

Summary
Cell isolation from complex mixtures is a key step in many clinical and research applications, but standard isolation methods may affect the cell’s biology and are difficult to reverse. Here, we present a method to isolate and restore cells to their native state using an aptamer that binds epidermal growth factor receptor (EGFR+)cells and a complementary antisense oligonucleotide to reverse binding.
For complete details on the use and execution of this protocol, please refer to Gray et al.1
Subject areas: Cell Biology, Cell isolation, Molecular/Chemical Probes, Biotechnology and bioengineering
Graphical abstract
Highlights
-
•
High-purity isolation of specific cell types and viable cells
-
•
Cells are returned to their native state post-purification
-
•
Adaptable for use with different aptamer-antidote pairs
-
•
Adaptable for use with blood, homogenized tissue, or other complex cell mixtures
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Cell isolation from complex mixtures is a key step in many clinical and research applications, but standard isolation methods may affect the cell’s biology and are difficult to reverse. Here, we present a method to isolate and restore cells to their native state using an aptamer that binds epidermal growth factor receptor (EGFR+) cells and a complementary antisense oligonucleotide to reverse binding.
Before you begin
Aptamers are small, single-stranded nucleic acid ligands that bind targets with high affinity and specificity (2,3 and reviewed in4,5,6). As RNA or DNA ligands, aptamers function by folding into unique three-dimensional structures that confer target binding.7,8 Aptamers are selected through a process known as Systematic Evolution of Ligands by EXponential enrichment (SELEX), whereby a randomized RNA or DNA library is repetitively bound to a target of interest (often a protein or cell type) until target-specific aptamers are identified.2,9,10,11,12 Additionally, as aptamers are single-stranded oligonucleotides, they can be reversed using oligonucleotide ‘antidotes’ complementary to a portion of the aptamer sequence.9,13,14 The antidote anneals to its complementary region on the aptamer, unfolding the aptamer and disrupting its function.
A number of different aptamers are reported in the literature to bind to cell surface ligands (15reviewed in10), and there are aptamer databases such as the Apta-Index™ maintained by Aptagen™.16 However, reported aptamers should be evaluated to ensure that they specifically bind to target cell types as subsequent studies have shown that not all reported aptamers reproducibly bind their cellular target.17 Therefore, before an aptamer can be effectively applied to isolate specific cell types from blood or other complex cell suspensions, aptamer binding should be validated on a cell surface target. An aptamer with true target specificity will bind in a sequence (and structure)-dependent manner which can be evaluated by including a control, non-targeted aptamer in all tests to ensure that the aptamer binding is not merely a nonspecific interaction; a specific aptamer will distinctly bind its target when compared to a control aptamer. Care should also be taken to ensure that aptamer binding is not merely a non-specific charge-based interaction. This can be evaluated by incubating cells with a non-specific negatively charged blocking agent, such as competitor DNA in the form of salmon sperm DNA (ssDNA), to reduce any charge-based binding. In the presence of competitor DNA, non-specific charge-based binding will be abrogated, and any remaining binding interactions will be due solely to the specific aptamer sequence and structure.
The below protocol for an “aptamer cell staining assay” can be used to verify aptamer specificity and includes the use of a non-targeting control aptamer as well as the non-specific DNA competitor, ssDNA, to block non-specific charge interactions. Before testing, aptamers should be synthesized (or purchased) with a biotin tag using the same sugar chemistry previously reported for that particular aptamer. An aptamer reported as DNA should only be made as DNA, and a modified RNA aptamer should be made using the exact same 2′ sugar modifications previously reported. Unmodified (non-2′ sugar modified) RNA aptamers are not recommended for cell purification as they are not stable enough to function well under the assay conditions. Additionally, before using aptamers for cell purification, antidotes complementary to a portion of the aptamer sequence must be designed and screened for their ability to reverse aptamer binding to its target receptor on cells as described in the “antidote screen” section below.
Institutional permissions
Human whole blood was collected from healthy volunteers according to a protocol approved by the Institutional Review Board of Duke University Medical Center.
Preparation of medias and buffers
Timing: 30 min–1 h
Aptamer-based Magnetic Activated Cell Sorting (aptamer-MACS) is adaptable to a wide range of cell types and corresponding cell surface receptor targeting aptamers. For best results, more sensitive cell types should be isolated using wash and blocking buffers based on the target cell’s preferred media whereas more robust cell types can be isolated using a phosphate buffered saline (PBS)-based wash and blocking buffers.
-
1.
Before performing the assays described below, prepare stock of 10× SB1T buffer (see Table 1) and order or synthesize biotin-modified cell surface receptor targeting and nontargeting control aptamers.
Note: Aliquot and store at −20°C for up to 1 year.
-
2.
Prepare stocks of PBS+ or Media+ by adding bovine serum albumin (BSA) to either PBS or media at a final concentration of 1% (see Table 2). Buffers should be kept at 4°C and handled with aseptic technique.
-
3.Prepare a stock solution of 10 mg/mL salmon sperm DNA (ssDNA):
-
a.Weigh out 100 mg powdered ssDNA.
-
b.Resuspend powdered ssDNA in 10 mL RNase and DNase-free water.
-
c.Vortex the solution until no particles are visible.
-
d.Filter the solution through a 0.22 μm filter.
-
a.
Note: Aliquot and store at −20°C for up to 1 year.
-
4.Prepare Blocking PBS+ or Blocking Media+:
-
a.For Blocking PBS+:
-
i.Add BSA to PBS to a final concentration of 1%.
-
ii.Add ssDNA from the 10 mg/mL aliquots prepared in step 1 to a final concentration of 1 mg/mL (see Table 2).
-
i.
-
b.For Blocking Media+:
-
i.Add BSA to a serum-free formulation of the recommended media for targeted cell type, to a final concentration of 1%.
-
ii.Add ssDNA from the 10 mg/mL aliquots prepared in step 1 to a final concentration of 1 mg/mL (see Table 2).
-
i.
-
a.
Note: Blocking PBS+ and Blocking Media+ should both be kept at 4°C and handled under standard aseptic conditions. They should be prepared fresh for each experiment.
Table 1.
10× SB1T buffer
| Reagent | Final concentration (mM) | Volume to add |
|---|---|---|
| HEPES (1 M) | 400 | 400 μL |
| NaCl (5 M) | 1250 | 250 μL |
| KCl (1 M) | 50 | 50 μL |
| MgCl (1 M) | 10 | 10 μL |
| CaCl (1 M) | 10 | 10 μL |
| Tween 20 | 0.5% total volume | 50 μL |
| ddH2O | N/A | 9.23 mL |
| Total | N/A | 10 mL |
Table 2.
Cell isolation buffers
| Buffer Name | Buffer composition |
|---|---|
| Media+ | Preferred media for target cell type + 1% BSA |
| Blocking Media+ | Preferred media for target cell type + 1% BSA + 1 mg/mL ssDNA |
| PBS+ | PBS with calcium and magnesium + 1% BSA |
| Blocking PBS+ | PBS with calcium and magnesium + 1% BSA + 1 mg/mL ssDNA |
Aptamer cell staining assay
Many aptamers have been selected on substrates other than cells (e.g., purified proteins) or on a different cell type than the intended cell target. A flow cytometry assay using the target cell type or a representative cell line expressing the surface biomarker of interest can be an effective tool to determine whether the selected aptamer will be an effective cell separation ligand for this bead-based method. To verify that the aptamer does not nonspecifically bind cells, a cell line that does not express the target receptor should be tested as a negative control. As an additional control, it is important to use a nontargeting control aptamer such as a nonbinding mutant version of the targeting aptamer, scrambled version of the targeting aptamer or control oligonucleotide known not to bind cells such as the C36 control oligonucleotide.18
-
5.At least 2 different cell lines should be chosen to verify specific aptamer binding.
-
a.A positive control cell line should express the cell surface target of interest.
-
b.A negative control cell line that does not express the target.
-
a.
-
6.
Grow at least 500,000 cells of each cell type under appropriate conditions.
-
7.Calculate the amounts of each reagent needed to prepare aptamer cell staining solutions so that 100 μL final volume is made with a final aptamer concentration of 250 nM:
-
a.Determine the amount of aptamer needed
-
b.Calculate the volume of PBS with Ca2+ and Mg2+ (PBS+/+) needed to dilute the aptamer at least 2-fold.
-
c.Determine the amount of streptavidin dye needed to bind the biotinylated aptamer at a 2:1 M ratio of biotinylated aptamer to streptavidin dye.
-
d.Calculate amount of 10 mg/mL ssDNA stock to add to make the final concentration 1 mg/mL ssDNA.
-
e.Finally, determine the volume of PBS+ or Media+ needed to reach the final staining volume of 100 μL. For an example, see Table 3.
-
a.
-
8.For adherent cells:
-
a.Aspirate media and wash 3 times with PBS to remove residual media.
-
b.Detach cells using normal trypsinization conditions recommended for the cell type.
-
c.When cells are fully detached, quench the trypsin by adding an equal volume of complete cell growth media.
-
a.
Alternatives: If trypsin would interfere with the binding of aptamers to receptors, alternative methods such as incubation with EDTA can be used.
-
9.Resuspend and count the suspension or trypsinized adherent cells:
-
a.Centrifuge cells at 300 × g for 5 min at 4°C
-
b.Remove supernatant and resuspend in Blocking PBS+ or Blocking Media+.
-
c.Count cells using a hemocytometer or cell counter.
-
a.
-
10.
For each cell type, make 3 aliquots of 150,000 cells in 200 μL each. Keep on ice. For each cell type, these three aliquots will become the unstained cells control, cells stained with the nontargeting control aptamer, and cells stained with the cell targeting aptamer.
-
11.Denature aptamer by heating:
-
a.Dilute both the targeting and control aptamers at least 2-fold in PBS+/+.
-
b.Vortex each aptamer sample thoroughly.
-
c.Spin the aptamer samples down using a benchtop microcentrifuge.
-
d.Incubate both samples at 65°C for 5 min.
-
e.Allow the samples to equilibrate to room temperature for 5 min (Table 3, Solution 1).
-
a.
-
12.Prepare aptamer-dyes:
-
a.Combine biotinylated targeting and control aptamers with a streptavidin-fluorescent dye, each at a 2:1 M ratio of aptamer to streptavidin dye.
-
b.Vortex for at least 15 s then spin down using a benchtop microcentrifuge.
-
c.Incubate for 20 min in the dark at room temperature. (Table 3, Solution 2).
-
d.Add ssDNA to the aptamer-dye mixtures to a final concentration of 1 mg/mL.
-
e.Complete the aptamer-dye solution by bringing the staining solution to a final volume of 100 μL by adding PBS+ or Media+.
-
f.Pipette up and down to mix, then incubate in the dark at room temperature for 10 min (Table 3, Solution 3).
-
a.
-
13.Stain cells with aptamer-dye solution:
-
a.Pellet cell aliquots by centrifuging at 300 × g at 4°C for 5 min.
-
b.Remove supernatant gently by aspirating with a pipette.
-
c.Add appropriate aptamer staining solutions to cells and incubate at 4°C for 30 min. Protect from light.
-
a.
-
14.
Following incubation, centrifuge cells at 300 × g at 4°C for 5 min to pellet. Then remove supernatant.
-
15.Wash the cells, performing all steps at 4°C:
-
a.Resuspend cell pellet in 300 μL PBS+ or Media+.
-
b.Centrifuge cells at 300 × g for 5 min to pellet cells again.
-
c.Repeat steps a. and b. two times.
-
d.Resuspend each sample in 150 μL PBS+.
-
a.
-
16.
Measure cell fluorescence using a flow cytometer, making sure to collect data in the channel that most closely matches the emission peak of the dye and gating on single, live cells (Figure 1). If staining and washing have been performed correctly, cells incubated with the nontargeting control aptamer should have low fluorescence on all cell types, with levels similar to the cells alone (see Figure 2 for an example). A good candidate for aptamer MACS will result in high fluorescence of the target cell types expressing the receptor of interest. See Figure 2A for an example of high-quality staining with the E07 EGFR-targeting aptamer19,20 vs. the C36 nontargeting control aptamer/oligo. A good candidate for aptamer MACS will also have low staining on nontarget cells that do not express the receptor of interest – similar to the nontargeting control aptamer/oligo (see Figure 2B for an example).
CRITICAL: PBS+ or Media+ must contain biotin to quench the remaining free biotin binding sites in the streptavidin dye. Although PBS does not contain biotin, some media formulations already contain biotin. If using PBS or a media formulation that does not include biotin, add free biotin to the PBS+ or Media + to a concentration of 250 nM.
Note: In general, aptamer concentrations in this assay can range from as low as 50 nM to 1 μM, although at high concentrations, non-specific cell binding interactions can emerge, making the non-specific control aptamer treatment an even more important control.17 If the initial aptamer concentration doesn’t work, consider performing a concentration curve experiment by selecting at least 5 concentrations ranging from 50 nM to 1 μM and assessing an effective staining concentration by flow cytometry analysis. Mean fluorescence of events in the gate for single cells can be plotted against staining concentration to assess what concentration of aptamer will yield strong staining without oversaturating the cells.
Table 3.
Example 250 nM aptamer staining solution
| Solution 1 | ||
|---|---|---|
| Reagent (concentration) | Final concentration | Volume to add |
| Biotinylated aptamer (10 μM) | 250 nM | 2.5 μL |
| PBS +/+ | N/A | 2.5 μL |
| 1. Denature aptamer structure by heating for 5 min at 65°C. 2. Allow to fold by cooling at room temperature for 3 min. 3. Add in the reagent below to make Solution 2. | ||
| Solution 2 | ||
|---|---|---|
| Reagent (concentration) | Final Concentration | Volume to add |
| Streptavidin dye (5 μM) | 125 nM | 2.5 μL (0.5 M equivalent compared to aptamer) |
| 1. After adding the streptavidin dye, incubate in the dark at room temperature for 20 min. 2. Add in the reagents listed below to make the final aptamer staining solution, Solution 3 (total volume of Solutions 1 + 2 + 3 = 100 μL). | ||
| Solution 3 | ||
|---|---|---|
| Reagent (concentration) | Final Concentration | Volume to add |
| ssDNA (10 mg/mL) | 1 mg/mL | 10 μL |
| PBS+ or Media+ | N/A | 82.5 μL |
| 1. After adding ssDNA and PBS+ or Media+, incubate in the dark at room temperature for 10 min (this ensures that any remaining biotin binding sites on the streptavidin dye are quenched). | ||
Figure 1.

Example gating on A431 cells to exclude debris and clumped cells from analysis
(A) Example of how to draw a gate around cells and not debris, based on appearance on a side scatter area (SSC-A) vs. forward scatter area (FSC-A) plot.
(B) After making the gate for cells, make a second gate as a subset of the ‘cells’ gate to include only singlets. This can be done by plotting events in the ‘cells’ subgroup onto a SSC-A vs. side scatter height (SSC-H) plot and then drawing a gate around the cluster of events that most closely matches the slope of the ‘1:1 line’ where SSC-A and SSC-H are close to equal. Singlet cells will be parallel with the 1:1 line, with smaller clusters outside the main cluster representing doublets or larger cell clusters. To gate these out, create a trapezoidal gate around the population of single cells but excluding outliers.
(C) An example of gating data from single cells on viability, as described in quality control of eluted cells.
Figure 2.

Example flow cytometry data comparing representative cell lines expressing or not expressing the receptor of interest using targeting and control nontargeting aptamers
(A) Example of an aptamer staining result for a target cell type expressing a receptor of interest. EGFR-expressing A431 cells were treated with 250nM of the C36 nontargeting aptamer (blue) or the E07 EGFR-targeting aptamer (red). Unstained cells are shown in green.
(B) Example of a staining result for a cell type not expressing the receptor of interest. Jurkat cells, which do not express EGFR, were treated with 250nM of the C36 nontargeting control aptamer or the E07 EGFR-targeting aptamer. These results are consistent with the prediction that E07 will bind EGFR-expressing A431 cells but not the Jurkat cells that do not produce EGFR. In the EGFR-expressing A431 cells, the E07 aptamer staining (red) has a strikingly stronger signal than the C36 or stain-free controls (green). However, in the Jurkat cells, E07 staining is as low as staining with the C36 nontargeting control aptamer and the cells alone.
Antidote screen
Aptamers can be reversed by complexing them with short complementary oligonucleotide “antidotes” that are antisense to a portion of the aptamer sequence.8,13,14 These antidotes reverse the aptamer’s binding to the target by hybridizing with a complementary region of the aptamer, changing its conformation from an active to an inactive structure. If such an antidote cannot be found in the literature, then antidotes can be created by designing a panel of candidate oligonucleotides and evaluating their ability to reverse binding of the dye-conjugated aptamer.
-
17.
Design a panel of 10- to 12-nucleotide-long DNA or 2′ O-methyl RNA oligonucleotides complementary to the aptamer, spanning the length of the aptamer in a stepwise fashion (see example in Figure 3 and Table 4).
-
18.For suspension cells, skip to step 3. For adherent cells, plate 75,000–100,000 cells/well in a 24-well plate (tissue-culture treated) in 300 μL of recommended complete cell culture media per well. Include the following wells as controls:
-
a.cells alone.
-
b.nonbinding aptamer/oligo.
-
c.fluorescently-labeled aptamer.
-
a.
-
19.
Remaining wells should be used for individual antidotes to test their ability to reverse aptamer binding. See Table 5 for an example 24-well plate setup corresponding to the E07 aptamer antidotes pictured in Figure 3.
-
20.
For suspension cells, plate 100,000 cells per well into a 96-well round bottom plate. For adherent cells, after 1–2 days, when the cells are confluent, remove cells from the 24 well plate by following standard trypsinization protocols for the cell type. Then dilute trypsinized cells at least 2-fold in complete media before replating into a 96-well round bottom plate, taking all of the cells from 1 well of the 24-well plate into 1 well of the 96-well plate.
-
21.To perform blocking incubation:
-
a.Spin cells down in the 96-well plate at 300 × g for 5 min.
-
b.Carefully remove media before resuspending cells in 190 μL of Blocking PBS+ or Media+ per well.
-
c.Incubate cells for 1 h at 37°C to block non-specific nucleic acid binding to cells.
-
a.
-
22.
During incubation, prepare enough 5 μM fluorescent aptamer solution to add 10 μL to all aptamer treatment wells. For an example staining solution, see Table 6 (Note, that this 5 μM solution is a 10× solution so the final aptamer staining concentration will be 500 nM).
-
23.Staining incubation:
-
a.After 1 h blocking incubation is complete, pellet cells by centrifuging at 300 × g for 5 min.
-
b.Remove supernatant and add fluorescent aptamer solutions to the cells.
-
c.Mix aptamer solution into cells by manually gyrating the 96-well plate on a flat surface for at least 5 s or gently pipetting up and down to mix.
-
d.Incubate for at least 30 min at 4°C in the dark.
-
a.
-
24.
During the aptamer incubation, prepare a 2.5 μM solution of each antidote in 200 μL Blocking PBS+ or Blocking Media+.
-
25.
After the cells have incubated with the aptamer solutions for 30 min, centrifuge the 96 well plate at 300 × g for 5 min to pellet the cells. If possible, do this centrifugation at 4°C. Then remove the aptamer solutions by gently aspirating, taking care to not disturb the cell pellets.
-
26.
Wash the cells 3 times with PBS+ or Media+, spinning down at 300 × g for 5 min to pellet cells in between washes. If possible, do these centrifugations at 4°C. After the final wash,
-
27.
resuspend the cells in 200 μL of the appropriate antidote solutions for each well.
-
28.
Incubate cells for 10 min at 37°C to allow antidote-aptamer hybridization.
-
29.
Following incubation, wash cells 3 times with 100 μL PBS, spinning down at 300 × g for 5 min in between washes to pellet the cells.
-
30.
Resuspend cells in PBS+ and measure fluorescence using a flow cytometer.
-
31.
Repeat screen as necessary until functional antidotes identified. A functional antidote will decrease aptamer signal down to the same level as the control, non-binding aptamer (Figure 4).
Note: If the first screen does not yield an effective antidote, increasing the length of antidotes to 15 nucleotides may improve their effectiveness.
Note: 2′O-methyl RNA antidotes will have increased stability and nuclease resistance compared to DNA antidotes. They will also likely have higher affinity binding to the aptamer than DNA antidotes.
Figure 3.

Example of antidotes tested against the E07 aptamer that binds human EGFR20,21
Colors correspond to the key in Table 4.
Table 4.
Example of an antidote panel designed against the E07 aptamer which targets human EGFR
| Antidote name | Sequence (5′–3′) |
|---|---|
| Antidote 1 (A1) | TACGGCGATTAAATC |
| Antidote 2 (A2) | TCTACGGCGATTAAA |
| Antidote 3 (A3) | TTTCTACGGCGATTA |
| Antidote 4 (A4) | GCTTTCTACGGCGAT |
| Antidote 5 (A5) | ATGCTTTCTACGGCG |
| Antidote 6 (A6) | ACATGCTTTCTACGG |
| Antidote 7 (A7) | TGACATGCTTTCTAC |
| Antidote 8 (A8) | TTTGACATGCTTTCT |
| Antidote 9 (A9) | GCTTTGACATGCTTT |
| Antidote 10 (A10) | CGGCTTTGACATGCT |
| Antidote 11 (A11) | TCCGGCTTTGACATG |
| Antidote 12 (A12) | GTTCCGGCTTTGACA |
Colors correspond to the portion of the aptamer that the antidote binds.1
Table 5.
An example 24-well plate setup for evaluating the ability of 12 candidate antidote oligonucleotides 1–12 (A1-A12) to reverse the binding of the E07 EGFR-targeting aptamer to EGFR on cells
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| A | Cells alone | Cells + C36 nontargeting control | Cells + E07 | Cells + A1 → E07 | Cells + A2 → E07 | Cells + A3 → E07 |
| B | Cells + A4 → E07 | Cells + A5 → E07 | Cells + A6 → E07 | Cells + A7 → E07 | Cells + A8 → E07 | Cells + A9 → E07 |
| C | Cells + A10 → E07 | Cells + A11 → E07 | Cells + A12 → E07 | |||
| D |
Table 6.
Example 5 μM Aptamer staining solution
| Solution 1 | ||
|---|---|---|
| Reagent (concentration) | Final concentration | Volume to add |
| Biotinylated aptamer (50 μM) | 5 μM | 2.5 μL |
| PBS +/+ | N/A | 2.5 μL |
| Fold for 5 min at 65°C, then cool at room temperature for 3 min. Then add the reagent below to make Solution 2. | ||
| Solution 2 | ||
|---|---|---|
| Reagent (concentration) | Final Concentration | Volume to add |
| Streptavidin dye (5 μM) | 125 nM | 2.5 μL (0.5 M equivalent compared to aptamer) |
| Incubate in the dark at room temperature for 20 min, and then add the following reagents to make Solution 3. (Total volume of Solutions 1 + 2 + 3 = 100 μL). | ||
| Solution 3 | ||
|---|---|---|
| Reagent (concentration) | Final Concentration | Volume to add |
| ssDNA (10 mg/mL) | 1 mg/mL | 10 μL |
| PBS+ or Media+ | N/A | 82.5 μL |
| Total | N/A | 100 μL |
Figure 4.

An example of the decrease in E07 binding to EGFR-rich A431 cells after incubation with 100-fold molar excess of antidote mA9
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Bovine Serum Albumin Fraction V (7.5% solution) (BSA) | Thermo Fisher Scientific | Cat# 15260037 |
| D-Biotin | Thermo Fisher Scientific | Cat# B20656 |
| Cell culture media recommended for target cell type | N/A | N/A |
| Dulbecco’s phosphate buffered saline (PBS) with MgCl2 and CaCl2 | Sigma | Cat# D8662-500ML |
| Dulbecco’s phosphate buffered saline (PBS) without MgCl2 or CaCl2 | Gibco | Cat# 14190-144 |
| eBioscience 10× RBC lysis buffer | Thermo Fisher Scientific | Cat# 00-4300-54 |
| Trypsin EDTA solution (1×) | Sigma | Cat# 25300-054 |
| Salmon Testes DNA, Sodium Salt (ssDNA) | Millipore Sigma | Cat# 262012-1GM |
| Critical commercial assays | ||
| Streptavidin Alexa Fluor 647 Conjugate | Thermo Fisher Scientific | Cat# S21374 |
| Hoescht 33342 Solution (20nM) | Thermo Fisher Scientific | Cat# 62249 |
| Zombie Violet™ Fixable Viability Kit | BioLegend | Cat# 423113 |
| Experimental models: Cell lines | ||
| Target cell line | Varies | N/A |
| Nontarget cell line | Varies | N/A |
| Oligonucleotides | ||
| Targeting aptamer with biotin modification | Varies | N/A |
| Nontargeting control aptamer with biotin modification | Varies | N/A |
| Software and algorithms | ||
| FlowJo 10.8 (or other flow cytometry analysis software) | FlowJo LLC | https://www.flowjo.com |
| Other | ||
| BD Vacutainer Buffered Sodium Citrate (9NC) Blood Collection Tubes | Becton, Dickinson and Company | Cat# 363083 |
| Corning® 50 mL Tube Top Vacuum Filter System, 0.22 μm Pore 13.6cm2 CA Membrane, Sterile, 12/Case | Corning | Cat# 430314 |
| Corning® cell strainer pore size 70 μm, white, sterile | Corning | Cat# CLS431751 |
| Dynabeads™ MyOne™ Streptavidin C1 Beads | Invitrogen | Cat# 65001 |
| DynaMagTM-2 Magnet | Thermo Fisher Scientific | Cat# 12321D |
Step-by-step method details
Aptamer-mediated isolation of cells from blood or other cell suspensions
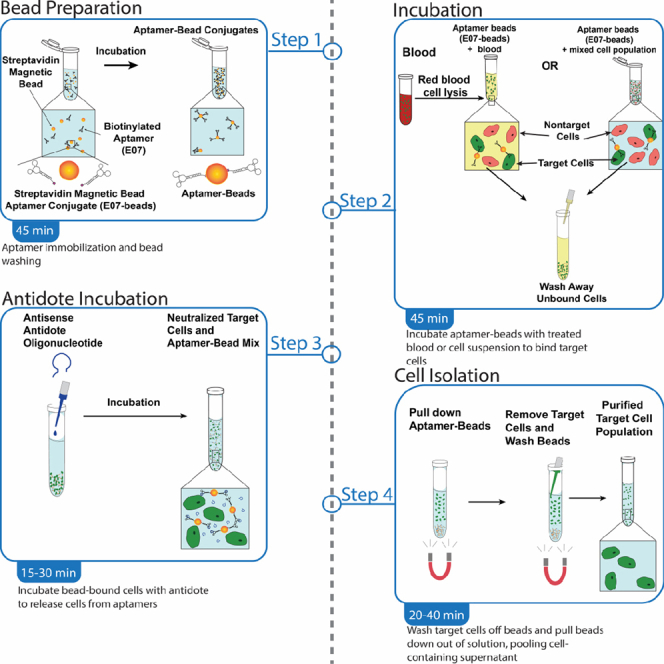
After verifying that the aptamer can selectively stain cells expressing the aptamer’s receptor target in an antidote-reversible manner, the aptamer-antidote pair can be used for aptamer-based magnetic-activated cell sorting (MACS) to isolate target cells from blood or other complex cell mixtures. Immobilizing biotinylated aptamers on streptavidin-coated magnetic beads enables binding and magnetic pulldown of target cells, which can be separated from complex cell mixtures by removing the supernatant containing nontarget cells. Washing the beads removes remaining weakly-bound nontarget cells and other particles. Subsequent incubation with antidote displaces bound target cells from the aptamer-coated beads, resulting in a population of label-free target cells in the supernatant. Isolated cells will be in a native, unlabeled state and can then be used for receptor signaling studies, sequencing, culturing or other downstream applications. See Figures 5, 6 and 7 for illustrated flowcharts.
Figure 5.

Illustration of the aptamer-bead preparation portion of the aptamer-MACS protocol
A solution of biotinylated aptamers is incubated with streptavidin-coated magnetic beads and allowed to incubate for 15 min.
Figure 6.

Illustration of the Cell Isolation portion of the aptamer-MACS protocol
Depending on the application, the sample containing the target cell type is pretreated with RBC Lysis Buffer in the case of blood or directly resuspended in blocking PBS+. After a 30-min incubation at 4°C, the target cells bound to the aptamer-coated beads are separated from the rest of the sample by repeatedly using a magnet to partition the target cell-coated aptamer-magnetic beads.
Figure 7.

Workflow of the Cell Binding and Antidote Treatment sections of the protocol
(Left-to-right) The antidote is added to the suspension and mixed by pipetting. Following a 10-min incubation at 37°C, the supernatant containing the target cell type is removed and the cells are washed to collect all of the target cells.
Aptamer-bead preparation
-
1.
Vortex the vial of Dynabeads™ MyOne™ Streptavidin C1 beads for 30 s and transfer 12 μL of beads to a 1.5 mL tube.
-
2.Wash beads:
-
a.Add 375 μL 1× SB1T buffer, placing the tube in the magnetic rack for 1–2 min to pellet the beads.
-
b.Remove the supernatant, taking care to aspirate carefully with the pipette, holding the tip to the bottom of the 1.5 mL tube and drawing the supernatant up gradually and steadily.
-
c.Remove the tube from the magnet and resuspend beads in 120 μL 1× SB1T buffer and pipetting up and down 3–5 times.
-
d.Place the tube on a magnet for 1–2 min to pull the beads out of solution.
-
e.Repeat steps b, c, and d 2 times, then aspirate the supernatant with a pipette.
-
a.
-
3.
Remove the tube from the magnet and resuspend the beads in 48 μL of 1× SB1T buffer + 1 mg/mL ssDNA (so that bead concentration is 2.5 μg/μL).
-
4.
Prepare targeting aptamer solution by resuspending 12.5 pmol of aptamer in 20 μL solution of PBS+/+. Vortex to mix.
-
5.Fold aptamer:
-
a.Denature aptamer solution by heating at 65°C for 5 min.
-
b.Let sit at room temperature for 3 min to allow the aptamer to renature and fold.
-
a.
-
6.Immobilize folded aptamers on streptavidin beads:
-
a.Vortex the beads for at least 30 s.
-
b.Add 40 μL of the bead solution to the renatured aptamer solution from step 5.
-
c.Vortex to mix.
-
d.Incubate at room temperature for 15 min with gentle rotation to allow the folded aptamers to bind to the surface of the beads.
-
a.
-
7.
Place tubes on the magnet for 2–3 min to separate the beads and supernatant and remove the supernatant by pipetting gently, taking care to avoid aspirating any beads with the pipette.
-
8.Wash the aptamer-coated beads 3 times:
-
a.Remove the tube containing the beads from the magnet rack.
-
b.Resuspend the beads with 50 μL 1× SB1 buffer + 1 mg/mL ssDNA.
-
c.Place the tube on the magnet for 2–3 min until the beads have been pulled out of solution.
-
d.Remove the supernatant gently with a pipette.
-
e.Repeat steps a, b, c, and d 2 more times.
-
f.remove the tube from the magnet and then resuspend in buffer.
-
a.
-
9.
After the 3rd wash in step 9, remove the tube from the magnet and resuspend beads in 40 μL of PBS+.
-
10.
Place beads on ice until it is time to incubate with cell sample of interest.
Alternatives: As a control for aptamer-specific isolation of cells, beads should also be coated with a control nontargeting aptamer following the same protocol described above for the receptor-specific aptamer.
Red blood cell (RBC) lysis
-
11.To isolate cells from whole blood:
-
a.Draw at least 1 mL of whole blood into a citrated collection tube to prevent clumping.
-
b.Combine 10 mL of 1× RBC Lysis Buffer with 1 mL of the citrated whole blood in a 15 mL conical and mix thoroughly by pipetting up and down 3–5 times.
-
c.Incubate the sample for 15 min at room temperature in a sample rotator. (See key resources table / manufacturer’s recommendations for more details).
-
a.
-
12.
Centrifuge the sample at 300–400 × g for 5 min at room temperature. The blood should separate into a dark-red pellet and a lighter supernatant.
-
13.
Remove supernatant with a pipette, taking care not to disturb the cell pellet.
-
14.
Wash cells by resuspending in 10 mL PBS +, centrifuging at 300–400 × g for 5 min to pellet and removing supernatant.
-
15.
After washing, resuspend pellet in 1 mL PBS+ and keep on ice.
-
16.
Immobilize aptamer-beads (prepared in “aptamer-bead preparation” section above) on the magnetic rack, remove supernatant and replace with cell mixture, mixing well by pipetting up and down. Then leave at 4°C for 30 min with gentle rotation to allow target cells to bind to aptamer-beads.
Alternatives: Depending on the amount of blood available, the volumes of aptamer-labeled beads and RBC Lysis Buffer can be scaled up or down in direct proportion to the volume of blood used.
Alternatives: This protocol can be adapted for liquid cell suspensions other than blood, provided the substance does not cause clumping of the beads. For isolation of target cells from other mixtures such as cultured cell suspensions or homogenized tissue samples, pellet 5 × 106 cells, resuspend in 1 mL Blocking media+ or Blocking PBS+, and add to magnet, as in steps 5 and 6 above, then continue to Cell Binding and Antidote Treatment section below.
Cell binding and antidote treatment
Note: The following steps should be performed at 4°C unless otherwise noted to ensure that the aptamer binds but does not internalize into cells.
-
17.
Place cell and bead mixture on the magnetic rack for 3–5 min to pellet the beads.
-
18.
Remove the supernatant by gently aspirating with a pipette.
-
19.Wash cell-bead mixture with 1 mL of Media+ or PBS+:
-
a.Resuspend cell-bead mixture in the buffer, placing the beads on the magnet for 3–5 min to pellet the beads and removing the supernatant to a 2nd tube.
-
b.To recover residual beads from the supernatant and optimize cell yield, place this 2nd tube on the magnetic rack and wait another 3–5 min for the beads to aggregate.
-
c.Remove the supernatant from the 2nd tube to a 3rd tube.
-
d.Place this 3rd tube on the magnetic rack and wait 3–5 min to again recover any residual beads from the supernatant.
-
a.
-
20.
Repeat the washing process from step 3 as needed until there are no beads visible in the supernatant.
-
21.
Repeat step 2 twice more to wash the cell-bead mixture,
-
22.
After the last wash, remove the tubes containing beads from the magnet and combine all the beads into a final volume of 40 μL of Media+ or PBS+ with a 100-fold molar excess of antidote (1.25 nmol antidote for the 12.5 pmol of aptamer on the beads). Pipette up and down to mix. Take care to avoid making bubbles.
-
23.
Place at 37°C with gentle rotation for 10 min to allow the antidote to bind to the aptamer and the cells to detach from the aptamer beads.
-
24.
Immediately place cells back at 4°C following antidote incubation. Then add 1 mL of Media+ or PBS+ buffer, mix by pipetting up and down and place tube on magnet for 3–5 min to draw beads out of solution, leaving detached cells in supernatant.
-
25.Collect the eluted target cells:
-
a.Gently aspirate the supernatant containing the detached target cells with a pipette and move it to a new Eppendorf tube.
-
b.Place the tube containing the supernatant on the magnet for 3–5 min to draw any beads that may remain;
-
c.Transfer the supernatant to a 2nd tube, and repeat steps a and b until there are no beads remaining in the supernatant.
-
a.
-
26.
Discard the tubes with the residual beads, and wash the original beads 2 more times with 1 mL of PBS+ using the same technique described in step 9 to collect cells washed off the beads
-
27.Pool the supernatants to collect all the eluted cells:
-
a.Combine the eluate from steps 9 and 10 into two Eppendorf tubes
-
b.Spin the tubes containing the pooled eluted cells down at 300 × g for 5 min
-
c.Gently remove the supernatant from the pellets of the purified target cells.
-
d.Resuspend cell pellets in Media+ or other desired buffer.
-
a.
-
28.
Assess recovery by flow cytometry and proceed to other downstream applications.
Quality control of eluted cells
If desired, a portion of the recovered cells can be evaluated to ensure that they are viable and to ensure the presence of a target biomarker via flow cytometry. Viability can be assessed via Hoescht 33342 staining, as described below, or by using another commercially available viability dye, such as the Zombie dyes from BioLegend™, following the manufacturer’s recommendations. Target biomarkers can also be assessed using biomarker-specific antibodies or biotinylated aptamers. Antibody staining for biomarkers can be performed according to manufacturer’s recommendations immediately downstream of the “cell binding and antidote treatment” section above. Aptamer staining can be performed as described in the “aptamer cell staining assay” section above.
-
29.
Stain a sample of eluted cells with appropriate targeting aptamer as described in aptamer cell staining assay or with corresponding biomarker-targeting antibody according to manufacturer’s recommendations.
-
30.
Prepare a solution of 1 ng/mL Hoescht 33342 staining solution in Hanks Balanced Salt Solution containing 1.3 Ca2+, 0.9 mM Mg2+, 1% BSA, and 0.1% sodium azide.
Alternatives: If the use of Hoescht 33342 would be impractical due to wavelength interference or some other reason, amine-reactive fluorescent dyes such as the Zombie™ line of dyes work well in conjunction with a targeting ligand conjugated to a noninterfering dye.
-
31.
Resuspend the cells eluted from the beads in cell binding and antidote treatment in 100 μL of the Hoescht staining solution.
-
32.
Assess sample purity and viability via flow cytometry, according to manufacturer specifications.
Note: In order to do a thorough evaluation of the viability and purity of eluate, it is recommended that you prepare a dead cell control of the same type as the target cell type. For most cell lines, incubating for 2 min in a solution of 70% ethanol in water should be enough to kill the cells. However, in applications where sufficient quantities of viable target cells cannot be obtained easily, such as the isolation of rare cell types, a similar immortalized cell line may be used for the dead cell control, or the dead cell control may be omitted entirely.
Expected outcomes
After washing, the isolated cells should be viable and free of beads. You should be able to resuspend them into solution easily with no clumping. Yield will be variable and dependent on the individual properties of the aptamer, the antidote and the target cell type. Cell types and viability of isolated cells can be determined using a combination of live-dead staining and the aptamer/cell-binding assay described in the before you begin section above.
Limitations
This protocol is intended for the isolation of viable cells without compromising the function of the cell surface target (e.g., a receptor) bound by a sorting ligand. Aptamers targeting internal structures are not suitable for this protocol, nor are aptamers targeting rapidly-internalizing cell-surface receptors. This protocol will not work with aptamers that cannot stably bind at 4°C. Recovery rates will vary based on several factors including the affinity of the aptamer for the receptor, the rarity of the target cell type, the affinity of the antidote for the aptamer and the biology of target cells.
Troubleshooting
Problem 1
Cells form insoluble fibrous or flakey clumps upon exposure to beads, which cannot be broken up by pipetting or straining.
Potential solution
Clumping due to avidity effects of beads on cells is irreversible, but can be prevented by using cells that are healthy and in log phase growth (i.e., not overconfluent) in the case of cultured cells or by straining cells with a 70 mm cell strainer before mixing them with beads. Note that straining cells may reduce the expected number of cells recovered. Decreasing the ratio of cells to beads or blood lysate to beads may also lower the chance of clumping.
Problem 2
Low efficiency of cell recovery.
Potential solution
Ensure that the aptamer stock is pure and fully biotinylated by checking purity via HPLC or denaturing polyacrylamide gel. After confirming that the aptamer is pure and properly biotinylated and that the aptamer functions under the conditions of this methodology, as outlined in aptamer-cell binding assay, above, try increasing the ratio of aptamer-coated beads to cells which may improve recovery efficiency. Experimenting with different concentrations of aptamer loaded onto the beads may also improve results.
Problem 3
Significant contamination of eluate with cells not expressing target receptor.
Potential solution
High concentrations of cells that do not express the surface receptor of interest in the final eluate could be due to a poorly-optimized ratio of beads to blood/cell suspension. Decreasing the ratio of beads to blood/cell suspension may help by providing less surface area for nontarget, unwanted cells to non-specifically adhere. Additional washes before antidote incubation may also help reduce contamination.
Problem 4
Significant contamination of eluate with beads.
Potential solution
If the final eluate still contains beads, simply apply the magnet again and remove the eluate to another tube, repeating as needed until beads are no longer present.
Problem 5
Cells have low viability post-elution.
Potential solution
If the cells taken from the final eluate have poor viability, making them unsuitable for downstream applications, a number of steps can be taken to decrease stressors to cells. Decreasing pipetting speed and number of washing steps may help preserve cells without affecting yield. In our experience, executing the protocol in a timely fashion, preferably with two people working together, gives the best results. Experimenting with different media types to better mimic the native environment of target cells may also improve viability of recovered cells.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bruce Sullenger (bruce.sullenger@duke.edu).
Materials availability
Duke University has applied for a patent on this technology.
Data and code availability
This study did not generate/analyze datasets and codes.
Acknowledgments
We thank Matt Levy’s lab, formerly at Albert Einstein College of Medicine, for use of their Expedite 8909 DNA synthesizer for aptamer synthesis and Amos Yan for suggestions and proofreading of this protocol. Funding sources: National Institutes of Health, USA R01-HL147147 (to B.A.S.).
Author contributions
M.D.R., B.P.G., and B.A.S. designed the experiments. M.D.R. and B.P.G. performed the experiments and analyses. M.D.R. wrote the manuscript. B.P.G. and B.A.S. conceived the project and edited the manuscript.
Declaration of interests
Duke University has applied for a patent on this technology.
Contributor Information
Martin D. Requena, Email: mdr45@duke.edu.
Bruce A. Sullenger, Email: bruce.sullenger@duke.edu.
References
- 1.Gray B.P., Requena M.D., Nichols M.D., Sullenger B.A. Aptamers as Reversible Sorting Ligands for Preparation of Cells in Their Native State. Cell Chem. Biol. 2020;27:232–244.e237. doi: 10.1016/j.chembiol.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conrad R.C., Giver L., Tian Y., Ellington A.D. In vitro selection of nucleic acid aptamers that bind proteins. Methods Enzymol. 1996;267:336–367. doi: 10.1016/s0076-6879(96)67022-0. [DOI] [PubMed] [Google Scholar]
- 3.Tuerk C., Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 4.Dollins C.M., Nair S., Sullenger B.A. Aptamers in immunotherapy. Hum. Gene Ther. 2008;19:443–450. doi: 10.1089/hum.2008.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nimjee S.M., White R.R., Becker R.C., Sullenger B.A. Aptamers as therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017;57:61–79. doi: 10.1146/annurev-pharmtox-010716-104558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou J., Satheesan S., Li H., Weinberg M.S., Morris K.V., Burnett J.C., Rossi J.J. Cell-specific RNA aptamer against human CCR5 specifically targets HIV-1 susceptible cells and inhibits HIV-1 infectivity. Chem. Biol. 2015;22:379–390. doi: 10.1016/j.chembiol.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long S.B., Long M.B., White R.R., Sullenger B.A. Crystal structure of an RNA aptamer bound to thrombin. RNA. 2008;14:2504–2512. doi: 10.1261/rna.1239308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunaratne R., Kumar S., Frederiksen J.W., Stayrook S., Lohrmann J.L., Perry K., Bompiani K.M., Chabata C.V., Thalji N.K., Ho M.D., et al. Combination of aptamer and drug for reversible anticoagulation in cardiopulmonary bypass. Nat. Biotechnol. 2018;36:606–613. doi: 10.1038/nbt.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nimjee S.M., Keys J.R., Pitoc G.A., Quick G., Rusconi C.P., Sullenger B.A. A novel antidote-controlled anticoagulant reduces thrombin generation and inflammation and improves cardiac function in cardiopulmonary bypass surgery. Mol. Ther. 2006;14:408–415. doi: 10.1016/j.ymthe.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Zhou J., Rossi J.J. Cell-type-specific, aptamer-functionalized agents for targeted disease therapy. Mol. Ther. Nucleic Acids. 2014;3:e169. doi: 10.1038/mtna.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan A., Levy M. Cell internalization SELEX: in vitro selection for molecules that internalize into cells. Methods Mol. Biol. 2014;1103:241–265. doi: 10.1007/978-1-62703-730-3_18. [DOI] [PubMed] [Google Scholar]
- 12.Pratico E.D., Sullenger B.A., Nair S.K. Identification and characterization of an agonistic aptamer against the T cell costimulatory receptor, OX40. Nucleic Acid Ther. 2013;23:35–43. doi: 10.1089/nat.2012.0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rusconi C.P., Scardino E., Layzer J., Pitoc G.A., Ortel T.L., Monroe D., Sullenger B.A. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature. 2002;419:90–94. doi: 10.1038/nature00963. [DOI] [PubMed] [Google Scholar]
- 14.Nimjee S.M., Dornbos D., 3rd, Pitoc G.A., Wheeler D.G., Layzer J.M., Venetos N., Huttinger A., Talentino S.E., Musgrave N.J., Moody H., et al. Preclinical development of a vWF aptamer to limit thrombosis and engender arterial recanalization of occluded vessels. Mol. Ther. 2019;27:1228–1241. doi: 10.1016/j.ymthe.2019.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou J., Soontornworajit B., Martin J., Sullenger B.A., Gilboa E., Wang Y. A hybrid DNA aptamer-dendrimer nanomaterial for targeted cell labeling. Macromol. Biosci. 2009;9:831–835. doi: 10.1002/mabi.200900046. [DOI] [PubMed] [Google Scholar]
- 16.LLC A. 2023. Apta-Index Aptamer Database.https://www.aptagen.com/apta-index/ [Google Scholar]
- 17.Kelly L., Maier K.E., Yan A., Levy M. A comparative analysis of cell surface targeting aptamers. Nat. Commun. 2021;12:6275. doi: 10.1038/s41467-021-26463-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magalhães M.L.B., Byrom M., Yan A., Kelly L., Li N., Furtado R., Palliser D., Ellington A.D., Levy M. A general RNA motif for cellular transfection. Mol. Ther. 2012;20:616–624. doi: 10.1038/mt.2011.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen C.H.B., Chernis G.A., Hoang V.Q., Landgraf R. Inhibition of heregulin signaling by an aptamer that preferentially binds to the oligomeric form of human epidermal growth factor receptor-3. Proc. Natl. Acad. Sci. USA. 2003;100:9226–9231. doi: 10.1073/pnas.1332660100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li N., Nguyen H.H., Byrom M., Ellington A.D. Inhibition of cell proliferation by an anti-EGFR aptamer. PLoS One. 2011;6:e20299. doi: 10.1371/journal.pone.0020299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powell Gray B., Kelly L., Ahrens D.P., Barry A.P., Kratschmer C., Levy M., Sullenger B.A. Tunable cytotoxic aptamer-drug conjugates for the treatment of prostate cancer. Proc. Natl. Acad. Sci. USA. 2018;115:4761–4766. doi: 10.1073/pnas.1717705115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze datasets and codes.