

Summary
Preculture is indispensable for achieving highly efficient non-homologous end joining (NHEJ)-based genome editing. Here, we present a protocol for optimizing genome editing conditions for murine hematopoietic stem cells (HSCs) and evaluating their function following NHEJ-based genome editing. We describe steps for sgRNA preparation, cell sorting, preculture, and electroporation. We then detail post-editing culture and transplanting of bone marrow. This protocol can be used to study genes related to HSC quiescence.
For complete details on the use and execution of this protocol, please refer to Shiroshita et al.1
Subject areas: CRISPR, Stem Cells
Graphical abstract
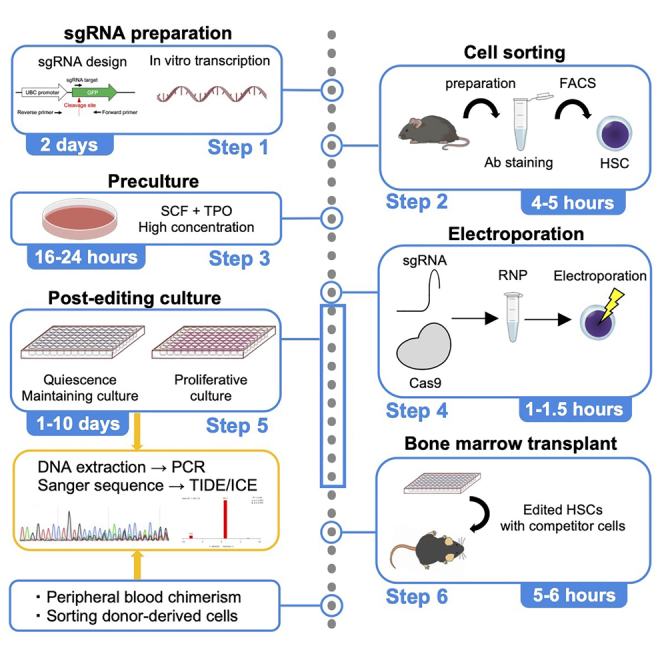
Highlights
-
•
Step-by-step protocol to evaluate the HSC function following gene editing
-
•
Optimize the post-editing culture condition for HSC quiescence
-
•
Regaining HSC quiescence following gene editing improves engraftment capacity
-
•
Our protocol is a useful tool for studying the gene related to HSC quiescence
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Preculture is indispensable for achieving highly efficient non-homologous-end-joining (NHEJ)-based genome editing. Here, we present a protocol for optimizing genome editing conditions for murine hematopoietic stem cells (HSCs) and evaluating their function following NHEJ-based genome editing. We describe steps for sgRNA preparation, cell sorting, preculture, and electroporation. We then detail post-editing culture and transplanting of bone marrow. This protocol can be used to study genes related to HSC quiescence.
Before you begin
Hematopoietic stem cells (HSCs) maintain lifelong hematopoiesis in the bone marrow. Most HSCs are quiescent in the cell cycle. Quiescent HSCs have high repopulation capacity by preventing proliferative stress and/or exhaustion. To investigate the regulators of HSC function, bone marrow transplantation (BMT) using HSCs from genetically engineered mouse models has been widely used. Although this approach clarifies several critical genes that regulate HSC function, making a genetically engineered mouse model is a time-consuming process and the number of target genes is limited. Furthermore, direct genetic engineering is required for translating findings from mouse models into human HSCs. Therefore, alternative tools are needed to study HSC function more quickly and easily than conventional mouse models.
Recently, we reported an ex vivo culture system that maintains the quiescence of HSCs for one month.2 This quiescence-maintaining culture is composed of physiological niche factors; high-concentration BSA, low concentration of cytokines, and hypoxia. Using this culture system, we tried to establish a novel in vitro tool to study the quiescence of HSCs following genome editing. Using the CRISPR-Cas9 system, we optimized the genome editing conditions to murine HSCs and showed that preculture before genome editing improved ribonucleoprotein (RNP) delivery into the nucleus. In addition, by combining the quiescence-maintaining culture with CRISPR-Cas9, we determined that the genome-edited HSCs could regain quiescence. The genome-edited HSCs in quiescence-maintaining culture maintained the original surface markers (CD150+CD48-Lin-Sca-1+c-Kit+: SLAM-LSK), fresh HSC-like transcriptional profile, and high engraftment capacity compared to genome-edited HSCs in conventional proliferative culture.1
The protocol below describes the specific steps for analyzing the repopulation capacity of HSCs following non-homologous end joining (NHEJ)-based genome editing. In this protocol, we used sgRNA targeting GFP in UBC-GFP mice for NHEJ-based editing efficiency, and the Ly5.1/Ly5.2 system and Sanger sequencing as the donor-derived chimerism. The following materials and reagents should be prepared before you begin.
Institutional permission
All experimental procedures involving animals should be performed in accordance with relevant national and institutional regulations and within dedicated experimental animal facilities. Please follow internal guidelines related to the purchase, housing, and breeding of experimental mice.
Setting up culture medium for quiescence-maintaining and proliferative conditions
Timing: 1–2 h
-
1.Prepare lipids.
-
a.Dissolve fatty acids (FA) salt and cholesterol (Sigma-Aldrich) in methanol separately in glass tubes with caps (Maruemu Corporation). FA includes only palmitic acid (Wako Pure Chemical Corporation) and oleic acid (Sigma-Aldrich).
-
b.Make the stock solution of FA and cholesterol (Sigma-Aldrich). The stock concentration of each lipid is as follows: 16 mg/mL of palmitic acid, 30 mg/mL of oleic acid, and 4 mg/mL of cholesterol.Note: Precise procedures for dissolving FA and cholesterol are described elsewhere.3
-
c.Set a water bath to 37°C.
-
d.Mix FA and/or cholesterol solutions in glass tubes. Final concentrations are 100 μg/mL palmitic acid, 100 μg/mL oleic acid, and 20 μg/mL cholesterol.
-
e.Blow air on the solution by using a 1 mL pipetor or pipette aid. If you are equipped with an N2 cylinder, blow nitrogen gas into the solution to avoid oxidation of unsaturated fatty acids by using a pipet. If nitrogen gas is not available, pass air through the solution by using a pipette aid.
-
f.Heat the glass tube with FA salt in the water bath at 37°C (up to 50°C is acceptable) until the methanol evaporates.
-
a.
-
2.Prepare 4% BSA medium.
-
a.Add 4% w/v of BSA (Sigma-Aldrich) to DMEM/F-12 medium.
-
b.Filter the medium using a 0.22 μm filter (Millipore).
-
c.Add medium containing 4% BSA to the glass tube which includes FA and cholesterol as prepared in the previous section (Prepare lipids).
-
d.Sonicate the medium until it is clear. We use a water bath-based sonicator. Avoid overheating the medium.
-
e.Adjust to pH 7.6 ± 0.1 using 1M NaOH (Wako Pure Chemical Corporation). We use approximately 70 μL for 1M NaOH for 10 mL of 4% BSA medium.
-
f.Add Insulin-Transferrin-Selenium-Ethanolamine (ITSX) mixture (Thermo Fisher Scientific) of 1/1000 to the total volume and 2-ME at a final concentration of 55 μM.
-
a.
BSA (4%) medium
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| DMEM/F12 | – | N/A | 9980 μL |
| BSA | – | 4% | 400 mg |
| Palmitic acid | 16 mg/mL | 100 μg/mL | 62.5 μL |
| Oleic acid | 30 mg/mL | 100 μg/mL | 33 μL |
| Cholesterol | 4 mg/mL | 20 μg/mL | 50 μL |
| 2-ME | 55 mM | 55 μM | 10 μL |
| IST mixture | N/A | N/A | 10 μL |
| Total | N/A | N/A | 10 mL |
Note: We generally purchase DMEM/F12 containing 4% BSA, FA, and cholesterol with adjusted pH from a custom medium supplier (e.g., GMEP). This might shorten the time to prepare the medium and minimize variation in composition experimental results.
Alternatives: We use DMEM/F12 medium without sodium pyruvate and with 1 mM sodium lactate. This might provide the best culture conditions after the quiescence-maintaining culture.
-
3.
Make a cytokine master stock for the quiescence-maintaining medium.
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| 4% BSA medium | – | – | 197.5 μL |
| Stem cell factor (SCF) | 20 ng/μL | 150 ng/mL | 1.5 μL |
| Thrombopoietin (TPO) | 20 ng/μL | 100 ng/mL | 1 μL |
| Total | N/A | N/A | 200 μL |
-
4.
Add the cytokines to the medium.
CRITICAL: Cytokines should be added immediately before use.
Quiescence-maintaining culture
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| 4% BSA medium | – | – | 9900 μL |
| Cytokine master stock | SCF 150 ng/mL TPO 100 ng/mL |
SCF 1.5 ng/mL TPO 1.0 ng/mL |
100 μL |
| Total | N/A | N/A | 10 mL |
Note: Cytokine concentration is one of the key determinants for a successful culture. We first prepare a medium containing 150 ng/mL SCF and 100 ng/mL TPO. The solution is diluted 100-fold in DMEM/F12 medium containing 4% BSA, FA, and cholesterol.
Proliferative culture
| Reagent | Stock concentration | Final concentration | Amount |
|---|---|---|---|
| 4% BSA medium | – | – | 9900 μL |
| Stem cell factor (SCF) | 20 ng/μL | 100 ng/mL | 50 μL |
| Thrombopoietin (TPO) | 20 ng/μL | 100 ng/mL | 50 μL |
| Total | N/A | N/A | 10 mL |
Alternatives: StemSpan SFEM-I and II (STEMCELL Technologies) are the media for proliferative conditions. In our experiences, CD150 expression is more reduced in the SFEM medium than in the 4% BSA medium when we perform the long-term ex vivo culture.
Synthesize single-guide RNA (sgRNA) in vitro
-
5.
Determine the target-specific crRNA sequence of your gene of interest (e.g., sgRNA targeting GFP: GGGCGAGGAGCTGTTCACCG).
-
6.
Design oligonucleotide DNA as template for in vitro transcription of sgRNA. Use the following primer sets; T7-sgRNA forward primer, sgRNA common primer, and sgRNA reverse primer.
T7-sgRNA forward primer:
5′-TTAATACGACTCACTATAX1X2X3X4X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20GTTTTAGAGCTAGAAATAGC-3’
sgRNA common primer: AAAAGCACCGACTCGGTGCC
sgRNA reverse primer: AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCT ATTTCTAGCTCTAAAAC
Note: The crRNA sequence of the gene of interest can be obtained using a web-based algorithm (e.g., benchling). To test the editing efficiency, we prepare 3-5 sgRNAs for each gene.
Note: To achieve optimal transcription using the T7 RNA promoter in the CUGA 7 enzyme reaction, add two guanines to the beginning of the target sequence if it does not already start with “GG”.
T7-sgRNA forward primer (if your selected sequence starts with “GG” ):
5′-TTAATACGACTCACTATAX1X2X3X4X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20GTTTTAGAGCTAGAAATAGC-3’
T7-sgRNA forward primer (if your selected sequence starts with “G” but not “GG”):
5′-TTAATACGACTCACTATAGX1X2X3X4X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20GTTTTAGAGCTAGAAATAGC-3’
T7-sgRNA forward primer (if your selected sequence does not start with “GG”):
5′-TTAATACGACTCACTATAGGX1X2X3X4X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20GTTTTAGAGCTAGAAATAGC-3′
Note: Do not include the PAM sequence (NGG).
-
7.
Perform PCR to amplify the sgRNA template following the recipe below.
ExTaq PCR for template DNA of sgRNA
| Reagent | Amount |
|---|---|
| T7-sgRNA Forward primer (50 μM) | 0.8 μL |
| sgRNA common primer (10 μM) | 4 μL |
| sgRNA reverse primer (0.1 μM) | 4 μL |
| 10× ExTaq buffer | 2 μL |
| 2.5 mM dNTP Mix | 1.6 μL |
| 5 U/μL ExTaq | 0.1 μL |
| ddH2O | 7.5 μL |
Note: The DNA template can be stored at −30°C.
PCR cycling conditions for ExTaq
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 3 min | 1 |
| Denaturation | 98°C | 30 s | 35 cycles |
| Annealing | 60°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final extension | 72°C | 3 min | 1 |
| Hold | 4°C | forever | |
-
8.
To synthesize sgRNA, incubate template DNA with CUGA7 Enzyme Solution at 37°C for 2 h.
sgRNA synthesis (CUGA7 system)
| Reagent | Amount |
|---|---|
| 5x Transcriptional buffer | 4 μL |
| 0.1 M DTT | 2 μL |
| NTP Mix | 6 μL |
| DNA template | 7 μL |
| CUGA7 Enzyme Solution | 1 μL |
-
9.
Add 2 μL of DNase I to remove template DNA (final volume 22 μL) and then place tube at 37°C for 15 min.
-
10.
Add 578 μL of gRNA Binding Buffer and thoroughly mix to avoid decreasing the sgRNA yield.
-
11.
Apply 600 μL of the mixture to a spin column and centrifuge at 13,000 × g for 1 min at 4°C.
-
12.
Wash the column by adding 750 μL of gRNA Wash Buffer and repeating the centrifugation step.
-
13.
Repeat the centrifugation step with the empty spin column.
-
14.
Apply 20 μL of RNase-free water to a spin column at 25°C for 3 min.
-
15.
To elute sgRNA, centrifuge the spin column at 13,000 × g for 1 min at 4°C.
-
16.
Determine the sgRNA concentration by using a NanoDrop Onec instrument (Thermo Fisher Scientific). Troubleshooting 1.
Note: We usually obtain the RNA solution at 3–5 μg/μL concentration.
-
17.
Dilute sgRNA with nuclease-free water to a concentration of 1.5 μg/μL (sgRNA stock) and cryopreserve at −80°C until use.
Alternatives: Chemically modified sgRNAs purchased from suppliers (e.g., Synthego, IDT) can also be used.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD4-PerCP-Cy5.5 (clone: RM4-5) | Tonbo Biosciences | Cat# 65-0042-U100; RRID: AB_2621876 |
| Anti-mouse CD8a-PerCP-Cy5.5 (clone: 53-6.7) | Tonbo Biosciences | Cat# 65-0081-U100; RRID: AB_2621882 |
| Anti-mouse B220-PerCP-Cy5.5 (clone: RA3-6B2) | Tonbo Biosciences | Cat# 65-0452-U100; RRID: AB_2621892 |
| Anti-mouse B220-APC (clone: RA3-6B2) | BioLegend | Cat# 103212; RRID: AB_312997 |
| Anti-mouse Ter-119-PerCP-Cy5.5 (clone: TER-119) | Tonbo Biosciences | Cat# 65-5921-U100 |
| Anti-mouse Gr1 (Ly-6G/6C)-PerCP-Cy5.5 (clone: RB6-8C5) | BioLegend | Cat# 108428; RRID: AB_893558 |
| Anti-mouse Gr1-PE-Cy7 (clone: RB6-8C5) | Tonbo Biosciences | Cat# 60-5931-U100; RRID: AB_2621870 |
| Anti-mouse Mac1 (CD11b)-PerCP-Cy5.5 (clone: M1/70) | Tonbo Biosciences | Cat# 65-0112-U100; RRID: AB_2621885 |
| Anti-mouse Mac1-PE-Cy7 (clone: M1/70) | Tonbo Biosciences | Cat# 60-0112-U100; RRID: AB_2621836 |
| Anti-mouse CD45.1-PE (clone: A20) | BD Biosciences | Cat# 553776; RRID: AB_395044 |
| Anti-mouse CD45.2-BV421 (clone: 104) | BD Biosciences | Cat# 562895; RRID: AB_2737873 |
| Anti-mouse Sca-1 (Ly-6A/E)-PE-Cy7 (clone: E13-161.7) | BioLegend | Cat# 122514; RRID: AB_756199 |
| Anti-mouse c-Kit (CD117)-APC-Cy7 (clone: 2B8) | BioLegend | Cat# 105826; RRID: AB_1626278 |
| CD117 MicroBeads Mouse | Miltenyi Biotec | Cat# 130-091-224 |
| Anti-mouse CD150-BV421 (clone: TC15-12F12.2) | BioLegend | Cat# 115926; RRID: AB_2562190 |
| Anti-mouse CD48-PE (clone: HM48-1) | BioLegend | Cat# 103406; RRID: AB_313021 |
| Fc-block (anti-mouse CD16/32) (clone: 2.4-G2) | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| Anti-mouse Flt3 (CD135)-APC (clone: A2F10) | BioLegend | Cat# 135310; RRID: AB_2107050 |
| Chemicals, peptides, and recombinant proteins | ||
| PBS | Nacalai Tesque | Cat# 14249-24 |
| DMEM/Ham’s-F12 medium | Nacalai Tesque | Cat# 11581-15 |
| StemSpan SFEM | STEMCELL Technologies | Cat# 09650 |
| StemSpan SFEM II | STEMCELL Technologies | Cat# 09655 |
| Insulin-Transferrin-Selenium-Ethanolamine (ISTX) 1000x | Thermo Fisher Scientific | Cat# 51500-056 |
| 2-Mercapto ethanol (2-ME) 1000x | Thermo Fisher Scientific | Cat# 21985-023 |
| Penicillin | Meiji Seika | PGLD755 |
| Streptomycin sulfate | Meiji Seika | SSDN1013 |
| Fetal bovine serum | Thermo Fisher Scientific | Cat# 10270-106 |
| Bovine serum albumin | Sigma-Aldrich | Cat# A4503-100G |
| Palmitic acid | Wako Pure Chemical Corporation | Cat# 165-00102 |
| Oleic acid | Sigma-Aldrich | Cat# O1383-1G |
| Cholesterol | Sigma-Aldrich | Cat# C3045-5G |
| Methanol | Nacalai Tesque | Cat# 21914-03 |
| Sodium hydroxide | Wako Pure Chemical Corporation | Cat# 191-01665 |
| Recombinant Murine SCF | PeproTech | Cat# 250-03 |
| Recombinant Human TPO | PeproTech | Cat# 300-18 |
| Propidium iodide | Life Technologies | Cat# P3566 |
| TrueCut Cas9 Protein v2 | Thermo Fisher Scientific | Cat# A36496 |
| Critical commercial assays | ||
| CUGA7 sgRNA Synthesis Kit | Nippon Gene | Cat# 314-08691 |
| TaKaRa Ex Taq | Takara Bio Inc | Cat# RR001A |
| Q5 High-Fidelity 2x Master Mix | New England Biolabs | Cat# M0492S |
| Wizard SV Gel and PCR Clean-Up System | Promega | Cat# A9282 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6JJmsSlc, 8–12 weeks old, male and female | Japan SLC, Inc. | http://www.jslc.co.jp/english/index2.htm |
| Mouse: C57BL/6J-Ly5.1, 8–12 weeks old, male and female | CLEA Japan, Inc | N/A |
| Mouse: C57BL/6-Tg(UBC-GFP)30Scha/J, 8–12 weeks old, male and female | The Jackson Laboratory | JAX stock #004353 |
| Oligonucleotides | ||
| sgRNA Common primer: AAAAGCACCGACTCGGTGCC |
Shiroshita et al.1 | N/A |
| sgRNA Reversed primer: AAAAGCACCGACTCGGTGCCA CTTTTTCAAGTTGATAACGGACT AGCCTTATTTTAACTTGCT ATTT CTAGCTCTAAAAC |
Shiroshita et al.1 | N/A |
| T7-sgRNA target Forward primer for GFP: ttaatacgactcactataGGGCGAGGAGCTGT TCACCGgttttagagctagaaatagc |
Shiroshita et al.1 | |
| UBC-GFP Forward primer: GTTCACCTTGATGCCGTTCT |
Shiroshita et al.1 | N/A |
| UBC-GFP Reverse and sequence primer: CACCCGTTCTGTTGGCTTAT | Shiroshita et al.1 | N/A |
| Software and algorithms | ||
| FlowJo version 10.7.2 | Tree Star | https://www.flowjo.com/solutions/flowjo |
| SnapGene | GLS Biotech | https://www.snapgene.com/ |
| Molecular Biology tool | Benchling | https://www.benchling.com |
| TIDE | Brinkman et al.4 | https://tide.nki.nl |
| ICE | Conant et al.5 | https://ice.synthego.com |
| Prism v7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| Neon Transfection System | Thermo Fisher Scientific | Cat# MPK5000 |
| Neon Transfection System 10 μL Kit | Thermo Fisher Scientific | Cat# MPK1096 |
| NanoDrop Onec Microvolume UV-Vis Spectrophotometer | Thermo Fisher Scientific | Cat# ND-ONEC-W |
NaOH and methanol are stored at 25°C. Cas9 protein, FA including palmitic acid and oleic acid, and cholesterol are stored at −30°C. gRNA is stored at −80°C. Antibodies, buffers, and other reagents can be stored at 4°C.
The maximum time for storage of gRNA and Cas9 is one year. Other reagents’ maximal storage time is as instructed by the manufacturer.
Step-by-step method details
Mouse bone marrow preparation
These steps describe the preparation of HSC from bone marrow.
-
1.
Prepare the adequate number of human ubiquitin C promoter (UBC)-GFP transgenic mice to obtain the HSCs you need.
Note: Approximately 2,000–3,000 HSCs can be obtained from two femurs, two tibias, and two pelvises from 8 to 14-week-old male mice when a strict gating strategy is applied for cell sorting. As for culture, a total of 500 HSCs per well in a 96-well culture plate is suitable for analysis.
Optional steps: CD45.1 congenic mice or other genetically-modified mice can be used according to your experimental purpose.
-
2.
Euthanize mice by CO2 euthanasia or cervical dislocation.
-
3.
Sterilize the dissection area, dissection kit, and the fur of the mice with 70% ethanol.
-
4.
Cut the skin of the abdomen and strip off the skin to the ankles.
-
5.
Cut off the foot from the tibia just above the ankle (Figure 1A) and strip off the triceps muscle and cut off the Achilles tendon (Figure 1B).
-
6.
Cut the patella tendon and remove the quadriceps femoris and the biceps femoris muscles from the femur (Figure 1C and 1D), and then dislocate the hind limb from the hip joint.
-
7.
Dislocate femurs and tibiae at the patella and resect the pelvic bone (Figure 1E).
-
8.
Remove residual muscles and connective tissues entirely on a paper towel using scissors (Figure 1F), then place the bones into 20–30 mL of ice-cold PBS + 2% FCS in a 10 cm Petri dish (Greiner).
-
9.
Flush the femurs, tibiae, and pelvis with PBS + 2% FCS using a 21-gauge needle (Terumo) and a 10 mL syringe (Terumo) to collect the bone marrow plug and disperse the plug by refluxing through the needle (Figure 1G).
-
10.
Transfer the cell suspension to a 50 mL conical tube.
-
11.
Centrifuge the supernatant at 400 × g for 5 min at 4°C.
-
12.
Discard the supernatant by inverting the tube quickly and remove the droplets on the rim with a paper towel.
-
13.
Add 5 mL per mouse of lysis buffer (0.17 M NH4Cl, 1 mM EDTA, 10 mM NaHCO3) and incubate on ice for 5 min.
-
14.
Add 2 volumes of PBS + 2% FCS and centrifuge at 400 × g for 5 min at 4°C.
-
15.
Discard the supernatant and resuspend cells in 10–20 mL of PBS + 2% FCS.
-
16.
Filter cell suspension through a 40 μm nylon mesh (BD Biosciences), and centrifuge at 400 × g for 5 min at 4°C.
-
17.
Discard the supernatant and resuspend cells in 80 μL/mouse of PBS + 2% FCS.
-
18.
Transfer the cell suspension to a 1.5 mL tube.
-
19.
Add anti-CD16/32 antibody for Fc-receptor block (2 μL/mouse) and incubate for 5 min at 4°C
-
20.
Add anti-c-Kit magnetic beads (Miltenyi) at a 1/10 v/v ratio and incubate for 15 min at 4°C in the dark.
-
21.
Add 500 μL to 1 mL of PBS + 2% FCS and centrifuge at 400 × g for 5 min at 4°C. Discard the supernatant.
-
22.
Repeat step 20.
-
23.
Resuspend cells with 2 mL of PBS + 2% FCS and filter the suspension with a 40 μm filter to avoid clogging the column and transfer to a 5 mL tube.
Alternatives: Resuspend the filtered cell suspension with 5 mL of PBS + 2% FCS and transfer to a 15 mL tube when the number of mice is greater.
-
24.
Enrich c-Kit positive cells using Auto-MACS Pro (Miltenyi) with the Possel-d2 program.
Alternatives: If Auto-MACS Pro is not available, Manual-MACS is an alternative method for cell separation
-
25.
Transfer the cell suspension of the positive fraction (2 mL) into two 1.5 mL tubes separately, centrifuge at 340 × g for 5 min, and discard the supernatant.
-
26.
Centrifuge the isolated cells once at 340 × g for 5 min at 4°C and aspirate the supernatant.
Figure 1.

Cell isolation from mouse bone marrow
(A–G) Cutting site is marked by red arrows (A–E). After removing the muscles and connected tissues (F), flush the femurs, tibiae, and pelvis with PBS + 2% FCS using a 21-gauge needle (G).
Antibody staining
These steps describe antibody staining of HSC.
-
27.
Label cells with an antibody cocktail as follows: lineage markers (CD4, CD8a, Gr-1, Mac-1, Ter-119, and B220)-PerCP-Cy5.5, c-Kit-APC-Cy7, Sca-1-PE-Cy7, CD150-BV421, CD48-PE, and Flt3-APC. Use 0.25 μL of antibody per mouse and mix with 20 μL PBS + 2% FCS. Incubate cells for 30 min at 4°C in the dark.
-
28.
Add 1 mL of PBS +2% FCS and centrifuge at 340 × g for 5 min at 4°C. Discard the supernatant.
-
29.
Resuspend cells in 0.5–2 mL of PBS + 2% FCS + 0.1% propidium iodide, , and transfer to a 5 mL tube.
Sorting of HSCs
These steps describe the sorting of HSC.
-
30.
Load the sample tube on the FACS Aria IIIu and set gates as shown in Figure 2.
-
31.
Sort cells into a 1.5 mL tube containing 500 μL of culture medium with 4% w/v BSA.
-
32.
Centrifuge sorted cells at 340 × g for 5 min at 4°C, then discard the supernatant and aspirate the residue carefully not to disturb the pellet.
-
33.
Keep the cell pellet on ice until culture.
Figure 2.

Gating strategy of HSCs
Representative flow cytometric plot for sorting HSCs from UBC-GFP mice. HSCs were defined as CD150+CD48-Flt3- LSK cells. Multipotent progenitors (MPPs) were sub-fractionated into MPP1 (CD150-CD48-Flt3- LSK), MPP2 (CD150+CD48+Flt3- LSK), MPP3 (CD150-CD48+Flt3- LSK), and MPP4 (Flt3+ LSK).
Preculture before genome editing
These steps describe preculture before genome editing of HSC.
-
34.
Prepare the 4% BSA medium or StemSpan supplemented with 50 ng/mL of SCF and TPO (preculture medium).
Optional: Separate some freshly-isolated HSCs and culture them under quiescence-maintaining conditions as a control of genome editing efficiency and transplantation.
-
35.
Dissolve the cell pellet in preculture medium at a density lower than 1.0×105/mL (maximum 2.0×104 cells in 200 μL of preculture medium).
-
36.
Incubate in 20% O2 and 5% CO2 for 16–24 h.
Note: Preculture improves RNP delivery into the nucleus.1 A shorter preculture time (< 16 h) and lower cytokine concentrations can lead to lower editing efficiency. A longer preculture time (> 24 h) does not improve editing efficiency and may promote differentiation of HSCs.
NHEJ-based genome editing with CRISPR-Cas9
These steps describe gene editing procedure of HSC.
-
37.
Prepare the post-editing culture medium as described in “setting up culture medium for quiescence-maintaining and proliferative conditions”.
-
38.Prepare the RNP complex.
-
a.Thaw the sgRNA stock on ice.
-
b.Incubate Cas9 protein (Thermo Fisher Scientific, TrueCut Cas9 Protein v2) and sgRNA (sgRNA stock) for 5–10 min at 25°C.
Reagent Stock concentration Final concentration Amount TrueCut Cas9 Protein v2 5 μg/μL 0.5 μg/μL 0.6 μL sgRNA stock 1.5 μg/μL 0.5 μg/μL 2.0 μL T-buffer N/A N/A 3.4 μL Total N/A N/A 6 μL -
c.Keep the RNP complex on ice until use.
-
a.
-
39.
Harvest precultured HSCs from the plate and centrifuge at 340 × g for 5 min at 4°C. Discard the supernatant.
-
40.
Resuspend cells in 30 μL of Buffer T.
-
41.
Add 6 μL of RNP to yield a total volume of 36 μL.
-
42.
Gently pipet the mixture of cells and RNPs with a 10 μL Neon Tip.
-
43.
Perform electroporation three times using the Neon Transfection System (Thermo Fisher Scientific). The electroporation condition is as follows: 1700 V, 20 ms, and one pulse for mouse HSPCs. The stop time between each electroporation is from 30 s to 1 min.
Note: A Neon Tip can be used for three serial electroporations without losing editing efficiency in our experience.
-
44.
Transfer 500 (-1000) electroporated HSCs directly into the post-editing culture medium (quiescence-maintaining or proliferative culture conditions).
Note: For testing multiple post-editing conditions, harvest electroporated cells into the 4% BSA medium and transfer them to multiple culture wells with a 20 μL pipette or 8-channel pipette.
-
45.
Place the culture plate in a humidified multi-gas incubator (Astec) at 37°C under 1% O2 and 5% CO2 conditions. Troubleshooting 2.
Evaluation of NHEJ-based genome editing efficiency
These steps describe how to evaluate the NHEJ-based genome editing efficiency.
-
46.Extract genomic DNA from genome-edited cells 2–3 days after electroporation.
-
a.Harvest edited cells from the culture well and centrifuge at 340 × g for 5 min at 4°C. Discard the supernatant. For DNA extraction, we usually prepare at least 10,000 cultured cells following gene editing.
-
b.Resuspend pellet in 100 μL of QuickExtract DNA Extraction Solution (Epicentre).
-
c.Keep the tube at 65°C for 5–10 min and then at 98°C for 5–10 min.
-
d.Cool the tube at 25°C.
-
a.
-
47.
Perform PCR using Q5 High-Fidelity DNA Polymerase (New England BioLabs), UBC-GFP Forward primer (GTTCACCTTGATGCCGTTCT), and UBC-GFP Reverse primer (CACCCGTTCTGTTGGCTTAT).
Figure 3.

Primer design for evaluating genome editing efficiency
(A) Design of sequence primer sets for sgRNA targeting of the UBC-GFP locus.
(B) Representative Sanger sequencing results of non-edited and GFP-edited cells.
(C) Results of a TIDE assay.
(D) Results of ICE analysis.
(E) Genome editing efficiency based on ICE positively correlated with GFP expression levels. The donor-derived peripheral blood cells from individual recipient mice in two independent transplant experiments were used for ICE analysis (mean ± SD, n = 10).
(F) The ratio of genome editing efficiency at the transplant to genome editing efficiency at four months after transplant. The same data set is used as in Figure 2E (mean ± SD, n = 10).
PCR reaction master mix
| Reagent | Amount |
|---|---|
| DNA template | 1 μL |
| Q5 High-Fidelity DNA Polymerase | 12.5 μL |
| UBC-GFP Forward Primer (5μM) | 2.5 μL |
| UBC-GFP Reverse Primer (5μM) | 2.5 μL |
| ddH2O | 6.5 μL |
PCR cycling conditions
| Steps | Temperature | Time | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 30 s | 1 |
| Denaturation | 98°C | 10 s | 35 cycles |
| Annealing | 67°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final extension | 72°C | 2 min | 1 |
| Hold | 4°C | forever | |
-
48.
Perform electrophoresis using the PCR products to check whether the PCR products are of the expected size.
-
49.
Purify PCR products using Wizard SV Gels and the PCR Clean-Up System (Promega) following the manufacturer’s instructions.
-
50.
Perform Sanger sequencing of each purified PCR product obtained from non-edited (control) and edited cells. Use a reverse primer as the sequencing primer.
-
51.
Perform TIDE analysis (https://tide.nki.nl) using both non-edited and edited Sanger sequence ab1 files (Figure 3).4 Troubleshooting 3 and 4.
Alternatives: For surface marker genes (e.g., CD45) or reporter genes (e.g., GFP), the knock-out levels can be easily evaluated using flow cytometry.
Alternatives: You can also use ICE (https://ice.synthego.com) to calculate the editing efficiency.5 A representative ICE result is shown in Figure 3.
Bone marrow transplantation using genome-edited HSCs
These steps describe the procedure of bone marrow transplantation.
The protocol below describes transplantation after long-term ex vivo proliferative and quiescence-maintaining cultures. The following groups should be prepared as controls; fresh HSCs, mock-electroporated HSCs, and cultured HSCs without genome editing. More edited HSCs should be prepared than transplanted HSCs to evaluate the editing efficiency at transplantation.
-
52.
Harvest some HSCs that were electroporated together with HSCs for transplantation to evaluate editing efficiency at the transplant 3 days after electroporation. See “evaluation of NHEJ-based genome editing efficiency”.
Note: Editing efficiency at transplantation is critical for confirming that transplanted cells were efficiently edited as well as for precisely interpreting post-transplant chimerism. For this purpose, donor-derived genome-edited HSCs are prepared for the number of recipient mice plus one or two.
-
53.
On the day before transplantation, lethally irradiate C57BL/6-Ly5.1 congenic recipient mice (8.5 Gy using an MBR-1520R instrument from Hitachi Power Solutions, 125 kV, 10 mA, 0.5 mm Al filter, and 0.2 mm Cu filter).
-
54.
Prepare competitor cells from CD45.1 congenic mice as a step of “ mouse bone marrow collection”.
-
55.
Count the number of competitor bone marrow mononuclear cells (BMMNC) after filtration.
-
56.
Harvest genome-edited HSCs.
Note: Because HSCs may attach to the culture well in the long-term culture (> 7 days), complete harvest of edited HSCs from the culture well is important.
Alternative: You can also perform transplantation on the day of genome editing. We prepare 500 genome-edited HSCs and 5.0×105 competitor cells for one recipient. Genome-edited HSCs 2–3 h after the proliferative condition (50–100 ng/mL of SCF and TPO) to promote cell recovery from electroporation.
-
57.
Centrifuge at 340 × g for 5 min at 4°C. Discard the supernatant.
-
58.
Prepare the cell mixture for one recipient by mixing 1000 genome-edited HSCs (equivalent number after the electroporation) and 5.0×105 competitor cells in 200 μL of PBS + 2% FCS.
-
59.Retro-orbitally transplant cells.
-
a.Anesthesia with sevoflurane.
-
b.Pipetting the cell mixture again.
-
c.Aspirate 200 μL of cell mixture into an insulin needle-syringe.
-
d.Insert the needle at an angle of approximately 30° into the medical canthus. Be careful not to damage the cornea.
-
e.Slowly and smoothly inject the cell mixture.
-
f.Slowly and smoothly withdrawn the needle.
-
a.
-
60.
Check the full recovery of transplanted mice from anesthesia.
Analysis of donor-derived genome-edited chimerism in peripheral blood
These steps describe how to analyze the donor-derived and genome-edited chimerism.
-
61.
Collect the peripheral blood at 1, 2, 3, and 4 months after transplantation. Before collecting the peripheral blood, anesthetize the mice by sevoflurane inhalation.
-
62.
Collect 40–80 μL of peripheral blood from the retro-orbital sinus using heparinized glass capillary tubes and suspend the sample in 1 mL of PBS + heparin (1 U/mL) in 1.5 mL tubes.
-
63.
Centrifuge the blood suspension at 340 × g for 3 min at 4°C. Discard the supernatant and resuspend the pellet in 1 mL of PBS + 1.2% w/v dextran (200 kDa) for 45 min at25°C.
Alternatives: To remove red blood cells, you can use perform two red blood cell lysis as a time saving method.
-
64.
Transfer the supernatant to another 1.5 mL tube and centrifuge at 340 × g for 3 min.
-
65.
For red blood cell lysis, resuspend the cells in 0.17 M NH4Cl for 5–10 min.
-
66.
Centrifuge the cell suspension at 340 × g for 3 min at 4°C. Resuspend the cells in 50 μL of staining buffer containing 0.3 μL of anti-mouse Fc-block. Incubate the sample at 4°C for 5 min.
-
67.
Make the following antibody premixture for surface marker staining: CD45.1-PE, CD45.2-BV421, CD4-PerCP-Cy5.5, CD8-PerCP-Cy5.5, B220-APC, Gr-1-PE-Cy7, and Mac-1-PE-Cy7. Use 0.3 μL of each antibody per sample.
-
68.
Add the premixed antibodies and incubate the cells at 4°C for 15 min.
-
69.
Wash once with 1 mL of PBS + 2% FCS and centrifuge at 340 × g for 5 min at 4°C.
-
70.
Resuspend cells in 200 μL of PBS + 2% FCS + 0.1% propidium iodide and and transfer to a 5 mL tube.
-
71.
Set up the flow cytometer (FACS Aria IIIu).
-
72.
Record data of donor-derived chimerism and sort donor-derived (Ly5.2+) cells.
-
73.
Extract DNA from donor-derived cells and perform TIDE analysis. See the section “evaluation of NHEJ-based genome editing efficiency”.
Note: Theoretically, the average editing efficiency from each recipient mouse is not changed compared to the editing efficiency at the transplant if the target genes are not functional (e.g., EGFP) (Figure 3). The decrease in donor-derived genome-edited chimerism compared to the control group suggests that target genes are functional for HSC function.
-
74.
Export the data in FCS format for analysis using software such as FlowJo.
-
75.
Calculate the donor-derived genome-edited chimerism of each recipient mouse using the following formula:
Note: Genome editing efficiency is evaluated by one of the following: TIDE, ICE, and FACS-based protein expression levels (e.g., GFP). We confirmed that GFP expression levels can reflect the genome editing efficiency, suggesting that we can use the frequency of GFP-negative cells as the editing efficiency (Figure 3).
Expected outcomes
An example of primer design is shown in Figure 3. To induce a highly efficient knock-out of GFP, the targeting locus of crRNA is located near the transcription start point (ATG). As for sequencing primer sets, the forward and reverse primers are designed at 400 bp away from the expected cleavage site (Figure 3A). The additional insertion near the expected cleavage site in the Sanger sequence can be detected (Figure 3B). TIDE assays using these Sanger sequence files show high levels of insertion-deletion (Figure 3C). ICE analysis using the same files also shows similar editing efficiency (Figure 3D). We confirmed a positive correlation between the genome-editing efficiency based on ICE and the GFP expression levels, suggesting that we can evaluate NHEJ-based genome-editing efficiency based on GFP expression levels (Figure 3E). In addition, we confirmed that genome editing efficiency at transplant is well maintained at four months after transplant by evaluating the ratio of genome editing efficiency at each time point(Figure 3F).
Representative results of the chimerism of genome-edited HSCs after a long-term ex vivo culture are shown in Figure 4. We prepared three HSC groups; HSCs in quiescence-maintaining culture (Culture group), genome-edited HSCs in quiescence-maintaining culture (CRISPR-quiescent), and genome-edited HSCs in proliferative culture (CRISPR-proliferative). Cultured HSCs are transplanted 10 d after electroporation. The gating strategy of peripheral blood cells is shown in Figure 4A. Chimerism of donor-derived genome-edited HSCs in quiescence-maintaining culture is considerably higher than in proliferative culture (Figure 4B).
Figure 4.

Analysis of donor-derived genome edited chimerism
(A) Gating strategy for analyzing peripheral blood cell chimerism.
(B) Donor-derived chimerism in the primary transplantation 10 days after genome editing (n = 6–10 from two independent transplantations) HSCs in quiescence-maintaining culture (black), GFP-edited HSCs in quiescence-maintaining culture (blue), and GFP-edited HSCs in proliferative culture (red). The Turkey–Kramer multiple comparisons test was used. ∗∗∗p < 0.001.
Limitations
The peripheral blood chimerism of the CRISPR-quiescent group is higher than that of the CRISPR-proliferative group but lower than that of the culture group. However, donor-derived HSC chimerism did not differ between the culture group and the quiescence-maintaining culture group (data not shown). Based on the changes in the chimerism, the quiescence-maintaining condition should be refined in a future study to improve the in vivo function of genome-edited HSCs.
Our protocol used HSCs defined by SLAM-LSK markers, but they contained a non-HSC population. Introduction of EPCR, which can prospectively isolate true HSCs, may improve the repopulation capacity.6
We were able to restore quiescence to NHEJ-edited mouse HSCs as well as HDR-edited mouse HSCs and NHEJ-edited human HSCs by integrating quiescence-maintaining culture conditions to the post-editing culture in the original study.1 In a future study, transplantation using homology-directed repair (HDR)-edited HSCs and NHEJ-edited human HSCs will be evaluated.
Troubleshooting
Problem 1: Low yield of sgRNA
Related to Step 16 in “before you begin”.
Potential solution
In vitro transcription of sgRNA can fail because of multiple factors.
-
•
Use reagents without RNase (e.g., RNase-free water, new PCR reagent for sgRNA production)
-
•
Check the T7-sgRNA forward primer starting with “GG”.
-
•
Completely mix the gRNA Binding Buffer before applying it to the column.
Problem 2: Low viability in the quiescence-maintaining culture
Related to Step 45 in “ NHEJ-based genome editing by CRISPR-Cas9”.
Potential solution
High BSA is required for adequate supplementation of FAs and cholesterol. Lower cytokine concentration has the advantage of maintaining the quiescence of HSCs, but extremely low concentration (e.g., SCF < 0.5 ng/mL) could lead to cell death.
-
•
Fully dissolve the BSA, FAs, and cholesterol in the medium
-
•
Check the final cytokine concentration of SCF and TPO. See also the section on “setting up culture medium for quiescence-maintaining and proliferative conditions” before you begin.
-
•
Transfer the edited cells to the post-editing culture medium as soon as possible. Longer exposure to the T-buffer may decrease cell viability during genome editing procedures.
Problem 3: Editing efficiency cannot be evaluated using TIDE/ICE
Related to Step 51 in “evaluation of NHEJ-based genome editing efficiency”.
Potential solution
-
•
Check the design of the PCR primer sets. Forward and reverse primers should be located 150–200 bp away from the expected cleavage site. We recommend at least 200 bp away from the cleavage site.
-
•
Design nested primers for sequencing. This might help to avoid non-target amplification in PCR.
-
•
Check whether the PCR amplicon has the expected size using gel electrophoresis. PCR failure and non-target amplification can be detected. If PCR fails, optimizing PCR conditions or using other PCR enzymes may be required.
-
•
Check the quality of the Sanger sequencing data. Low quality sequencing results lead to background noise and affect the interpretations.
-
•
Use next-generation sequencing (NGS) because some editing outcomes (e.g., large deletions) are not considered in TIDE/ICE.
Problem 4: Low editing efficiency
Related to Step 51 in “evaluation of NHEJ-based genome editing efficiency”.
Potential solution
-
•
Perform the preculture before genome editing for the optimal period (16–24 h).
-
•
Prepare a new sgRNA when you use old sgRNA stock or sgRNA stock that has been repeatedly frozen and thawed. Both may reduce the quality or amount of sgRNA.
-
•
Completely remove any bubbles within the Neon Tip before electroporation. If macro bubbles are present within the Tip, you will see sparks after performing electroporation. This impairs cell viability as well as editing efficiency. Efficient electroporation occurs when microbubbles are present within the tip and there are no sparks. However, in this case, editing efficiency is also significantly reduced.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Keiyo Takubo (keiyot@gmail.com).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
We thank all the authors of the original study1 from which this protocol was generated. We thank M. Haraguchi and S. Tamaki for technical assistance and suggestions. K.S. was supported in part by a KAKENHI grant from the Ministry of Education, Culture, Sports, Science and Technology (MEXT)/Japan Society for the Promotion of Science (JSPS) (21J11016). H.K. was supported in part by a KAKENHI grant from MEXT/JSPS (19K17847 and 21K08431) and a grant from the National Center for Global Health and Medicine. K.T. was supported in part by KAKENHI grants from MEXT/JSPS (20K21621, 21H02957, and 22K19550), grants from the National Center for Global Health and Medicine, a grant from the Japan Health Research Promotion Bureau, Japan Agency for Medical Research and Development grants (JP18ck0106444, JP18ae0201014, JP20bm0704042, and JP20gm1210011), a grant from the Takeda Science Foundation, Kaketsuken Grant for Young Researchers, and the MEXT Joint Usage/Research Center Program at the Advanced Medical Research Center, Yokohama City University.
Author contributions
K.S. and H.K. performed experiments and analyzed the data. K.S., H.K., and K.T. wrote the manuscript. K.T. conceived the project and supervised the research.
Declaration of interests
The authors declare no competing interests.
Data and code availability
This study did not generate or analyze any datasets.
References
- 1.Shiroshita K., Kobayashi H., Watanuki S., Karigane D., Sorimachi Y., Fujita S., Tamaki S., Haraguchi M., Itokawa N., Aoyoama K., et al. A culture platform to study quiescent hematopoietic stem cells following genome editing. Cell Rep. Methods. 2022;2:100354. doi: 10.1016/j.crmeth.2022.100354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kobayashi H., Morikawa T., Okinaga A., Hamano F., Hashidate-Yoshida T., Watanuki S., Hishikawa D., Shindou H., Arai F., Kabe Y., et al. environmental optimization enables maintenance of quiescent hematopoietic stem cells ex vivo. Cell Rep. 2019;28:145–158.e9. doi: 10.1016/j.celrep.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi H., Takubo K. Protocol for the maintenance of quiescent murine hematopoietic stem cells. STAR Protoc. 2020;1:100078. doi: 10.1016/j.xpro.2020.100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conant D., Hsiau T., Rossi N., Oki J., Maures T., Waite K., Yang J., Joshi S., Kelso R., Holden K., et al. Inference of CRISPR edits from Sanger trace data. CRISPR J. 2022;5:123–130. doi: 10.1089/crispr.2021.0113. [DOI] [PubMed] [Google Scholar]
- 6.Kent D.G., Copley M.R., Benz C., Wöhrer S., Dykstra B.J., Ma E., Cheyne J., Zhao Y., Bowie M.B., Zhao Y., et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood. 2009;113:6342–6350. doi: 10.1182/blood-2008-12-192054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate or analyze any datasets.