

Abstract
Cell survival largely depends on the faithful maintenance of genetic material since genomic DNA is constantly exposed to genotoxicants from both endogenous and exogenous sources. The evolutionarily conserved base excision repair (BER) pathway is critical for maintaining genome integrity by eliminating highly abundant and potentially mutagenic oxidized DNA base lesions. BER is a multistep process, which is initiated with recognition and excision of the DNA base lesion by a DNA glycosylase, followed by DNA end processing, gap filling and finally sealing of the nick. Besides genome maintenance by global BER, DNA glycosylases have been found to play additional roles, including preferential repair of oxidized lesions from transcribed genes, modulation of the immune response, participation in active DNA demethylation and maintenance of the mitochondrial genome. Central to these functions is the DNA glycosylase NEIL2. Its loss results in increased accumulation of oxidized base lesions in the transcribed genome, triggers an immune response and causes early neurodevelopmental defects, thus emphasizing the multitasking capabilities of this repair protein. Here we review the specialized functions of NEIL2 and discuss the consequences of its absence both in vitro and in vivo.
Keywords: DNA glycosylase NEIL2, BER, TC-BER, Cancer, Inflammation, Demethylation
1. Introduction
The integrity of cellular DNA is continuously challenged by exogenous and endogenous insults (Vermeij et al., 2014). Exogenous sources such as environmental toxicants, ultraviolet (UV) light and ionizing radiation induce a wide variety of DNA lesions (Hoeijmakers, 2001). Reactive oxygen species (ROS) generated endogenously as byproducts of aerobic metabolism or during inflammatory responses also directly modify DNA. These modifications include DNA base oxidation and DNA strand-breaks (both single- and double-strand), which are major threats to the integrity of the genome (Lindahl, 1993). It has been estimated that >10000 base lesions are generated in each cell per day during cellular metabolism and by spontaneous chemical reactions. If left unrepaired or repaired with errors, these DNA base lesions are a major source of mutations, which are implicated in various diseases including aging, neurodevelopmental disorders and cancer (De Bont and van Larebeke, 2004). Thus repair of base lesions in DNA is critical for maintaining genome integrity and species survival.
BER is the major pathway for repair of oxidized DNA bases and single-strand breaks with blocked termini (Wallace et al., 2012). This pathway is initiated with excision of the damaged base by a DNA glycosylase. Five oxidized DNA base-specific glycosylases have been characterized in mammalian cells, namely NTHL1, OGG1, NEIL1, NEIL2 and NEIL3. After excision of the oxidized bases, all these glycosylases cleave the DNA backbone by their intrinsic AP lyase activities (Fig. 1). The preferred substrate for NTHL1 and OGG1 is oxidized pyrimidines and purines, respectively. Both of these enzymes use β-elimination as the catalytic mechanism and generate 3-αβ unsaturated aldehyde and 5′-P termini (Mullins et al., 2019). However, the NEILs (NEIL1–3) have a broad range of substrates and excise both damaged or ring opened oxidized purines and pyrimidines. While NEIL3 incises damaged bases by β-elimination, both NEIL1 and NEIL2 instead possess a β,δ-elimination activity, generating a one-nucleotide gap with 3′-P and 5′-P termini (Dou et al., 2003). The repair is completed in several sub-sequent steps that begin with the processing of 3′-ends, either by APE1 (3′-ab unsaturated aldehyde) or PNKP (3′-P) to generate 3′-OH ends (Wiederhold et al., 2004). DNA repair synthesis is carried out primarily by DNA polymerase β, and finally the nick is sealed by DNA ligase IIIα/XRCC1 (Tomkinson and Sallmyr, 2013). The NEILs are distinct from NTHL1 and OGG1 in that they preferentially excise base lesions from single-stranded DNA including bubble and fork DNA structures that mimic transcription and replication intermediates, respectively. This property is the basis for proposing preferential repair of oxidized bases during transcription or replication (Hazra and Mitra, 2006). The S-phase specific activation of NEIL1 and NEIL3 suggest their involvement in repair of base lesions from template DNA prior to replication (Hazra et al., 2002a,b; Hedge et al., 2013; Zhou et al., 2017). By contrast, NEIL2’s expression is cell cycle independent, and it is involved in repair of oxidized bases from transcribed regions, as reviewed here (Hazra et al., 2002a,b; Banerjee et al., 2011). However, several recent findings attest to its having additional functions, which are also addressed in the following section.
Fig. 1.
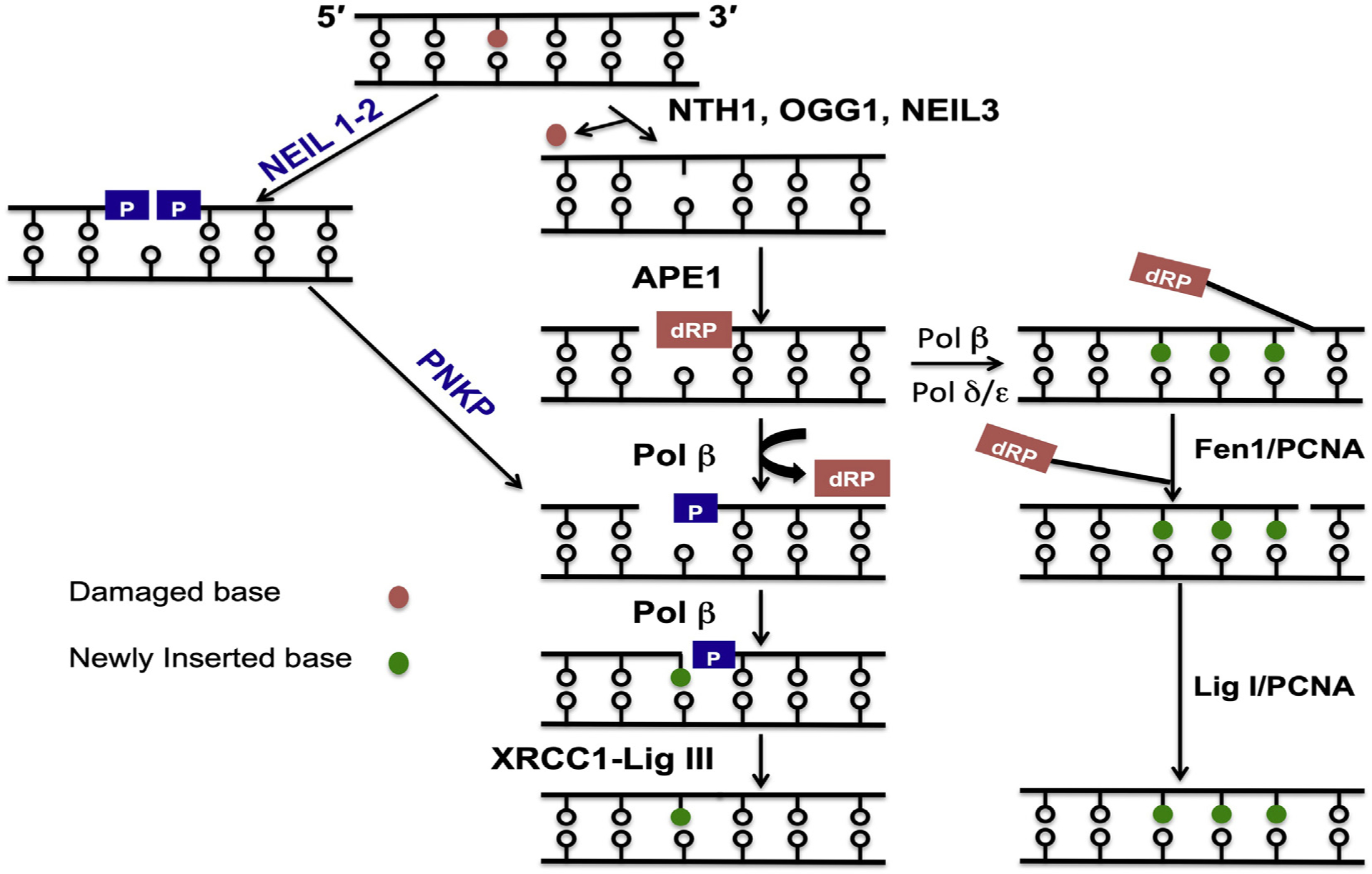
Simplified model for short-patch BER. DNA glycosylase excise the damaged base by cleaving the N-glycosydic bond leaving AP site which is cleaved by AP endonuclease to create single-strand break containing 5′-dRP moiety. DNA polymerase beta remove’s the 5′-dRP moiety and simultaneously fills the gap. In contrast, BER initiated by NEIL 1–2 glycosylases create single-strand gap containing 3′- and 5′-phosphate ends. The 3′-phosphate is removed by PNKP before insertion a nucleotide by pol beta. Finally, the nick is sealed by XRCC1-LigIIIa complex to complete the short-patch BER. In some cases Pol beta is unable to remove the 5′-dRP. moiety, then a polymerase switch to polymerase δ/ε occur followed by 2–8 nt gap filling creating 5′-flap structure incised by Fen1/PCNA. The remaining nick is sealed by Lig I in association with PCNA to complete the long-patch BER.
2. Specialized functions of NEIL2
NEIL’s functions are generally required to protect cells against the accumulation of oxidative DNA damage in the genome to maintain genomic stability. Recent findings from several laboratories have indicated that the NEILs are multifunctional DNA repair proteins as demonstrated by their involvement in diverse cellular processes. This review will primarily focus on the multifunctional roles of NEIL2, which depend on its ability to recognize damaged bases within unique DNA structures and which appear to modulate various cellular processes (Fig. 2), and will discuss the consequences of NEIL2’s loss or functional impairment.
Fig. 2.
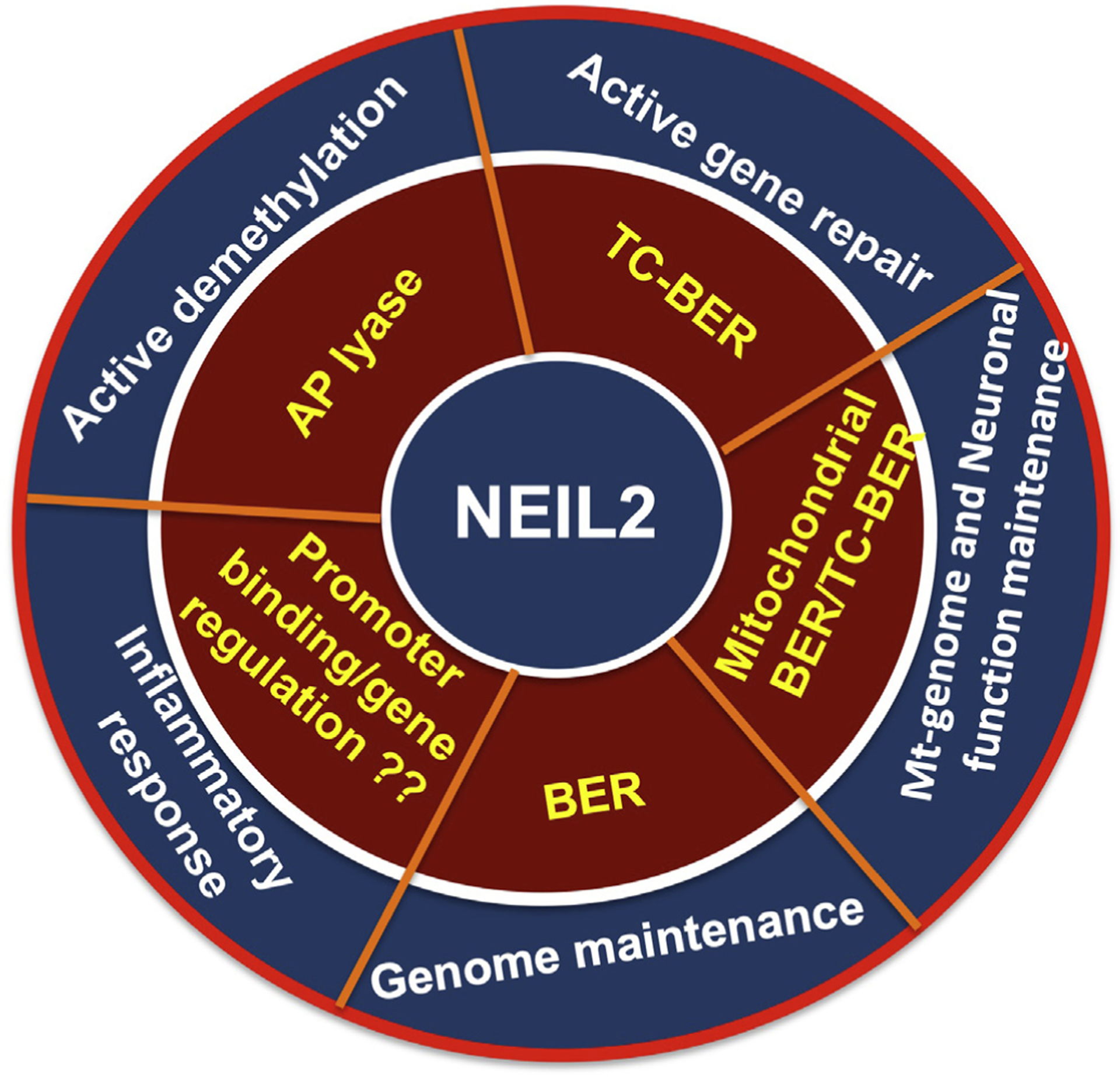
Cellular processes associated with NEIL2 and the respective activity. Inner circle depicts the NEIL2 activity associated with the cellular processes as shown outer circle.
2.1. Transcription-coupled repair
Transcription-coupled repair (TCR) refers to the preferential repair of lesions from the transcribed strands of active genes (Hanawalt and Spivak, 2008). TCR was first discovered by Hanawalt’s group as a sub-pathway of nucleotide excision repair (NER), which is initiated when elongating RNAPIIo is blocked at a DNA lesion. Helix distorting bulky DNA adducts induced by environmental carcinogens, UV irradiation or intracellular metabolites can block the forward translocation of RNAPIIo causing it to stall at the site (Gregersen and Svejstrup, 2018). Arrested RNAPIIo at such lesions serves as a signal to initiate TC-NER for efficient recovery of RNA synthesis and cell survival (Lans et al., 2019; Gregersen and Svejstrup, 2018; Hanawalt and Spivak, 2008). Premutagenic oxidized DNA base lesions are abundant; hence repair of such lesions would be important for the cells (particularly neuronal cells) to avoid generation of mutant transcripts and/or transcriptional mutagenesis. The possibility of preferential repair of oxidative base modifications via the BER pathway from the transcribed strand of active genes remained unexplored for a long time. Many oxidative modifications of DNA bases cause only minor helix distortions that do not pose strong blocks to RNAPII elongation as examined using in vitro transcription systems, although some were reported to block RNAPII elongation to different extents depending on the transcription system used (Kuraoka et al., 2003; Charlet-Berguerand et al., 2006). However, these blocks were alleviated and the lesions bypassed at the expense of transcriptional mutagenesis by the action of transcription elongation factors when the reaction was carried out with cell free nuclear extracts (Charlet-Berguerand et al., 2006; Kuraoka et al., 2007). Transcriptional mutagenesis via bypassing the oxidized DNA lesions during transcription in mammalian cells has also been reported previously (Saxowsky et al., 2008). Transcriptional mutagenesis would be an undesirable outcome, and hence accurate repair of functional genes is critical to prevent mutant RNA synthesis. There are >20 different oxidized DNA lesions induced by ROS (Cadet et al., 2003), but the extent to which oxidative base damage can affect the rate of transcription is not fully understood and depends on several factors including the nature of the lesion, transcription system used and sequence context. It is known that guanine is highly vulnerable to oxidation due to having a low redox potential, causing the formation of abundant 8-oxoG lesions. These do not pose a strong block to RNAPII transcription in vitro (Kuraoka et al., 2003). However, 8-oxoG is more prone to further oxidation resulting in the formation of several nonbulky hydantoin lesions, including 5-guanidinohydantoin (Gh) and spiroiminodihydantoin (Sp) (Steenken et al., 2000; Cadet et al., 2017). These lesions have been detected in mouse tissues with induced bacterial infection but at lower frequency than 8-oxoG (Mangerich et al., 2012). However, unlike 8-oxoG, both Gh and Sp lesions cause significant distortions of DNA duplexes and are potent blocks to RNAPII-mediated transcription elongation (Oh et al., 2020; Jin et al., 2013; Kolbanovskiy et al., 2017; Hailer et al., 2005). Hence, it is possible that the highly mutagenic Sp and Gh lesions are repaired via the TC-BER pathway.
The neurodevelopmental disorder Cockayne syndrome (CS) is characterized by defects in TCR. However, defects in TC-NER cannot explain the onset of CS as the absence of core NER factor XPA does not cause CS (Lans et al., 2019). Since elongating RNAPIIo stalls at a variety of DNA lesions including certain oxidative DNA lesions (Oh et al., 2020) and also at BER intermediates, abasic sites and single-strand breaks (Tornaletti et al., 2006; Kitsera et al., 2011; Menoni et al., 2018), it is possible that defects in TC-BER may underlie the onset of CS. Therefore, a detailed mechanistic understanding of TCR of oxidative DNA damage may help to understand the basis for the onset of CS. Spivak’s group reported TCR of 8-oxoG lesions that required actively transcribing RNAPII along with OGG1, XPA and CSB, a factor regulating TCR with diverse types of lesions, and proposed a model that TCR of oxidized lesions is initiated by BER and resolved by NER (Guo et al., 2013). Live cell imaging studies demonstrated the recruitment of CSB to 8-oxoG lesions in a transcription-dependent but NER-independent fashion. Although the recruitment of OGG1 glycosylase to the site of 8-oxoG is independent of CSB or active transcription, the recruitment of the downstream BER-scaffolding protein XRCC1 does rely on CSB and transcription (Menoni et al., 2018). Their model for TCR of oxidized DNA lesions is based on stalling of RNAPII at BER-generated single-strand breaks (SSB), followed by CSB recognition of the stalled polymerase and recruitment of XRCC1 for coordinating the late steps of BER through its protein-protein interactions.
Based on its ability to remove lesions from single-stranded and bubble DNA and its association with RNAPII (Banerjee et al., 2011) we hypothesized that NEIL2 is a candidate DNA glycosylase for initiation of oxidized lesion repair via the TC-BER pathway. Using an in vitro reconstituted transcription coupled-repair system with purified proteins and a mammalian promoter-based plasmid DNA containing a single base lesion placed in the transcribed or non-transcribed strand, we characterized NEIL2-initiated repair of an oxidized base. We found that NEIL2 can initiate the repair of mutagenic 5-OHU, a cytosine oxidation product, in a transcription-dependent manner when the lesion was placed in the transcribed strand but not the non-transcribed strand, supporting the possibility of a similar mechanism in cells to prevent transcriptional mutagenesis at oxidized base lesions (Banerjee et al., 2011). Consistent with this in vitro biochemical study, increased accumulation of oxidized base damage in transcribed genes in different organs of Neil2-KO mice as they aged further highlights the biological importance of NEIL2 in protecting transcribed regions for long-term genome maintenance (Chakraborty et al., 2015). To provide insight into the mechanism, we have investigated the protein-protein interactions of NEIL2 and found that it interacts with RNAPII, CSB and downstream BER factors, including PNKP, Polb, and Lig IIIa (Chakraborty et al., 2015). Furthermore, CSB, which is essential for TCR and which causes CS when defective, has been shown to physically interact with and stimulate NEIL2’s catalytic activity in a transcription bubble-mimic DNA with a defined oxidized lesion (Aamann et al., 2014). We recently purified a NEIL2 complex from cell nuclear extracts and found its association with RNAPII, CSB, XPG, TFIIH and downstream BER factors, with the association being significantly reduced in XPG-deficient cells (Sarker et al., unpublished data). We have further shown that XPG stimulates NEIL2’s catalytic activity on a bubble DNA containing a defined oxidative lesion (Sarker et al., unpublished data). The association of NEIL2 with RNAPII and TFIIH was significantly reduced following exposure of the cells to 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole (DRB), a transcription inhibitor. The cellular NEIL2 complex contains PNKP but not APE1 or XPA, supporting the specificity of the complex formation (Das et al., 2006; Wiederhold et al., 2004). These unpublished data are consistent with a model that NEIL2 recognizes and incises oxidized lesions within a transcription bubble, generating breaks with 3′-P ends that cause the RNAPIIo to stall, thus masking the repair intermediate. The stalled RNAPII would then be recognized by CSB and XPG (Sarker et al., 2005), which recruit TCR and BER factors to form a dynamic complex poised to repair the lesion on the transcribed strand. The model further proposes that changes in spatio-temporal positioning of the factors within the complex takes place, resulting in backtracking or remodeling of the stalled RNAPIIo, so that the downstream BER proteins get access to repair the lesion and finally transcription is resumed (Fig. 3).
Fig. 3.
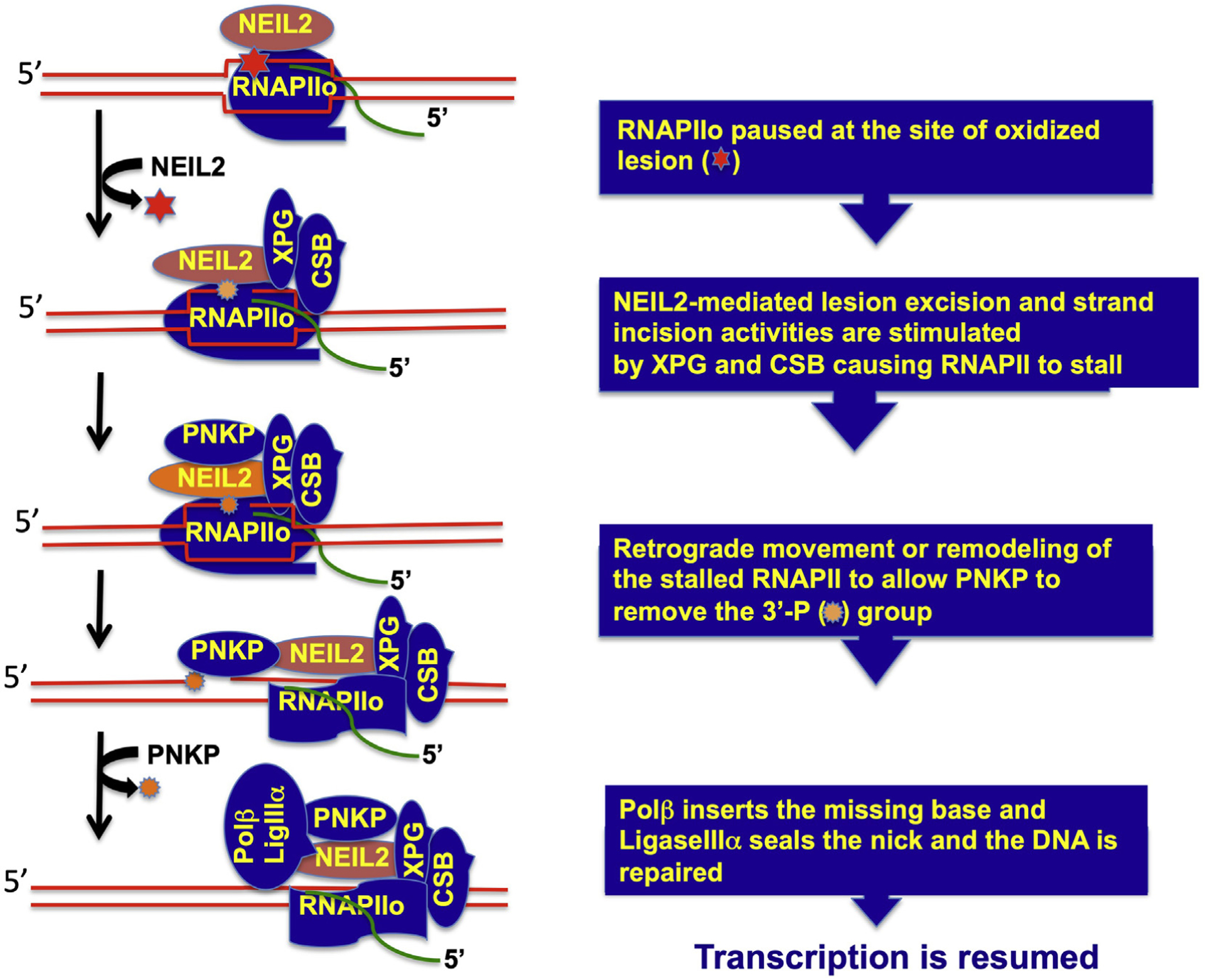
Simplified model for TC-BER.
2.2. Inflammatory responses
Overlapping substrate specificities of multiple DNA glycosylases raises the question of whether they have other functions in the cells besides BER. Recently, it was shown that OGG1 has a distinct role in pro-inflammatory gene expression via modulation of nuclear factor kappa-light chain-enhancer of activated B cells (NF-kB) pathway (Ba and Boldogh, 2018). This is achieved due to binding of OGG1 with high affinity to its cognate 8-oxoG lesion in the guanine rich promoter region of the pro-inflammatory genes followed by assembly of the transcription machinery. When mice are challenged with inflammatory agents, up-regulation of pro-inflammatory genes is significantly reduced in either Ogg1-deficient mice or mice treated with a small molecule inhibitor of OGG1, leading to significantly decreased inflammation (Visnes et al., 2018; Li et al., 2012). These results are consistent with the observation that higher OGG1 levels may favor inflammation. Among different base modifications from oxidative stress following inflammatory stimuli, only 8-oxoG is specifically recognized by OGG1. However, cytosine in GC-rich promoters is also likely to be oxidized and will induce the generation of 5-OHU, which is preferentially recognized by NEIL2. Thus, NEIL2 is a candidate for recognition of oxidized cytosine in promoters, resulting in modulation of transcription. In sharp contrast to Ogg1-null mice, Neil2-deficient mice have increased susceptibility to inflammation following challenge with pro-inflammatory mediators, highlighting the importance of NEIL2 in protecting mammals from the development of inflammation (Chakraborty et al., 2015). However, the mechanistic basis for the induction of inflammation in Neil2-null mice remains unknown. It is possible that oxidative base modifications at gene promoters play a role, such that binding of NEIL2 to the modified bases precludes binding of transcription factors such as NF-kB, thus limiting pro-inflammatory gene expression and hence limiting the inflammatory response.
Many pathological conditions and infection with bacteria and viruses can induce inflammation and cause the accumulation of oxidative DNA damage through induction of ROS. It has recently been reported that Helicobacter pylori infection-induced accumulation of oxidative DNA damage is associated with increased production of pro-inflammatory mediators, and this response is significantly pronounced in Neil2-deficient mouse cells compared to wild type cells (Sayed et al., 2020). Furthermore, Helicobacter pylori infected Neil2-KO mice developed higher inflammation in the stomach, in contrast to Ogg1-null mice that are resistant to inflammation, suggesting that Neil2 can suppress inflammation whereas Ogg1 elicits inflammation (Sayed et al., 2020) in mice. However, the mechanistic details of the interplay between the opposing inflammatory responses of the two DNA glycosylases in the cells remain to be established.
Similar to bacterial infection, viral infection-induced inflammation may also be modulated by OGG1 and NEIL2. Viral infections are associated with neutrophilic infiltration in the lung, which is a major source of ROS at the site of injury (Knaapen et al., 2006). In addition to killing viruses, ROS damage cellular DNA and can induce the inflammatory response, with increased production of pro-inflammatory mediators. OGG1-mediated immune response may play a role in elevated production of pro-inflammatory molecules. These responses can be alleviated by using pharmacological inhibitors that may prevent OGG1 from engaging damaged DNA. It is also possible that activator-mediated enhanced binding of NEIL2 to the damaged base in the pro-inflammatory gene promoter will reduce inflammatory response by reducing the production of inflammatory molecules. An OGG1 inhibitor, TH5487, has recently been developed and demonstrated to selectively prevent OGG1 from binding to the damaged DNA both in vitro and in cells, with a decrease in pro-inflammatory gene expression (Visnes et al., 2018). Furthermore, reduced neutrophil infiltration in mouse lungs following challenge with TNF-alpha demonstrated the therapeutic effectiveness of this inhibitor to various inflammatory diseases. The mechanistic basis for the inflammatory response induced by NEIL2 deficiency and the development of NEIL2 activators to suppress inflammation remain areas for future investigation.
2.3. Active DNA demethylation
DNA methylation is an epigenetic mark that is important for embryonic development. Cytosine at its 5′-position is methylated at approximately 80–90% of CpG sites. BER is important for maintaining cytosine methylation status at the CpG sites and is important for epigenetic gene regulation. Deamination of 5-methyl cytosine (5 mC), predominantly in the context of CpG sites, also has potential for miscoding, leading to transition mutations (Nablel et al., 2012). But how CpG sites are kept under control from the detrimental consequences of mutations while maintaining epigenetic regulation by DNA methylation has been an intense area of investigation for the last few years. It is now clear that TDG is the major DNA glycosylase that plays complex roles in active DNA demethylation via the BER pathway to avoid mutations at the CpG sites (Kohli and Zhang, 2013). Targeted inactivation of TDG causes embryonic lethality and developmental abnormalities in mice associated with promoter hypermethylation (Cortellino et al., 2011; Cortazar et al., 2011). Importantly, knock-in mice with catalytically inactive TDG manifest similar embryonic lethality and persistent hypermethylation, suggesting an active enzymatically-driven process for DNA demethylation and viability. This complex demethylation process is restricted to specific genes and involves oxidation of 5 mC by Ten-eleven translocation (TET) family of dioxygenases. TETs progressively oxidize 5 mC to 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC), followed by TDG-mediated excision of the oxidized product and subsequent downstream BER to restore unmodified cytosine (He et al., 2011; Maiti and Drohat, 2011). TDG is a monofunctional glycosylase that cleaves the N-glycosidic bond between the base and the sugar, resulting in the generation of an abasic (AP) site followed by processing with the AP endonuclease 1 (APE1)-mediated BER sub-pathway to complete repair (Bellacosa and Drohat, 2015). Recently, NEIL glycosylases have been implicated in TET-mediated DNA demethylation by co-operating with TDG. TDG has low turnover due to extremely high affinity to its product AP site. NEIL1 and NEIL2 were found to be recruited by TDG and to stimulate its 5-fC and 5-caC excision activity followed by displacement from the AP site (Schomacher et al., 2016). NEILs then cleave the AP site via their associated AP-lyase activity, thus allowing TDG to overcome its product inhibition and accelerate 5-fC and 5-caC repair (Fig. 4). However, the NEILs themselves showed no excision activity per se of 5-fC and 5-caC from either double or singlestranded DNA. Nonetheless, deficiency of NEIL2 causes increased accumulation of genomic 5-fC and 5-caC and reduced gene expression, consistent with gene-specific promoter CpG methylation in NEIL2 down-regulated cells (Schomacher et al., 2016). Importantly, deficiency of APE1, the enzyme that is capable of displacing TDG from AP sites, causes only marginally increased level of TET-induced 5-fC and 5-caC lesions in the genomic DNA. Together these results strongly suggest a requirement for NEIL1 or NEIL2, but not APE1, in the TET-TDG mediated demethylation process. Notably, NEIL2 was shown to associate with TET and to be recruited to the site of demethylation (Muller et al., 2014), but whether NEIL2 co-ordinates the demethylation cascade to limit TET activity is unclear.
Fig. 4.
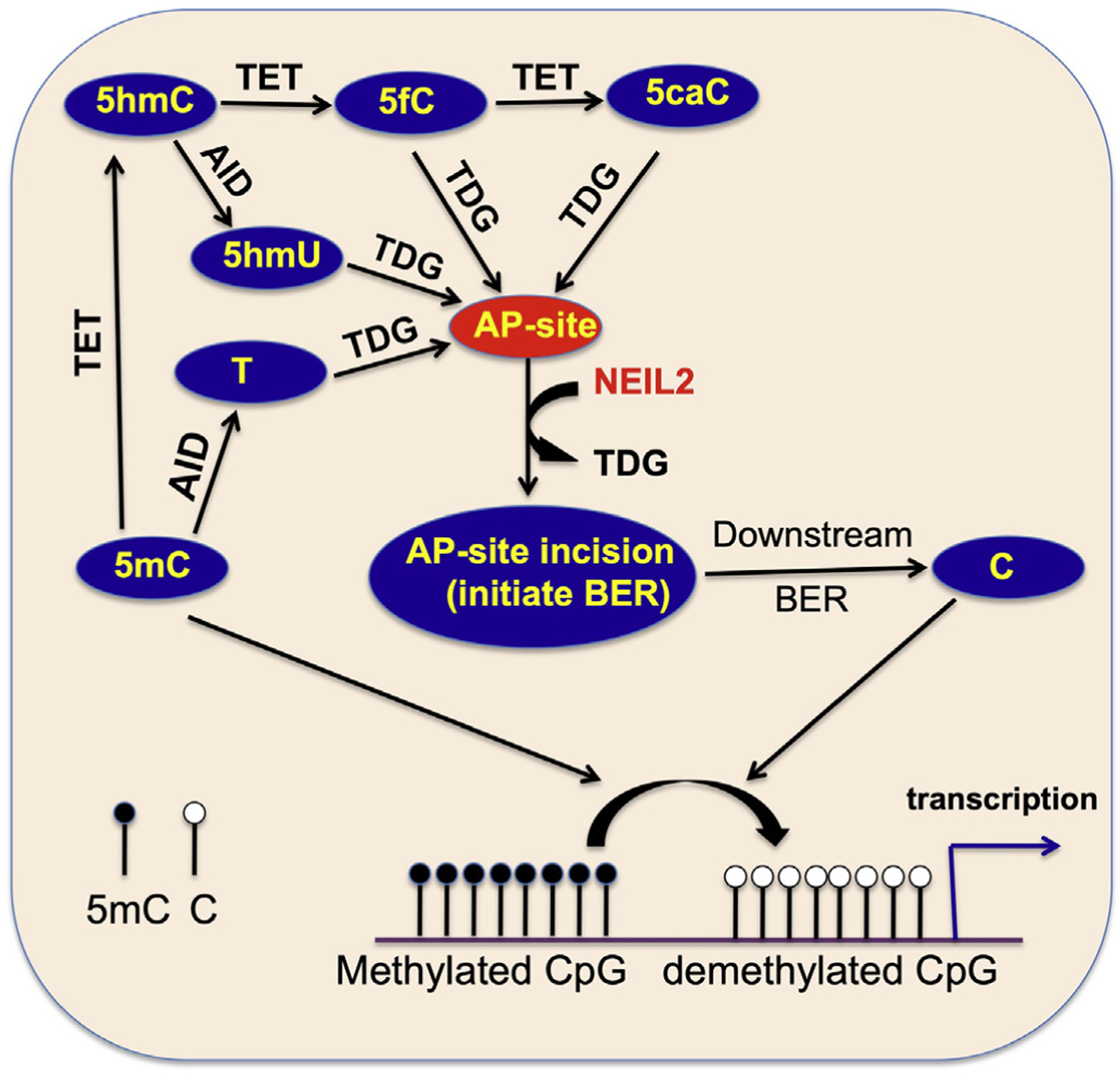
NEIL2 involvement in active DNA demethylation pathway.
NEIL2 may also be involved in processing of AP sites during TET-TDG mediated demethylation specifically in the context of the gene body. Transcribing RNAPII is known to stall at AP sites, but it only transiently pauses at 5-fC and 5-caC, causing a reduced rate of transcription with increased mis-incorporation (Kellinger et al., 2012). A genome wide study showed a correlation between 5-fC sites and RNAPII peaks, presumably reflecting paused sites in the gene body (Raiber et al., 2012). Furthermore, 5-fCs are also enriched at the CpG promoters, which correspond to transcriptionally active genes. Since TDG is actively involved in the removal of 5-fC from CpGs both in the promoters and in gene bodies, it is possible that TDG-NEIL2 cross-talk is necessary for protecting CpGs from hypermethylation to maintain proper methylation status that is necessary for gene activation. In that case, NEIL2 would not only be important for preferential repair of oxidized lesions from transcribed genes but also for processing AP sites during TET-TDG mediated gene activation.
2.4. Aberrant NEIL2 promotes cancer development
BER prevents mutations by eliminating small base modifications, and defects in BER have been associated with various diseases including neurodevelopmental disorders, aging and cancer. By analyzing large datasets from the Catalogue Of Somatic Mutations In Cancer (COSMIC; Forbes et al., 2015), NEIL2 was found among the top 20 DNA repair genes that most frequently displayed copy number loss concordance with down-regulation (Chae et al., 2016). This observation suggests that NEIL2 may play a tumor suppressive role and could potentially be used as a biomarker for genomic instability. We recently have reported the level of NEIL2 in a lung adenocarcinoma cancer tissue microarray and found that NEIL2 protein levels were significantly low in 50% of cancer tissues, with only a few cases in which NEIL2 was higher or comparable to the normal (Sarker at al., 2014). Furthermore, knockdown of NEIL2 leads to a modest increase in spontaneous DNA damage and a more dramatic increase in the accumulation of oxidative damage following exposure of cells to second hand tobacco smoke. Downregulation of NEIL2 causes a 6–7 fold increase in spontaneous mutation frequency (Dey et al., 2012). Together these results suggest that lower NEIL2 levels are associated with lung tumor development through accumulation of oxidative DNA damage. While searching for mutations in the coding region of NEIL2, we found that one specific NEIL2 variant rs8191664 (R257L) was significantly more common in lung and cervical carcinoma patients (Dey et al., 2012; Ye et al., 2020). Further biochemical studies showed that this variant has a poor total BER activity due to its lack of association with downstream repair proteins, suggesting that fully functional NEIL2-mediated repair completion is important for protection against lung carcinogenesis (Dey et al., 2012). The crystal structure of unliganded NEIL2 was reported to have an unusual open conformation distinct from NEIL1 and NEIL3 orthologs, with the structure providing insight into its substrate diversity (Eckenroth et al., 2021; Tsutakawa and Sarker, 2021). Activity analysis of the cancer-associated variants of NEIL2 suggests that they involve structural changes which impact enzymatic activity, with significant implications for BER.
Recently, a large genome wide association study (GWAS) analyzed the lifetime risk of cancer development in carriers of germ-line mutations in the BRCA1 and BRCA2 genes. They found that not all BRCA carriers develop cancer and that the age of disease onset is highly variable, suggesting that some genetic, environmental or other unknown modifying factors may influence the risk of developing breast/ovarian cancer. Their analysis identified a variant of NEIL2 (rs804271) that was strongly associated with increased breast cancer risk in BRCA2 mutation carriers (Osorio et al., 2014). This rs804271 variant is localized within the NEIL2 promoter region and is associated with significant transcriptional up-regulation that correlated with increased protein expression (Benítez-Buelga et al., 2017). The accumulation of DNA single-strand breaks by aberrant NEIL2 over-expression in BRCA2 mutated cells may lead to increased accumulation of DSBs with a resultant increase in genomic instability, thus contributing to increased risk of breast carcinogenesis. Recent studies also demonstrated that elevated NEIL2 levels in breast cancer cells are associated with APOBEC3-mediated mutagenesis and induction of DSBs, likely by perturbing the classical BER pathway (Shen et al., 2020) through outcompeting APE1. This can explain how DNA repair dysregulation contributes to the APOBEC3-mediated mutator phenotype observed in some cancers. A homozygous NEIL2 variant rs804270 located in the 5′-UTR promoter region enriched in cervical carcinoma is associated with reduced mRNA and protein expression (Ye et al., 2020). Inter-individual variations of NEIL2 transcription (up to 63-fold) were observed in a population based study and two variants, ss74800505 and rs8191518, located in the 5′ promoter region close to the transcription start site exist at a relatively high frequency and are associated with significant reduction in NEIL2 expression (Kinslow et al., 2008). Individuals carrying these variants will have reduced NEIL2 levels, with increased likelihood of accumulating genomic damage upon exposure to oxidants. Thus this population group is likely to have increased cancer susceptibility.
Together these results suggest that the expression level of NEIL2 is tightly regulated in the cell to maintain genomic integrity, as either over- or under-expression can cause genomic instability, leading to the development of tumors. Not only NEIL2 but also other DNA glycosylases display similar phenotypes. We recently reported that overexpression of NTHL1 is associated with increased DSBs, replication stress, and micronuclei formation, all of which are hallmarks of genomic instability (Limpose et al., 2018). Of further interest, NTHL1 deficiency due to biallelic germline NTHL1 mutations underlies the mutational process found in multiple malignancies including high breast cancer incidence (Grolleman et al., 2019), consistent with NTHL1 expression also being tightly control.
Why, then, are mice deficient in oxidized base specific DNA glycosylases viable, with no overt phenotype? As described earlier, we found increased accumulation of oxidative DNA damage with age in various organs in Neil2-KO mice. Using embryonic fibroblasts (MEFs) from Neil2 -KO mice, we have reported increased telomere loss and chromosomal aberrations (Chakraborty et al., 2015) both of which are hallmarks of genomic instability. These observations suggest that oxidative damage in the genome alone is not sufficient for cancer development and that a promoting process or impairment of back-up pathways will be necessary. Consistent with this notion, Ogg1−/− (Osterod et al., 2001) mice do not develop tumors despite increased accumulation of highly mutagenic 8-oxoG lesions in their genome. However, deletion of a parallel repair pathway in Ogg1−/− Myh −/− double knockout mice causes increased tumor incidence in the lung, lymphomas and ovary compared to single knockout mice (Xie et al., 2004). Similarly, NEIL1 deficiency alone is not sufficient to induce cancer but Nth1−/− Neil1−/− double knockout mice developed tumors in the lung and liver compared to single knockout mice, suggesting that damage accumulation may underlie the development of cancer (Chan et al., 2009). Crossing of Neil2−/− with other repair-compromised mice such as Brca2−/− mice to compare if NEIL2 loss influences the risk of developing cancer awaits further follow-up study
2.5. NEIL2 in mitochondrial genome maintenance
In addition to their role in energy production, mitochondria are also involved in several other cellular processes including apoptosis, cell cycle regulation and immune responses (Shaughnessy et al., 2014). The mitochondrial genome is the major target for somatic mutation due to the increased accumulation of oxidative DNA damage because of continuous attack by endogenous ROS generated at close proximity. Many lines of evidence indicate that both oxidative stress and the resulting mutations in mitochondrial DNA contribute to various diseases and/or pathologies (Shaughnessy et al., 2014). Unlike the NER pathway, which mainly repairs bulky lesions in the nuclear genome, the BER pathway is important for the maintenance of both nuclear and mitochondrial genomes (Hu et al., 2005). DNA glycosylases play an important role in mitochondrial DNA repair, since targeting DNA glycosylases to mitochondria leads to enhanced repair and increased survival following exposure of cells to oxidative stress (Rachek et al., 2002). At the organismal level, deficiency of NEIL1 glycosylase in mice has been reported to cause a combination of clinical manifestations known as metabolic syndrome in human (Vartanian et al., 2006). This syndrome was attributed to increased ROS-induced damage accumulation and deletions in the mitochondrial genome in the Neil1−/− mice. Since Neil1 initiates repair of lesions from single-stranded or bubble DNA, this loss may impair mitochondrial DNA replication and disruption of energy homeostasis, leading to impaired metabolism.
NEIL1 and NEIL2 localize both in the nucleus and mitochondria. Depletion of NEIL2 causes increased accumulation of oxidized bases in the nuclear and mitochondrial DNA, suggesting that NEIL2 is involved in both the nuclear and mitochondrial genome maintenance (Mandal et al., 2012). As mentioned earlier, NEIL2-KO mice apparently do not show any overt phenotypes, but Neil2 deficiency in frog leads to face and skull deformities (Han et al., 2019). These phenotypes are due to reduced production of cranial neural crest cells (cNCC) that give rise to bones and facial cartilage which are the backbone of face and skull formation in animals. Neural crest cells are a multipotent population that arise from the embryonic ectoderm germ layer and extensively migrate to various locations where they differentiate into different cell types in the embryo. When NEIL1 or NEIL2 is missing, these cells lose their ability to become cNCC, suggesting differentiation defects during embryonic development in the absence of NEIL’s function in those animals. These NCC differentiation defects caused by NEIL2 (or NEIL1) deficiency are due to the inability to protect against oxidative damage particularly in the mitochondrial genome rather than nuclear genome, thus connecting mitochondrial BER with neural cell differentiation. The mitochondrial DNA damage accumulation in NEIL2 deficient neural crest cells triggers the TP53-mediated DNA damage response, causing intrinsic apoptosis and thus impairing cNCC specification. Deficiency in NEIL3, which is not found in mitochondria, had no effect on cranial neural crest cell development. TDG-deficiency also did not affect cranial neural crest cell development, arguing against active DNA demethylation as the cause of the differentiation defect (Han et al., 2019). These results suggest that when stem cells, such as neural crest cells, turn into cNCCs, NEIL2 is important for protecting mitochondrial DNA during embryonic development. The NEIL’s are required for proper craniofacial formation through the development of cranial neural crest cells, and defects in NEIL2 leads to aberrant cranial neural crest cells development in frogs. This defect may underlie congenital craniofacial malformations, which account for one-third of all congenital birth defects in humans (Neben et al., 2016).
3. Future challenges
Work from several laboratories, including ours, has demonstrated the involvement of NEIL2 in multiple cellular processes through protein-protein interactions and recognition and repair of oxidative lesions. NEIL2-mediated preferential repair of oxidative lesions from the transcribed strand can protect cells from the deleterious consequences of transcriptional mutagenesis or recovery of stalled RNAPII to resume transcription by TC-BER. Although TC-BER has been a topic of debate for quite some time, the functional association of NEIL2 with CSB and RNAPII (Aamann et al., 2014; Banerjee et al., 2011; Chakraborty et al., 2015) and the recruitment of CSB and XRCC1 (Menoni et al., 2018) to the site of oxidative DNA damage in a transcription-dependent manner indicate that NEIL2 and its associated protein complexes function in repair of oxidative lesions in the transcribed regions. We have recently isolated a large cellular NEIL2 complex consisting of RNAPII, XPG, CSB, TFIIH and the downstream BER proteins following oxidative stress and shown that formation of the complex depends on active transcription, supporting the idea that NEIL2 mediates a TC-BER process in the cell (Sarker et al., unpubl.). Defects in TC -NER cannot explain the onset of Cockayne syndrome (CS), leaving open other possibilities. Our demonstration of association of the TCR protein CSB with the NEIL2 glycosylase is consistent with the possibility that deficiency in TC-BER contributes to CS phenotypes. However, many questions still remain. Is the mechanism of TC-NER initiation distinct from that for TC-BER, since not all oxidative lesions pose a potent block to RNAPII transcription? Does CSB discriminate TC-NER from TC-BER? Do post-translational modifications of CSB play a role, since a ubiquitination site in CSB has been implicated as specific for oxidative DNA damage repair but not TC-NER (Ranes et al., 2016)? The next obvious question is whether NEIL1 or other DNA glycosylases are also involved in TC-BER. Functional interaction of NEIL1 with CSB and RNAPII imply a role of NEIL1 in repair of oxidative DNA lesions in association with transcription (Muftuoglu et al., 2009), but this needs to be addressed in future studies.
A major question in this field is why organisms need five DNA glycosylases with overlapping substrate specificities. Their diverse subcellular distribution enables us to predict the functional importance. Subcellular fractionation studies demonstrated that NTHL1 is primarily localized to the nucleus (Limpose et al., 2018). However, a significant amount of NEIL2 is present in the cytoplasm. This is consistent with the report that defects in NCC differentiation caused by NEIL deficiency are due to the inability to protect against oxidative damage in the mitochondrial genome rather than the nuclear genome (Han et al., 2019). In fact, co-localization of NEIL2 and PNKP with the mitochondrion-specific protein MT-CO2 and their functional importance in repair of oxidative DNA lesions and SSBs in the mitochondrial genome were documented (Mandal et al., 2012). Whether NEIL’s are the major DNA glycosylases to protect the mitochondrial genome against oxidative damage via NEIL mediated TC-BER remains to be elucidated.
Funding
This work was supported by Grant 26IR-0017 to AHS from the University of California Tobacco Related Disease Research Program (UC-TRDRP), by the NIH/NCI grant P01 CA092584 to Priscilla K. Cooper and R01 NS073976 (to TH); and R01HL145477 (TH and Sanjiv Sur); W81XWH–18–1–0743 (to Sanjiv Sur and TH).
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Aamann MD, Hvitby C, Popuri V, Muftuoglu M, Lemminger L, Skeby CK, Keijzers G, Ahn B, Bjørås M, Bohr VA, Stevnsner T, 2014. Cockayne syndrome group B protein stimulates NEIL2 DNA glycosylase activity. Mech Ageing Dev. Jan 135, 1–14. 10.1016/j.mad.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee D, Mandal SM, Das A, Hegde ML, Das S, Bhakat KK, Boldogh I, Sarker PS, Mitra S, Hazra TK, 2011. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem 286 (8), 6006–6016. 10.1074/jbc.M110.198796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba X, Boldogh I, 2018. 8-Oxoguanine DNA glycosylase 1: beyond repair of the oxidatively modified base lesions. Redox Biol 14, 669–678. 10.1016/j.redox.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellacosa A, Drohat AC, 2015. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. Published online 2015 May 1 DNA Repair (Amst). Aug 32, 33–42. 10.1016/j.dnarep.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benítez-Buelga C, Baquero JM, Vaclova T, Fernández V, Martín P, Inglada-Perez L, Urioste M, Osorio A, Benítez J, 2017. Genetic variation in the NEIL2 DNA glycosylase gene is associated with oxidative DNA damage in BRCA2 mutation carriers, 8 Oncotarget 23 (70), 114626–114636. 10.18632/oncotarget.22638. eCollection 2017 Dec 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J, Douki T, Gasparutto D, Ravanat JL, 2003. Oxidative damage to DNA: formation, measurement and biochemical features. Mutat. Res 531, 5–23. [DOI] [PubMed] [Google Scholar]
- Cadet J, Davies JA, Medeiros MH, Mascio P Di Wagner JR, 2017. Formation and repair of oxidatively generated damage in cellular DNA. Free Radic. Biol. Med 107, 13–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae YK, Anker JF, Carneiro BA, Chandra S, Kaplan J, Kalyan A, Santa-Maria CA, Platanias LC, Giles FJ, 2016. Genomic landscape of DNA repair genes in cancer. Oncotarget 7 (17), 23312–23321. 10.18632/oncotarget.8196. Apr 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Wakamiya M, Venkova-Canova T, Pandita RJ, Aguilera-Aguirre L, Sarker AH, Singh DK, Hosoki K, Wood TG, Sharma G, Cardenas V, Sarkar PS, Sur S, Pandita TK, Boldogh I, Hazra TK, 2015. Neil2-null mice accumulate oxidized DNA bases in the transcriptionally active sequences of the genome and are susceptible to innate inflammation, 24636–48 J. Biol. Chem Oct 9 290 (41). 10.1074/jbc.M115.658146. Epub 2015, Aug 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MK, Ocampo-Hafalla MT, Vartanian V, Jaruga P, Kirkali G, Koenig KL, Brown S, Lloyd RS, Dizdaroglu M, Teebor GW, 2009. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. Jul 4 DNA Repair 8 (7), 786–794. 10.1016/j.dnarep.2009.03.001. Epub 2009, Apr 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlet-Berguerand N, Feuerhahn S, Kong SE, Ziserman H, Conaway JW, Conaway R, Egly JM, 2006. RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J 25, 5481–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortellino S, et al. , 2011. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazar D, et al. , 2011. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470, 419–423. [DOI] [PubMed] [Google Scholar]
- Das D, et al. , 2006. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: evidence for a repair complex in human cells. Dec 9 DNA Repair 5 (12), 1439–1448. 10.1016/j.dnarep.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey S, Maiti AK, Hegde ML, Hegde PM, Boldogh I, Sarkar PS, Abdel-Rahman SZ, Sarker AH, Hang B, Xie J, Tomkinson AE, Zhou M, Shen B, Wang G, Wu C, Yu D, Lin D, Cardenas V, Hazra TK, 2012. Increased risk of lung cancer associated with a functionally impaired polymorphic variant of the human DNA glycosylase NEIL2. DNA Repair 11 (6), 570–578. 10.1016/j.dnarep.2012.03.005. June 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bont R, van Larebeke N, 2004. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis, May 19 (3), 169–185. 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- Dou H, Mitra S, Hazra TK, 2003. Repair of Oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem 12 (50), 49679–49684. 10.1074/jbc.M308658200, 278. [DOI] [PubMed] [Google Scholar]
- Eckenroth BE, Cao V, Averill AM, Dragon JA, Doublié S, 2021. Unique structural features of mammalian NEIL2 DNA glycosylase prime its activity for diverse DNA substrates and environments. Jan 7 Structure 29 (1), 29–42. 10.1016/j.str.2020.08.001. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, et al. , 2015. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res 43 10.1093/nar/gku1075. Database issue, D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen LH, Svejstrup JQ, 2018. The cellular response to transcription-blocking DNA damage. Trends in Biochemical Sciences, May 43 (5), 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grolleman JE, et al. , 2019. Mutational signature analysis reveals NTHL1 deficiency to cause a multi-tumor phenotype, 35 Canc. Cell 11 (2), 256–266. 10.1016/j.ccell.2018.12.011. e5. [DOI] [PubMed] [Google Scholar]
- Guo J, Hanawalt PC, Spivak G, 2013. Comet-FISH with strand-specific probes reveals transcription-coupled repair of 8-oxoGuanine in human cells. Nucleic Acids Res 41, 7700–7712. 10.1093/nar/gkt524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailer KM, Slade PG, Martin BD, Rosenquist TA, Sugden KD, 2005. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair 4 (1), 41–50. 10.1016/j.dnarep.2004.07.006. Jan 2. [DOI] [PubMed] [Google Scholar]
- Han D, Schomacher L, Schüle KM, Mallick M, Musheev MU, Karaulanov E, Krebs L, von Seggern A, Niehrs C, 2019. NEIL1 and NEIL2 DNA glycosylases protect neural crest development against mitochondrial oxidative stress. Elife. Sep 30 (8), e49044. 10.7554/eLife.49044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G, 2008. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol, Dec 9 (12), 958–970. 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra S, Izumi T, 2002a. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem Aug 23 277 (34), 30417–30420. 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S, 2002b. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Mar 19 Proc. Natl. Acad. Sci. U.S.A 99 (6), 3523–3528. 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra TK, Mitra S, 2006. Purification and characterization of NEIL1 and NEIL2, members of a distinct family of mammalian DNA glycosylases for repair of oxidized bases. Methods Enzymol. 408, 33–48. 10.1016/S0076-6879(06)08003-7. [DOI] [PubMed] [Google Scholar]
- He YF, et al. , 2011. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Hegde PM, Bellot LJ, Mandal SM, Hazra TK, Li GM, Boldogh I, Tomkinson AE, Mitra S, 2013. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Aug 13 Proc. Natl. Acad. Sci. U.S.A 110 (33), E3090e–E3099. 10.1073/pnas.1304231110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH, 2001. Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374. [DOI] [PubMed] [Google Scholar]
- Hu J, de Souza-Pinto NJ, Haraguchi K, Hogue BA, Jaruga P, Greenberg MM, Dizdaroglu M, Bohr VA, 2005. Repair of formamidopyrimidines in DNA involves different glycosylases:role of the OGG1, NTH1 and NEIL1 enzymes. J. Biol. Chem 280, 40544–40551. 10.1074/jbc.M508772200. Dec 9. [DOI] [PubMed] [Google Scholar]
- Jin Q, Fleming AM, Ding Y, Burrows CJ, White HS, 2013. Structural destabilization of DNA duplexes containing single-base lesions investigated by nano-pore measurement. Biochemistry 52 (45), 7870–7877. 10.1021/bi4009825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellinger MW, Song CX, Chong J, Lu XY, He C, Wang D, 2012. 5-formylcytosine and 5- carboxylcytosine reduce the rate and substrate specificity of RNA Polymerase II transcription. Nat. Struct. Mol. Biol 19, 831–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinslow CJ, El-Zein RA, Hill CE, Wickliffe JK, Abdel-Rahman SZ, 2008. Single nucleotide polymorphisms 5’ upstream the coding region of the NEIL2 gene influence gene transcription levels and alter levels of genetic damage. Genes Chromosomes Cancer. Nov 47 (11), 923–932. 10.1002/gcc.20594. [DOI] [PubMed] [Google Scholar]
- Kitsera N, Stathis D, Luhnsdorf B, Muller H, Carell T, Epe B, Khobta A, 2011. 8-Oxo-7,8 dihydroguanine in DNA does not constitute a barrier to transcription, but is converted into transcription-blocking damage by OGG1. Nucleic Acids Res 39, 5926–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaapen Ad M., Gungor N, Schins F, A RP, Borm PJ, Van Schooten FJ, 2006. Neutrophils and respiratory tract DNA damage and mutagenesis: a review. Mutagenesis 21 (4), 225–236. [DOI] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y, 2013. TDG and the dynamics of DNA demethylation. Nature 502, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbanovskiy M, Chowdhury MA, Nadkarni A, Broyde S, Geacintov NE, Scicchitano DA, Shafirovich V, 2017. The non-bulky DNA lesions spiroiminodihydantoin and 5-guanidinohydantoin significantly block human RNA polymerase II elongation in vitro. Biochemistry 56 (24), 3008–3018. 10.1021/acs.biochem.7b00295. June 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraoka I, Endou M, Yamaguchi Y, Wada T, Handa H, Tanaka K, 2003. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J Biol Chem,Feb 28 278 (9), 7294–7299. 10.1074/jbc.M208102200. Epub 2002 Dec 3. [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Suzuki K, Ito S, Hayashida M, Kwei JS, Ikegami T, Handa H, Nakabeppu Y, Tanaka K, 2007. RNA polymerase II bypasses 8-oxoguanine in the presence of transcription elongation factor TFIIS. Jun 1 DNA Repair 6 (6), 841–851. 10.1016/j.dnarep.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Lans H, Hoeijmakers JH, Vermeulen W, Marteijin JA, 2019. The DNA damage response to transcription stress. Nat Rev Mol Cell Biol, Dec 20 (12), 766–784. 10.1093/nar/gkz977. [DOI] [PubMed] [Google Scholar]
- Li G, Yuan K, Yan C, Fox J 3rd, Gaid M, Breitwieser W, Bansal AK, Zen H, Gao H, Wu M, 2012. 8-Oxoguanine-DNA Glycosylase 1 deficiency modifies allergic airway inflammation by reulating STAT6 and IL-4 in cells and in mice. Free Radic. Biol. Med 52 (2), 392–401. 10.1016/j.free-radbiomed.2011.10.490. Jan 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpose KL, Trego KS, Li Z, Leung SW, Sarker AH, Shah JA, Ramalingam SS, Werner EM, Dynan WS, Cooper PK, Corbett AH, Doetsch PW, 2018. Overexpression of the base excision repair NTHL1 glycosylase causes genomic instability and early cellular hallmarks of cancer. May 18 Nucleic Acids Res 46 (9), 4515–4532. 10.1093/nar/gky162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, 1993. Instability and decay of the primary structure of DNA. Nature 362, 709–715. [DOI] [PubMed] [Google Scholar]
- Maiti A, Drohat AC, 2011. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem 286, 35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal SM, Hegde ML, Chatterjee A, Hegde PM, Szczesny S, Banerjee D, Boldogh I, Gao R, Falkenberg M, Gustafsson CM, Sarkar PS, Hazra TK, 2012. Role of human DNA glycosylase nei-like 2 (NEIL2) and single strand break repair protein polynucleotide kinase 3′-phosphatase in maintenance of mitochondrial genome. Published online 2011 Nov 30 J Biol Chem, Jan 20 287 (4), 2819–2829. 10.1074/jbc.M111.272179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangerich A, et al. , 2012. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. U.S.A 109, E1820–E1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menoni H, Wienholz F, Theil AF, Janssens RC, Lans H, Campalans A, Radicella JP, Marteijn JA, Vermeulen W, 2018. The transcription-coupled DNA repair-initiating protein CSB promotes XRCC1 recruitment to oxidative DNA damage. Nucleic Acids Res 46, 7747–7756. 10.1093/nar/gky579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M, de Souza-Pinto NC, Dogan A, Aamann M, Stevnsner R, Rybanska I, Kirkali G, Dizdaroglu M, Bohr VA, 2009. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J. Biol. Chem 284 (14), 9270–9279. 10.1074/jbc.M807006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Bauer C, Siegl M, Rottach A, Leonhardt H, 2014. TET-mediated oxidation of methylcytosine causes TDG or NEIL glycosylase dependent gene reactivation. Nucleic Acids Res 42, 8592–8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins EA, Rodriguez AA, Bradley NP, Eichman BF, 2019. Emerging Roles of DNA glycosylases and the base excision repair pathway. Trends Biochem. Sci 44(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel CS, Manning SA, Kohli RM, 2012. The curious chemical biology of cytosine: deamination, Methylation, and Oxidation as modulators of genomic potential. ACS Chem. Biol 7, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neben CL, Roberts RR, Dipple KM, Merrill AE, Klein OD, 2016. Modeling craniofacial and skeletal congenital birth defects to advance therapies. Hum. Mol. Genet 25 (R2), R86–R93. 10.1093/hmg/ddw171, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Fleming AM, Xu J, Chong J, Burrows CJ, Wang D, 2020. RNAP polymerase II stalls on oxidative DNA damage via a torsion-latch mechanism involving lone pair- and CH- interactions. Proc. Natl. Acad. Sci. U. S. A 117, 9338–9348. April 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio A, Milne RL, Kuchenbaecker K, Vaclova T, Benitez J, 2014. DNA glycosylases involved in base excision repair may Be associated with cancer risk in BRCA1 and BRCA2 mutation carriers. PLoS Genet. 10 (April 4), e1004256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterod M, Hollenbach S, Hengstler JG, Barnes DE, Lindahl T, Epe B, 2001. Age-related and tissue specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis 22, 1459–1463. [DOI] [PubMed] [Google Scholar]
- Rachek LI, Grishko VI, Musiyenko SI, Kelley MR, LeDoux SP, Wilson GL, 2002. Conditional targeting of the DNA repair enzyme hOGG1 into mitochondria, 277 J Biol Chem Nov 22 (47), 44932–44937. 10.1074/jbc.M208770200. [DOI] [PubMed] [Google Scholar]
- Raiber EA, Beraldi D, Ficz G, Burgess HE, Branco MR, Murat P, Oxley D, Booth MJ, Reik W, Balasubramaniam S, 2012. Genome-wide distribution of 5-formylcytosine in embryonic stem cells is associated with transcription and depends on thymine DNA glycosylase, 13 Genome Biol 17 (8), R69. 10.1186/gb-2012-13-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranes M, Boeing S, Wang Y, Wienholz F, Menoni H, Walker J, Encheva V, Chakravarty P, Mari PO, Stewart A, Giglia-Mari G, Snijders AP, Vermeulen W, Svejstrup JQ, 2016. A ubiquitylation site in Cockayne syndrome B required for repair of oxidative DNA damage, but not for transcription-coupled nucleotide excision repair, 44 Nucleic Acids Res 20 (11), 5246–5255. 10.1093/nar/gkw216. Epub 2016 Apr 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker AH, Chatterjee A, Willams M, Lin S, Havel C, Jacob P, Boldogh I, Hazra TK, Talbot P, Hang B, 2014. NEIL2 Protects against oxidative DNA damage induced by sidestream smoke in human cells. PloS One 9 (3), e90261. 10.1371/journal.pone.0090261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, Cooper PK, 2005. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol Cell 20, 187–198. [DOI] [PubMed] [Google Scholar]
- Saxowsky TT, Meadows KL, Klungland A, Doetsch PW, 2008. 8-oxoguanine-mediated transcriptional mutagenesis causes ras activation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A 105 (48), 18877–18882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed IM, Sahan AZ, Venkova T, Chakraborty A, Mukhopadhyan D, Bimczok D, Beswick EJ, Reyes VE, Pinchuk I, Sahoo D, Ghosh P, Hazra TK, Das S, 2020. Helicobacter pylori infection down-regulates the DNA glycosylase NEIL2, resulting in increased genome damage and inflammation in gastric epithelial cells, 2020 Jun 9:jbc.RA119 J. Biol. Chem, 009981. 10.1074/jbc.RA119.009981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomacher L, Han D, Musheev MU, Arab K, Kienhofer S, Seggern AV, Niehrs C, 2016. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat. Struct. Mol. Biol 23 (2), 116–124. 10.1038/nsmb.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, Van Houten B, Mostoslavsky R, Bultman SJ, Baccarelli AA, Begley TJ, Sobol RW, Hirschey MD, Ideker T, Santos JH, Copeland WC, Tice RR, Balshaw DM, Tyson FL, 2014. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect. Dec 122 (12), 1271–1278. 10.1289/ehp.1408418. Epub 2014 Aug 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Chapman JH, Custance MF, Tricola GM, Jones CE, Furano AV, 2020. Perturbation of base excision repair sensitizes breast cancer cells to APOBEC3 deaminase-mediated mutations. Jan 6 Elife 9, e51605. 10.7554/eLife.51605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenken S, Jovanovic SV, Bietti M, Bernhard K, 2000. The trap depth (in DNA) of 8-oxoG-7,8-dihydro-22’deoxyguanosine as derived from electron-transfer equilibria in aqueous solution. J. Am. Chem. Soc 122, 2373–2374. [Google Scholar]
- Tomkinson AE, Sallmyr A, 2013. Structure and function of the DNA ligases encoded by the mammalian LIG3 gene. Dec 1 Gene 531 (2), 150–157. 10.1016/j.gene.2013.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S, Maeda LS, Hanawalt PC, 2006. Transcription arrest at an abasic site in the transcribed strand of template DNA. Chem. Res. Toxicol 19, 1215–1220. [DOI] [PubMed] [Google Scholar]
- Tsutakawa SE, Sarker AH, 2021. Breaking the rules: protein sculpting in NEIL2 regulation. Structure 29 (1), 1–2. 10.1016/j.str.2020.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, Ballinger SW, Corless CL, McCullough AK, Lloyd RS, 2006. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase, 2006 Feb 7 Proc. Natl. Acad. Sci. U. S. A 103 (6), 1864–1869. 10.1073/pnas.0507444103. Epub 2006 Jan 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij WP, Hoeijmakers JH, Pothof J, 2014. Aging: not all DNA damage is equal. Curr. Opin. Genet. Dev 26, 124–130. [DOI] [PubMed] [Google Scholar]
- Visnes T, et al. , 2018. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 362, 834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SS, Murphy DL, Sweasy JB, 2012. Base excision repair and cancer. Dec 31 Canc. Lett 327 (1–2), 73–89. 10.1016/j.canlet.2011.12.038. Epub 2012 Jan 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederhold L, et al. , 2004. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell 15 (2), 209–220. 10.1016/j.molcel.2004.06.003. Jul 23. [DOI] [PubMed] [Google Scholar]
- Xie Y, Yang H, Cunanan C, Okamoto K, Shibata D, Pan J, Barnes DE, Lindahl T, Mcllhatton M, Fishel R, Miller JH, 2004. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Canc. Res 64 (9), 3096–3102. 10.1158/0008-5472.can-03-3834. May 1. [DOI] [PubMed] [Google Scholar]
- Ye F, Liu J, Wang H, chen X, cheng Q, chen H, 2020. Cervical carcinoma risk associate with genetic polymorphisms of NEIL2 gene in Chinese population and its significance as predictive biomarker. Sci. Rep 10, 5136. 10.1038/s41598-020-62040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Chan J, Lambelé M, Yusufzai T, Stumpff J, Opresko PL, Thali M, Wallace SS, 2017. NEIL3 repairs telomere damage during S phase to secure chromosome segregation at mitosis. Cell Rep Aug 29 20 (9), 2044–2056. 10.1016/j.celrep.2017.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]