

Abstract
Herein we describe the research and development of the process for the scale-up total synthesis of largazole, a potent class I selective histone deacetylase (HDAC) inhibitor, a potential anticancer agent and also useful for the treatment of other disorders where transcriptional reprogramming might be beneficial. The synthetic route and conditions for each fragment and final product were modified and optimized to make them suitable for larger scale synthesis. With the process we developed, hundreds of grams of each fragment and decagrams of final productlargazole were synthesized in good to excellent yields. The final target largazole was obtained in 21% overall yield over eight steps based on the longest sequence with over 95% HPLC purity.
Keywords: largazole, anticancer, histone deacetylase (HDAC) inhibitor, scalable process, total synthesis, process chemistry
Graphical Abstract
INTRODUCTION
Largazole is a macrocyclic depsipeptide (Figure 1) marine natural product. It was isolated from a cyanobacterium of genus Symploca by our research group in 20081 (re-classified as a new genus, Caldora penicillata).2 Largazole possesses highly differential growth inhibitory activity towards cancer cells attributed to its potent class I histone deacetylase (HDAC) inhibitory activity.1,3 It is an antiproliferative natural product with selectivity for cancer cells over normal cells.4 The low-nanomolar anticancer agent largazole is at the preclinical stage and shows promising in vivo efficacy.3c,5 The structure was determined by extensive use of 1D and 2D NMR coupled with mass spectrometry and enantioselective analysis of chemical degradation products.1 Hong and Luesch then devised a total chemical synthetic method for largazole,3a providing enough research grade largazole material to establish the mode of action.
Figure 1.
The chemical structure of largazole and largazole thiol
Largazole is a pro-drug and liberates largazole thiol that can complex Zn2+ in the active site of class I HDACs (HDAC1, 2, 3 and 8) (Figure 1, HDAC1 IC50 0.4 nM);3a and it is the most potent known class I HDAC inhibitor, supported by multiple independent groups’ findings.3b,3d Structure–activity relationship (SAR) studies were carried out in due course.3c Largazole has some structural similarity to FK228,3c which is an approved HDAC inhibitor (second after pan-HDAC inhibitor Vorinostat, i.e., SAHA) that requires metabolic activation by disulfide reduction.6 Various other HDAC inhibitors are in various phases of clinical trials.7 The main problems in HDAC inhibitor development include the issue of achieving isoform selectivity, potential risk for cardiovascular side effects and inadequate stability/bioavailability affecting their PK/PD properties.8
While there are several other HDAC inhibitors in development, largazole has several key differentiating factors that warrant further development of this novel class I selective HDAC inhibitor. In addition to potent and selective inhibition of class I isoforms of HDAC (particularly HDAC1, HDAC2 and HDAC3) and inhibition of tumor growth in a mouse xenograft colon cancer model, largazole has shown a collateral benefit of osteogenic activity in mice and rabbits.9 It also displayed beneficial activity in liver cirrhosis models10 as well as excellent synergy with dexamethasone for the treatment of triple-negative breast cancer in a mouse model.11 The pharmacokinetics and bioavailability of largazole have been tested in rats,12 and alternative prodrug strategies12b and zinc-binding groups have also been explored,13 which led to the modulation of the activity profiles. We have demonstrated that largazole has limited oral bioavailability.12b Due to largazole’s unique chemical structure and potential as a drug candidate, numerous other groups have targeted largazole for synthesis,3b,14 or reported additional analogues,12,13,15 or identified further biological properties.16
The largazole skeleton is also a “privileged” scaffold that as a starting point for structure-activity relationship studies to optimize HDAC isoform specificity, which makes it distinct from other HDAC inhibitors in development. In order to conduct further preclinical development studies, the pre-requisite step is to develop a synthetic process that will allow the manufacture of adequate amount of largazole for formulation development, toxicity studies and clinical supply. However, in all published papers,3,12–15 only several milligrams (7.5 – 22 mg) of largazole were synthesized. So far, there is no protocol published for the larger-scale synthesis of largazole. Consistent with our initial synthesis,3a we envisioned a strategy that enables the late stage incorporation of the thioester moiety to allow flexibility for alternative prodrug strategies. Here we report the development of a scalable and practical process for the total synthesis of largazole on decagram scale.
RESULTS AND DISCUSSION
Retrosynthetic Analysis.
The late stage incorporation of the thioester moiety (Figure 2)3a was adopted by other groups14a,14b,15d for small scale synthesis. From the retrosynthetic analysis (Figure 2), largazole (1) can be fused from thioester 2 and cyclic depsipeptide core 3 by olefin cross-metathesis reaction. Compound 3 is obtained from linear compound 4 by macrocyclization, while 4 could be dissected into four basic units: alcohol 9, Fmoc-Val-OH, cyano-substituted thiazole 8 and α-methyl cysteine 7. The building blocks 7, 8, 9, Val and 2, and the incorporation of thioester unit at late stage would be advantageous for “combinatorial” preparation of versatile analogues in HTS screening by expansion of variability of each building block.17 Our investigation results show that this strategy is convenient for scale-up synthesis and the synthesis of diverse analogues.
Figure 2.
Retrosynthetic analysis
Synthesis of fragment 2.
The fragment thioester 2 was synthesized as depicted in Scheme 1.14a,14b Thiooctanoic acid 13 (102 g, 90%) was prepared from one-pot reaction of thioacetamide 10 (53.0 g, 709.4 mmol) with n-octanoyl chloride 11 (115.4 g, 709.4 mmol) via unstable intermediate 1-acylthio-ethaniminium chloride.14a,18 In order to alleviate by-product, n-octanoic acid, equal molar of 10 and 11 should be used. Appropriate concentration of aq. NaOH (i.e., 10%) and its hydrolysis time (i.e., 30 min) were also important to decrease acid by-product. Pure 13 was obtained by simple distillation of crude product under vacuum with heating. The condenser in the distillation apparatus must have a larger diameter ID (≥10 mm) to avoid condensing and clogging by mono-sulfur on the wall. It seemed that thioacid 13 was not stable, so it should be used for the next step as soon as possible (e.g., within several days). Thioester 2 (122 g, 90%) was easily obtained by the reaction of 13 (101 g, 631 mmol) with 1-bromo-3-butene 14 (65 mL, 631 mmol) in the presence of anhydrous K2CO3 in dried acetone.
Scheme 1.
Synthesis of thioester 2
Synthesis of segment 7.
The synthetic procedures of α-methyl-cysteine 7 was shown in Scheme 2.14e,15d Using Seebach procedure,19 thiazolidine 17 was formed by intermolecular condensation of pivalaldehyde 15 (258 g, 3.0 mol) and (R)-cysteine methyl ester hydrochloride 16 (257 g, 1.5 mol). An inlet thermometer must be used to monitor the temperature of the reaction (34–37 °C) because of pentane’s high volatility. Another critical aspect is that an effective recirculating coolant should be used (10–15°C) in condenser. Over-refluxing and ineffective condensing would make pentane escape from the reaction and the apparatus would be at risk of exploding. Usually, a balloon filled with nitrogen was used to alleviate the pressure in the reactor. A Dean-Stark trap was used to collect the produced water, and its theoretical volume (mL) was used as the criteria of completion of reaction (18–22 h). The N-formylation of thiazolidine (17) using Pattenden method20 provided intermediate 18 (300 g, 90%), which was purified firstly by large column and then recrystallized from Et2O/Hexane. Caution: respirator and gloves must be worn during operation since a large volume of corrosive formic acid was used in the reaction. The α-methylation reaction of 18 was the key reaction, in which fresh LDA was made in situ and the temperature must be controlled at –78 ºC. Measures to prevent fire and explosion must be considered because a large volume of n-butyllithium solution in hexane was used in the reaction. This step was very sensitive to humidity and temperature. Aldehyde 18 (300 g, 1.3 mol) was used to provide methylated product 20 (183 g, 58%) in two steps. The hydrolysis of 20 to α-methyl-Cys 7 in the final step was simple but tricky in this large-scale reaction: usually, a one-time treatment with 5 M aq. HCl under refluxing for 3 days did not hydrolyze 20 completely; the crude product from the first-time reaction must be re-treated by refluxing fresh 5 M aq. HCl for the second time. Finally, 7 was obtained in quantitative yield (130 g, 100%) with high purity (Scheme 2).
Scheme 2.
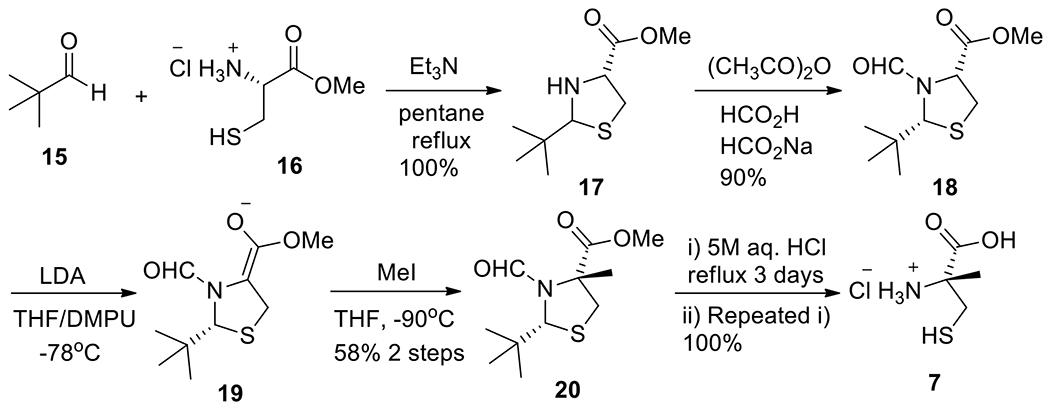
Synthesis of segment 7
Synthesis of segments 8 and 5.
The final adopted protocol for the synthesis of nitrile 8 and thiazoline carboxylic acid 5 is shown in Scheme 3a. Thiazole ester 23 was synthesized by modified Hantzsch’s “one-pot” protocol.14b,14e,21 Starting from commercially available thioamide 21 (80.7g, 424.3 mmol) and ethyl bromopyruvate 22 (91.1g, 980 mmol), thiazole ester 23 (95 g) was obtained in 74% yield after purification. Sequential aminolysis of thiazole ester 23 (89 g, 292.7 mmol) with ammonia gas followed by dehydration with TFAA provided nitrile 8 (68 g, 97.2%). Utilizing a mixture of phosphate buffer (pH 5.95) and methanol in the presence of NaHCO3,14a,14b,15j,22 the condensation of 8 (77.3 g, 323.3 mmol) with 7 (105.0 g, 614 mmol) provided pure thiazoline-thiazole 5 (115.4 g, 100%). This route was highly efficient for the synthesis of fragment 8 as well as 5.
Scheme 3.
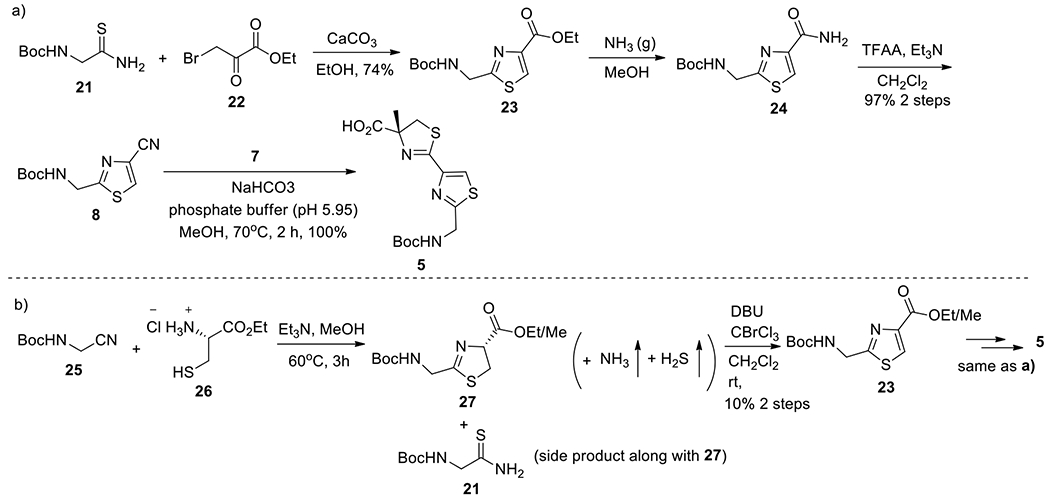
Synthesis of Segments 8 and 5
For the three-step reaction sequence from 21 to 8, the overall yield was 72%. However, another alternative route to synthesize 23 (and then 8), depicted in Scheme 3b,14a was of very low efficiency for the large-scale reaction. From compound 25 to 8, for hundred-gram scale, the overall yield was 10% for four steps, but this number was 35% for medium scale (30 grams). The low yield for the route shown in Scheme 3b resulted from the side reaction of condensation of 25 with 26 (step 1). Ammonia (NH3) and hydrogen sulfide (H2S) were the gases generated by the condensation. For small scale reactions, NH3 and H2S could escape efficiently from the reaction mixture due to small volume and thus did not have much effect on the reaction; however, for the large-scale reaction, NH3 and H2S could not move out efficiently because of the large volume, which caused starting material 25 undergo aminolysis and sulfonation to give compound 21.21 Additionally, the reaction mixture produced by the route in Scheme 3b was difficult to separate.
The synthesis of 5 was non-problematic. The mixture of α-methyl-Cys 7 (105 g) and nitrile 8 (77.3 g) was refluxed in MeOH in the presence of phosphate buffer (pH 5.95) for 2 h, which provided 5 smoothly in 100% yield (115 g).
Synthesis of fragments 9 and 6.
We followed Scheme 4a14a–c,15d to synthesize fragment (S)-9, and further fragment 6. In the first step (aldol reaction), LDA (1.1 eq.) was freshly prepared in situ from n-butyllithium (n-BuLi) and diisopropylamine (iPr2NH) in THF. The aldol reaction of acrolein 29 (119.6g/142.5 mL, 2.133 mol) at –78 °C with enolate derived from tert-butylacetate 28 (247.8 g/286.1 mL, 2.133 mol) in the presence of LDA provided allylic alcohol (RS)-9 (255 g, 70%), in which the yield was lower than medium-scale reaction (acrolein 37.8 g/45 mL, 92% yield). This reaction must be carried out in a well-ventilated fume hood since acrolein is very toxic as well as volatile; both reaction time and work-up time should be minimized because the propensity of the product to dehydrate. Enzymatic resolution of alcohol 9 (83.3 g, 484 mmol) with vinyl acetate 30 by amano lipase PS provided acetate 31 (41g, 41%) with excellent enantioselectivity (95% ee). Pentane is very volatile and minimizing its escape from the reaction system was helpful to obtain the product in good yield.
Scheme 4.
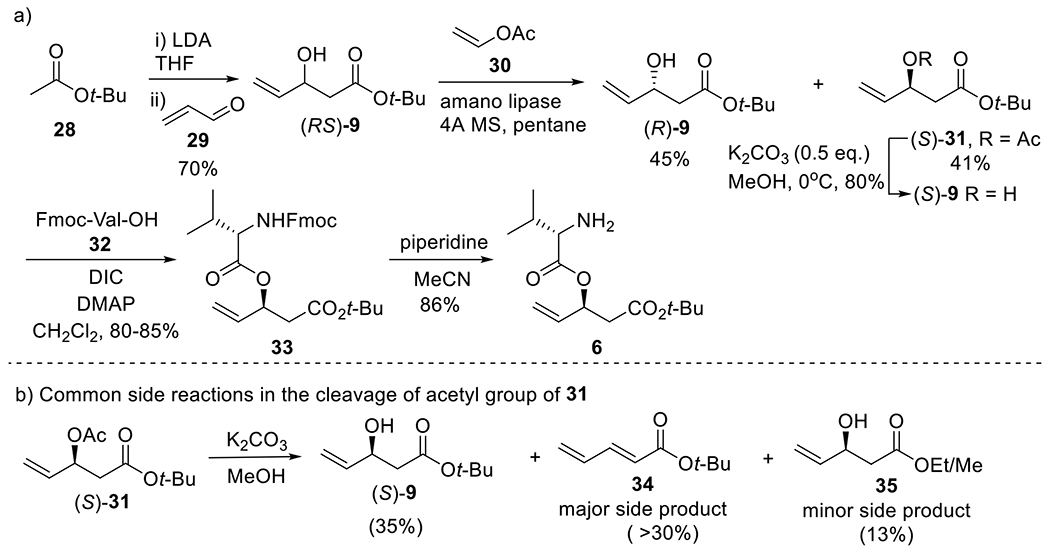
Synthesis of fragments (S)-9 and 6
The subsequent saponification of 31 (81.70 g, 381.60 mmol) with K2CO3 (26.36 g, 190.8 mmol, 0.5 eq.) in MeOH (800 mL) provided alcohol (S)-9 in very good yield (52.56 g, 80%). However, due to the strong propensity of elimination of 9 to the conjugated diene (34, Scheme 4b), mild conditions are required for this reaction. K2CO3 is a good choice to alleviate this side reaction. With the reported condition of 2 eq. K2CO3 in MeOH at 0 ºC (30 min),14a,15d 61% yield was obtained for medium scale reaction (30 g 31), but only 35% yield for the larger scale reaction (70 g scale 31) (Scheme 4b). The unsatisfactory yields resulted from the side reactions: elimination (to 34) and transesterification (to 35). Ethyl ester was one of the products of transesterification because ethanol is present as stabilizer in commercial MeOH. However, the Me/Et ester side product could be used to prepare the final product, largazole, too. Multiple equivalents of K2CO3 (e.g., 2 eq. or more) increased both side reactions. In our optimized condition, the equivalent of K2CO3 was reduced to 0.5 eq.; the yield improved to 80% and both side reactions were suppressed significantly. The lower reaction temperature (0 °C or −10 °C) also improved the yield; however, an even lower temperature (e.g., −30 °C or less) could prolong the reaction time.
Using DIC as coupling reagent, the reaction of alcohol (S)-9 (50.38 g, 293.3 mmol) with Fmoc-Val-OH 32 (119 g, 352.0 mmol) provided ester 33 (112.0 g, 80%), which was treated with piperidine in MeCN to give fragment 6 (50.4 g, 86%) (Scheme 4a).15d Since decagrams of DMAP were used, the crude product 33 must be purified as soon as possible; long time storage (e.g., 3 days or more) would lead to side reactions and would then require cumbersome purification. In the preparation of free amine 6, we chose MeCN as the solvent15d rather than DMF,14a because fulvalene derivatives derived from Fmoc group precipitated due to low solubility in MeCN; the precipitate was removed simply by filtration from the reaction mixture. In addition, MeCN is more easily evaporated than DMF.
Synthesis of 4, 3 and 1.
The synthesis of cyclic core 3 and final product 1 is depicted in Scheme 5.14a,14b,15d The linear compound 4 was prepared by standard coupling method. Acid 5 (50 g, 140.05 mmol) was coupled with amine 6 (38 g, 140.05 mmol) by HATU (64 g, 168.07 mmol) in DMF (350 mL) in presence of DIEA (73 mL, 420.16 mmol) to provide 4 (82.1 g, 94%). The tert-butyl ester and Boc group of 4 were cleaved simultaneously by TFA with triethylsilane (TES) as scavenger, and the crude product was used in the next step without purification.
Scheme 5.
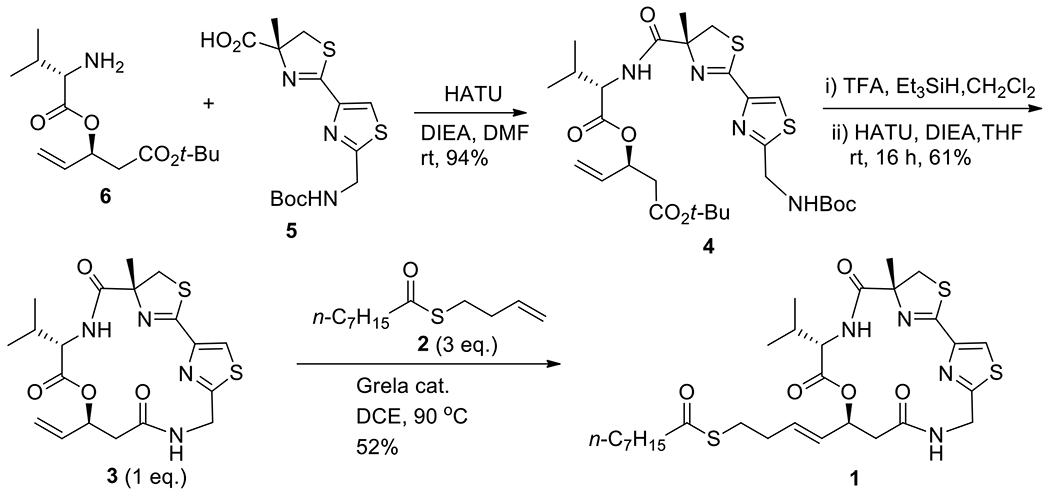
Synthesis of precursors 4 and 3 and of target 1
The macrocyclization of 4 was carried out in three parallel reactions in anhydrous THF (0.004 M, 3 × 10 L). HATU (3 × 50.5 g) was chosen as coupling reagent, and DIEA (3 × 70 mL) was used as base. For each reaction, the solution of 4 in THF (1 L) was added dropwise to the solution of HATU and DIEA in THF (9 L) at 0 °C over 16 h. Finally, the lactam 3 was obtained 35 g in 61% yield. The choice of THF as solvent, rather than CH2Cl2, was made because the TFA salt of deprotected 4 has better solubility in THF. An inlet thermometer must be used to monitor the reaction temperature (0 °C).
The cross-metathesis reaction to give the final product was carried out by three parallel reactions, in which Grela catalyst was used for the coupling.14a The mixture of cyclic core 3 (3 × 7.5 g, 3 × 17.2 mmol) and thioester 2 (3 × 12.8 g, 3 × 60 mmol) was mixed in 1,2-dichloroethane (3 × 220 mL) in a pressure flask (500 mL) and refluxed for 24 h. Crude product 1 was initially purified by normal-phase chromatography silica gel column; however, the HPLC purity was only about 80% and the product color was brownish (from catalyst). A second purification by C18 cartridge was carried out to provide 1 (16.7 g, 52%) in over 95% HPLC purity as a colorless solid (Figure 7). The 1H NMR and 13C NMR spectra are identical to those of natural largazole.
CONCLUSION
Largazole is a promising epigenetic anticancer agent targeting class I HDACs. In this paper, we initiated the development of process chemistry for the preparation of decagram quantities of largazole. The protocols in this publication were confirmed to represent a practical, reliable, efficient and scalable process for the synthesis of largazole on multi-gram scale with an overall yield of 21% for the longest linear sequence (8 steps). The protocols for each intermediate were also optimized and are suitable for the preparation on hundred-gram scale. Some apparent advancements are emphasized here: 1) “re-treatment” process ensures the high purity of α-MeCys HCl salt (7); 2) for the synthesis of nitrile 8 on large scale, the “thioamide” method was confirmed to be suitable (Scheme 3a); in contrast, the “aminoacetonitrile” method failed (Scheme 3b); 3) in the preparation of allyl alcohol ester (S)-9, a lower equivalent (0.5 eq.) of K2CO3 markedly increased product yield; 4) slow addition of starting material in the preparation of cyclic core 3 provided good yield; 5) pressure reaction in 1,2-dichloromethane with heating gave good yield in the final cross-metathesis reaction; 6) the additional purification by C18 cartridge removed impurity from catalyst to provide colorless product; this method could be readily applied to large scale purification. Overall, the process reported here provides an important reference for the preparation of largazole on large scale, which must advance the comprehensive studies of largazole, including additional pre-clinical and then clinical investigations.
EXPERIMENTAL SECTION
General Procedures.
All commercial reagents were used without further purification unless otherwise noted. Anhydrous solvents were purchased from Sigma-Aldrich and used directly; other solvents were used from bottles directly if no special note. All reactions were performed in heat-gun dried flasks (400 °C under reduced pressure) under an inert atmosphere of anhydrous N2 unless otherwise noted. Thin layer chromatography was performed on EMD silica gel 60Å F254 glass plates. Flash column chromatography was performed with Sorbent Technologies Inc’s 230–400 mesh silica gel. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury 400 MHz or Aligent VNMR 600 MHz spectrometer as indicated in the data list. HPLC analyses were carried out at λmax 220 nm or 254 nm using column Synergi 4u Hydro-RP 250 mm × 4.6 mm × 4 μm on Shimadzu SPD-M20A-PDA system. HPLC purity is reported in area %. Chemical shifts for proton nuclear magnetic resonance (1H NMR) spectra are reported in parts per million relative to the signal residual CDCl3 at 7.26 ppm. Chemicals shifts for carbon nuclear magnetic resonance (13C NMR) spectra are reported in parts per million relative to the center line of the CDCl3 triplet at 77.16 ppm. The abbreviations s, d, dd, ddd, dddd, t, q, br and m stand for the resonance multiplicity singlet, doublet, doublet of doublets, doublet of doublet of doublets, doublet of doublet of doublet of doublets, triplet, quartet, broad and multiplet, respectively.
Thiooctanoic acid (13).
Octanoylchloride (11) (121.1 mL, 709.4 mmol) was added to the solution of thioacetamide (10) (53.3 g, 709.4 mmol) in anhydrous benzene (450 mL). After the reaction mixture was stirred at 30 °C for 4 h, it was cooled down with an ice-water bath, and then 10% NaOH solution (530 mL) was added. The biphasic mixture was stirred at room temperature for another 30 min and then acidified with 1M KHSO4 to pH 2–3. The acidified solution was extracted with ethyl acetate (3 × 500 mL), dried over anhydrous MgSO4 and the solvent was evaporated in vacuo to give crude product. The crude product was distilled under high vacuum to give thiooctanoic acid (13) (102 g, 90% yield) as orange liquid.
S-Butyl-3-enyl octanethioate (2).
Anhydrous K2CO3 (175.3 g, 1.268 mol) and 1-bromo-3-butene (14) (65.0 mL, 640.4 mmol) were added sequentially to the solution of 13 (101.0 g, 631 mmol) in anhydrous acetone (1 L). The reaction mixture was stirred at room temperature for 1 h, then was diluted with 1 L pentane, filtered through Celite and concentrated in vacuo. The crude product was purified by flash chromatography column on silica gel to give pure product 2 (122 g, 90% yield, 99.5% HPLC purity) (eluted by 3% ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3): δ 5.76 (dddd, J = 17.0 Hz, 10.6, 6.4, 6.4 Hz, 1H), 5.09–5.00 (m, 2H), 2.92 (t, J = 7.2 Hz, 2H), 2.52 (t, J = 7.4 Hz, 2H), 2.31 (q, J = 7.2 Hz, 2H), 1.67–1.60 (m, 2H), 1.34–1.20 (m, 8H), 0.86 (t, J = 6.3 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 199.6, 136.2, 116.5, 44.2, 33.8, 31.7, 29.0, 29.0, 28.1, 25.8, 22.7, 14.2 ppm. HRMS (ESI) m/z calcd for C12H22OS (M+H)+ 215.1464, found 215.1457.
(2R,4R)-Methyl-2-tert-butyl-1,3-thiazolidine-3-formyl-4-carboxylate (18).
To the solution of (R)-cysteine methyl ester hydrochloride (16) (257.24 g, 1.499 mol) in pentane (1.5 L) was added pivalaldehyde (15) (325.5 mL, 2.997 mol) and triethylamine (230 mL, 1.649 mol) and the reaction mixture was refluxed for 24 hours. The water of dehydration (about 27 mL) from the reaction was continuously removed by a Dean-Stark trap. Then the reaction mixture was cooled down to room temperature and filtered. The filtered solid was washed with diethyl ether and the filtrate was combined and evaporated to give the crude 17 (304.4 g), which was used in the next step without further purification.
All of crude 17 (304.4 g) and sodium formate (116.0 g, 1.649 mol) were dissolved in formic acid (2.4 L). The resulting mixture was cooled to 0–5 °C, then acetic anhydride (435 mL, 1.499 mol) was added dropwise. The reaction mixture was stirred overnight at room temperature, then the solvents were evaporated under reduced pressure, the resulting residue was neutralized with saturated NaHCO3 solution (to pH 4–5) and extracted with diethyl ether (3 × 1 L). The organic layers were combined and dried over anhydrous Mg2SO4 and filtered. After removal of the solvent, the resulting crude was purified by flash chromatography on silica gel (eluted by 30–50% ethyl acetate in hexane). The collected fractions were evaporated and recrystallized from diethyl ether–hexanes (1:1, v/v) to afford N-formyl thazolidine 18 (300 g, 90% yield, 97% HPLC purity) as colorless crystals. 1H NMR (400 MHz, CDCl3, mixture of conformers (7:1), major): δ 8.35 (s, 1H), 4.89 (t, J = 8.0 Hz, 1H), 4.74 (s, 1H), 3.77 (s, 3H), 3.34–3.24 (m, 2H), 1.03 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3, mixture of conformers (7:1), major): δ 170.1, 162.8, 75.3, 61.6, 52.8, 38.7, 33.0, 26.5 ppm. HRMS (ESI) m/z calcd for C10H17NO3S (M+H)+ 232.1002, found 232.1001.
(2R,4R)-Methyl-2-tert-butyl-3-formyl-4-methyl-1,3-thiazolidine-4-carboxylate (20).
To the stirred solution of diisopropylamine (275.2 mL, 1.947 mol) in anhydrous THF (4.5 L) at −78 °C (acetone-dry ice bath) was added the solution of n-butyllithium (1.6 M) in hexanes (900 mL, 1.428 mol) dropwise over 1.5 h under a nitrogen atmosphere. DMPU (900 mL) was added in one portion. After the reaction mixture was stirred at −78 °C for l h, it was cooled to −90 °C (acetone-dry ice-liquid nitrogen bath). The solution of 18 (300 g, 1.298 mol) was added dropwise in THF (200 mL) over 1 h. The reaction mixture was stirred at −90 °C for 1 h, then MeI (97.0 mL, 1.558 mol) was added dropwise over 45 min. The reaction mixture was stirred at −90 °C for 2 h, then warmed to room temperature, concentrated in vacuo, partitioned between brine (1.6 L) and diethyl ether (2.0 L), and extracted with diethyl ether (2 × 800 mL). The combined organic layer was dried over anhydrous MgSO4, solvent was evaporated in vacuo and the crude product was purified by chromatography column on silica gel (eluted by 20–50% ethyl acetate in n-hexanes). The collected fractions were concentrated and washed with n-hexanes to give the thiazolidine 20 (183.3 g, 57.6% yield, 96% HPLC purity) as a colorless solid. 1H NMR (400 MHz, CDCl3, mixture of conformers (7:3)), major conformer: δ 8.28 (s, 0.7H), 4.66 (s, 0.7H), 3.76 (s, 2.1H), 3.31 (d, J = 12.0 Hz, 0.7H), 2.72 (d, J = 12.0 Hz, 0.7H), 1.75 (s, 2.1H), 1.06 (s, 6.3H) ppm; minor conformer: δ 8.40 (s, 0.3H), 5.30 (s, 0.3H), 3.82 (s, 0.9H), 3.63 (d, J = 12.0 Hz, 0.3H), 2.86 (d, J = 12.0 Hz, 0.3H), 1.78 (s, 0.9H), 0.95 (s, 2.7H) ppm. 13C NMR (100 MHz, CDCl3, mixture of conformers (7:3)), major conformer: δ 172.2, 161.3, 74.5, 70.2, 52.9, 41.7, 39.6, 26.8, 20.8 ppm; minor conformer: δ 173.2, 163.0, 72.0, 70.0, 53.4, 42.3, 40.4, 28.3, 27.2 ppm. HRMS (ESI) m/z calcd for C11H19NO3S (M+H)+ 246.1158, found 246.1156.
(R)-α-Methylcysteine hydrochloride (7).
Thiazolidine 20 (183.0 g, 746.634 mmol) was dissolved in 5M hydrochloric acid (2.6 L). After the solution was heated under refluxing in atmosphere of nitrogen for 3 days, it was cooled down to room temperature and washed with ethyl acetate (3 × 500 mL), and then the aqueous layer was concentrated under reduced pressure to give the crude product; usually for large scale of this reaction, the starting material was hydrolyzed incompletely. The crude product was re-hydrolyzed using the same protocol as above. The second hydrolysis provided (R)-α-methylcysteine hydrochloride salt 7 (127.0 g, 99.5% yield, 95% HPLC purity) as a colorless solid. 1H NMR (400 MHz, D2O): δ 3.16 (d, J = 16.0 Hz, 1H), 2.88 (d, J = 16.0 Hz, 1H), 1.58 (s, 3H) ppm. 13C NMR (400 MHz, D2O): δ 173.0, 61.3, 30.1, 21.0 ppm. HRMS (ESI) m/z calcd for C4H10ClNO2S (M-HCl-H)− 134.0276, found 134.0283.
Ethyl 2-(t-butoxycarbonylaminomethyl)-thiazole-4-carboxylate (23).
Ethyl bromopyruvate (58.6 mL, 467.2 mmol) and calcium carbonate (42.42 g, 424.2 mmol) were added to the solution of 21 (80.66 g, 424.3 mmol) in absolute ethanol (1.1 L). The reaction mixture was stirred overnight at room temperature under nitrogen atmosphere and filtered. The filtrate was evaporated in vacuo. Sequentially, the concentrated residue was dissolved in ethyl acetate (1.5 L), washed with saturated NaHCO3 solution (3 × 500 mL) and water (3 × 500 mL), dried over anhydrous MgSO4, filtered and evaporated. The resulting crude was purified by flash chromatography on silica gel (eluted with 30-50% ethyl acetate in hexanes) to provide product 23 (95 g, 74% yield, 99.5% HPLC purity) as a colorless solid. 1H NMR (400 MHz, CDCl3): δ 8.11 (s, 1H), 5.31 (brs, 1H), 4.64 (d, J = 8.0 Hz, 2H), 4.41 (q, J = 8.0 Hz, 2H), 1.45 (s, 9H), 1.39 (t, J = 8.0 Hz, 3H) ppm. 13C NMR (400 MHz, CDCl3): δ 161.4, 155.7, 147.1, 128.0, 80.6, 61.7, 42.5, 28.4, 14.5 ppm. HRMS (ESI) m/z calcd for C12H18N2O4S (M+H)+ 287.1060, found 287.1057.
2-(t-butoxycarbonylaminomethyl)-thiazole-4-carboxamide (24).
Compound 23 (88.9 g, 292.3 mmol) was dissolved in anhydrous MeOH (650 mL). The resulting solution was saturated (bubbled) with ammonia (gas) for 10–15 min each day (total 3 days). After stirring for three days, the reaction solution was evaporated to dryness in vacuo to give the crude thiazole amide 24 as a colorless solid (80.5 g, 100% yield, 88% HPLC purity), which was used in the next step without further purification. 1H NMR (500 MHz, CDCl3): δ 8.07 (s, 1H), 7.13 (br s, 1H), 6.06 (br s, 1H), 5.41 (br s, 1H), 4.58 (d, J = 6.0 Hz, 2H), 1.46 (s, 9H) ppm. 13C NMR (125 MHz, CDCl3): δ 169.6, 163.1, 155.8, 149.3, 124.7, 80.7, 42.5, 28.5 ppm. HRMS (ESI) m/z calcd for C10H15N3O3S (M+H)+ 258.0907, found 258.0905.
2-(t-butoxycarbonylaminomethyl)-thiazole-4-carbonitrile (8).
To the solution of crude thiazole amide 24 (81.0 g, 315.05 mmol) in CH2Cl2 (700 mL) at 0 °C were added triethylamine (112.0 mL, 803.48 mmol) and trifluoroacetic anhydride (TFAA) (57 mL, 410.07 mmol). The reaction mixture was stirred for 1.5 h at the same temperature, then warmed to room temperature and stirred for additional 30 min. The reaction mixture was quenched with 500 mL water. The organic layer was separated and the water layer was extracted with CH2Cl2 (2 × 500 mL). The combined CH2Cl2 extracts were dried over anhydrous MgSO4, evaporated in vacuo, and purified by chromatography column on silica gel (eluted by 30–50% ethyl acetate in hexanes) to give the thiazole nitrile 8 (68.0 g, 97.3% yield 2 steps, 100% HPLC purity) as a light golden solid. 1H NMR (400 MHz, CDCl3): δ 7.95 (br s, 1H), 5.61 (br, 1H), 4.57 (d, J = 8.0 Hz, 2H), 1.42 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): δ 171.9, 155.8, 131.1, 126.3, 113.9, 80.8, 42.3, 28.3 ppm. HRMS (ESI) m/z calcd for C10H13N3O2S (M+Na)+ 262.0626 found 262.0623.
(R)-2-(2-t-Butoxycarbonylaminomethyl-thiazol-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxylic acid (5).
To a solution of 8 (77.26 g, 323.3 mmol) and 7 (105.0 g, 614.0 mmol) in MeOH (3.7 L) was added NaHCO3 (135.75 g, 1.6158 mol) and phosphate buffer (pH 5.95, 1.85 L) and the reaction was stirred at 70 °C for 2 h. Then the reaction was cooled to room temperature and evaporated to remove MeOH. To the residue was added water (2.5 L) and saturated aqueous NaHCO3 solution (1.5 L) and the mixture was stirred at room temperature for 30 min and extracted with EtOAc (3 × 800 mL). The aqueous layer was acidified with 1M KHSO4 solution to pH 2 and then extracted with EtOAc (3 × 4 L). The combined organic layers were washed with brine (3 × 2 L), dried with anhydrous MgSO4 and evaporated in vacuo to give product thiazole-thiazoline acid 5 (115.4 g, 100% yield, 99.7% HPLC purity). The product 5 did not need further purification and was pure enough for the next reaction. 1H NMR (400 MHz, CDCl3): δ 10.65 (br s, 1H), 7.7(s, 1H), 6.68 (br s, 0.26H), 5.85 (br s, 0.74H), 4.53 (m, 2H), 3.82 (d, J = 12.0 Hz, 1H), 3.22 (d, J = 12.0 Hz, 1H), 1.67 (s, 3H), 1.44 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): δ 175.8, 170.3, 164.6, 155.9, 147.7, 123.0, 84.2, 80.5, 42.3, 41.3, 28.4, 24.2 ppm. HRMS (ESI) m/z calcd for C14H19N3O4S2 (M+H)+ 358.0890, found 358.0879.
(RS)-tert-Butyl 3-hydroxypent-4-enoate ((RS)-9).
To the solution of diisopropylamine (331.6 mL, 2.346 mol) in THF (3.8 L) was added n-BuLi (1.466 L, 2.346 mol, 1.6 M in hexane) dropwise at 0 °C over 2 h. After the reaction mixture was stirred at 0 °C for 30 min, it was cooled down to −78 °C, and t-butyl acetate 28 (286.1 mL, 2.133 mol) was added dropwise for 2 h. After stirring for 1h at −78 °C, acrolein 29 (142.5 mL, 2.133 mol) was added and the mixture was stirred at −78 °C for 1 h, then warmed to room temperature and stirred for an additional 1 h. The reaction was quenched with saturated NH4Cl aqueous solution (2 L), then Et2O (2 L) was added and the organic phase was separated. The water phase was extracted with Et2O (2 × 1 L). The combined organic layers were dried over anhydrous MgSO4 and evaporated in vacuo. The residue was purified by flash chromatography column on silica gel (eluted with 15–30% EtOAc in hexanes) to afford (RS)-9 (255.0 g, 70% yield, 90.7% HPLC purity) as a colorless oil. 1H NMR (400 MHz, CDCl3, mixture of R and S configuration): δ 5.89–5.81 (m, 1H), 5.31–5.25 (m, 1H), 5.14–5.10 (m, 1H), 4.49–4.44 (m, 1H), 3.20 (br s, 1H), 2.51–2.46 (m, 1H), 2.44-2.38 (m, 1H), 1.44 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): δ 171.8, 139.0, 115.2, 81.5, 69.1, 42.2, 28.2 ppm. HRMS (ESI) m/z calcd for C9H16O3 (M+H)+ 173.1172, found 173.1175.
(S)-tert-Butyl 3-acetoxypent-4-enoate ((S)-31).
To a solution of (RS)-9 (249.15 g, 1.448 mol) in pentane (5.0 L) was sequentially added amano lipase (149.5 g), 4 Å molecular sieves (239.0 g), and vinyl acetate (133.5 mL, 1.448 mol). After the reaction was stirred at 30 °C for 24 h, it was filtered through an Celite pad, the solids were washed with diethyl ether, the filtrates were combined and the solvent was evaporated. The residue was purified by column chromatography on silica gel (eluted by 10–20% ethyl acetate in hexanes) to afford 146.0 g (45% yield, 85% HPLC purity) (S)-31 as colorless oil. 1H NMR (400 MHz, CDCl3): δ 5.80 (ddd, J = 16.8, 10.4, 6.4 Hz, 1H), 5.61-5.55 (m, 1H), 5.28 (dt, J = 17.2, 1.2 Hz, 1H), 5.18 (dt, J = 10.4, 1.2 Hz, 1H), 2.58 (dd, J = 15.2, 8.0 Hz, 1H), 2.50 (dd, J = 15.2, 5.6 Hz, 1H), 2.03 (s, 3H), 1.41 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): δ 169.9, 169.1, 135.2, 117.4, 81.1, 71.2, 40.8, 28.1, 21.1 ppm. HRMS (ESI) m/z calcd for C11H18O4 (M+H)+ 215.1278, found 215.1279.
(R)-tert-Butyl 3-hydroxypent-4-enoate ((R)-9).
During the purification of (S)-31, (R)-9 was separated as well (102 g, 41% yield, 95.3% HPLC purity). 1H NMR (400 MHz, CDCl3): δ 5.87 (ddd, J = 17.2, 10.4, 5.2 Hz, 1H), 5.33 (dt, J = 17.2, 1.6, 1H), 5.14 (dt, J = 10.8, 1.2, 1H), 4.52 – 4.46 (m, 1H), 3.14 (d, J = 4.8, 1H), 2.51 (dd, J = 16.0, 3.6 Hz, 1H), 2.43 (dd, J = 16.4, 8.4, 1H), 1.48 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): δ 171.7, 138.9, 115.1, 81.4, 69.0, 42.0, 28.1 ppm. HRMS (ESI) m/z calcd for C9H16O3 (M+H)+ 173.1172, found 173.1173.
(S)-tert-Butyl 3-hydroxypent-4-enoate ((S)-9).
K2CO3 (26.36 g, 190.8 mmol) was added to the solution of compound (S)-31 (81.7 g, 381.60 mmol) in anhydrous MeOH (800 mL) at 0 °C. After stirring at 0 °C for 30 min, the resulting mixture was filtered off through Celite under reduced pressure. The filtrate was diluted with water (800 mL), concentrated to remove most of methanol and extracted with EtOAc (2 × 800 mL). The combined EtOAc layer was washed with H2O (500 mL) and brine (500 mL), dried over anhydrous MgSO4, evaporated and purified by flash chromatography on silica gel (eluted by 15–30% EtOAc in hexanes) to afford allylic alcohol (S)-9 (52.56 g, 80%, 89% HPLC purity) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 5.86 (ddd, J = 17.2, 10.4, 5.2 Hz, 1H), 5.29 (dt, J = 17.2, 1.6, 1H), 5.13 (dt, J = 10.8, 1.2, 1H), 4.47 (br m, 1H), 3.16 (br s, 1H), 2.50 (dd, J = 16.4, 4.0 Hz, 1H), 2.42 (dd, J = 16.0, 8.0, 1H), 1.45 (s, 9H) ppm. 13C NMR (100 MHz, CDCl3): δ 171.9, 139.0, 115.3, 81.6, 69.0, 42.2, 28.2 ppm. HRMS (ESI) m/z calcd for C9H16O3 (M+H)+ 173.1172, found 173.1172.
t-Butyl (S)-3-((S)-2-(9H-fluoren-9-ylmethoxycarbonylamino)-3-methylbutanoyloxy)-pent-4-enoate (33).
To the solution of allylic alcohol (S)-9 (50.38 g, 293.32 mmol) and Fmoc-L-Val (119.42 g, 352.0 mmol) in anhydrous CH2Cl2 (1.5 L) at 0 °C was added the solution of DMAP (35.84 g, 293.32 mmol) and DIC (54.46 mL, 352.0 mmol) in anhydrous CH2Cl2 (200 mL). After stirring at −5 to −10 °C for 30 min, the reaction mixture was warmed to room temperature and stirred for additional 1.5 h, then it was quenched with phosphate buffer (pH 7) and extracted with CH2Cl2 (2 × 700 mL). The combined CH2Cl2 layer was dried over anhydrous MgSO4, concentrated in vacuo, and purified by flash chromatography on silica gel (eluted by 20-30% ethyl acetate in hexanes) to afford compound 33 (112.0 g, 80% yield) as a thick colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 7.6 Hz, 2H), 7.61 (d, J = 7.6 Hz, 2H), 7.40 (dd, J = 7.2, 7.2 Hz, 2H), 7.32 (dd, J = 7.6, 7.6 Hz, 2H), 5.83 (ddd, J = 16.8, 10.4, 6.4 Hz, 1H), 5.69 (dt, J = 6.8, 6.8 Hz, 1H), 5.38-5.32 (m, 2H), 5.24 (d, J = 10.8 Hz, 1H), 4.39 (d, J = 7.2 Hz, 2H), 4.33 (dd, J = 9.2, 4.4 Hz, 1H), 4.23 (dd, J = 7.2, 7.2 Hz, 1H), 2.67 (dd, J = 15.6, 8.0 Hz, 1H), 2.56 (dd, J = 15.6, 6.0 Hz, 1H), 2.24-2.16 (m, 1H), 1.43 (s, 9H), 0.98 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 171.1, 168.7, 156.3, 144.0, 143.9, 141.4, 134.7, 127.8, 127.2, 127.2, 125.2, 120.1, 120.1, 118.6, 81.4, 72.4, 67.1, 59.0, 47.3, 40.6, 31.5, 28.1, 19.1, 17.4 ppm. HRMS (ESI) m/z calcd for C29H35NO6 (M+NH4)+ 511.2803, found 511.2791.
tert-Butyl (S)-3-((S)-2-amino-3-methylbutanoyloxy)-pent-4-enoate (6).
To the solution of compound 33 (96.40 g, 195.43 mmol) in MeCN (1.25 L) was added piperidine (77.2 mL, 781.72 mmol). The reaction mixture was stirred for 20 min at room temperature under nitrogen atmosphere and much colorless solid appeared. The suspending solid was filtered off with a vacuum funnel. The filtrate was concentrated in vacuo, and purified by flash chromatography on silica gel (EtOAc/hexanes 1:1 to 2:1) to give the free amine 6 (45.6 g, 86 % yield, 81% HPLC purity) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 5.80 (ddd, J = 17.2, 10.4, 6.4 Hz, 1H), 5.63 (dt, J = 6.8, 6.8 Hz, 1H), 5.31 (d, J = 17.2 Hz, 1H), 5.19 (d, J = 10.4 Hz, 1H), 3.23 (d, J = 4.8 Hz, 1H), 2.60 (dd, J = 15.2, 8.0 Hz, 1H), 2.51 (dd, J = 15.6, 5.6 Hz, 1H), 2.05–1.97 (m, 1H), 1.46 (br s, 2H), 1.41 (s, 9H), 0.95 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 174.6, 169.0, 135.1, 118.0, 81.2, 71.5, 59.9, 40.7, 32.0, 28.1, 19.5, 17.0 ppm. HRMS (ESI) m/z calcd for C14H25NO4 (M+H)+ 272.1856, found 272.1850.
Open chain precursor for macrocyclization (4).
Acid 5 (52.0 g, 145.62 mmol) and HATU (67.7 g, 178.02 mmol) were dissolved in anhydrous DMF (270 mL). To the solution was added DIEA (75.0 mL, 431.38 mmol) dropwise over 20 min. After the mixture was stirred for 20 min at room temperature under nitrogen atmosphere, amine 6 (38.0 g, 140.13 mmol) in anhydrous DMF (80 mL) was added. When the reaction mixture was stirred for 1h at room temperature, it was quenched with phosphate buffer (pH 7.0, 400 mL), and extracted with EtOAc (3 × 500 mL). The combined organic layer was washed with water (3 × 200 mL) and brine (200 mL), dried with anhydrous MgSO4, evaporated and purified by flash chromatography column on silica gel (eluted by 25–50% EtOAc in hexanes) to provide open chain precursor 4 (82.1 g, 94.0% yield, 100% HPLC purity) as a thick liquid. 1H NMR (400 MHz, CDCl3): δ 7.93 (s, 1H), 7.19 (d, J = 9.2 Hz, 1H), 5.79 (ddd, J = 17.2, 10.4, 6.8 Hz, 1H), 5.64 (dt, J = 6.8, 6.8 Hz, 1H), 5.44 (br s, 1H), 5.32 (dd, J = 17.2, 1.2 Hz, 1H), 5.20 (dd, J = 10.4, 1.2 Hz, 1H), 4.60 (d, J = 6.4 Hz, 2H), 4.49 (dd, J = 9.2, 4.8 Hz, 1H), 3.74 (d, J = 11.6 Hz, 1H), 3.30 (d, J = 11.6 Hz, 1H), 2.64 (dd, J = 15.6, 7.6, 1H), 2.52 (dd, J = 15.6, 6.0 Hz, 1H), 2.19–2.11 (m, 1H), 1.43 (s, 9H), 1.40 (s, 9H), 0.84 (d, J = 6.4 Hz, 3H), 0.78 (d, J = 6.8, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 174.5, 170.5, 170.0, 168.7, 163.3, 155.7, 148.7, 134.7, 121.4, 118.5, 85.2, 81.4, 80.5, 72.2, 56.9, 42.4, 41.5, 40.6, 31.2, 28.4, 28.1, 24.8, 19.2, 17.5 ppm. HRMS (ESI) m/z calcd for C28H42N4O7S2 (M+H)+ 611.2568, found 611.2555.
Macrocyclic core (3).
Triethylsilane (TES) (26.5 mL, 165.92 mmol), trifluoroacetic acid (TFA) (305 mL) was sequentially added to the solution of 4 (81.0 g, 132.73 mmol) in anhydrous CH2Cl2 (760 mL) at 0 °C. After stirring for 4 h at 0 °C, the reaction mixture was diluted with toluene (700 mL) and evaporated, then it was azeotroped again with toluene (3 × 200 mL) and dried with an oil pump for 2 h to give the free amino acid intermediate as TFA salt (120.0 g), which was used in the next step without further purification.
The macrocyclization was carried out in three parallel reactions. The free amino acid intermediate TFA salt from 4 was divided into three equal portions (3 × 40.0 g) and used for three parallel reactions, respectively. The following description was based on one of the three parallel reactions. The above TFA salt from 4 (40.0 g, ~44.24 mmol) in anhydrous THF (1.0 L) was added dropwise (~20 h) to the solution of HATU (50.48 g, 132.73 mmol) and DIEA (69.23 mL, 398.2 mmol) in anhydrous THF (8.5 L) at 0 °C via an addition funnel. The resulting mixture was stirred at 0 °C for 8 h and room temperature for an additional 15 h. The solvent of the reaction mixture was evaporated in vacuo. The resulting residue was dissolved in EtOAc (1.5 L), washed with water (2 × 300 mL), brine (300 mL), dried over anhydrous MgSO4 and evaporated in vacuo to remove the solvent. The resulting residue was purified by flash chromatography column on silica gel (eluted by mixture of EtOAc/hexane/MeOH, 10:10:1 to 10:10:1.5, v/v/v). The collected product fractions were evaporated to dryness and washed with a mixture of EtOAc/hexane (1:1, v/v) to give pure macrocyclic core 3 (12.0 g, total 36.0 g from three parallel reactions, 61% yield, 92% HPLC purity). 1H NMR (400 MHz, CDCl3): δ 7.73 (s, 1H), 7.15 (d, J = 9.2 Hz, 1H), 6.56 (dd, J = 9.2, 3.2 Hz, 1H), 5.80 (ddd, J = 16.8, 10.4 Hz, 6.0, 1H), 5.66–5.61 (m, 1H), 5.38 (dt, J = 17.2, 1.2 Hz, 1H), 5.25–5.18 (m, 2H), 4.57 (dd, J = 9.6, 3.2 Hz, 1H), 4.20 (dd, J = 18.0, 3.6 Hz, 1H), 3.99 (d, J = 11.2 Hz, 1H), 3.23 (d, J = 11.6 Hz, 1H), 2.81 (dd, J = 16.8, 10.8 Hz, 1H), 2.67 (dd, J = 16.4, 2.8 Hz, 1H), 2.12–2.01 (m, 1H), 1.81 (s, 3H), 0.64 (d, J = 6.8 Hz, 3H), 0.47 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3): δ 173.6, 169.3, 168.9, 168.0, 164.6, 147.4, 134.7, 124.3, 117.9, 84.3, 72.3, 57.6, 43.3, 41.1, 40.0, 34.2, 24.2, 18.9, 16.6 ppm. HRMS (ESI) m/z calcd for C19H24N4O4S2 (M+H)+ 437.1311, found 437.1302.
Largazole (1):
The cross-metathesis reaction for largazole was carried out in parallel reactions due to volume limit of available pressure flask. The following description was based on one of the parallel reactions. The Grela catalyst (1.7 g, 2.53 mmol) was added to the solution of alkene 2 (12.86 g, 60.1 mmol) and the cyclic core 3 (7.5 g, 17.2 mmol) in anhydrous 1,2-dichloroethane (DCE) (220 mL) in a one-neck pressure flask. The mixture was briefly degassed with nitrogen, stirred for 15 h at 90 °C, concentrated in vacuo and purified by flash chromatography column on silica gel (eluted by mixture of EtOAc/hexane/MeOH, 10:10:0.5 to 10:10:1.2, v/v/v) and then re-purified by Biotage Ultra SNAP C18 cartridge with Isolera (MeCN/H2O, MeCN 22% to 100% in 6 CV, then 100% MeCN in 7 CV) to provide largazole 1 (total 5.56 g, 52% yield, major fraction (~ 5.0 g) 95.2% HPLC purity and a minor fraction (~0.5 g) 95.8% HPLC purity) as a colorless foamy solid. We carried out three batches of this reaction and obtained 12.0 g largazole. [α]25D: +55 (c 0.21, MeOH) (lit1 [α]20D: +22 (c 0.1, MeOH); lit3a [α]27D: +38.9 (c 0.027, MeOH); lit14f [α]20D: +24 (c 0.4, MeOH); lit14b [α]D: +40.5 (c 0.1, MeOH); lit14a [α]20D: +25.2 (c 0.33, CHCl3); lit14c [α]23D –24 (c 0.13, CHCl3); lit14h [α]23D: +18 (c 0.06, CHCl3)). 1H NMR (600 MHz, CDCl3) δ: 7.74 (s, 1H), 7.12 (d, J = 9.0 Hz, 1H), 6.52 (dd, J = 9.6, 3.6 Hz, 1H), 5.82 (dtd, J = 15.6, 6.6, 1.2 Hz, 1H), 5.62 (ddd, J = 10.2, 6.6, 2.4 Hz,1H), 5.48 (ddt, J = 15.6, 7.2, 1.2 Hz, 1H), 5.24 (dd, J = 17.4, 9.6 Hz, 1H), 4.57 (dd, J = 9.6, 3.6 Hz, 1H), 4.23 (dd, J = 17.4, 3.0 Hz, 1H), 4.00 (d, J = 11.4 Hz, 1H), 3.24 (d, J = 11.4 Hz, 1H), 2.86 (t, J = 7.2 Hz, 2H), 2.83 (dd, J = 16.8, 10.8 Hz, 1H), 2.64 (dd, J = 16.2, 2.4 Hz, 1H), 2.49 (t, J = 7.2 Hz, 2H), 2.27 (dt, J = 6.6, 6.9 Hz, 2H), 2.10 – 2.03 (m, 1H), 1.83 (s, 3H), 1.63–1.58 (m, 2H), 1.29–1.22 (m, 8H), 0.84 (t, J = 6.6 Hz, 3H), 0.65 (d, J = 6.6 Hz, 3H), 0.47 (d, J = 7.2 Hz, 3H) ppm. 13C NMR (150 MHz, CDCl3): δ 199.4, 173.6, 169.5, 168.9, 168.0, 164.6, 147.5, 132.7, 128.4, 124.3, 84.4, 72.1, 57.8, 44.2, 43.4, 41.1, 40.4, 34.2, 32.3, 31.7, 28.9, 28.0, 25.7, 24.2, 22.6, 19.0, 16.7, 14.1 ppm. HRMS (ESI) m/z calcd for C29H42N4O5S3 (M+H)+ 623.2390, found 623.2379.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health, National Cancer Institute (NCI) grant R43CA195895, and the Debbie and Sylvia DeSantis Chair Professorship.
ABBREVIATION
- Ac
acetyl
- Boc
tert-butoxycarbonyl
- tBu
tert-butyl
- Cys
cysteine
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene
- DCE
1,2-dichloroethane
- DIEA
N,N-Diisopropylethylamine
- DIC
N,N′-Diisopropylcarbodiimide
- DMAP
4-(dimethylamino)pyridine
- DMF
dimethylformamide
- DMPU
1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone
- Et
ethyl
- Fmoc
9-fluorenylmethoxycarbonyl
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HDAC
histone deacetylase
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- HTS
high throughput screening
- IC50
the half maximal inhibitory concentration
- ID
internal diameter
- LDA
lithium diisopropylamine
- Me
methyl
- PK
pharmacokinetic modeling
- PD
pharmacodynamic modeling
- iPr
isopropyl
- NMR
nuclear magnetic resonance
- SAR
structure-activity relationship
- TES
triethylsilane
- TFA
trifluoroacetic acid
- TFAA
trifluoro acetic anhydride
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- Val
valine
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Selected HPLC profiles (PDF), product largazole picture, 1H and 13C NMR spectra for all compounds
The authors declare the following competing financial interest(s): H.L. and P.R.C. are co-founders of Oceanyx Pharmaceuticals, Inc., which is negotiating licenses for largazole and related patents and patent applications.
REFERENCES
- (1).Taori K; Paul VJ; Luesch HJ Am. Chem. Soc 2008, 130, 1806–1807. [DOI] [PubMed] [Google Scholar]
- (2).(a) Salvador-Reyes LA; Engene N; Paul VJ; Luesch HJ Nat. Prod 2015, 78, 486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Engene N; Tronholm A; Salvador-Reyes LA; Luesch H; Paul VJ J Phycol. 2015, 51, 670–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Ying Y; Taori K; Kim H; Hong J; Luesch HJ Am. Chem. Soc 2008, 130, 8455–8459. [DOI] [PubMed] [Google Scholar]; (b) Bowers A; West N; Taunton J; Schreiber SL; Bradner JE; Williams RM J. Am. Chem. Soc 2008, 130, 11219–11222. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu YX; Salvador LA; Byeon S; Ying Y; Kwan JC; Law BK; Hong J; Luesch HJ Pharmacol. Exp. Ther 2010, 335, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hong J; Luesch H Nat. Prod. Rep 2012, 29, 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yu M; Salvador LA; Sy SKB; Tang Y; Singh RSP; Chen Q-Y; Liu Y; Hong J; Derendorf H; Luesch H Mar. Drugs 2014, 12, 1623–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Salvador LA, Luesch H HDAC inhibitors and other histone modifying natural products as emerging anticancer agents. In: Koehn FE, editor. Natural Products and Cancer Drug Discovery. New York: Springer; 2013. p. 59–95. [Google Scholar]
- (6).(a) Lansigan F; Foss FM Drugs 2010, 70, 273–286. [DOI] [PubMed] [Google Scholar]; (b) Marks PA; Breslow R Nat Biotechnol. 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- (7).(a) Paris M; Porcelloni M; Binaschi M; Fattori DJ Med. Chem 2008, 51, 1505–1529. [DOI] [PubMed] [Google Scholar]; (b) Senese S; Zaragoza K; Minardi S; Muradore I; Ronzoni S; Passafaro A; Bernard L; Draetta GF; Alcalay M; Seiser C; Chiocca S Mol Cell Biol. 2007, 27, 4784–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Thaler F; Minucci S Expert Opin Drug Discov. 2011, 6, 393–404. [DOI] [PubMed] [Google Scholar]
- (9).Lee SU; Kwak HB; Pi SH; You HK; Byeon SR; Ying Y Luesch H; Hong J, Kim SH ACS Med Chem Lett. 2011, 2, 248–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liu Y; Wang Z; Wang J; Lam W; Kwong S; Li F; Friedman SL; Zhou S; Ren Q; Xu Z; Wang X; Ji L; Tang S; Zhang H; Lui EL; Ye T Liver International 2013, 33, 504–515. [DOI] [PubMed] [Google Scholar]
- (11).Law ME; Corsino PE; Jahn SC; Davis BJ; Chen S; Patel B; Pham K; Lu J; Sheppard B; Norgaard P; Hong J; Higgins P; Kim J-S; Luesch H; Law BK Oncogene 2013, 32, 1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Kim B; Park H; Salvador LA; Serrano PE; Kwan JC; Zeller SL; Chen Q-Y; Ryu S; Liu Y; Byeon S; Luesch H; Hong J Bioorg. Med. Chem. Lett 2014, 24, 3728–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Salvador LA; Park H; Al-Awadhi FH; Liu Y; Kim B; Zeller SL; Chen Q-Y; Hong J; Luesch H ACS Med. Chem. Lett 2014, 5, 905–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Su J; Qiu Y; Mac K; Yao Y; Wang Z; Li X; Zhang D; Tu Z; Jiang S Tetrahedron 2014, 70, 7763–7769. [Google Scholar]; (b) Kim B; Ratnayake R; Lee H; Shi G; Zeller SL; Li C; Luesch H; Hong J Bioorg. Med. Chem 2017, 25, 3077–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Seiser T; Kamena F; Cramer N Angew. Chem., Int. Ed 2008, 47, 6483–6485. [DOI] [PubMed] [Google Scholar]; (b) Nasveschuk CG; Ungermannova D; Liu XD; Phillips AJ Org. Lett 2008, 10, 3595–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh AK; Kulkarni S Org. Lett 2008, 10, 3907–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang B; Forsyth CJ Synthesis 2009, 2873–2880. [Google Scholar]; (e) Wang B; Huang P-H; Chen C-S; Forsyth CJ J. Org. Chem 2011, 76, 1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Xiao Q; Wang LP; Jiao XZ; Liu XY; Wu QA; Xie PJ Asian Nat. Prod. Res 2010, 12, 940–949. [DOI] [PubMed] [Google Scholar]; (g) Ren Q; Dai L; Zhang H; Tan W; Xu Z; Ye T Synthesis 2008, 2379–2383. [Google Scholar]; (h) Zeng X; Yin B; Hu Z; Liao C; Liu J; Li S; Li Z; Nicklaus MC; Zhou G; Jiang S Org. Lett 2010, 12, 1368–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Bowers AA; West N; Newkirk TL; Troutman-Youngman AE; Schreiber SL; Wiest O; Bradner JE; Williams RM Org. Lett 2009, 11, 1301–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ying Y; Liu YX; Byeon SR; Kim H; Luesch H; Hong J Org. Lett 2008, 10, 4021–4024. [DOI] [PubMed] [Google Scholar]; (c) Souto JA; Vaz E; Lepore I; Poppler AC; Franci G; Alvarez R; Altucci L; de Lera AR J. Med. Chem 2010, 53, 4654–4667. [DOI] [PubMed] [Google Scholar]; (d) Guerra-Bubb JM; Bowers AA; Smith WB; Paranal R; Estiu G; Wiest O; Bradner JE; Williams RM Bioorg. Med. Chem. Lett 2013, 23, 6025–6028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Clausen DJ; Smith WB; Haines BE; Wiest O; Bradner JE; Williams RM Bioorg. Med. Chem 2015, 23, 5061–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Bhansali P; Hanigan CL; Perera L; Casero RA Jr; Tillekeratne LM V. Eur. J. Med. Chem 2014, 86, 528–541. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Almaliti J; Al-Hamashi AA; Negmeldin AT; Hanigan CL; Perera L; Pflum MKH; Casero RA Jr.; Tillekeratne LMV J. Med. Chem 2016, 59, 10642–10660. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Chen F; Chai H; Su M-B; Zhang Y-M; Li J; Xie X; Nan H-J ACS Med. Chem. Lett 2014, 5, 628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Bhansali P; Hanigan CL; Casero RA Jr.; Tillekeratne LMV J. Med. Chem 2011, 54, 7453–7463. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Li X; Tu Z; Li H; Liu C; Li Z; Sun Q; Yao Y; Liu J; Jiang S ACS Med. Chem. Lett 2012, 4, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Reddy DN; Ballante F; Chuang T; Pirolli A; Marrocco B; Marshall GR J. Med. Chem 2016, 59, 1613–1633. [DOI] [PubMed] [Google Scholar]
- (16).(a) Zhou H; Jiang S; Chen J; Ren X; Jin J; Su SB Eur. J. Pharmacol 2014, 740, 619–626. [DOI] [PubMed] [Google Scholar]; (b) Decroos C; Clausen DJ; Haines BE; Wiest O; Williams RM; Christianson DW Biochemistry 2015, 54, 2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pilon JL; Clausen DJ; Hansen RJ; Lunghofer PJ; Charles B; Rose BJ; Thamm DH; Gustafson DL; Bradner JE; Williams RM Cancer Chemother Pharmacol. 2015, 75, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wu L-C; Wen Z-S; Qiu Y-T; Chen X-Q; Chen H-B; Wei M-M; Liu Z; Jiang S; Zhou G-B ACS Med. Chem. Lett 2013, 4, 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Seiple IB; Zhang Z; Jakubec P; Langlois-Mercier A; Wright PM; Hog DT; Yabu K; Allu SR; Fukuzaki T; Carlsen PN; Kitamura Y; Zhou X; Condakes ML; Szczypinski FT; Green WD; Myers AG Nature 2016, 533, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Toriyama M; Kamijo H; Motohashi S; Takido T; Itabashi K Phosphorus, Sulfur, and Silicon 2003, 178, 1661–1665. [Google Scholar]
- (19).Jeanguenat A; Seebach DJ Chem. Soc. Perkin Trans 1 1991, 2291–2298. [Google Scholar]
- (20).Pattenden G; Thom SM; Jones MF Tetrahedron 1993, 49, 2131–2138. [Google Scholar]
- (21).Aihara K; Kano Y; Shiokawa S; Sasaki T; Setsu F; Sambongi Y; Ishii M; Tohyama K; Ida T; Tamura A; Atsumi K; Iwamatsu K Bioorg. Med. Chem 2003, 11, 3475–3485. [DOI] [PubMed] [Google Scholar]
- (22).Bergeron RJ; Wiegand J; McManis JS; McCosar BH; Weimar WR; Brittenham GM; Smith RE J. Med. Chem 1999, 42, 2432–2440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.