

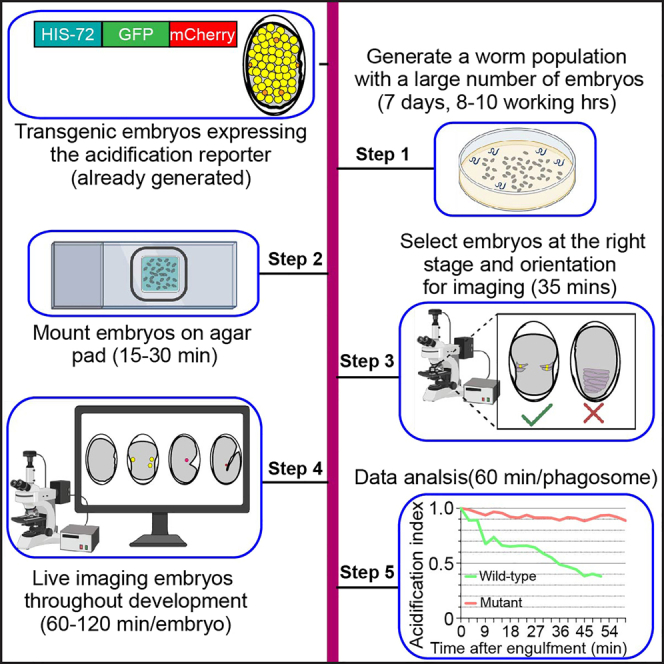
Summary
In metazoans, the acidification of the phagosomal lumen is essential for the efficient degradation of cargoes. Here, we present a protocol for measuring the rate of acidification inside phagosomal lumen containing apoptotic cells in living C. elegans embryos. We describe steps for generating a worm population, selecting embryos, and mounting embryos on agar pads. We then detail live imaging of embryos and data analysis. This protocol is applicable to any organism in which real-time fluorescence imaging can be performed.
For complete details on the use and execution of this protocol, please refer to Pena-Ramos et al. (2022).1
Subject areas: Cell Biology, Developmental Biology, Microscopy, Model Organisms
Graphical abstract
Highlights
-
•
Protocol for determining the rate of acidification inside phagosomal lumen in C. elegans
-
•
An acidification reporter based on the differential acid resistance of GFP and mCherry
-
•
Step-by-step protocol for time-lapse recording conducted in developing embryos
-
•
Detailed description of data quantification using the SoftWorx 5.5 software
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
In metazoans, the acidification of the phagosomal lumen is essential for the efficient degradation of cargoes. Here, we present a protocol for measuring the rate of acidification inside phagosomal lumen containing apoptotic cells in living C. elegans embryos. We describe steps for generating a worm population, selecting embryos, and mounting embryos on agar pads. We then detail live imaging of embryos and data analysis. This protocol is applicable to any organism in which real-time fluorescence imaging can be performed.
Before you begin
Background and rationale of this protocol
Phagocytosis is a “cell-eat-cell” event in which phagocytes engulf and degrade invading pathogens, dying cells, or damaged neuronal processes.2 The engulfed objects are confined inside phagosomes, which are membrane vacuoles, and are gradually degraded.2 The degradation of phagosomal cargoes relies on the fusion of intracellular organelles to phagosomes and the acidification of the phagosomal lumen.3 During the phagosome maturation process, the pH value of the lumen reduces from ∼6.5 to ∼4.5, due to the function of the vacuolar ATPase (V-ATPase) on the phagosomal membrane.4 The rate of phagosomal acidification thus is an important indication of the efficiency of phagosome maturation. Previously, a few methods were established for measuring the acidification status of the phagosomal lumen inside mammalian phagocytes.5,6,7,8 These methods require the incubation of the fluorescently-labeled cargoes with phagocytes in culture, an action not feasible for the whole-animal assay such as our assay in C. elegans embryos. Here we describe a novel protocol for measuring the rate of the acidification of the phagosomal lumen. This assay relies on the in vivo expression of pH-sensitive fluorescent proteins and the real-time imaging of the phagosomal lumen. It measures the phagosomal acidification status in live cells inside developing embryos.
During C. elegans embryonic development, 113 somatic cells undergo apoptosis and are swiftly engulfed and degraded by neighboring cells.9,10,11 The identity and location of the apoptotic cells, and the timing of death are all rigidly reproduced in each embryo, allowing the real-time imaging of the engulfment and degradation of apoptotic cells with known identities.9 We routinely image many dynamic events of three phagosomes that each contains the apoptotic cells C1, C2, and C3, respectively, which undergo apoptosis at approximately 330-min post-1st cleavage.12 Apoptotic cells C1, C2, and C3 are each engulfed and degraded by a particular ventral hypodermal cell (Figures 1B and 1C). The protocol below describes the specific steps for measuring the acidification of the C1, C2, and C3 phagosomes in the developing C. elegans embryo.
Figure 1.

The strategy of the quantitative phagosomal lumen acidification assay
(A) Domain structure of the HIS-72::GFP::mCherry reporter construct. The pKa value of each individual fluorescent protein is indicated.
(B) A diagram illustrating the three phagosomes that contain cell corpses C1, C2, and C3, with which we monitor the acidification process, at ∼330 min post the 1st embryonic division. Both the positions of C1, C2, and C3 (brown dots) and the identities of their engulfing cells are shown.
(C) An image showing how C1, C2, and C3 (arrowheads) can be distinguished under DIC objectives. The scale bar is 5 μm.
(D and E) Time-lapse imaging series of phagosomes (white arrowheads in DIC images) of wild-type and cup-5 mutant embryos expressing Phis-72his-72::gfp::mCherry. Open white arrows depict the nuclei of engulfed cell corpses, labeled with both the GFP and mCherry markers. Reduction of the GFP signal intensity over time is indicative of phagosome acidification. ‘0 min’ is when a phagosome is just sealed. Scale bars are 2 μm.
(F) A diagram illustrating where the intensities of the mCherry and GFP signals are measured in the center of a phagosome.
(G) The acidification index curves of two phagosomes (Y-axis) over time (in the 3 min interval) (X-axis) in embryos with the labeled genotypes. ‘0 min’ indicates the moment when a phagosome is just sealed. The data of the wild-type and cup-5 (n3265) are from D-E, respectively.
(B, F, D, E, G) are adapted from Figure 1 (B and C) and Figure 13 (A, B, and D) of Pena-Ramos et al.1
Our acidification reporter for the phagosomal lumen is a HIS-72 (histone H3.3)::GFP::mCherry fusion protein (Figure 1A), which is nuclei-localized and is expressed in all cells, including cells that undergo programmed cell death.13 Because the fluorophore within GFP is sensitive to acidic pH (pKa = 6.0),14 the GFP signal diminishes inside the phagosomal lumen, which is undergoing acidification. On the other hand, mCherry (pKa<4.5) is resistant to acidic pH.15 Thus, the mCherry signal intensity serves as an internal reference to which the temporally reduced GFP signal is normalized. To quantify the rate of acidification, the GFP and mCherry signal intensities in the center of the phagosomes that carry C3, C1, C2 are measured over time for at least 60 min (Figures 1B–1G). The GFP/mCherry intensity ratio in the phagosomal lumen at each time point is calculated and normalized to generate an “acidification index” to represent the acidification state. In a wild-type embryo, the acidification index of a typical C3 phagosome reduces from 1.0 (t = 0 min) to 0.4 (t = 51 min) (Figures 1D and 1G).1 When a cargo-specific reporter is used, this method can be used to measure the acidification index in phagosomes that carry all sorts of cargo. Assays based on pH-sensitive GFP and its variants have been used to measure the rate of acidification in the lumen of a broad range of intracellular organelles, including lysosomes16 and autophagosomes.17 Likewise, our method can be adapted to measure the acidification index in different kinds of intracellular vesicles. Moreover, this method is not limited to C. elegans. Any model system that allows the expression of a transgenic reporter and the real-time recording of subcellular events is suitable for this protocol.
Culture C. elegans to obtain embryos
Work time: 4–8 h, starting from seven days (Day -7) before the time-lapse recording experiment (Day 1).
-
1.
Prepare nematode growth media (NGM) as agar plates (Day -7). Seed these plates with the E. coli strain OP50 (Day -5). At least 5 plates are needed to propagate each strain.
-
2.
Make sure to use fresh M9 buffer that is not older than 2 months, preferably, <1 month old.
-
3.
Prepare 50 mL of fresh 4% agarose dissolved in autoclaved deionized water (diH2O) and let it solidify on the bench.
-
4.
Two or three days before the start of the experiment (Day -3 or Day -2), chunk 5 pieces of agar from a starved plate containing the strain of interest (ZH2059) and place each of them on an NGM plate seeded with OP50. Incubate the plates for 2–3 days at 22–23°C.
Note: Nematodes are normally raised at 22–23°C. If they are incubated at 15°C the waiting period is doubled due to the slower rate of development.
Institutional permissions (if applicable)
Institutional permissions are not required for working with C. elegans.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| DeltaVision Immersion oil N = 1.516 | Cytiva | Cat#:29162940 |
| High vacuum grease | Fisher Scientific | Cat#:14-635 |
| Agarose | Fisher Scientific | Cat#: BP160-500 |
| KH2PO4 | EMD Millipore | Cat#: PX1565 |
| K2HPO4 | EMD Millipore | Cat# PX1570 |
| Na2HPO4 | EMD Millipore | Cat#: 567550 |
| NaCl | Fisher Scientific | Cat#: S271-500 |
| CaCl2 | Fisher Scientific | Cat#: 10043-52-4 |
| MgSO4 | Fisher Scientific | Cat#: M65-500 |
| Cholesterol | EMD Millipore | Cat#: CAS 57-88-5 |
| Bacto Peptone | Fisher Scientific | Cat#: 211677 |
| Granulated Agar | Fisher Scientific | Cat#: BP97445 |
| Experimental models: Organisms/strains | ||
| C. elegans Strain ZH2059: unc-76(e911)V; enEx979 [punc-76(+), Phis-72his-72::GFP::mCherry] | Zhou Lab | ZH2059 |
| Software and algorithms | ||
| SoftWoRx 5.5 | GE Healthcare, Inc. | NA |
| Microsoft Excel | Microsoft, Inc | NA |
| Prism GraphPad | Dotmatics, Inc | NA |
| Other | ||
| Microscope slides | Premiere | Cat#: 9101 |
| Coverslips (22 × 22 mm) | Fisher Scientific | Cat#: 12-542B |
| Pasteur pipette | Fisher Scientific | Cat# 13-678-20B |
| Platinum wire | Alfa Aesar | Cat# 10287 |
Materials and equipment
Major equipment
To visualize C. elegans adults and embryos and to prepare a slide with embryo samples we use a Nikon SMZ 645 Stereo Microscope. For collecting embryos onto a slide, this protocol uses a homemade worm pick. Using a diamond pen, we cut half of the tip of a glass Pasteur pipette off. We then insert a platinum wire that is approximately 5cm in length to the now shortened tip of the Pasteur pipette, leaving 4cm outside the pipette. Using a Bunsen burner and a pair of metal tweezers we seal the glass tip. Alternatively, a commercially available worm pick from Genesee Scientific (Cat#: 59-AWP-B) can be used instead of the homemade pick.
For real-time imaging of developing C. elegans embryos, this protocol utilizes a DeltaVision Deconvolution Imaging System that includes an inverted microscope (Olympus IX70) equipped with 20×, 63×, and 100× Uplan Apo objectives, a Differential Interference Contrast (DIC) imaging apparatus, a motorized stage (X, Y, and Z-axis), and a Photometrics CoolSnap HQ2 digital camera. For fluorescent imaging, two sets of fluorescent filters (Chroma Inc) are used, the GFP filter (excitation wavelength 475/28 nm; emission wavelength 525/50 nm), and the mCherry filter (excitation wavelength 575/25 nm; emission wavelength 632/60 nm). The temperature of the room hosting the DeltaVision microscope is kept at 20°C. Alternatively, an environmental chamber from GE Healthcare can be used to maintain the required temperature if the room air conditioning is unstable.
In addition to the DeltaVision Deconvolution Imaging System, other imaging systems, as long as they cause no or minimal photodamage to embryonic development during real-time imaging, can be used to perform this protocol. These include, but not limited to the spinning disk confocal imaging system such as the PerkinElmer Spinning Disk Confocal Microscope or the Laser sheet confocal imaging system.
4% Agarose solution (per 50 mL)
| Reagent | Final concentration | Amount |
|---|---|---|
| Agarose | 4% | 2.0 g |
| dH2O | N/A | 50 mL |
We microwave the solution in a 200 mL flask until the agarose is completely dissolved. The solution can be stored at room temperature with a piece of aluminum foil as a cover. To reuse the agar solution, melt the solidified solution for 35 s in a microwave oven.
M9 Buffer (per Liter)
| Reagent | Final concentration | Amount |
|---|---|---|
| Na2HPO4 | 40.9 mM | 5.8 g |
| NaCl | 8.5 mM | 0.5 g |
| NH4Cl | 18.7 mM | 1.0 g |
| KH2PO4 | 22.0 mM | 3.0 g |
| dH2O | N/A | 1 L |
We aliquoted the solution into ten 100 mL capped bottles and autoclaved them for 40 min. Once sterilized, the solution can be stored at room temperature for three months.
NGM plates (per Liter)
| Reagent | Final concentration | Amount |
|---|---|---|
| CaCl2 | 1.0 mM | 0.111 g |
| NaCl | 51.3 mM | 3.0 g |
| NH4Cl | 18.7 mM | 1.0 g |
| KH2PO4 | 19.9 mM | 2.7 g |
| K2HPO4 | 5.1 mM | 0.89 g |
| MgSO4 | 1.0 mM | 0.12 g |
| Cholesterol | 130 μM | 5 mg |
| Bacto Peptone | 2.5 g/L | 2.5 g |
| Granulated Agar | 15 g/L | 15 g |
| dH2O | N/A | 1 L |
Prepare stock solutions of 1 M CaCl2, 1M MgSO4, and 1M KPO4 buffer pH6.0 108 3g KH2PO4 and 35.6 g K2HPO4 in 1 L diH2O solution). Autoclave. Prepare 5 mg/mL cholesterol in ethanol. Filter sterilize. Store all of the above solutions at room temperature. In a flask, weight 15 g granulated agar, 2.5 g of Bacto peptone, and 3.0 g of NaCl, add 1 L of diH2O, mix well, autoclave. When the autoclaved solution is cooled to 55°C, add 1 mL 1 M CaCl2, 1M MgSO4, 25 mL 1 M KPO4 buffer PH6.0, and 1 mL 5 mg/mL cholesterol in ethanol to the flask. Mix well. Pour the liquid to 6 cm petri dishes. This media can be stored in a closed plastic box at 22°C–24°C for one month.
Step-by-step method details
Mount embryos for imaging
Timing: 15–30 min
This step will prepare a slide ready for time-lapse observations on Day 1. The goal of this step is to transfer embryos from our experimental plates prepared two days before to a slide that contains an agarose pad with M9 buffer. After the coverslip is placed on the samples, vacuum grease is used to seal the edges of the cover slip to keep the moisture of the microenvironment where the embryos are placed. This setting generates a microenvironment that permits C. elegans embryos to develop while being imaged under the microscope.
-
1.
Use a microwave oven to melt the 4% agarose solution.
-
2.
Dispense between 100–300 μL of the melted agarose solution to the center of a slide.
Note: The melted solution is very sticky, pipette it gently; this will avoid generating bubbles.
-
3.
Immediately flatten the agarose drop by placing another glass slide perpendicular to the one holding the drop and press gently.
-
4.
Let it stand for 2 min for the agar to solidify completely.
-
5.
Carefully separate the two slides by sliding one against the other.
-
6.
Using a glass slide as a blade, cut the agar pad into an approximately 12 × 12 mm square.
-
7.
Place 3 μL of M9 buffer at the center of the agar pad.
-
8.
Under a stereotype microscope and a worm pick, collect between 50-80 embryos with a worm pick and transfer them into the drop of M9 in the agarose pad.
Note: Our protocol does not required synchronization of hermaphrodites. On every agar pad we collect between 50 to 80 embryos from a mixed staged worm plate. The number of embryos ensures that at least some of them will be at the right stage and orientation for live imaging.
-
9.
Squeeze a thin line of high vacuum grease around the agarose pad.
-
10.
Place a cover slip over the grease and with the tip of a P10 pipette press the corners first and then the remaining grease to seal the slide.
CRITICAL: When transferring embryos from the plate to the agar pad, make an effort to carry as little bacteria as possible. An excessive amount of bacteria in the sample mix will consume large quantities of oxygen and cause the arrest of embryonic development. See Troubleshooting Problem 2.
Time-lapse recording of the acidification status of the C1, C2, and C3 phagosomes
The goal of this step is to monitor the acidification process of phagosomes in developing embryos. We measure the acidification index of engulfed apoptotic cells C1, C2, and C3 over time by following the Phis-72 his-72::gfp::mCherry reporter, which labels the nuclei of C1, C2, and C3 as well as nuclei of all other cells in the embryo. C1, C2, and C3 are identified by their fixed positions on the ventral surface of an embryo (Figures 1B and 1C) (See “Notes” below for how to identify the embryos with the ventral surface facing the objective). The 0 min time point is determined by the appearance of the ¨bottom-like¨ structure under the DIC optics (Figures 1C, 1D, and 1E). At each time point, the mCherry and GFP signal intensities are measured at the center of the phagosome (Figures 1D and 1E). The acidification index at a particular time point is defined as the ratio of GFP/mCherry signals over the GFP/mCherry ratio at the 0 min time point (T0) (Figure 1G). The acidification index of 1.0 indicates no phagosome acidification compared to the T0 value.
-
11.
Set up the microscope parameters (5 min).
| Parameter | Exposure time | Filter intensity |
|---|---|---|
| DIC | 0.01 s | 32% |
| mCherry | 0.08 s | 50% |
| GFP | 0.08 s | 2% |
-
12.
Under 20× objective, identify the embryos that carry the fluorescent reporter (which are GFP+ and mCherry+) and at the mid-embryonic developmental stage (Figure 1B) (15 min).
-
13.
Under the 100× objective (Numerical aperture 1.35), identify embryos that are between 320-330 min post-1st cleavage (the 1st embryonic cell division) (Figure 2).
Note: It is imperative that the embryos to be imaged are with their ventral side facing the microscope objectives. The arrangement pattern of the hypodermal cells, which are highlighted in (Figures 2B and 2D) can be used as a guide to determine the orientation and stage of the embryo (Figure 2). The hypodermal cells on the ventral side of an embryo appear to extend towards the center of an embryo in symmetric pairs (Figures 2A and 2B), whereas those on the dorsal side of an embryo line up in a column (Figures 2C and 2D).
-
14.
Using the ¨point mark¨ and the ¨position visiting¨ functions of the Softworx 5.5 software to mark and visit the embryos to be recorded. We usually record 3 or fewer embryos in the same experiment (20 min).
-
15.
Define the Z-position where the z section recording should start. The serial z section recording is performed from the embryo’s ventral surface and proceeds to the center of the embryo (5 min for each embryo).
-
16.
To cover the depth of three phagosomes (C1, C2, and C3) and providing enough depth even when the focal planes alter over time during the recording period, 12–16 sections of 0.5 μm Z-interval are recorded.
-
17.
Start recording when the embryo reaches 320 min post-1st embryonic cell division.
Note: Usually, we set our time interval of the recording to 3 minutes for 60–120 min or until the embryo reaches the 1.5-fold stage (Figure 3). (60–120 min for each embryo)
Figure 2.

Distinct morphological features allow the identification of the ventral and dorsal sides of an embryo
DIC images of an embryo at one moment between 320-330 min post the 1st cleavage (the first embryonic cell division). Scale bars are 10 mm.
(A and B) The same image of the ventral side of an embryo.
(C and D) The same image of the dorsal side of the same embryo.
(B, D) The ventral (B) and dorsal (D) hypodermal cells, the distinct organization patterns of which allow the viewer to distinguish the ventral from the dorsal surfaces, are highlighted in purple.
Figure 3.

Time-lapse DIC images of a C. elegans embryo from the initiation of the ventral enclosure to the 1.5-fold stage
Arrows indicate apoptotic cells C1, C2 or C3. “0 min” is the time point when C1 is first observed under the DIC optics in the time-lapse recording process. The scale bar is 5 μm.
Expected outcomes
A successful real-time imaging series tracking the acidification of C1, C2, and C3 phagosomes in the wild-type background will observe a progressive reduction of the GFP signal and the constant mCherry signal over time. Quantitatively, this progression is reflected by the reduction of the acidification index (the quantification of the acidification index is described in the following section) from 1.0 at T0 min to 0.4 at T51 min. In mutants that bear mutations in lysosomal acidification and lysosomal biogenesis such as the cup-5 (n3264) mutations,1,18 the acidification index at T51 min is > 0.6 (Figures 1D–1G), demonstrating the effectiveness of this assay in detecting acidification defect.1
Quantification and statistical analysis
This section gives instructions on how to measure the reporter signal intensities and calculate the acidification index. An example set of data are presented in Table 1.
Table 1.
Example of raw data measured on a wild-type C1 phagosome over time
| File name | Cell corpse | Time point | Z section | IntGFP (arbitrary digital value) | IntmCherry (arbitrary digital value) | (IntGFP/IntmCherry) | AI |
|---|---|---|---|---|---|---|---|
| ZH2059_1011202 | C1 | 0 | 7 | 3510 | 2576 | 1.363 | 1.000 |
| 3 | 6 | 3615 | 2980 | 1.213 | 0.890 | ||
| 6 | 5 | 3599 | 2966 | 1.213 | 0.891 | ||
| 9 | 6 | 2770 | 3020 | 0.917 | 0.673 | ||
| 12 | 5 | 2987 | 2979 | 1.003 | 0.736 | ||
| 15 | 6 | 2916 | 3240 | 0.900 | 0.661 | ||
| 18 | 7 | 2890 | 3253 | 0.888 | 0.652 | ||
| 21 | 6 | 2961 | 3301 | 0.897 | 0.658 | ||
| 24 | 6 | 3198 | 3561 | 0.898 | 0.659 | ||
| 27 | 8 | 3310 | 3777 | 0.876 | 0.643 | ||
| 30 | 9 | 3165 | 3945 | 0.802 | 0.589 | ||
| 33 | 8 | 3080 | 4103 | 0.751 | 0.551 | ||
| 36 | 8 | 2937 | 4417 | 0.665 | 0.488 | ||
| 39 | 8 | 2968 | 4652 | 0.638 | 0.468 | ||
| 42 | 9 | 2974 | 4926 | 0.604 | 0.443 | ||
| 45 | 10 | 2806 | 5380 | 0.522 | 0.383 | ||
| 48 | 9 | 2945 | 5374 | 0.548 | 0.402 | ||
| 51 | 9 | 2730 | 5267 | 0.518 | 0.380 |
Data quantification is conducted using the SoftWoRx 5.5 software. The results are annotated in an Excel spreadsheet.
-
1.
Open your image file. Form your recording files by identifying the¨0¨min time point when a phagosome is just sealed. This is determined by the appearance of the ¨button-like¨ structure at the expected position of an embryo (Figure 1C).
-
2.
Define the most ventral section of a Z-stack.
Note: In each time point, the Z focal plane may change; always choose the Z-section in which the center of a phagosome is in focus.
-
3.
Go to “Tool”, select “Data Inspection” from the dropdown manual.
-
4.
Select a square of 3 × 3 pixels, which is 0.158 μm2.
-
5.
For each time point, measure the total mCherry and GFP intensities (IntmCherry and IntGFP, which are arbitrary digital values) inside the 3 × 3 square by placing the area frame generated in step 3 at the center of the phagosome (Figure 1F) and read the total arbitrary intensity values.
-
6.
The acidification index (AI) at a particular time point (Tn) is defined as:
Limitations
To determine the “0 min” moment of phagosome sealing, our protocol relies on the appearance of the distinct button-like morphology of the apoptotic cells C1, C2, and C3 observed under DIC objectives. While the appearance of the button-like morphology correlates well with the engulfment of the cell corpse, which takes only 4–6 min, the precise moment of pseudopod sealing can be more accurately determined by a pseudopod reporter. This assay can be further modified by co-expressing a pseudopod reporter Pced-1 PH(PLCδ)::gfp19 with Phis-72 his-72::gfp::mCherry in the same C. elegans strains. The PH domain::GFP fusion protein is enriched on pseudopodal membrane through association with PtdIns(4,5)P2 during engulfment and forms a GFP+ ring around the phagosome once the pseudopods seal.19 It thus allows accurate determination of the “0 min” moment. The PH::GFP signal can be easily distinguished from the HIS-72::GFP signal as the former reporter is localized to the plasma membrane whereas the latter one is localized to the nucleus.
The Phis-72 his-72::gfp::mCherry reporter used here is specifically suitable for tracking phagosomes that contain apoptotic cells. To quantify the acidification state of phagosomes that contain other kinds of cargoes, different cargo proteins tagged with the GFP-mCherry fusion reporter need to be expressed in the sample of interest. Alternatively, a phagosomal lumen protein can be tagged with the GFP-mCherry fusion reporter. Candidates of such proteins can be found among lysosomal luminal proteins, as lysosomes fuse to a phagosome and deposit their luminal proteins into the phagosomal lumen.
This assay requires real-time recording of the phagosome maturation process, as the process of acidification at different time points is measured by comparing to that at the “0 min” time point. In other animals and or systems, real-time imaging might not be feasible.
Alternative strategy
Alternative to measuring the GFP and mCherry signal intensities using the SoftWorx 5.5 software, publicly available image analysis software such as ImageJ, an image analysis software developed in the National Institute of Health and is freely available to the public, can also be used for the image quantification.
Troubleshooting
Problem 1
Having difficulty finding embryos at the wanted stage.
Despite that the embryos are collected from a mixed-staged population of C. elegans, some times embryos at between 320-330 min post 1st-cleavage are hard to find in a population of embryos.
Potential solution
If the majority of embryos are of younger stages, we mark between 10-15 young embryos using the ¨point mark¨ function of the Softworx 5.5 software to mark each embryo. In regular time interval such as every 20 min, we use the ¨position visiting¨ function of Softworx 5.5 to observe whether any embryos reach the 320–330 min stage and have their ventral surfaces facing the objective. Once we find the right embyos, we start the recording. If, on the other hand, the majority of embryos are older than 320–330 min stage, abort the experiment and find a new source of embryos. In practice, we pick 20 transgenic hermaphrodites at L4 stage to a plate. After 24 h, we collect embryos for imaging. Within this time frame, at least 50% of the embryos should be younger than 320–330 min stage. Time-lapse recording of the acidification status of the C1, C2, and C3 phagosomes-Day1.
Problem 2
Arresting or slowing down of embryonic development during the real-time recording process:
During the real-time recording period, improper settings of the recording will result in the slowing down or arrest of embryonic development. If that occurs, the defect might affect the progress of phagosomal acidification as well.
Evidence of embryonic developmental slowdown or arrest:
In a wild-type C. elegans embryo, the time span between the “0 min” time point for the nascent phagosomes containing C1, C2, and C3, which is at ∼330 min-post first embryonic cleavage, to the 1.5-fold stage (∼420 min-post first embryonic cleavage) is 90 min (Figure 3). If within 90 min, the embryo that is under recording has not reached 1.5-fold stage, it indicates a slowdown of embryonic development. If the morphology of an embryo under the DIC optics remains unchanged, it indicates an embryonic developmental arrest.
Potential solutions
There are two major reasons for abnormal embryonic development during the real-time recording process. The first is the insufficient amount of oxygen inside the embryo chamber. This could be due to that, when preparing the slide, a large amount of E.coli is transferred from the NGM plate to the agar pad on the glass slide. The bacteria will compete with C. elegans embryos for oxygen consumption and a large amount of bacteria will deprive oxygen of embryos. E.coli is visible under DIC optics. If too much E. coli is observed on the slide, abort the recording and prepare another slide, avoiding carrying too much bacteria when transferring the embryos. Mount embryos for imaging - Day1.
The second common reason for the stall of embryonic development is photodamage, when embryos are exposed to too much UV light. In this case, the neutral density filters of the GFP and mCherry channels can be adjusted to allow minimum UV light exposure. Our imaging system is equipped with multiple neutral density filters, the minimal level of light passing through is 2%. In addition, the exposure time can be shortened from the initial setting of 0.08 s to 0.04, 0.02, or even 0.01 s, depending on the signal intensity. Time-lapse recording of C1, C2, and C3 phagosomes acidification-Day1.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Zheng Zhou (zhengz@bcm.edu).
Materials availability
The C. elegans strain ZH2059 and the acidification reporter plasmid Phis-72 his-72::gfp::mCherry are both generated in the Zheng Zhou Lab and are available upon request.
Acknowledgments
This work is supported by NIH R01GM067848.
Author contributions
O.P.-R. wrote the initial draft of the manuscript, created the figures, and participated in the revision. Z.Z. participated in the discussion on deciding the outline of the manuscript and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Data and code availability
The Dataset based on which Figure 1G is generated is available in ref.1
References
- 1.Peña-Ramos O., Chiao L., Liu X., Yu X., Yao T., He H., Zhou Z. Autophagosomes fuse to phagosomes and facilitate the degradation of apoptotic cells in Caenorhabditis elegans. Elife. 2022;11:e72466. doi: 10.7554/eLife.72466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lancaster C.E., Ho C.Y., Hipolito V.E.B., Botelho R.J., Terebiznik M.R. Phagocytosis: what’s on the menu? 1. Biochem. Cell. Biol. 2019;97:21–29. doi: 10.1139/bcb-2018-0008. [DOI] [PubMed] [Google Scholar]
- 3.Levin R., Grinstein S., Canton J. The life cycle of phagosomes: formation, maturation, and resolution. Immunol. Rev. 2016;273:156–179. doi: 10.1111/imr.12439. [DOI] [PubMed] [Google Scholar]
- 4.Vieira O.V., Botelho R.J., Grinstein S. Phagosome maturation: aging gracefully. Biochem. J. 2002;366:689–704. doi: 10.1042/BJ20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deretic V., Singh S., Master S., Harris J., Roberts E., Kyei G., Davis A., de Haro S., Naylor J., Lee H.H., et al. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol. 2006;8:719–727. doi: 10.1111/j.1462-5822.2006.00705.x. [DOI] [PubMed] [Google Scholar]
- 6.Sokolovska A., Becker C.E., Stuart L.M. Measurement of phagocytosis, phagosome acidification, and intracellular killing of Staphylococcus aureus. Curr Protoc Immunol. 2012 doi: 10.1002/0471142735.im1430s99. [DOI] [PubMed] [Google Scholar]
- 7.Colas C., Menezes S., Gutiérrez-Martínez E., Péan C.B., Dionne M.S., Guermonprez P. An improved flow cytometry assay to monitor phagosome acidification. J. Immunol. Methods. 2014;412:1–13. doi: 10.1016/j.jim.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Nunes P., Guido D., Demaurex N. Measuring phagosome pH by ratiometric fluorescence microscopy. J. Vis. Exp. 2015:e53402. doi: 10.3791/53402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sulston J.E., Schierenberg E., White J.G., Thomson J.N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 10.Reddien P.W., Horvitz H.R. The engulfment process of programmed cell death in Caenorhabditis elegans. Annu. Rev. Cell Dev. Biol. 2004;20:193–221. doi: 10.1146/annurev.cellbio.20.022003.114619. [DOI] [PubMed] [Google Scholar]
- 11.Lu N., Zhou Z. Membrane trafficking and phagosome maturation during the clearance of apoptotic cells. Int. Rev. Cell Mol. Biol. 2012;293:269–309. doi: 10.1016/B978-0-12-394304-0.00013-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu N., Yu X., He X., Zhou Z. Detecting apoptotic cells and monitoring their clearance in the nematode Caenorhabditis elegans. Methods Mol. Biol. 2009;559:357–370. doi: 10.1007/978-1-60327-017-5_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ooi S.L., Priess J.R., Henikoff S. Histone H3.3 variant dynamics in the germline of Caenorhabditis elegans. PLoS Genet. 2006;2:e97. doi: 10.1371/journal.pgen.0020097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsien R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 15.Shaner N.C., Campbell R.E., Steinbach P.A., Giepmans B.N., Palmer A.E., Tsien R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 16.Ponsford A.H., Ryan T.A., Raimondi A., Cocucci E., Wycislo S.A., Fröhlich F., Swan L.E., Stagi M. Live imaging of intra-lysosome pH in cell lines and primary neuronal culture using a novel genetically encoded biosensor. Autophagy. 2021;17:1500–1518. doi: 10.1080/15548627.2020.1771858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manil-Segalen M., Lefebvre C., Jenzer C., Trichet M., Boulogne C., Satiat-Jeunemaitre B., Legouis R. The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev. Cell. 2014;28:43–55. doi: 10.1016/j.devcel.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 18.Treusch S., Knuth S., Slaugenhaupt S.A., Goldin E., Grant B.D., Fares H. Caenorhabditis elegans functional orthologue of human protein h-mucolipin-1 is required for lysosome biogenesis. Proc. Natl. Acad. Sci. USA. 2004;101:4483–4488. doi: 10.1073/pnas.0400709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen Q., He B., Lu N., Conradt B., Grant B.D., Zhou Z. Phagocytic receptor signaling regulates clathrin and epsin-mediated cytoskeletal remodeling during apoptotic cell engulfment in C. elegans. Development. 2013;140:3230–3243. doi: 10.1242/dev.093732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The Dataset based on which Figure 1G is generated is available in ref.1