

Abstract
Two borane-functionalized bidentate phosphine ligands that vary in tether length have been prepared to examine cooperative metal-substrate interactions. Ni(0) complexes react with aryl azides at low temperature to form structurally unusual κ2-(N,N)-N3Ar adducts. Annealing these adducts affords products of N2 extrusion and in one case, a Ni-imido compound that is capped by the appended borane. Reactions with 1-azidoadamantane (AdN3) provide a distinct outcome, where a proposed nickel imido intermediate activates the sp2 C-H bonds of arenes, even in the presence of benzylic C-H sites. Combined experimental and computational mechanistic studies demonstrate that the unique reactivity is a consequence of Lewis-acid induced polarization of the Ni–NR bond, potentially providing a synthetic strategy for chemoselective reaction engineering.
Graphical Abstract
INTRODUCTION
In addition to steric/electronic perturbations that can be made to a metal’s primary coordination sphere, introduction of acidic/basic groups within a metal’s secondary coordination sphere can be used to augment metal-based reactivity preferences.1–3 Secondary coordination sphere engineering is receiving increased attention from the synthetic community, largely to facilitate small molecule activation (H2, O2, N2, CO2, CO, oxyanions, etc.).4–9 Within the context of small molecule functionalization, secondary sphere acidic groups can modify the electronic structure of metal-coordinated intermediates that ultimately can introduce divergent reactivity for complexes with/without secondary sphere acids.6–7, 10–22 Exogenous borane Lewis acids have been shown to influence substrate binding/activation, although strong Lewis acids are typically required.23–24 One strategy to lower the acidity required for substrate binding is to decrease the entropic penalty of Lewis acid/substrate binding. A highlight of this approach was work by Miller and Bercaw, who demonstrated that a borane-tethered monodentate phosphine ligand enabled Re-mediated CO reductive coupling.8,25 Related examples with bidentate phosphines are rare, and multi-borylated bis(phosphino)ethanes were recently reported by Drover and co-workers26–29 as a strategy to promote hydride transfer.27
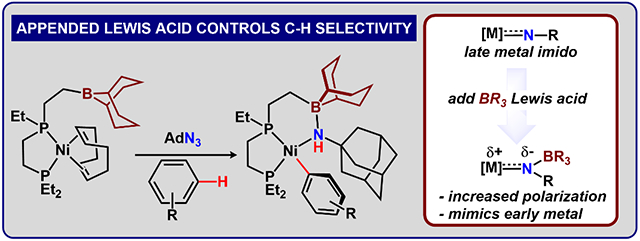
Transition metal imido complexes are important intermediates in nitrogen fixation,30–31 nitrene transfer,32–34 and C-H bond amination.35–38 Although metal imidos are less common among the late metals such as nickel, Hillhouse and coworkers made pioneering contributions using sterically encumbered bidentate phosphine (dtbpe: tBu2PCH2CH2PtBu2)39–40 and N-heterocyclic carbene (NHC) ligands41. These imides can activate the C-H bonds of phenylacetylene42 or ethylene,41 while they are stable in aromatic solvents (benzene or toluene). Related work by groups of Warren43 as well as Betley44 reported several formally Ni(III) imides using β-diketiminate or dipyrrin ligands whose electronic structures can also be described as Ni(II) iminyls. These compounds have been applied in stoichiometric45 and catalytic46–47 C-H bond aminations, and importantly, both systems activate toluene and related substrates at the homolytically weak benzylic position. By contrast, heterolytic C-H activation pathways are most commonly observed in early metal imido complexes, which are more polarized than late metal analogues, and less prone to react through homolytic pathways.48–50 We hypothesized that the addition of a suitable borane Lewis acid to a nickel imido unit might similarly polarize the Ni-NR bond, providing an avenue to access unique reactivity.51–52
Our group has recently explored a series of ligands containing tunable secondary coordination sphere environments with nitrogen-based and NHC ligands7, 15, 19, 53–56 and our previous studies showed that the appended 9-BBN (BBN = borabicyclo[3.3.1]nonyl) group provides moderate Lewis acidity, enabling unique reactivity that is facilitated by reversible borane-substrate bond formation.19 Our prior efforts demonstrated that, when coordinated to low valent metals, depe (depe = 1,2-bis(diethylphosphino)ethane) provides sufficient electron density to activate nitrogenous substrates, without precluding interactions to a Lewis acid.7,15 Herein, we establish the synthesis and reactivity of depe derivatives containing a single appended Lewis acidic borane (Figure 1) to evaluate the extent that acidic groups modulate the reactivity of late transition metal (Ni) imidos.57–58
Figure 1.

C-H bond activation reactivity triggered by previously reported Ni imido complexes as well as that reported in this work.
RESULTS AND DISCUSSION
Synthesis of borane-functionalized bisphosphine ligands.
Key to our synthetic approach was access to Et2PCH2CH2P(Et)(Cl). Because this compound was not cleanly formed from reactions between Cl2PCH2CH2PCl2 (A) and 3 equiv. EtMgBr, we adapted a method previously used to prepare polymer-supported phosphines.59 As shown in Figure 2, the reaction of A with 1 equiv. iPr2NH in the presence of 1 equiv. Et3N generated a mixture of Cl2PCH2CH2P(Cl)(NiPr2) (B) and (iPr2N)(Cl)PCH2CH2P(Cl)(NiPr2), from which B was isolated in 41% yield (31P NMR: 191.7 for -PCl2 and 130.7 ppm for -P(Cl)(NiPr2) group). The reaction of B with 3 equiv. EtMgBr afforded C (31P NMR: 35.2 ppm for -P(Et)NiPr2 and −18.9 ppm for -PEt2). Subsequent deprotection using HCl (2 eq)60 revealed a new 31P NMR resonance at 116.7 ppm, consistent with a -P(Et)(Cl) group, and after workup, afforded D as a white solid in 53 % yield.61–62
Figure 2.

Synthesis of 9-BBN functionalized bisphosphine ligands 1a and 1b.
To install terminal olefins amenable to hydroboration, D was treated with vinylmagnesium bromide, affording E in 66% yield (allylmagnesium bromide afforded F in 75% yield). The 31P NMR spectrum of E contained two resonances at −19.4 and −20.0 ppm63–64 (−19.0 and −23.9 ppm for F). Hydroboration of E and F with 9-H-BBN in benzene at 130 °C afforded 1a and 1b as colorless oils. In contrast to the precursor, 31P NMR spectra of 1a exhibited resonances far removed from each other at −25.9 ppm and −2.3 ppm. The 11B NMR spectrum exhibited a resonance at −8.1 ppm (−21.8/8.5 ppm for 31P NMR and −2.0 ppm for 11B NMR of 1b), consistent with the coexistence of a free phosphine group and a quenched P→B interaction (note that 1a may exist as a higher order aggregate, but for clarity, we have depicted one structure that satisfies NMR data).65–66
Metalation with nickel.
Metalation of 1a with NiCl2(dme) afforded 2a as a yellow solid in 85% yield (Figure 3). The 31P NMR spectrum of 2a exhibited two resonances at 78.2 and 76.1 ppm, similar to that of (depe)NiCl2 (76.9 ppm), and consistent with bidentate coordination.67 The 11B NMR spectrum exhibited a resonance at 85.5 ppm, consistent with boron in a trigonal environment. Metalation of 1b with NiCl2(dme) gave similar results, with two 31P NMR resonances at 76.4 and 73.2 ppm as well as a 11B NMR resonance at 87.6 ppm. Reduction of 2a (or 2b) with Mg in the presence of 10 equiv. of cyclooctadiene (COD) afforded the Ni(0) complex 3a as an oil in 95% yield (or 3b in 91% yield) as assessed by NMR spectroscopy (Figure 3). The 31P NMR spectrum of 3a featured resonances at 52.3 and 49.2 ppm (49.1 and 46.9 ppm for 3b), while the 11B NMR spectrum contained a broad resonance at 87.7 ppm (88.0 ppm for 3b), consistent with boron in a trigonal environment.
Figure 3.
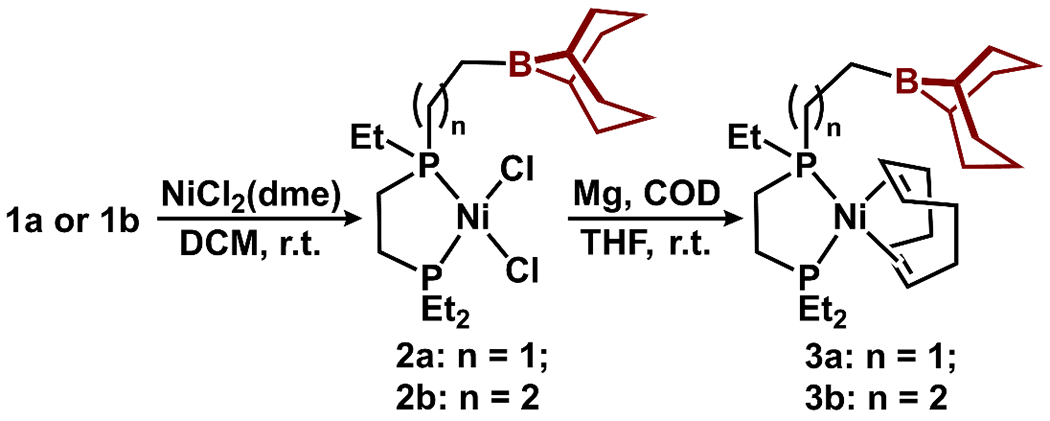
Synthesis of Ni(0)-COD complexes 3a and 3b based on ligands 1a and 1b.
Group transfer from azides to low valent metal centers is a common strategy to prepare metal imido complexes, including related Ni=NR compounds reported by Hillhouse.40 Prior to N2 extrusion, several metal azide complexes, either in η1-Nγ or η2-Nβ/Nγ coordination modes, have been isolated as intermediates.68–69 To access nickel imido complexes with an appended borane and study their subsequent reactivity, we evaluated reactions between 3 and representative aryl and alkyl azides.
Synthesis of monomeric Ni(0)-azide complexes.
We evaluated reactions between 3 and two different aryl azides (MesN3 or DmpN3, Mes = 2,4,6-Me3C6H2, Dmp = 2,6-Mes2C6H3). Solutions of aryl azide were slowly added to frozen solutions of 3 in toluene-d8 and the reaction progress was assessed by 31P NMR spectroscopy. Upon warming to −30 °C, 3 transformed into a new compound, 4, with concurrent generation of free COD (based on 1H and 31P NMR spectroscopy), and was tentatively assigned as a Ni(0) azide species. We found that the tether length to the appended borane dramatically influenced stability. While 4a and 4c, with two methylene units (–CH2CH2–), were stable at −35 °C for >12 h, 4b, with three methylene units (–CH2CH2CH2–), started to decompose as it was generated (ca. −30 °C). The 31P NMR spectrum of 4 exhibits two major resonances in a 1:1 ratio (4a: 76.5 and 47.6 ppm, JP-P = 21.9 Hz; 4b, 57.5 and 52.9 ppm, JP-P = 21.8 Hz; 4c, 62.3 and 39.3 ppm, JP-P = 26.3 Hz). Although the thermal instability of 4 hampered isolation in bulk quantities, crystals suitable for X-ray diffraction studies were obtained from diethyl ether solutions at −35 °C.
The solid-state structures of 4b and 4c revealed Ni-(N3Ar) complexes, where the azide unit is κ2-coordinated through the α- and γ-N atoms. This coordination mode is supported by an interaction between the γ-N atom and the appended boron (Figure 4. B-N3 bond length = 1.591(3) Å and ∑Bα= 325.17(19)° for 4b). In 4b, the N-N bond lengths lie between single and double bonds, with the N1-N2 bond (1.378(3) Å) longer than the N2-N3 bond (1.276(3) Å). The Ni-N1 and Ni-N3 distances range between 1.87 - 1.94 Å, which is consistent with single bonds. 4c exhibited a similar structure, although the data quality did not allow reliable discussion of metrical parameters (see SI section 6.2 for more details). Complexes 4b and 4c represent the first structurally characterized examples of monometallic complexes with a κ2(N,N) Ni(II) triazenido binding mode. Importantly, this bonding mode has been proposed by Hillhouse and co-workers as transition state en route to Ni(II) imidos from Ni(0) azide complexes.40,70–72 Azide coordination modes through a single nitrogen (Nγ), or two nitrogen atoms (either η2-Nβ/Nγ or κ2-Nα/Nγ) have previously been calculated to be within 15 kcal/mol for several (PP)Ni(RN3) species (PP = bisphosphine ligands) and η2-Nβ/Nγ coordination is the most common and stable.73–74 The secondary sphere borane modulates the binding mode to prefer κ2-Nα/Nγ over η2-Nβ/Nγ by providing significant stabilization through a B-N interaction (24.1 kcal/mol for the 2 carbon tether and 20.7 kcal/mol for the 3 carbon tether based on DFT calculations, see xyz file in SI). These results demonstrate that the Lewis acid serves an integral role to favor this unusual azide binding mode
Figure 4.
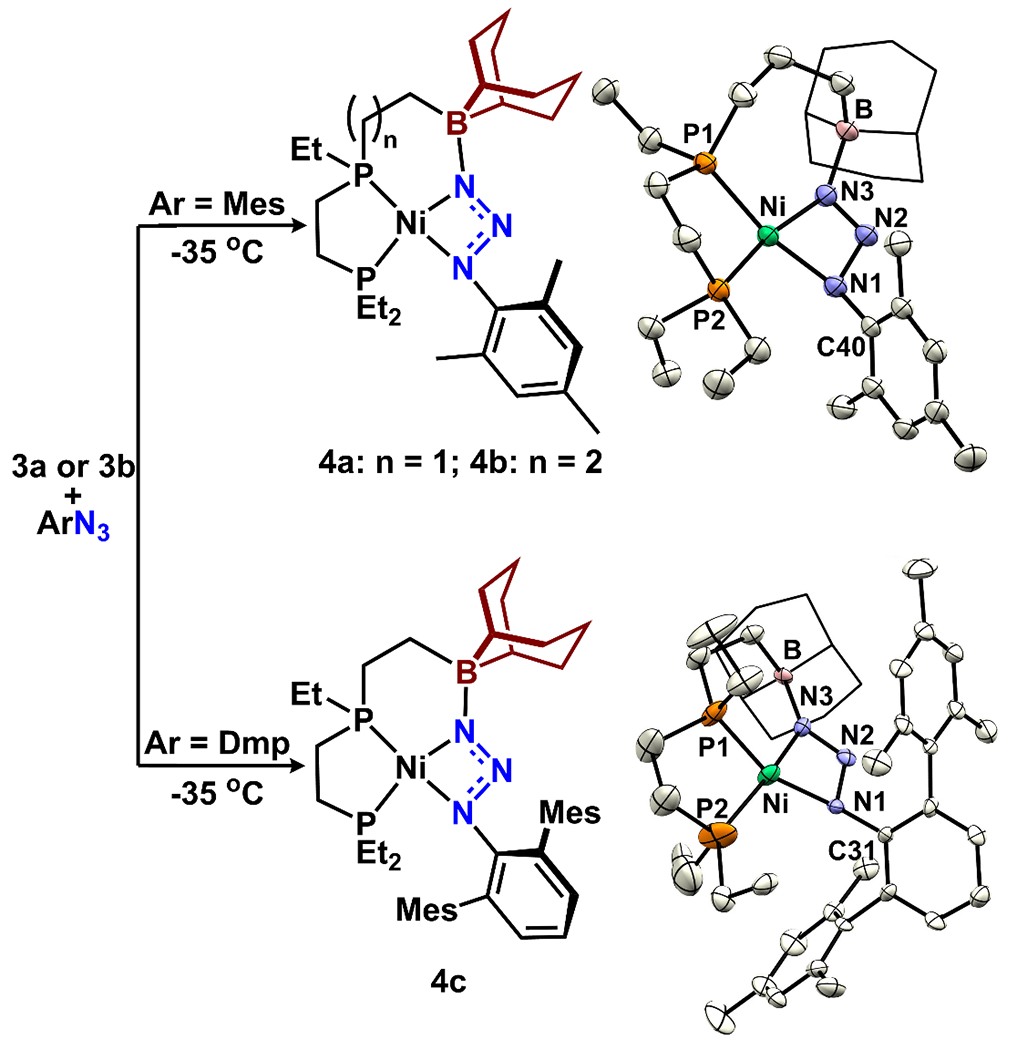
Generation of Ni(0)-azide monomer complexes, and the X-ray structures of 4b with 50% probability ellipsoids and 4c with 30%. The solvent (diethyl ether) and all H atoms were omitted and the 9-BBN is displayed in wireframe for clarity. Note that compounds 4 are only stable at temperatures <−35 °C.
Generation of Ni(II) imido complexes.
Although monomeric Ni(0) azide complexes, such as 4a and 4b can be generated at low temperature, they underwent subsequent thermally-induced N2 extrusion at higher temperatures. For example, the thermal decomposition of 4a generated a major product 5a in ca. 75% chemical conversion (vs. free COD; see SI section 2.15). The 31P NMR spectrum exhibited resonances at 85.0 and 45.6 ppm, while the 11B NMR spectrum exhibited a single sharp resonance at −8.4 ppm, consistent with boron in a tetrahedral environment. The Ni=NR unit of 5a was tentatively confirmed by its reactions with CO or tBuNC, which formed MesNCO or tBuNCNMes, as assessed by GCMS analysis and 13C NMR spectroscopy (see SI section 2.16). Although solutions of 5a decomposed in less than 1 day to give unidentified mixtures, single crystals of 5a were grown by slow vapor diffusion of hexane into a concentrated solution of 5a in toluene at −35 °C. Analysis of single crystal X-Ray Diffraction (scXRD) of 5a revealed a Ni imido species with a B–NAr interaction (Figure 5). The Ni-N bond length (1.897(2) Å) is longer than that in Hillhouse’s imido complex [(dtbpe)Ni(NMes)] (1.703(4) Å) but comparable to those in a tetranuclear Ni imido cluster [(iPr3P)4Ni4(μ4-NCH2Ph)] (1.87-1.90 Å).75 Similar to redistributing charge over more than one metal site, we propose that the B-N interaction (B-N bond length 1.567(3) Å) elongates the Ni–N bond in a manner that resembles a bridging mode. The Ni-N bond length may reflect the continuum between nickel imido resonance structures.76–77 The structure of 5a also features a π-arene interaction (Ni-C40 and Ni-C41 bond lengths = 2.038(2) Å and 2.114(2) Å, respectively). This interaction imposes a bent Ni-N1-C40 bond (bond angle 75.40(14)°), which is distinct from that in Hillhouse’s imido complex (bond angle = 180°).40 Another notable difference between complex 5a and Hillhouse’s (dtbpe)Ni=NAr complex is the coordination number at Ni: the latter is 3-coordinate. In contrast to the geometry imposed by steric interactions, we attribute the different local geometry in 5a primarily to the tridentate chelate formed by the B-N bond, which positions the imido N linearly to one phosphine (P2-Ni-N bond angle: 164.61(7)°).
Figure 5.

Generation of Ni(II) imido complex 5a and Ni(II) borylamide complex 5b, and X-ray structures (50% probability ellipsoids, selected H atoms omitted).
We found that the tether length to the appended borane has large effects on reactions with aryl azides. Although 4b exhibited a similar thermal stability profile to 4a, the reaction products following N2 extrusion were unique. ScXRD experiments of 5b indicated that rather than a nickel imido complex, B–C cleavage occurred, forming a nickel alkyl borylamide complex (Figure 5). In 5b, the boron and nitrogen are both trigonal planar (∑Bα = 359.99°, ∑Nα = 359.84°), which is similar to a previous reported Ni(II) borylamide complex (∑N1α = 358.61° and ∑N2α = 358.40°).78 Formation of 5b is consistent with a Ni intermediate that is unstable with respect to the B-C bond. Thus, we propose that ligand 1a provides better stabilization of a Ni imido species compared with ligand 1b. Following these observations, subsequent reactions with azides were only pursued using 3a.
In order to provide a sterically encumbered environment surrounding the imido moiety, we evaluated reactions with dimesitylphenyl (Dmp) azide. When a toluene-d8 solution of 4c was warmed above 0 °C, a complex mixture formed that slowly transformed into major product 5c (42% isolated yield) after 16 h at room temperature, as assessed by 31P NMR spectroscopy (62.7 and 60.5 ppm). An X-ray diffraction study revealed a Ni(II) amido, rather than imido, with a cyclometalated mesityl ligand (Figure 6). Although a Ni(II) imido [(PPB)Ni=NDmp] may be implicated as an intermediate during the transformation of 4c to 5c, it was not isolable. Similar Ni-mediated benzylic C-H activation of the Dmp group has been reported by Hillhouse and co-workers following single-electron oxidation of an NHC-supported nickel amide.79 In the solid-state structure of 5c, the Ni-amido unit is coordinated to the appended borane (B-N1 bond length: 1.669(4) Å; ∑Bα = 327.6(3)°). The Ni-N bond length (2.032(3) Å) is ca. 0.135 Å longer than that in complex 5a, and similar to those in related Ni-amido complexes that feature Lewis acid interactions to the amido nitrogen atoms.80
Figure 6.
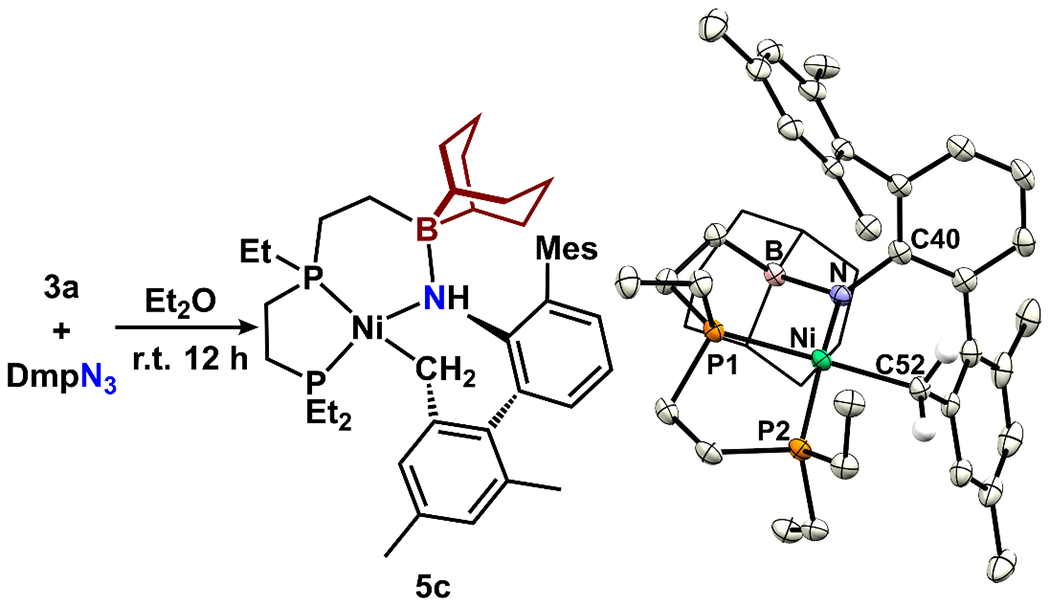
Generation of Ni(II) benzyl amide complex 5c as well as its structure (50% probability ellipsoids, selected H atoms omitted).
Studies toward the generation and reactivity of a Ni(II) imido complex with an adamantyl substituent.
To provide a Ni=NR unit that is able to react with exogenous substrates, we targeted an organoazide that cannot interact with nickel through a π system, and is unlikely to undergo intramolecular H-atom transfer. We selected AdN3 for subsequent reactivity studies because it fulfills both of these criteria. When we allowed 3a and AdN3 to react in benzene, the 31P NMR spectrum of the product exhibited resonances at 46.8 and 42.7 ppm (JP-P = 25.8 Hz). Following isolation as a yellow solid in 65% yield, a scXRD experiment was performed, enabling assignment as a Ni phenyl amido complex 6a (Figure 7). Similar to 5c, the amido unit features a B-N interaction (1.658(4) Å) with the appended borane Lewis acid. To interrogate the intermediate formed with AdN3, we quenched the reaction of 3a and AdN3 with tBuNC in THF solvent and observed tBuNCNAd (see SI, section 2.25). This reactivity profile is consistent with imide character of the intermediate derived from AdN3. Collectively, the formation of 6a may indicate that a transiently formed nickel imido species undergoes subsequent C-H bond activation of a benzene solvent molecule.
Figure 7.

Arene activations by proposed Ni(II) imido complexes as well as the structures of 6a and 6b (only one isomer was shown) with 50% probability ellipsoids, with selected H atoms omitted.
The reactivity observed with benzene is atypical for late metal imidos, which most commonly activate C-H bonds that have lower BDE values.37–38, 44–45, 81 We hypothesized that we might capitalize on this result, perhaps by favoring distinct regioselectivity to activate sp2− rather than sp3 C-H bonds. Toluene and m-xylene contain both weak benzylic sp3 (C-H BDE: ca. 90 kcal/mol) and strong sp2 C-H bonds (~20 kcal/mol higher).82 When complex 3a was allowed to react with AdN3 in toluene at room temperature for 16 h, we observed a mixture of products, as assessed by 31P NMR spectroscopy. To clarify the composition, we quenched the reaction with CH3COOD and analyzed the deuterium position by 1H NMR spectroscopy. From this experiment, we found that the reaction afforded a 1.7 : 1 ratio of m- and p-CH3C6H4D (See SI section 2.22), with no ortho-deuterated products. Importantly, we did not observe any C-H activation products of the sp3 C-H bonds. Based on these results, we assign the 31P NMR spectrum as two isomers of meta and one of para with resonances between 40-45 ppm.83 The structural assignment was validated by scXRD (Figure 7). The bond metrics of 6b are similar to those of 6a.84 Similar to toluene, we found that reactions with m-xylene, 3a, and AdN3 generated the meta-activation product, which was isolated in 51% yield. Distinct from toluene, we observed only one pair of doublets in the 31P NMR spectrum. Complex 6c was characterized by NMR (1H, 13C, 31P, 11B) spectroscopy and scXRD (see SI sections 2.24 and 6.8). The regioselectivity profile of these reactions is distinct from known monometallic nickel imide complexes.44–47,85–86 The high BDE values of arene sp2 C-H bonds likely preclude a homolytic mechanism, therefore we experimentally investigated the C-H bond activation step.
To provide a mechanistic understanding for the observed C-H bond regioselectivity, we performed a series of experimental and computational studies. Most late transition metal imido complexes operate through H atom transfer (HAT) that exhibit large primary KIE values (>5-1387–88), while a 1,2-addition mechanism has been proposed by a dinickel imido complex which exhibits a similar sp2 C-H regioselectivity (KIE = 4.389 and 5.190). We performed parallel kinetic experiments with C6H6 and C6D6 and product formation was tracked at 10°C using 31P{1H} inverse-gated NMR spectroscopy to measure the initial rates. These experiments afforded a kH/kD value of 3.22, which is within a reasonable range for a classical primary KIE and suggestive of little tunneling.
To examine the charge transfer properties of the transition state of C-H activation, we performed a Hammett-type study. To simplify analysis, we used 1,3-disubstituted arenes because they: a) form a single C-H activation product (vide supra), and b) ensure that primarily inductive substituent effects contribute to the reaction. Solutions of 3a in THF were mixed with 1 equiv. of AdN3 and a 1,3-disubstituted arene in 1:1 volumetric ratio (see SI section 3.1). Spanning 2σm-values from 0.86 to −0.42, we observed a linear correlation (R2=0.94) with positive slope, where arenes with electron-withdrawing substituents proceed with faster rates (Figure 8). These results are similar to those from studies that proposed a σ-bond metathesis or oxidative hydrogen migration mechanism in aromatic C-H activation.91–92
Figure 8.

Hammett-type plot of observed initial rate of C-H activation on 1,3-disubstituted arenes against 2σm.
We assessed whether an intramolecular, compared to intermolecular, Lewis acid is required to mediate the C-H bond activation pathway. Through a series of control reactions, we repeated conditions noted above that afforded C-H activation of toluene, then assessed deuterium incorporation after quenching with CD3COOD. To probe the requirement of the borane Lewis acid, we independently prepared a borane-free variant, (depe)Ni(COD) (3c), as a suitable control complex because of the similar steric and electronic properties to 3a (31P NMR spectra of 3c and 3a are similar; 49.8 ppm vs. 49.1 and 46.9 ppm, respectively). To probe the requirement of the tethered Lewis acid, we prepared a borane with a similar steric and electronic profile as that found in 3a. 9-octyl-9-BBN exhibits similar steric and electronic environments (11B NMR: 88.1 ppm vs. 87.7 ppm for 3a). When each control reaction was subjected to identical conditions, we observed distinct results. Compound 3a afforded aryl C-H activation (Figure 9). In contrast, we observed no deuterium incorporation when (depe)Ni(COD) was used in place of 3a, either with or without exogenous 9-octyl-9-BBN Lewis acid (Figure 9). In both cases, we found that 31P NMR resonances attributable to (depe)Ni(COD) decrease after introducing AdN3. These results are consistent with either: 1) no formation of a Ni=NAd intermediate with a depe ligand, or 2) a Ni=NAd intermediate that does not react with toluene. The latter point is consistent with prior reports where a related compound, (dtbpe)Ni=NAd, is stable to benzene-d6.40,93 Importantly, these control experiments demonstrate that the unique C-H bond reactivity of 3a is derived from the intramolecular borane within the ligand scaffold.
Figure 9.

2H NMR spectra of toluene-d8, C-H activation experiments with 3a, 3c, 3c in conjunction with 9-octyl-9-BBN.
To further clarify the role(s) that the appended Lewis acid serves to modify the electronic properties of the intermediates across the C-H activation pathway, we performed DFT computational analysis (PBE-D3/6-31G(d,p)//SDD(Ni)). Geometry optimization of a truncated nickel imido species (tBu instead of Ad) converged to structure 7a (Figure 10, bottom center). In the absence of an added solvent molecule, the optimized structure has a T-shaped geometry (P1-Ni-N angle = 164.7°).94–96 The Ni-N bond length (1.776Å) is longer than prior reported Ni-imido complexes (1.673(4) Å in (dtbpe)Ni(NAd)),40 and closer to that of a cationic nickel amido complex reported by Hillhouse (Ni-N = 1.771(4)Å).97 Furthermore, the optimized structure has a Ni-N-C bond angle of 117.4°. Strongly bent imido ligands have been attributed to: 1) the chelate effect, 2) lone pair character, or 3) triplet nitrene contribution on the nitrogen atom.76,98–99 We sought to investigate the contribution(s) of the appended borane to influence both the geometry and the electronic structure of the nickel imido moiety.
Figure 10.
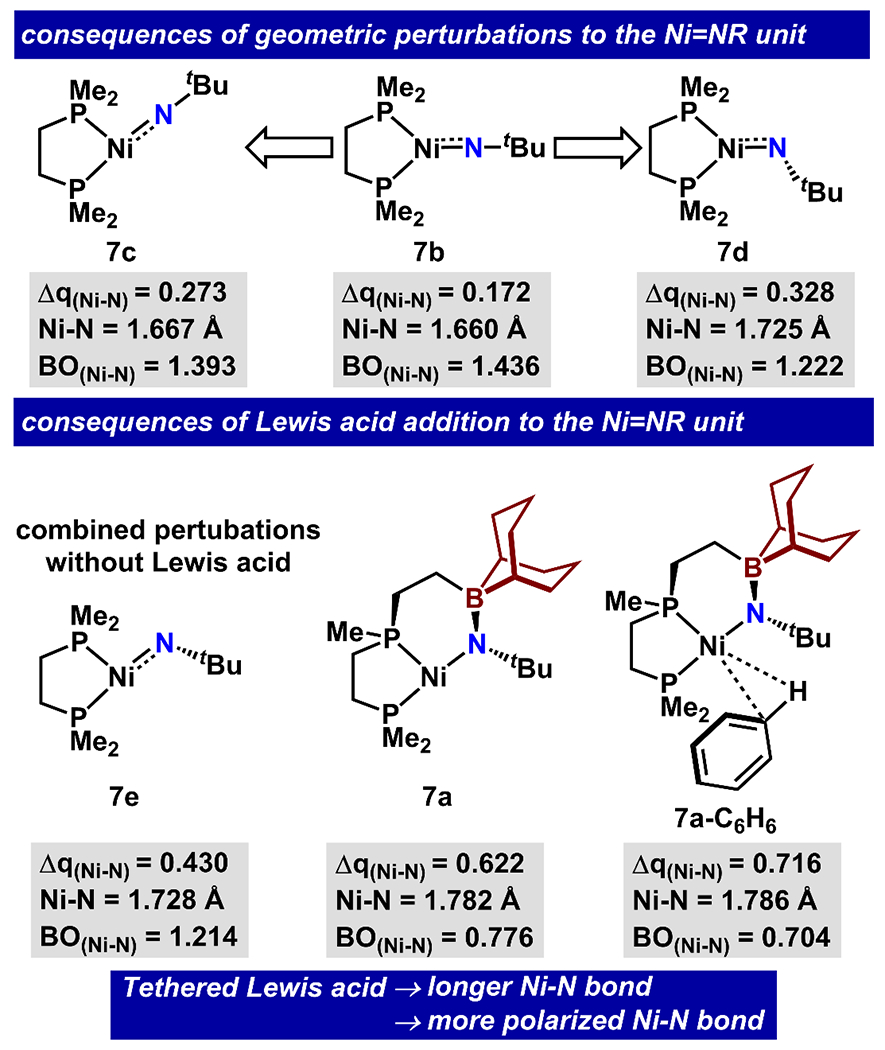
NBO analysis of (dmpe)NiNtBu (7b) and 7a with a comparison in structural parameters such as natural charge difference between Ni and N (Δq(Ni-N)), Ni-N bond length and order (BO(Ni-N)).
Given the atypical primary sphere environment of the nickel imido unit in 7a, we probed whether the appended Lewis acid primarily serves a structural role to perturb the primary sphere geometry by forming a chelate to the imido unit. We undertook a Walsh-type analysis to examine the relationship between P1-Ni-N/Ni-N-C angles and the structural and electronic parameters of the nickel imido unit, separately from the influence of Lewis acid. We performed relaxed potential energy surface scans with constraints on P1-Ni-N (7b to 7c; from 134° to 165°) and Ni-N-C (7b to 7d from 171° to 117°) angles. Finally, we optimized a structure with identical P1-Ni-N and Ni-N-C angles to 7a (7e). During these angular perturbations, we observed modest polarization and lengthening of Ni-N bond (see Table S16–S18). 7e exhibits a larger natural charge difference between nickel and nitrogen (Δq(Ni-N) of 0.430) than 7b (Δq(Ni-N) = 0.172). Similarly, the Ni-N bond elongates from 1.660 Å to 1.728 Å while the Wiberg bond order decreases from 1.436 to 1.214. Collectively, these results show that modest changes on the Ni imido unit are imposed as a result of purely structural changes to the Ni-primary sphere geometry.
We found that the electronic and structural parameters of 7a were influenced by the addition of a borane Lewis acid to a greater extent than could be imposed by the primary sphere modifications above. We compared the computed structures of the T-shaped Ni imido unit where an appended BBN Lewis acid is either present (7a) or absent (7e). When the BBN Lewis acid is present, the Δq(Ni-N) increases to 0.622 (larger than 7e by 0.192). Concomitant with this polarization are changes to the Ni-N bond (lengthening from 1.728 Å (7e) to 1.782 Å) and the Wiberg bond order (decreasing from 1.214 (7e) to 0.776). We also calculated a solvent-coordinated intermediate to provide an approximation of 7a in benzene solution (7a-C6H6). The solvent adduct optimized as a σ complex slightly higher in energy than 7a (6.8 kcal/mol) with a more polarized (Δq(Ni-N = 0.716) and slightly lengthened Ni-N bond (1.786 Å). Both 7a and 7a-C6H6 exhibit the longest Ni-N bonds in this series, which is consistent with an increase in N-basicity.100 Analysis on (dmpe)NiNMes and a truncated model of 5a show a consistent pattern in bond polarization (see Table S19). Overall, results of the calculations support our proposal that the Lewis acid redistributes charge at the nickel imido unit, rendering the nitrogen more basic. This proposal is further bolstered by related work where alkali metal additives were shown to both lengthen and polarize the Fe-N bond of an iron bis(NHC) imido complex.52
Following C-H bond activation, we evaluated whether a subsequent C-N bond could be induced to form via reductive elimination, using 6a as a model compound. In contrast to previous related reports that showed thermal- and oxidation-induced reductive elimination, we did not observe any reductive elimination products when 6a was: 1) heated to 110 °C, 2) treated with I2 and FcPF4 oxidants, or 3) combined with a π-accepting ligand such as CO. We propose that the B–N interaction between the BBN group and amide prevents reductive elimination in this system. To overcome this challenge, we hypothesized that a strong Lewis base might competitively bind the BBN Lewis acid, thus rendering the Ad-NH unit available for C-N reductive elimination. When 6a was treated with a combination of 9-azajulolidine and sodium t-pentoxide101 we observed the reductive elimination product, N-phenyl-N-adamantyl amine in 26% yield (Figure 9).102–103 These results represent a proof of principle that reductive elimination can be induced using a suitable set of reagents.
CONCLUSION
In summary, we have reported the preparation of a bidentate phosphine containing a single appended borane Lewis acid. This ligand complements the suite of commonly used bidentate phosphine ligands, such as depe, and it’s fully borylated counterparts.26–28 The single secondary sphere borane provides access to unique reactivity for reactions between Ni(0) and organoazides. Although κ2(N,N)-coordinated Ni(0) azide complexes were stabilized by both ligands 1a and 1b, only the scaffold of 1a was stable during subsequent reactions, illustrating a clear effect of Lewis acid tether length. Nickel azide complexes supported by 1a afford Ni imido complexes that exhibit diffeent reactivity profiles when the N-substituent is either aryl or alkyl. The aryl products were isolable with mesityl, while bulkier substituents underwent intramolecular C-H activation. The intermediate derived from AdN3 underwent 1,2-addition reactions with exogenous arenes: a reaction pathway atypical of late metal imidos. For all compounds featuring a Ni-NR unit, we observed Lewis acid/base interactions to the nitrogen atom. Through experimental and computational analyses, these secondary B–N interactions impart three important consequences during reactions of Ni(0) with organoazides, they: 1) improve stability of reaction intermediates, 2) influence the geometry by forming chelates, and 3) polarize the Ni–NR bond, which enables a rare example of monometallic late metal sp2 C-H activation favored over benzylic sp3 C-H bonds. These conclusions contribute several important principles that can be used to provide regioselectivity control over C-H bond activation pathways at first row, late-metals. Efforts to apply these principles to related organometallic reactions and ultimately develop catalytic versions, including C-H bond amination are currently underway in our lab.
Supplementary Material
Figure 11.

Base-induced reductive elimination of 6a.
ACKNOWLEDGMENT
We thank Prof. Dominik Munz of Saarland University, Prof. Yang Wang of University of Chinese Academy of Sciences, Prof. John Anderson of the University of Chicago, and Dr. Weiqing Mao of Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) for insightful discussions.
Funding Sources
This work was supported by the NIGMS of the NIH under Award 1R01GM111486-01A1, 1R35GM136360-01 (to N.K.S.), the National Natural Science Foundation of China (Nos. 22001249) and the Chinese Academy of Sciences (to J.C.), and the Fundamental Research Funds for the Central Universities (to J.C. and C.Z.). The X-ray diffractometers at UM were funded by the NSF (CHE 1625543).
Footnotes
Supporting Information. Synthetic and computational details (PDF). Crystallographic information (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Zhao M; Wang HB; Ji LN; Mao ZW Insights into metalloenzyme microenvironments: biomimetic metal complexes with a functional second coordination sphere. Chem. Soc. Rev 2013, 42 (21), 8360–8375. [DOI] [PubMed] [Google Scholar]
- 2.Elsby MR; Baker RT Strategies and mechanisms of metal–ligand cooperativity in first-row transition metal complex catalysts. Chem. Soc. Rev 2020, 49 (24), 8933–8987. [DOI] [PubMed] [Google Scholar]
- 3.Trouve J; Gramage-Doria R Beyond hydrogen bonding: recent trends of outer sphere interactions in transition metal catalysis. Chem. Soc. Rev 2021, 50 (5), 3565–3584. [DOI] [PubMed] [Google Scholar]
- 4.Drover MW A guide to secondary coordination sphere editing. Chem. Soc. Rev 2022, 51 (6), 1861–1880. [DOI] [PubMed] [Google Scholar]
- 5.Shook RL; Borovik AS Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem 2010, 49 (8), 3646–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ford CL; Park YJ; Matson EM; Gordon Z; Fout AR A bioinspired iron catalyst for nitrate and perchlorate reduction. Science 2016, 354 (6313), 741–743. [DOI] [PubMed] [Google Scholar]
- 7.Geri JB; Shanahan JP; Szymczak NK Testing the Push-Pull Hypothesis: Lewis Acid Augmented N2 Activation at Iron. J. Am. Chem. Soc 2017, 139 (16), 5952–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller AJM; Labinger JA; Bercaw JE Reductive Coupling of Carbon Monoxide in a Rhenium Carbonyl Complex with Pendant Lewis Acids. J. Am. Chem. Soc 2008, 130 (36), 11874–11875. [DOI] [PubMed] [Google Scholar]
- 9.Nichols AW; Machan CW Secondary-Sphere Effects in Molecular Electrocatalytic CO2 Reduction. Front. Chem 2019, 7, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook SA; Borovik AS Molecular Designs for Controlling the Local Environments around Metal Ions. Acc. Chem. Res 2015, 48 (8), 2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hale LVA; Szymczak NK Hydrogen Transfer Catalysis beyond the Primary Coordination Sphere. ACS Catal. 2018, 8 (7), 6446–6461. [Google Scholar]
- 12.Zurakowski JA; Austen BJH; Drover MW Exterior decorating: Lewis acid secondary coordination spheres for cooperative reactivity. Trends Chem. 2022, 4 (4), 331–346. [Google Scholar]
- 13.Churchill MR; Wasserman HJ; Holmes SJ; Schrock RR Coupling of methylidyne and carbonyl ligands on tungsten. Crystal structure of W(η2-HC≡COAlCl3)(CO)(PMe3)3Cl. Organometallics 1982, 1 (5), 766–768. [Google Scholar]
- 14.Protasiewicz JD; Masschelein A; Lippard SJ Kinetic, spectroscopic, and structural evidence for carbene-carbyne intermediates in carbyne/CO coupling. J. Am. Chem. Soc 1993, 115 (2), 808–810. [Google Scholar]
- 15.Shanahan JP; Szymczak NK Hydrogen Bonding to a Dinitrogen Complex at Room Temperature: Impacts on N2 Activation. J. Am. Chem. Soc 2019, 141 (21), 8550–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ménard G; Stephan DW Room Temperature Reduction of CO2 to Methanol by Al-Based Frustrated Lewis Pairs and Ammonia Borane. J. Am. Chem. Soc 2010, 132 (6), 1796–1797. [DOI] [PubMed] [Google Scholar]
- 17.Sampson MD; Kubiak CP Manganese Electrocatalysts with Bulky Bipyridine Ligands: Utilizing Lewis Acids To Promote Carbon Dioxide Reduction at Low Overpotentials. J. Am. Chem. Soc 2016, 138 (4), 1386–1393. [DOI] [PubMed] [Google Scholar]
- 18.Marberger A; Ferri D; Elsener M; Kröcher O The Significance of Lewis Acid Sites for the Selective Catalytic Reduction of Nitric Oxide on Vanadium-Based Catalysts. Angew. Chem. Int. Ed 2016, 55 (39), 11989–11994. [DOI] [PubMed] [Google Scholar]
- 19.Kiernicki JJ; Zeller M; Szymczak NK Hydrazine Capture and N-N Bond Cleavage at Iron Enabled by Flexible Appended Lewis Acids. J. Am. Chem. Soc 2017, 139 (50), 18194–18197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buss JA; VanderVelde DG; Agapie T Lewis Acid Enhancement of Proton Induced CO2 Cleavage: Bond Weakening and Ligand Residence Time Effects. J. Am. Chem. Soc 2018, 140 (32), 10121–10125. [DOI] [PubMed] [Google Scholar]
- 21.Simonneau A; Etienne M Enhanced Activation of Coordinated Dinitrogen with p-Block Lewis Acids. Chem. Eur. J 2018, 24 (48), 12458–12463. [DOI] [PubMed] [Google Scholar]
- 22.Abucayon EG; Khade RL; Powell DR; Zhang Y; Richter-Addo GB Lewis Acid Activation of the Ferrous Heme–NO Fragment toward the N–N Coupling Reaction with NO To Generate N2O. J. Am. Chem. Soc 2018, 140 (12), 4204–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becica J; Dobereiner GE The roles of Lewis acidic additives in organotransition metal catalysis. Org. Biomol. Chem 2019, 17 (8), 2055–2069. [DOI] [PubMed] [Google Scholar]
- 24.Wang C; Xi Z Co-operative effect of Lewis acids with transition metals for organic synthesis. Chem. Soc. Rev 2007, 36 (9), 1395–1406. [DOI] [PubMed] [Google Scholar]
- 25.Miller AJM; Labinger JA; Bercaw JE Homogeneous CO Hydrogenation: Ligand Effects on the Lewis Acid-Assisted Reductive Coupling of Carbon Monoxide. Organometallics 2010, 29 (20), 4499–4516. [Google Scholar]
- 26.Drover MW; Dufour MC; Lesperance-Nantau LA; Noriega RP; Levin K; Schurko RW Octaboraneyl Complexes of Nickel: Monomers for Redox-Active Coordination Polymers. Chem. Eur. J 2020, 26 (49), 11180–11186. [DOI] [PubMed] [Google Scholar]
- 27.Zurakowski JA; Bhattacharyya M; Spasyuk DM; Drover MW Octaboraneyl [Ni(H)(diphosphine)2]+ Complexes: Exploiting Phosphine Ligand Lability for Hydride Transfer to an [NAD]+ Model. Inorg. Chem 2021, 60 (1), 37–41. [DOI] [PubMed] [Google Scholar]
- 28.Zurakowski JA; Austen BJH; Dufour MC; Spasyuk DM; Nelson DJ; Drover MW Lewis Acid-Promoted Oxidative Addition at a [Ni0(diphosphine)2] Complex: The Critical Role of a Secondary Coordination Sphere. Chem. Eur. J 2021, 27 (64), 16021–16027. [DOI] [PubMed] [Google Scholar]
- 29.Zurakowski JA; Austen BJH; Brown KR; Drover MW Bis(1-bora-4-phosphorinane) ring closure at Cp*M (M = Fe, Co) complexes. Chem. Commun 2022, 58 (15), 2500–2503. [DOI] [PubMed] [Google Scholar]
- 30.Burford RJ; Fryzuk MD Examining the relationship between coordination mode and reactivity of dinitrogen. Nat. Rev. Chem 2017, 1 (4), 13. [Google Scholar]
- 31.Nishibayashi Y Recent Progress in Transition-Metal-Catalyzed Reduction of Molecular Dinitrogen under Ambient Reaction Conditions. Inorg. Chem 2015, 54 (19), 9234–9247. [DOI] [PubMed] [Google Scholar]
- 32.Waterman R; Hillhouse GL Group Transfer from Nickel Imido, Phosphinidene, and Carbene Complexes to Ethylene with Formation of Aziridine, Phosphirane, and Cyclopropane Products. J. Am. Chem. Soc 2003, 125 (44), 13350–13351. [DOI] [PubMed] [Google Scholar]
- 33.Cramer SA; Jenkins DM Synthesis of Aziridines from Alkenes and Aryl Azides with a Reusable Macrocyclic Tetracarbene Iron Catalyst. J. Am. Chem. Soc 2011, 133 (48), 19342–19345. [DOI] [PubMed] [Google Scholar]
- 34.Beaumier EP; Pearce AJ; See XY; Tonks IA Modern applications of low-valent early transition metals in synthesis and catalysis. Nat. Rev. Chem 2019, 3 (1), 15–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berry JF Terminal nitride and imido complexes of late transition metals. Comments Inorganic Chem. 2009, 30 (1-2), 28–66. [Google Scholar]
- 36.Ray K; Heims F; Pfaff FF Terminal Oxo and Imido Transition-Metal Complexes of Groups 9-11. Eur. J. Inorg. Chem 2013, 2013 (22-23), 3784–3807. [Google Scholar]
- 37.Davies HML; Manning JR Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451 (7177), 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ju M; Schomaker JM Nitrene transfer catalysts for enantioselective C–N bond formation. Nat. Rev. Chem 2021, 5 (8), 580–594. [DOI] [PubMed] [Google Scholar]
- 39.Mindiola DJ; Hillhouse GL Terminal Amido and Imido Complexes of Three-Coordinate Nickel. J. Am. Chem. Soc 2001, 123 (19), 4623–4624. [DOI] [PubMed] [Google Scholar]
- 40.Waterman R; Hillhouse GL η2-Organoazide Complexes of Nickel and Their Conversion to Terminal Imido Complexes via Dinitrogen Extrusion. J. Am. Chem. Soc 2008, 130 (38), 12628–12629. [DOI] [PubMed] [Google Scholar]
- 41.Laskowski CA; Miller AJM; Hillhouse GL; Cundari TR A Two-Coordinate Nickel Imido Complex That Effects C–H Amination. J. Am. Chem. Soc 2011, 133 (4), 771–773. [DOI] [PubMed] [Google Scholar]
- 42.Mindiola DJ; Waterman R; Iluc VM; Cundari TR; Hillhouse GL Carbon–Hydrogen Bond Activation, C–N Bond Coupling, and Cycloaddition Reactivity of a Three-Coordinate Nickel Complex Featuring a Terminal Imido Ligand. Inorg. Chem 2014, 53 (24), 13227–13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kogut E; Wiencko HL; Zhang LB; Cordeau DE; Warren TH A terminal Ni(III)-imide with diverse reactivity pathways. J. Am. Chem. Soc 2005, 127 (32), 11248–11249. [DOI] [PubMed] [Google Scholar]
- 44.Dong YY; Lukens JTT; Clarke RM; Zheng SL; Lancaster KM; Betley TA Synthesis, characterization and C-H amination reactivity of nickel iminyl complexes. Chem. Sci 2020, 11 (5), 1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiese S; McAfee JL; Pahls DR; McMullin CL; Cundari TR; Warren TH C-H Functionalization Reactivity of a Nickel-Imide. J. Am. Chem. Soc 2012, 134 (24), 10114–10121. [DOI] [PubMed] [Google Scholar]
- 46.Dong Y; Clarke RM; Porter GJ; Betley TA Efficient C–H Amination Catalysis Using Nickel-Dipyrrin Complexes. J. Am. Chem. Soc 2020, 142 (25), 10996–11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong Y; Lund CJ; Porter GJ; Clarke RM; Zheng S-L; Cundari TR; Betley TA Enantioselective C–H Amination Catalyzed by Nickel Iminyl Complexes Supported by Anionic Bisoxazoline (BOX) Ligands. J. Am. Chem. Soc 2021, 143 (2), 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolczanski PT Activation of Carbon–Hydrogen Bonds via 1,2-RH-Addition/-Elimination to Early Transition Metal Imides. Organometallics 2018, 37 (4), 505–516. [Google Scholar]
- 49.Webb JR; Burgess SA; Cundari TR; Gunnoe TB Activation of carbon–hydrogen bonds and dihydrogen by 1,2-CH-addition across metal–heteroatom bonds. Dalton Trans. 2013, 42 (48), 16646–16665. [DOI] [PubMed] [Google Scholar]
- 50.Lu E; Chu J; Chen Y Scandium Terminal Imido Chemistry. Acc. Chem. Res 2018, 51 (2), 557–566. [DOI] [PubMed] [Google Scholar]
- 51.Alkali metal ions have been shown to polarize iron imido species, see ref 52.
- 52.Gao Y; Pink M; Smith JM Alkali Metal Ions Dictate the Structure and Reactivity of an Iron(II) Imido Complex. J. Am. Chem. Soc 2022, 144 (4), 1786–1794. [DOI] [PubMed] [Google Scholar]
- 53.Kiernicki JJ; Shanahan JR; Zeller M; Szymczak NK Tuning ligand field strength with pendent Lewis acids: access to high spin iron hydrides. Chem. Sci 2019, 10 (21), 5539–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiernicki JJ; Zeller M; Szymczak NK Requirements for Lewis Acid-Mediated Capture and N-N Bond Cleavage of Hydrazine at Iron. Inorg. Chem 2019, 58 (2), 1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kiernicki JJ; Norwine EE; Zeller M; Szymczak NK Tetrahedral iron featuring an appended Lewis acid: distinct pathways for the reduction of hydroxylamine and hydrazine. Chem. Commun 2019, 55 (79), 11896–11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shanahan JP; Moore CM; Kampf JW; Szymczak NK Modulation of H+/H− exchange in iridium-hydride 2-hydroxypyridine complexes by remote Lewis acids. Chem. Commun 2021, 57 (88), 11705–11708. [DOI] [PubMed] [Google Scholar]
- 57.A recent paper calculated substrate/borane interactions with a single pendent borane as a model for an octaborylated bidentate phosphine. See ref 58.
- 58.Facchinato D; Zurakowski JA; Drover MW Rhodium disulfur and dioxygen complexes: examination of boron secondary coordination sphere effects. J. Coord. Chem 2022, Ahead-of-print, 1-11. doi: 10.1080/00958972.2022.2067989. [DOI] [Google Scholar]
- 59.Li GY; Fagan PJ; Watson PL Versatile approaches to the polymer-supported synthesis of bidentate phosphorus-containing ligands. Angew. Chem. Int. Ed 2001, 40 (6), 1106–1109. [DOI] [PubMed] [Google Scholar]
- 60.McEwen WE; Janes AB; Knapczyk JW; Kyllingstad VL; Shiau WI; Shore S; Smith JH Role of Through Space 2p-3d Overlap in the Alkylation of Phosphines. J. Am. Chem. Soc 1978, 100 (23), 7304–7311. [Google Scholar]
- 61.We propose the high melting point of D is due to the contribution of ionic form D’. Similar R3P-R2P+ interaction has been reported for penta-phenylphosphinophosphonium salts. See ref 62.
- 62.Burford N; Cameron TS; Ragogna PJ; Ocando-Mavarez E; Gee M; McDonald R; Wasylishen RE Phosphine Ligand Exchange at a Phosphine Lewis Acceptor: The First Structural Characterization of Homoleptic Phosphinophosphonium Salts. J. Am. Chem. Soc 2001, 123 (32), 7947–7948. [DOI] [PubMed] [Google Scholar]
- 63.For comparasion, the 31P NMR of PEt3 shows at −20.0 ppm, while that of Et2P(CH=CH2) show at −21.2 ppm. See ref 64.
- 64.Askham FR; Stanley GG; Marques EC A new type of transition-metal dimer based on a hexaphosphine ligand system: Co2(CO)4(eHTP)2+ (eHTP = (Et2PCH2CH2)2PCH2P(CH2CH2PEt2)2). J. Am. Chem. Soc 1985, 107 (25), 7423–7431. [Google Scholar]
- 65.A related report showed a similar interaction. A doubly Lewis-acid-functionalized phosphine MeP(CH2CH2CH2(9-BBN))2 has been reported and the 31P rand 11B NMR resonance consistent with one tetra- and one tricoordinate borane. See ref 66.
- 66.Korte LA; Blomeyer S; Peters JH; Mix A; Neumann B; Stammler HG; Mitzel NW Dynamic Exchange in Intramolecular Lewis Pairs with Multiple Lewis-Acidic Functions. Organometallics 2017, 36 (3), 742–749. [Google Scholar]
- 67.Lanni EL; Locke JR; Gleave CM; McNeil AJ Ligand-Based Steric Effects in Ni-Catalyzed Chain-Growth Polymerizations Using Bis(dialkylphosphino)ethanes. Macromolecules 2011, 44 (13), 5136–5145. [Google Scholar]
- 68.Reinholdt A; Kwon S; Jafari MG; Gau MR; Caroll PJ; Lawrence C; Gu J; Baik M-H; Mindiola DJ An Isolable Azide Adduct of Titanium(II) Follows Bifurcated Deazotation Pathways to an Imide. J. Am. Chem. Soc 2022, 144 (1), 527–537. [DOI] [PubMed] [Google Scholar]
- 69.Cenini S; Gallo E; Caselli A; Ragaini F; Fantauzzi S; Piangiolino C Coordination chemistry of organic azides and amination reactions catalyzed by transition metal complexes. Coord. Chem. Rev 2006, 250 (11-12), 1234–1253. [Google Scholar]
- 70.We note two structurally reported examples of this binding mode for bimetallic complexes. See ref 71 and 72.
- 71.Ni C; Ellis BD; Long GJ; Power PP Reactions of Ar’CrCrAr’ with N2O or N3(1-Ad): complete cleavage of the Cr-Cr quintuple interaction. Chem. Commun 2009, (17), 2332–2334. [DOI] [PubMed] [Google Scholar]
- 72.Wu B; Sanchez RH; Bezpalko MW; Foxman BM; Thomas CM Formation of Heterobimetallic Zirconium/Cobalt Diimido Complexes via a Four-Electron Transformation. Inorg. Chem 2014, 53 (19), 10021–10023. [DOI] [PubMed] [Google Scholar]
- 73.Cundari TR; Morello GR A Computational Study of Metal-Mediated Decomposition of Nitrene Transfer Reagents. J. Org. Chem 2009, 74 (15), 5711–5714. [DOI] [PubMed] [Google Scholar]
- 74.Cundari TR; Pierpont AW; Vaddadi S Computational study of methane functionalization by a multiply bonded, Ni-bis(phosphine) complex. J. Organomet. Chem 2007, 692 (21), 4551–4559. [Google Scholar]
- 75.Shoshani MM; Beck R; Wang X; McLaughlin MJ; Johnson SA Synthesis of Surface-Analogue Square-Planar Tetranuclear Nickel Hydride Clusters and Bonding to μ4-NR, -O and -BH Ligands. Inorg. Chem 2018, 57 (5), 2438–2446. [DOI] [PubMed] [Google Scholar]
- 76.Grünwald A; Anjana SS; Munz D Terminal Imido Complexes of the Groups 9–11: Electronic Structure and Developments in the Last Decade. Eur. J. Inorg. Chem 2021, 2021 (40), 4147–4166. [Google Scholar]
- 77.Grünwald A; Goswami B; Breitwieser K; Morgenstern B; Gimferrer M; Heinemann FW; Momper DM; Kay CWM; Munz D Palladium Terminal Imido Complexes with Nitrene Character. J. Am. Chem. Soc 2022, 144 (20), 8897–8901. [DOI] [PubMed] [Google Scholar]
- 78.Chen H; Bartlett RA; Olmstead MM; Power PP; Shoner SC Series of two-coordinate and quasi-two-coordinate transition-metal complexes: synthesis, structural, and spectroscopic studies of sterically demanding borylamide ligands -NRBR’2 (R = Ph, R’ = Mes, Xyl; R = R’ = Mes), their lithium salts, Li(Et2O)2NRBR’2, and their transition-metal derivatives, M(NPhBMes2)2 (M = Cr, Co, Ni), Co(NPhBXyl2)2 and M(NMesBMes2)2 (M = Cr → Ni). J. Am. Chem. Soc 1990, 112 (3), 1048–1055. [Google Scholar]
- 79.Laskowski CA; Morello GR; Saouma CT; Cundari TR; Hillhouse GL Single-electron oxidation of N-heterocyclic carbene-supported nickel amides yielding benzylic C–H activation. Chem. Sci 2013, 4 (1), 170–174. [Google Scholar]
- 80.Pinkowicz D; Birk FJ; Magott M; Schulte K; Dunbar KR Systematic Study of Open-Shell Trigonal Pyramidal Transition-Metal Complexes with a Rigid-Ligand Scaffold. Chem. Eur. J 2017, 23 (15), 3548–3552. [DOI] [PubMed] [Google Scholar]
- 81.Hennessy ET; Liu RY; Iovan DA; Duncan RA; Betley TA Iron-mediated intermolecular N-group transfer chemistry with olefinic substrates. Chem. Sci 2014, 5 (4), 1526–1532. [Google Scholar]
- 82.Xue X-S; Ji P; Zhou B; Cheng J-P The Essential Role of Bond Energetics in C–H Activation/Functionalization. Chem. Rev 2017, 117 (13), 8622–8648. [DOI] [PubMed] [Google Scholar]
- 83.We propose that the two isomers of 6b-meta are due to restricted rotation of the tolyl ligand, surrounded by sterically encumbering BBN and adamantyl amide.
- 84.In scXRD data, three isomers - two isomers of meta and one of para - are present. An extended analysis is available in the SI.
- 85.The selectivity resembles that of Ni-mediated arene C-H activation in hydroarylation. See work by ref 86.
- 86.Saper NI; Ohgi A; Small DW; Semba K; Nakao Y; Hartwig JF Nickel-catalysed anti-Markovnikov hydroarylation of unactivated alkenes with unactivated arenes facilitated by non-covalent interactions. Nat. Chem 2020, 12 (3), 276–283. [DOI] [PubMed] [Google Scholar]
- 87.Du Y-D; Xu Z-J; Zhou C-Y; Che C-M An Effective [FeIII(TF4DMAP)Cl] Catalyst for C–H Bond Amination with Aryl and Alkyl Azides. Org. Lett 2019, 21 (4), 895–899. [DOI] [PubMed] [Google Scholar]
- 88.King ER; Hennessy ET; Betley TA Catalytic C–H Bond Amination from High-Spin Iron Imido Complexes. J. Am. Chem. Soc 2011, 133 (13), 4917–4923. [DOI] [PubMed] [Google Scholar]
- 89.Powers IG; Andjaba JM; Zeller M; Uyeda C Catalytic C(sp2)–H Amination Reactions Using Dinickel Imides. Organometallics 2020, 39 (21), 3794–3801. [Google Scholar]
- 90.Powers IG; Kiattisewee C; Mullane KC; Schelter EJ; Uyeda CA 1,2-Addition Pathway for C(sp2)–H Activation at a Dinickel Imide. Chem. Eur. J 2017, 23 (32), 7694–7697. [DOI] [PubMed] [Google Scholar]
- 91.DeYonker NJ; Foley NA; Cundari TR; Gunnoe TB; Petersen JL Combined Experimental and Computational Studies on the Nature of Aromatic C–H Activation by Octahedral Ruthenium(II) Complexes: Evidence for σ-Bond Metathesis from Hammett Studies. Organometallics 2007, 26 (26), 6604–6611. [Google Scholar]
- 92.Note that C-H activation of benzene proceeded much faster than any substituted arenes. We propose that, compared to other arenes, C-H activation of benzene is kinetically more favorable due to having six times more C-H bonds, in addition to the absence of steric hinderance from substituents. Therefore, the outlier presented by benzene was not included for the Hammett-type plot.
- 93.Note that (dtbpe)Ni(NAd) reacts with the sp C-H bond of phenylacetylene, see ref 42.
- 94.Related T-shaped nickel imido and bis(amido) complexes have been reported, see ref 95 and 96.
- 95.Lipschutz MI; Yang X; Chatterjee R; Tilley TD A Structurally Rigid Bis(amido) Ligand Framework in Low-Coordinate Ni(I), Ni(II), and Ni(III) Analogues Provides Access to a Ni(III) Methyl Complex via Oxidative Addition. J. Am. Chem. Soc 2013, 135 (41), 15298–15301. [DOI] [PubMed] [Google Scholar]
- 96.Reckziegel A; Battistella B; Werncke CG On the Synthesis of a T-Shaped Imido Nickel Complex and Trigonal Amido Nickel Complexes. Eur. J. Inorg. Chem 2022, 2022 (10), e202101102. [Google Scholar]
- 97.Iluc VM; Miller AJM; Anderson JS; Monreal MJ; Mehn MP; Hillhouse GL Synthesis and Characterization of Three-Coordinate Ni(III)-Imide Complexes. J. Am. Chem. Soc 2011, 133 (33), 13055–13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.For more detailed discussion on linear vs. bent geometry on imido ligand, see review by Grünwald et al. (ref 76).
- 99.Grünwald A; Orth N; Scheurer A; Heinemann FW; Pöthig A; Munz D An Isolable Terminal Imido Complex of Palladium and Catalytic Implications. Angew. Chem. Int. Ed 2018, 57 (49), 16228–16232. [DOI] [PubMed] [Google Scholar]
- 100.Cowley RE; Eckert NA; Vaddadi S; Figg TM; Cundari TR; Holland PL Selectivity and Mechanism of Hydrogen Atom Transfer by an Isolable Imidoiron(III) Complex. J. Am. Chem. Soc 2011, 133 (25), 9796–9811. [DOI] [PubMed] [Google Scholar]
- 101.Simon CM; Dudra SL; McGuire RT; Ferguson MJ; Johnson ER; Stradiotto M Identification of a Nitrenoid Reductive Elimination Pathway in Nickel-Catalyzed C–N Cross-Coupling. ACS Catal. 2022, 12 (2), 1475–1480. [Google Scholar]
- 102.McGuire RT; Lundrigan T; MacMillan JWM; Robertson KN; Yadav AA; Stradiotto M Mapping Dual-Base-Enabled Nickel-Catalyzed Aryl Amidations: Application in the Synthesis of 4-Quinolones. Angew. Chem. Int. Ed 2022, 61 (13), e202200352. [DOI] [PubMed] [Google Scholar]
- 103.Lundrigan T; Tassone JP; Stradiotto M Nickel-Catalyzed N-Arylation of Amides with (Hetero)aryl Electrophiles by Using a DBU/NaTFA Dual-Base System. Synlett 2021, 32 (16), 1665–1669 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.