (although not exclusively) asymptomatic carriers. 4 Clinical symptoms include muscle weakness and degeneration in early childhood; the diagnosis of DMD was based on the results of genetic analysis by multiplex ligation‐dependent probe amplification (MLPA) and direct sequencing of the DMD gene. 3 By the age of 12, individuals with DMD are typically non‐ambulant. They require a wheelchair 2 and commonly develop additional orthopedic problems such as scoliosis. 5 This, in turn, can lead to chest cavity reduction and respiratory dysfunction. 2 The development of cardiomyopathy in young adulthood may cause additional breathing difficulties. 6 As a result, for most individuals with DMD, the average life expectancy is approximately 30 years. 3
Until now, many available treatments (e.g., surgery for spinal deformity, cardiac failure management, respiratory support, physiotherapy) addressed only the symptoms of DMD and were able to slow the course of the disease but could not halt the progressive muscle loss. 7 The only accepted disease‐modifying treatment has been the use of corticosteroids, which can suppress muscle inflammation; however, the efficacy of corticosteroid treatment is limited, and side effects can be considerable. 8 , 9
Based on the clear need for improved disease‐modifying treatments for DMD, several radical therapies have been studied in recent years, including gene therapy and stem cell transplantation. 10 Exon‐skipping therapy, which induces an in‐frame mutation in mature messenger RNA (mRNA) with sequence‐specific antisense oligonucleotides (ASOs), is of particular interest to DMD researchers and clinicians. 7 , 11 Exon‐skipping aims to mask specific exons in the DMD gene transcripts, thereby overcoming the out‐of‐frame mutation that underlies DMD and restoring the expression of a shorter but functional dystrophin protein. 12 , 13 , 14 In‐frame deletions result in the production of truncated dystrophin, which leads to a less severely affected Becker muscular dystrophy (BMD). Several exon‐skipping therapeutic options have now been approved for use in the United States (US), although their availability elsewhere differs. Eteplirsen (Exondys 51) is an exon 51‐skipping ASO that was approved to treat patients with DMD by the Food and Drug Administration (FDA) in the US in 2016 15 ; however, it has not yet been approved by the European Medicines Agency for use in Europe. Golodirsen (Vyondys 53) is an exon 53‐skipping ASO approved by the FDA in 2019. 16 Viltolarsen (NS‐065/NCNP‐01; Viltepso) is another exon 53‐skipping ASO, and this treatment was approved in 2020 by both the FDA 17 and the Japanese Pharmaceuticals and Medical Devices Agency. 18
Viltolarsen (NS‐065/NCNP‐01) was discovered and developed by the National Center of Neurology and Psychiatry (NCNP) in collaboration with Nippon Shinyaku Co., Ltd., Tsukuba, Ibaraki. In preclinical studies, this morpholino oligomer was found to promote skipping of exon 53 in a dose‐dependent manner and restore dystrophin protein levels in patient‐derived cells. 19 Based on the reported locations of mutations in the DMD gene, it was anticipated that NS‐065/NCNP‐01 could benefit the 6%–9.4% of patients with DMD amenable to exon 53 skipping, 20 and early‐stage clinical development began in 2016. 21 Based on positive phase II data, 22 the FDA granted conditional approval to viltolarsen in 2020, pending the ongoing phase III RACER53 trial. 23
Subsequently, the NCNP researchers and Nippon Shinyaku Co., Ltd. are collaborating to develop another novel morpholino oligomer, NS‐089/NCNP‐02, which induces exon 44 skipping to correct the open reading frame. This exon was selected because the neighboring exon 45 is the single exon most commonly deleted 24 ; in theory, in patients with exon 45 mutations, skipping of exon 44 should restore the open reading frame to allow translation of a partially functional dystrophin protein and result in less severe disease. 24 It will be anticipated that NS‐089/NCNP‐02 could benefit the 7%–11% of patients with DMD amenable to exon 44 skipping. 25 To verify the exon‐skipping efficiency of NS‐089/NCNP‐02 in cells derived from DMD patients, primary fibroblasts were obtained from patients with DMD amenable to exon 44 skipping. After patient fibroblasts were transfected with the human MYOD gene to induce differentiation into myotubes, the myotubes were treated with NS‐089/NCNP‐02 in the presence of Endo‐Porter as a delivery agent, and the effects of exon 44 skipping were measured by RT‐PCR 2 days after the start of treatment. We confirmed NS‐089/NCNP‐02 induced efficient exon 44 skipping in cells from a patient with a deletion of exons 45 or exons 45–54 (i.e., a complete inability to produce functional dystrophin; NCNP and Nippon Shinyaku Co., Ltd., data on file). Based on these results, there is an expectation that NS‐089/NCNP‐02 could induce dystrophin expression and suppress disease progression in patients amenable to exon 44 skipping (under review).
For trials of radical therapies in DMD, there is still an ongoing debate about the best method for showing clinical benefit. 7 , 26 In general, the usefulness of an exon‐skipping drug should be based on demonstrating its exon‐skipping efficiency and its effect on dystrophin restoration. Thus, we planned this phase I/II study to undertake an early‐phase clinical evaluation of NS‐089/NCNP‐02 in a limited number of patients. One of the specific objectives of this study was to assess the potential utility of NS‐089/NCNP‐02 as a drug for the treatment of patients with DMD by evaluating its safety and pharmacokinetics (PK; under review). Additionally, we aimed to identify markers predictive of therapeutic efficacy and determine the optimal dosing for future studies.
2. METHODS/DESIGN
2.1. Study design and setting
This is an open‐label, dose‐escalation, two‐center phase I/II trial. The study is registered at clinicaltrials.gov under the identifier NCT04129294 and with the University hospital Medical Information Network as UMIN000038505. The study has been conducted at the NCNP (Tokyo, Japan) since December 2, 2019, and at Kagoshima University Hospital (Kagoshima, Japan) since January 2021, and the study was completed in May 31, 2022. The study site will be responsible for compensation in the event of a health injury that occurs to a subject as a result of participating in the present study. Any protocol modifications or amendments will require approval from the Institutional Review Board.
The trial was designed in two parts. Part 1 is a stepwise dose‐finding stage (4 weeks). Part 2 is a 24‐week evaluation period based on the dosages determined during Part 1. Interim assessments by the safety monitoring committee are planned to decide whether to transition between dose levels in Part 1 and proceed from Part 1 to Part 2; the relevant criteria are described in Table 1.
TABLE 1.
Criteria to determine whether to proceed to the next dose level and study part.
| Procedures for transition | Criteria to be considered |
|---|---|
| Transition of dose level in Part 1 | |
|
(1) An ADR of Grade ≥3 in at least 1 subject (2) Any of the following criteria in at least 2 subjects
a
|
| Transition from Part 1 to Part 2 | |
|
The determination will be made considering the following items:
|
Abbreviations: ADR, adverse drug reaction; AE, adverse event; ALT, alanine aminotransferase; HPF, high power field; LPF, low power field; P/C, protein/creatinine; PK, pharmacokinetic; PT‐INR, prothrombin time‐international normalized ratio; RBC, red blood cell; ULN, upper limit of normal.
Baseline values, and other clinical findings and laboratory test data, will be taken into consideration by the Safety Monitoring Committee when evaluating the relevance of ADRs.
2.2. Study population
Patients enrolled in the study will be assigned subject identification codes to ensure subject confidentiality, and these codes will be used in case report forms and all documents related to the conduct of the study.
The study population will include ambulant patients (although non‐ambulant patients may be enrolled depending on enrollment status) with DMD, presence of an out‐of‐frame deletion (confirmed by MLPA), and a dystrophin mutation amenable to exon 44 skipping (confirmed by direct sequencing). The key inclusion criteria are male patients who are ≥4 and <17 years of age at the time of obtaining informed consent and who have a life expectancy of ≥1 year; presence of an out‐of‐frame deletion (confirmed by MLPA) that can be restored to an in‐frame deletion by exon 44 skipping of the DMD gene; ability to undergo muscle biopsy (i.e., lacking severe atrophy of the biceps brachii muscle or tibialis anterior muscle); corrected QT interval (QTc) < 450 ms on standard 12‐lead electrocardiogram (ECG; based on Fridericia's formula), or QTc < 480 ms for patients with bundle branch block; and systemic corticosteroid therapy (if any) started ≥6 months prior to enrollment with the dosage unchanged within 3 months of enrollment.
The study exclusion criteria include participation in any other clinical trial designed to restore the expression of dystrophin protein or related substances, or receipt of any other investigational drug within 3 months before the start of administration of NS‐089/NCNP‐02; impaired respiratory function to the extent that continuous use of a ventilator (excluding non‐invasive positive pressure ventilation while sleeping) is necessary; a forced vital capacity of <50% of the predicted value (based on the reference values for spirometry in Japanese children 27 ), a left ventricular ejection fraction of <40%, or fractional shortening of <25% by ECHO; presence of any immunodeficiency, autoimmune disease, active or uncontrolled infection, cardiomyopathy, hepatic disease, renal disease, a positive test result for hepatitis B virus surface antigen, hepatitis C virus antibody, or human immunodeficiency virus antibody; any recent (within 3 months of enrollment) or planned surgery; a history of serious hypersensitivity reactions to pharmacologic drugs; and any patient unable to comply with the study procedures due to cognitive challenges or any patient whose safety might be compromised by study participation.
2.3. Study treatment
During the study, NS‐089/NCNP‐02 is planned to be administered at four dose levels (1.62 mg/kg once weekly [QW], 10 mg/kg QW, 40 mg/kg QW, and 80 mg/kg QW). Dosing Level 1 (1.62 mg/kg QW) was selected as the first dose based on the results of animal toxicity studies (NCNP and Nippon Shinyaku Co., Ltd., data on file). Dosing Level 4 (80 mg/kg QW) was calculated based on a combination of data from the toxicity studies in animals (NCNP and Nippon Shinyaku Co., Ltd., data on file) and PK inferences from the related morpholino oligomer viltolarsen (NS‐065/NCNP‐01) administered at a dose of 80 mg/kg in humans. 21 The area under the plasma concentration–time curve from 0 to 24 h (AUC0‐24) when viltolarsen was administered at 80 mg/kg in humans did not exceed that of NS‐089/NCNP‐02 administered at the maximum tolerated dose in animals. It was, therefore, considered possible to administer NS‐089/NCNP‐02 at a dose of up to 80 mg/kg QW. NS‐089/NCNP‐02 will be administered intravenously over 1 h (±10 min).
2.4. Study procedures
In Part 1 of the study, Cohort 1 (three patients) will initially receive NS‐089/NCNP‐02 at dosing Level 1 (1.62 mg/kg QW) for 2 weeks and at dosing Level 3 thereafter (40 mg/kg QW) for 2 weeks (Figure 1). Cohort 2 (three patients) will initially receive NS‐089/NCNP‐02 at dosing Level 2 (10 mg/kg QW) for 2 weeks and at dosing Level 4 thereafter (80 mg/kg QW) for 2 weeks. In Part 2 of the study, two different doses of NS‐089/NCNP‐02 (determined from the results of Part 1) will be intravenously administered QW for 24 weeks. Patients who have completed Part 1 are allowed to participate in Part 2. Muscle biopsies will be performed twice, at the beginning of Part 1 and at the end of Part 2.
FIGURE 1.
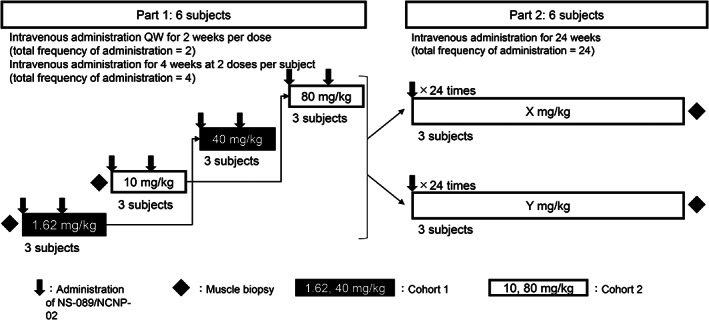
Study design and flow. In Part 1, the dose of NS‐089/NCNP‐02 will be escalated in a stepwise manner from Level 1 (1.62 mg/kg QW) to Level 4 (80 mg/kg QW). A total of four doses will be intravenously administered to two cohorts consisting of three subjects each, and each cohort will receive two doses. In Part 2, two doses of NS‐089/NCNP‐02 (determined from the results of Part 1) will be intravenously administered once weekly for 24 weeks. QW, once weekly.
If a patient discontinues, an additional subject will be added to evaluate three patients at each dose level. The second patient at each dose level in Part 1 will start receiving NS‐089/NCNP‐02 after the second administration of NS‐089/NCNP‐02 in the first patient. An interval of at least 4 weeks was mandated between dose levels in each cohort.
Physiotherapy or exercise therapy regimens must be continued without change between 90 days before starting the treatment and the end of the treatment period at Part 2; no new therapies can be initiated during the study period. Pre‐existing concomitant drugs (including steroids) may be used between enrollment and the end of the treatment period at Part 2. Other investigational drugs, adenosine–triphosphate products, idebenone, and other coenzyme Q10 products will be prohibited.
The schedule of observations and examinations is shown in Figure 2.
FIGURE 2.

Study observations and examinations: implementation schedules. (A) Part 1; (B) Part 2. aPatients who complete Level 1 or 2 and proceed to Level 3 or 4 will not have a pre‐treatment observation period prior to the administration at Level 3 or 4 but will have their height/weight measured during the period between Days −7 and −1 before administration of NS‐089/NCNP‐02. bConsent may be obtained before the day of interim enrollment. cInterim enrollment. After obtaining informed consent, the investigator or sub‐investigator will confirm that the patient is a candidate for exon 44 skipping treatment and enroll the patient. The results of exon 44 sequence analysis shall be confirmed before formal enrollment. dFormal enrollment. Patients will be enrolled after confirming examination results obtained within 14 days before the start of administration of NS‐089/NCNP‐02. Formal enrollment on the day before the first administration may be accepted. eTo be performed at the end of administration and at 2 h after the end of administration. fTo be performed at the following times: 30 min after the start of administration on Day 1; at the end of administration on Day 1; at 1, 2, 4, and 6 h after the end of administration on Day 1; at the end of administration on Day 8; and at 1 h after the end of administration on Day 8. gFor Part 1, the results of hepatitis B antigen, hepatitis C antibody, and human immunodeficiency virus antibody tests must be confirmed by the day of formal enrollment; such tests will not be performed post‐treatment for Part 1 and pre‐treatment for Part 2. hTo be performed at Levels 3 and 4. iMuscle and skin samples will be taken simultaneously with muscle biopsy. Urine samples will be collected at Part 2, if it could not be collected in a pre‐treatment observation period at Part 1. jTo be performed after implementing motor function assessment and confirming the muscle tissue to be sampled by MRI. In addition, this is to be performed before the start of administration (pre‐treatment for Part 1). Subjects will be admitted at the time of muscle biopsy. kIn Part 1, the motor function assessment will be performed between Days −28 and −1. In Parts 1 and 2, the 2‐min walk test will be conducted on a different day from other motor function parameters. lTo be performed 30 min after the start of administration, at the end of administration, and at 15 min, and 1, 2, 4, and 8 h after the end of administration. mTo be performed 23 h after the end of administration. nTo be performed before administration. oTo be performed on Day169 or 176. pTo be performed at the end of administration and 1 h after the end of administration. qMuscle sample is collected at the same time as muscle biopsy. ECG, electrocardiogram; MRI, magnetic resonance imaging.
2.5. Study endpoints
The primary (safety) endpoints are the results of physical examinations, vital signs, 12‐lead ECG, echocardiography tests, and adverse event (AE) reporting. Measures include DMD gene tests (MLPA analysis), exon 44 sequencing analysis (by polymerase chain reaction using genome DNA derived from peripheral venous blood), hematology, blood chemistry, urinalysis, cytokine/complement levels (Table 2), immunology tests, and pulmonary function tests. AEs and adverse drug reactions (ADRs) will be coded using the Medical Dictionary for Regulatory Activities version 23.1. Blood samples, muscle tissues, dermal fibroblasts, and urine‐derived cells collected will be anonymized and stored at a genetic testing laboratory or NCNP with no time limit.
TABLE 2.
Laboratory test measures.
| Test | Parameter |
|---|---|
| Hematology | RBC, hemoglobin, hematocrit, reticulocytes, MCV, MCH, MCHC, WBC, platelet count, WBC differential (neutrophil, eosinophil, basophil, monocyte, lymphocyte), fibrinogen, aPTT, and PT‐INR |
| Blood chemistry | Na, K, Ca, Cl, inorganic P, BUN, creatinine, cystatin C, AST, ALT, GGT, ALP, haptoglobin, LDH, CK/CK‐MB/CK‐MM, total bilirubin, direct bilirubin, cardiac troponin T, BNP, total protein, albumin, A/G ratio, total cholesterol, triglyceride, blood glucose, uric acid, and CRP |
| Urinalysis | Glucose, occult blood, urobilinogen, specific gravity, osmotic pressure, sediment (red blood cell, white blood cell, cast), protein (CBB method) a , albumin a , NAG a , α1‐microglobulin a , creatinine a , Na a , K a , Cl a , inorganic P a , and β2‐microglobulin a |
| Cytokine/complement levels | IL‐6, TNF‐α, MCP‐1, CH50, C3, and C4 |
Abbreviations: A/G, albumin/globulin; ALP, alkaline phosphatase; ALT, alanine aminotransferase; aPTT, activated partial thromboplastin time; AST, aspartate aminotransferase; BNP, brain natriuretic peptide; BUN, blood urea nitrogen; C3/C4, complement proteins 3 and 4; CBB, Coomassie brilliant blue; CH50, 50% hemolytic complement; CK, creatine kinase (CK‐MB, heart isoenzyme; CK‐MM, muscle and heart isoenzyme); CRP, C‐reactive protein; GGT, gamma‐glutamyl transpeptidase; IL, interleukin; LDH, lactate dehydrogenase; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCP‐1, monocyte chemoattractant protein‐1; MCV, mean corpuscle volume; NAG, N‐acetyl‐β‐D‐glucosaminidase; PT‐INR, prothrombin time‐international normalized ratio; RBC, red blood cells; TNF, tumor necrosis factor; WBC, white blood cells.
Measurements were conducted using 24‐h pooled urine on Days 2, 4, and 11 of Part 1, and Days 2 and 163 of Part 2.
Planned secondary endpoints include dystrophin protein expression (by Western blot analysis), motor function assessment, exon 44 skipping efficiency, plasma and urinary NS‐089/NCNP‐02 concentrations, and changes in blood creatine kinase levels. Motor function assessments include the North Star Ambulatory Assessment score 28 ; the time to stand from supine test 29 ; the 10‐m walk/run test 30 ; the 6‐min walk test 31 ; the 2‐min walk test 32 ; the timed “up & go” test 33 ; quantitative muscle strength assessment (knee flexion/extension, elbow flexion/extension, and shoulder abduction); and grip strength, pinch strength, and upper limb performance assessed using the Performance of the Upper Limb module for DMD. 34
Additional study examinations included skeletal muscle magnetic resonance imaging (to determine the amount of muscle that can be biopsied); in vitro assays using MYOD‐transduced urine‐derived cells (MYOD‐UDCs), dermal MYOD‐fibroblasts, and myoblasts derived from the six enrolled subjects in the trial, evaluated by dystrophin RT‐PCR and Western blot analysis; and plasma and urinary PK parameters. The timings of the blood and urine collections are shown in File S1. NS‐089/NCNP‐02 concentration and PK parameters will be measured by high‐performance liquid chromatography with tandem mass spectrometry.
2.6. Statistical analysis
The sample size in each study part was set at six patients. This sample size was determined based on the feasibility of enrollment and motor function assessment at a single center. No statistical significance was considered for the determination of sample size. Data will be assessed in three analysis sets (safety, efficacy, and PK). The safety analysis set will include all patients treated at least once with NS‐089/NCNP‐02. The efficacy and PK analysis sets will include all patients enrolled, excluding those who have a serious eligibility violation and those who lack the applicable data.
Patient demographic data will be tabulated using summary statistics. The primary analysis (safety evaluation) will include tabulation of AEs and ADRs by number and frequency and graphical representation of changes from baseline in vital signs, mean laboratory test values, ECG, echocardiography, and pulmonary function parameters. The efficacy and PK analyses will include an itemization of motor function testing and calculation of NS‐089/NCNP‐02 exposure (including the maximum plasma concentration [C max] and AUC0‐24), half‐life, systemic clearance, and distribution volume. Analyses will also include tabulation of administration of NS‐089/NCNP‐02 (number of administrations and exposure level by dose in each of the study parts). Analyses will be conducted using SAS software version 9.4 (SAS Institute Inc., Cary, NC, USA) and Microsoft Excel 2016/2019.
2.7. Data control and dissemination
All data and processes will be audited by independent monitors. The study data will be published in peer‐reviewed journals, using anonymized information to preserve patient confidentiality.
3. DISCUSSION
This is an exploratory phase I/II study of NS‐089/NCNP‐02. The protocol is reported according to the Standard Protocol Items: Recommendation for Interventional Trials (SPIRIT) 2013 Checklist (File S2).
DMD is a disease with a significant impact on life expectancy, and although radical therapies are being investigated, these generally involve complex procedures and other challenges. Exon‐skipping therapy using ASOs is a treatment that shows promise in selected patients. This first‐in‐human study is expected to provide critical information for subsequent clinical development of NS‐089/NCNP‐02, with the ultimate aim of expanding the treatment armamentarium for affected patients.
In this study, the in vitro assay, which includes the differentiation of urine‐derived cells into myocytes, is based on the authors' recently published technique to create a novel DMD muscle cell model. 35 This method could be used to screen different ASOs prior to commencing clinical trials, 20 , 36 potentially streamlining the therapeutic development process and reducing the time to availability of new treatment strategies. Recently, we have reported an efficient cellular skeletal muscle modeling in DMD using MYOD‐UDCs obtained from DMD patients. 35 UDCs might be an ideal cellular model for neuromuscular diseases because they have the high proliferative ability and can be collected non‐invasively. However, only a few studies are conducted on MYOD‐UDCs, it is desirable to use them in parallel with the well‐characterized MYOD‐fibroblasts model. 37 Very recently, we, for the first time, evaluated the usefulness of MYOD‐UDCs from study subjects to evaluate the efficacy of NS‐089/NCNP‐02 in the DMD clinical trial. We confirmed the drug's dose‐dependent exon 44 skipping and dystrophin protein recovery in MYOD‐UDCs obtained from all the subjects (under review).
The modeling of truncated dystrophin protein lacking exons 44–45 or 45–46 predicts that both should result in comparable stable hybrid rod structure, suggesting that, in patients with exon 45 mutations, skipping of exon 44 or exon 46 should be excellent therapeutic strategies. 24 However, it is already known that the resulting protein is functional enough to maintain a mild BMD phenotype, which generally has mutations in the DMD gene that maintain the open reading frame, allowing the production of dystrophin proteins and thus are partially functional. 38 A documented example of this is the spontaneous skipping of exon 44 that occurs when the exons flanking it are deleted. 39 , 40 Importantly, deletions amenable to exon 44 skipping are usually associated with more dystrophin‐revertant fibers and milder DMD phenotypes. 41 , 42 , 43 As the stability of the dystrophin transcript is a key indicator of dystrophin expression, additional clinical exploration is warranted to discern whether transcript instability in DMD compared with BMD could be a potential biomarker of response to ASOs. 38
Potential study limitations include the small sample size and open‐label design, limiting the conclusions that can be drawn. Then, significant challenges about ASOs are the difficulty in selecting an optimal sequence for exon‐skipping drug, identifying biomarkers of treatment effect, large‐scale pharmaceutical synthesis, and pharmaceutical marketing. Other limitations are the requirement for more sensitive outcome measures to quantify disease severity and our current incomplete understanding of the long‐term natural history of DMD in ambulant patients with mutations amenable to exon 44 skipping.
The duration of the Part 2 is too short to identify potential efficacy, because the progression of DMD in exon 44‐skipped patients is relatively slow. Therefore, a long‐term extension study for 72 weeks is ongoing by our collaborator Nippon Shinyaku (NCT05135663).
In summary, in this phase I/II study, we plan to assess the safety and PK of the novel morpholino oligomer, NS‐089/NCNP‐02, which induces exon 44 skipping in the DMD gene. These data will allow us to determine the optimal dosage for future clinical development and provide the first indication of therapeutic efficacy. As such, the safety, efficacy, and dosing analyses of this study are critical precursors to undertaking later phase studies of NS‐089/NCNP‐02.
Trial status
The study was completed on May 31, 2022. The first patient was included on December 2, 2019. Enrollment is now finished, with the final patient's enrollment on March 24, 2020. The current article is based on protocol version 12.0, prepared on November 11, 2021.
AUTHOR CONTRIBUTIONS
TI, HK, YAs, HN, and YAo conceived and designed the study and were involved in protocol development. TI, YAs, and HN coordinated the regulatory aspects. ET, YSM, AI, CY, and SM were responsible for data acquisition. TI, HK, and EH were responsible for data analysis and interpretation. TI, HK, and YAo wrote the first draft of the manuscript. All other authors critically reviewed the manuscript for intellectual content. All authors read and approved the final version of the manuscript.
FUNDING INFORMATION
This research was supported by the Japan Agency for Medical Research and Development (Grant number 20lm0203086h0002) and an Intramural Research Grant (Grant number 2‐6) from the National Center of Neurology and Psychiatry. Nippon Shinyaku Co., Ltd. Kyoto Japan provided investigational drugs. The study was designed and conducted, analyzed, and interpreted by the investigators independently of Nippon Shinyaku Co., Ltd.
CONFLICT OF INTEREST STATEMENT
TI declares that there is no conflict of interest. HK has received grants from Taiho, Pfizer, Nippon Shinyaku, Daiichi Sankyo, Chugai, and PTC Therapeutics; and personal fees from Sarepta Therapeutics, Nippon Shinyaku, Daiichi Sankyo, Kaneka, and Astellas. YAs declares that there is no conflict of interest. HN has received grants from Pfizer, Nippon Shinyaku, Daiichi Sankyo, and Astellas. NM declares that there is no conflict of interest. ET has received grants from Nippon Shinyaku, Taiho, Takeda, and Daiichi Sankyo. YSM declares that there is no conflict of interest. AI declares that there is no conflict of interest. CY declares that there is no conflict of interest. SM declares that there is no conflict of interest. EH declares that there is no conflict of interest. YAo reports receiving grants for joint research and researchers from Nippon Shinyaku Co., Ltd.
ETHICS APPROVAL
Approval of the research protocol by an Institutional Reviewer Board: This study is being conducted in compliance with the ethical principles that have their origin in the Declaration of Helsinki and all applicable Japanese local and national regulatory laws. The protocol and related documentation were approved by the Institutional Review Board of the NCNP (approval number II‐012).
Informed Consent: Legal representatives for each patient provided written informed consent prior to study participation, and patients provided voluntary assent where possible.
Registry and the Registration No. of the study/trial: This study was registered at clinicaltrials.gov (NCT04129294) and with the University Hospital Medical Information Network (UMIN000038505).
Animal Studies: N/A.
Supporting information
File S1
File S1
ACKNOWLEDGMENTS
The authors would like to thank the patients and their families for participating in this study. The authors also thank Sally‐Anne Mitchell, PhD, of Edanz Pharma for providing medical writing services funded by Nippon Shinyaku Co., Ltd., Kyoto, Japan.
Ishizuka T, Komaki H, Asahina Y, Nakamura H, Motohashi N, Takeshita E, et al. Systemic administration of the antisense oligonucleotide NS‐089/NCNP‐02 for skipping of exon 44 in patients with Duchenne muscular dystrophy: Study protocol for a phase I/II clinical trial. Neuropsychopharmacol Rep. 2023;43:277–286. 10.1002/npr2.12335
Takami Ishizuka and Hirofumi Komaki shared first authorship.
DATA AVAILABILITY STATEMENT
Data are not included in this article for protocol but will be presented in a coming paper summarizing the result of this clinical trial. The datasets are available from the corresponding author upon reasonable request taking into account our patent strategy and its influence on a development of other exon‐skipping therapeutics by our competitors.
REFERENCES
- 1. Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S, Trifiro G. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta‐analysis. Orphanet J Rare Dis. 2020;15(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ryder S, Leadley RM, Armstrong N, Westwood M, de Kock S, Butt T, et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. 2017;12(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giliberto F, Radic CP, Luce L, Ferreiro V, de Brasi C, Szijan I. Symptomatic female carriers of Duchenne muscular dystrophy (DMD): genetic and clinical characterization. J Neurol Sci. 2014;336(1–2):36–41. [DOI] [PubMed] [Google Scholar]
- 5. Archer JE, Gardner AC, Roper HP, Chikermane AA, Tatman AJ. Duchenne muscular dystrophy: the management of scoliosis. J Spine Surg. 2016;2(3):185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Adorisio R, Mencarelli E, Cantarutti N, Calvieri C, Amato L, Cicenia M, et al. Duchenne dilated cardiomyopathy: cardiac management from prevention to advanced cardiovascular therapies. J Clin Med. 2020;9(10):3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Verhaart IEC, Aartsma‐Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. 2019;15(7):373–86. [DOI] [PubMed] [Google Scholar]
- 8. Miyatake S, Shimizu‐Motohashi Y, Takeda S, Aoki Y. Anti‐inflammatory drugs for Duchenne muscular dystrophy: focus on skeletal muscle‐releasing factors. Drug des Devel Ther. 2016;10:2745–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Werneck LC, Lorenzoni PJ, Ducci RD, Fustes OH, Kay CSK, Scola RH. Duchenne muscular dystrophy: an historical treatment review. Arq Neuropsiquiatr. 2019;77(8):579–89. [DOI] [PubMed] [Google Scholar]
- 10. Sun C, Shen L, Zhang Z, Xie X. Therapeutic strategies for Duchenne muscular dystrophy: an update. Genes. 2020;11(8):837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018;17(4):347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dzierlega K, Yokota T. Optimization of antisense‐mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 2020;27(9):407–16. [DOI] [PubMed] [Google Scholar]
- 13. Echevarria L, Aupy P, Goyenvalle A. Exon‐skipping advances for Duchenne muscular dystrophy. Hum Mol Genet. 2018;27(R2):R163–72. [DOI] [PubMed] [Google Scholar]
- 14. Nakamura A. Mutation‐based therapeutic strategies for Duchenne muscular dystrophy: from genetic diagnosis to therapy. J Pers Med. 2019;9(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. US Food and Drug Administration . FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. 19 September 2016. Accessed March 16, 2021. https://www.fda.gov/news‐events/press‐announcements/fda‐grants‐accelerated‐approval‐first‐drug‐duchenne‐muscular‐dystrophy
- 16. US Food and Drug Administration . FDA grants accelerated approval to first targeted treatment for rare Duchenne muscular dystrophy mutation. 12 December 2019. Accessed March 16, 2021. https://www.fda.gov/news‐events/press‐announcements/fda‐grants‐accelerated‐approval‐first‐targeted‐treatment‐rare‐duchenne‐muscular‐dystrophy‐mutation
- 17. US Food and Drug Administration . FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. 12 August 2020. Accessed March 16, 2021. https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐targeted‐treatment‐rare‐duchenne‐muscular‐dystrophy‐mutation
- 18. Dhillon S. Viltolarsen: first approval. Drugs. 2020;80(10):1027–31. [DOI] [PubMed] [Google Scholar]
- 19. Watanabe N, Nagata T, Satou Y, Masuda S, Saito T, Kitagawa H, et al. NS‐065/NCNP‐01: an antisense oligonucleotide for potential treatment of exon 53 skipping in Duchenne muscular dystrophy. Mol Ther Nucleic Acids. 2018;13:442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lim KRQ, Nguyen Q, Yokota T. Genotype‐phenotype correlations in Duchenne and Becker muscular dystrophy patients from the Canadian neuromuscular disease registry. J Pers Med. 2020;10(4):241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Komaki H, Takeshima Y, Matsumura T, Ozasa S, Funato M, Takeshita E, et al. Viltolarsen in Japanese Duchenne muscular dystrophy patients: a phase 1/2 study. Ann Clin Transl Neurol. 2020;7(12):2393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clemens PR, Rao VK, Connolly AM, Harper AD, Mah JK, Smith EC, et al. Safety, tolerability, and efficacy of viltolarsen in boys with Duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol. 2020;77(8):982–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. NS Pharma Inc . Study to assess the efficacy and safety of viltolarsen in ambulant boys with DMD (RACER53). ClinicalTrials.gov Identifier: NCT04060199. Accessed March 16, 2021. https://clinicaltrials.gov/ct2/show/NCT04060199
- 24. Findlay AR, Wein N, Kaminoh Y, Taylor LE, Dunn DM, Mendell JR, et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann Neurol. 2015;77(4):668–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Catherine LB, David S, Soledad M, Maria EF, Kyriaki K, Konstantina K, et al. The TREAT‐NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Straub V, Mercuri E. DMD outcome measure study group. Report on the workshop: meaningful outcome measures for Duchenne muscular dystrophy, London, UK, 30‐31 January 2017. Neuromuscul Disord. 2018;28(8):690–701. [DOI] [PubMed] [Google Scholar]
- 27. Takase M, Sakata H, Shikata M, Tatara K, Fukushima W, Yoshi T, et al. Preparation of spirometry standards for Japanese children (final report). J Jpn Soc Pediatr Resp Dis. 2008;19(2):164–76. [In Japanese]. [Google Scholar]
- 28. Scott E, Eagle M, Mayhew A, Freeman J, Main M, Sheehan J, et al. Development of a functional assessment scale for ambulatory boys with Duchenne muscular dystrophy. Physiother Res Int. 2012;17(2):101–9. [DOI] [PubMed] [Google Scholar]
- 29. Nesbitt D, Molina SL, Cattuzzo MT, Robinson LE, Phillips D, Stodden D. Assessment of a supine‐to‐stand (STS) task in early childhood: a measure of functional motor competence. J Mot Learn Devel. 2017;5:252–66. [Google Scholar]
- 30. Watson MJ. Refining the ten‐metre walking test for use with neurologically impaired people. Physiotherapy. 2002;88(7):386–97. [Google Scholar]
- 31. ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories . ATS statement: guidelines for the six‐minute walk test. Am J Respir Crit Care Med. 2002;166(1):111–7. [DOI] [PubMed] [Google Scholar]
- 32. Butland RJ, Pang J, Gross ER, Woodcock AA, Geddes DM. Two‐, six‐, and 12‐minute walking tests in respiratory disease. Br Med J. 1982;284(6329):1607–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Podsiadlo D, Richardson S. The timed “up & go”: a test of basic functional mobility for frail elderly persons. J Am Geriatr Soc. 1991;39(2):142–8. [DOI] [PubMed] [Google Scholar]
- 34. Mayhew A, Mazzone ES, Eagle M, Duong T, Ash M, Decostre V, et al. Development of the performance of the upper limb module for Duchenne muscular dystrophy. Dev Med Child Neurol. 2013;55(11):1038–45. [DOI] [PubMed] [Google Scholar]
- 35. Takizawa H, Hara Y, Mizobe Y, Ohno T, Suzuki S, Inoue K, et al. Modelling Duchenne muscular dystrophy in MYOD1‐converted urine‐derived cells treated with 3‐deazaneplanocin a hydrochloride. Sci Rep. 2019;9(1):3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sato M, Takizawa H, Nakamura A, Turner BJ, Shabanpoor F, Aoki Y. Application of urine‐derived stem cells to cellular modeling in neuromuscular and neurodegenerative diseases. Front Mol Neurosci. 2019;12:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takizawa H, Takeshita E, Sato M, Shimizu‐Motohashi Y, Ishiyama A, Mori‐Yoshimura M, et al. Highly sensitive screening of antisense sequences for different types of DMD mutations in patients' urine‐derived cells. J Neurol Sci. 2021;423:117337. [DOI] [PubMed] [Google Scholar]
- 38. Anthony K, Arechavala‐Gomeza V, Ricotti V, Torelli S, Feng L, Janghra N, et al. Biochemical characterization of patients with in‐frame or out‐of‐frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurol. 2014;71(1):32–40. [DOI] [PubMed] [Google Scholar]
- 39. Aartsma‐Rus A, Muntoni F. 194th ENMC international workshop. 3rd ENMC workshop on exon skipping: towards clinical application of antisense‐mediated exon skipping for Duchenne muscular dystrophy 8‐10 December 2012, Naarden, The Netherlands. Neuromuscul Disord. 2013;23(11):934–44. [DOI] [PubMed] [Google Scholar]
- 40. Dwianingsih EK, Malueka RG, Nishida A, Itoh K, Lee T, Yagi M, et al. A novel splicing silencer generated by DMD exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J Hum Genet. 2014;59(8):423–9. [DOI] [PubMed] [Google Scholar]
- 41. Brogna C, Coratti G, Pane M, Ricotti V, Messina S, D'Amico A, et al. Long‐term natural history data in Duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. PLoS One. 2019;14(6):e0218683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van den Bergen JC, Ginjaar HB, Niks EH, Aartsma‐Rus A, Verschuuren JJ. Prolonged ambulation in Duchenne patients with a mutation amenable to exon 44 skipping. J Neuromuscul Dis. 2014;1(1):91–4. [PubMed] [Google Scholar]
- 43. Wang RT, Barthelemy F, Martin AS, Douine ED, Eskin A, Lucas A, et al. DMD genotype correlations from the Duchenne registry: endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat. 2018;39(9):1193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1
File S1
Data Availability Statement
Data are not included in this article for protocol but will be presented in a coming paper summarizing the result of this clinical trial. The datasets are available from the corresponding author upon reasonable request taking into account our patent strategy and its influence on a development of other exon‐skipping therapeutics by our competitors.