

Abstract
Trans-amplifying RNA (taRNA) is a split-vector derivative of self-amplifying RNA (saRNA) and a promising vaccine platform. taRNA combines a non-replicating mRNA encoding an alphaviral replicase and a transreplicon (TR) RNA coding for the antigen. Upon translation, the replicase amplifies the antigen-coding TR, thereby requiring minimal amounts of TR for immunization. TR amplification by the replicase follows a complex mechanism orchestrated by genomic and subgenomic promoters (SGPs) and generates genomic and subgenomic amplicons whereby only the latter are translated into therapeutic proteins. This complexity merits simplification to improve the platform. Here, we eliminated the SGP and redesigned the 5′ untranslated region to shorten the TR (STR), thereby enabling translation of the remaining genomic amplicon. We then applied a directed evolution approach to select for faster replicating STRs. The resulting evolved STR (eSTR) had acquired A-rich 5′ extensions, which improved taRNA expression thanks to accelerated replication. Consequently, we reduced the minimal required TR amount by more than 10-fold without losing taRNA expression in vitro. Accordingly, eSTR-immunized mice developed greater antibody titers to taRNA-encoded influenza HA than TR-immunized mice. In summary, this work points the way for further optimization of taRNA by combining rational design and directed evolution.
Keywords: trans-amplifying RNA, alphaviral vector, transreplicon, vaccine, directed evolution
Graphical abstract
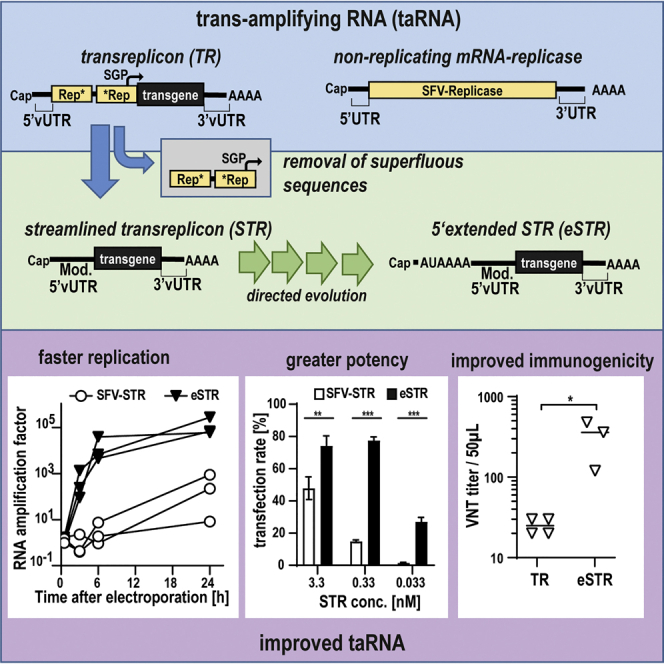
This work presents the improvement of trans-amplifying RNA (taRNA), a bipartite vector system aiming to accelerate RNA vaccine adaptation by using an mRNA-encoded alphaviral replicase that amplifies minimal amounts of antigen-coding transreplicon RNA. The transreplicon design was streamlined to improve taRNA safety, and a directed evolution approach increased its potency.
Introduction
When severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) began its global spread in early 2020, mRNA vaccines were ready to explode off the starting blocks, becoming a game changer in the COVID-19 pandemic. Moreover, the pressing need for very large numbers of vaccine doses has propelled the industrial scaleup of RNA production.1,2 Meanwhile, it became clear that SARS-CoV-2 evolves rapidly, leading to constant emergence of new variants and serotypes that more and more escape immunity.3 The continuous vaccine adaptation needed to keep pace with viral evolution requires an extraordinarily rapid production of large quantities of mRNA for billions of doses. Reducing the effective amount of antigen-coding RNA per dose is an obvious key to reaching this goal.
Self-amplifying RNA (saRNA) and trans-amplifying RNA (taRNA) appear suitable for achieving a significant dose reduction per vaccination.4,5,6,7 saRNA and the related taRNA are engineered from the single-stranded positively sensed RNA genome of alphaviruses, such as Semliki Forest virus (SFV), Sindbis virus (SINV), or Venezuelan equine encephalitis virus (VEEV). While the alphaviral structural genes are replaced with a desired antigen, these vector systems retain the genes encoding the four non-structural proteins (nsPs) that translate and then build the poly-enzyme complex called the replicase (Figure S1A). In essence, the replicase acts as an RNA-dependent RNA polymerase that recognizes terminal and internal conserved sequence elements (CSEs) of the replicating RNA. It transcribes full-length (genomic) and partial (subgenomic) copies of the transferred RNA in a CSE-controlled manner, leading to a massive amplification of the antigen-coding RNA (Figure S1B). Accordingly, it was shown that a much lower dose of saRNA compared with non-replicating mRNA was needed to induce protective immunity in preclinical vaccine experiments,8 and saRNA proved to be a promising vaccine platform against a variety of pathogens.9,10
However, saRNA is a very long molecule of about 10 kb, which challenges its production and may diminish the benefit of a potential dose reduction. The 4 nsP genes of the replicase, with its 7.5 kb open reading frame, are mainly responsible for the large vector size. With that in mind, and aiming to ease and accelerate RNA vaccine production, we recently described a split-vector derivative of saRNA, called taRNA (Figure S1A).6 taRNA employs two RNAs, a mid-sized conventional non-replicating mRNA that expresses replicase (nrRNA-replicase) and a short antigen-coding transreplicon (TR) that contains CSEs and is amplified by co-transfected replicase (Figure S1B illustrates saRNA and taRNA replication). We envision a future scenario in which the invariant nrRNA-replicase is stockpiled in large quantities, ready to be combined with low amounts of freshly produced and pandemic-specific TRs. The results of our proof-of-concept study were promising: taRNA comprising 20 μg nrRNA-replicase and only 50 ng antigen-coding TR was as immunogenic in mice as 20 μg conventional mRNA.6 Thus, although the total RNA amounts of mRNA and taRNA were identical, the antigen-coding TR was reduced to trace amounts, which supports the concept. In this work, we made further optimization for taRNA vectors with clinical application in mind, eliminating redundant sequence segments that do not contribute functionally.
We revisited the taRNA design and the expression level of the TR. First, we rationally simplified the TR structure by removing remnants of the replicase gene and the subgenomic transcript. Then, we applied a directed evolution strategy based on serial RNA transfer to select for a more rapidly amplifying TR. Both measures improved taRNA expression and immunogenicity. We suggest that the resulting simplified TR design is an ideal starting vector for upcoming taRNA optimization.
Results
Simplifying the TR improves taRNA performance
In the first generation of taRNA, the TR-RNA was engineered from SFV-saRNA by deleting 80% of the replicase-coding region. To conserve the ability of the TR to replicate, we kept a 221-nucleotide-long fragment of the nsP1 coding region overlapping with the 5′ CSE, including the replicase start codon, and 984 nucleotides of nsP4 comprising the subgenomic promoter (Figure 1A).6 With this design, replication of the TR in trans leads to new RNA copies equivalent to the saRNA genomic copy, with an identical sequence to the in-vitro-transcribed (IVT) template TR and a subgenomic transcript identical to the one made from saRNA (Figure S1B). Due to the conserved nsP1 start codon, we assumed that a truncated nsP1 peptide is translated from the genomic equivalents of the TR (Table S1; Figure 1B, left). For clinical application, it may be necessary to demonstrate that this peptide is not harmful. Further, the nsP4-derived region that we kept was much longer than needed for assuring its subgenomic promoter function.11 At the time, this crude design was successful, but for clinical application, the TR should optimally be devoid of unneeded regions and truncated open reading frames. The genomic RNA equivalent of the TR serves as the template for subgenomic RNA (sgRNA) synthesis but itself does not contribute to antigen translation. Here, we removed the lengthy nsP4-subgenomic promoter (SGP) sequence, thereby disabling subgenomic transcription and reducing the replication products to the genomic equivalent only (the replication process before and after the TR redesign is explained below and shown in Figure 1B). Furthermore, the original 5′ CSE region contains not only the replicase start codon in the second stem-loop structure of the SFV 5′ CSE12 but also a number of other AUG triplets (Figure S2). These putative start codons could either prevent translation of the transgene or, if in frame, result in fusions of nsP1-derived peptides to the transgene, introducing risk in clinical application (Figure 1A). We therefore aimed to mutate all AUGs in order to silence any unwanted translation and used RNA structure prediction tools to assess whether mutations would affect folding of the 5′ CSE. To conserve appropriate base pairing, we introduced compensating mutations into complementary strands within local double-stranded regions. Overall, the secondary structure of the resulting AUG-free 5′ CSEs was predictably well conserved (Figure S2). Jointly, this shortened the TR by approximately 1 kb and shifted the start codon of the transgene to the most upstream AUG. As a result of the redesign, the replication and translation of the short TR (STR) (Figure 1B right) is less complex compared with the former TR (Figure 1B left): first, since subgenomic transcription no longer occurs, STR replication is limited to de novo transcription of genomic RNA equivalents. Second, due to missing AUGs, the truncated nsP1 open reading frame (ORF) is no longer translated from the genomic RNA equivalents. Noteworthy of this system, IVT and capped STRs are translatable before being replicated (Figure 1B, right). In contrast, the former TR design required replication by replicase for sgRNA transcription and subsequent transgene translation (Figure 1B, left). Accordingly, we observed reporter gene expression in K562 cells upon co-transfection of in vitro capped STR with an nrRNA encoding inactive replicase, which was greatly enhanced when nrRNA encoding active replicase was co-transfected. In contrast, a TR was only expressed when combined with active replicase, except for low background translation (Figures 1C and S3A). Since it was previously described that the SFV replicase copies SINV-TR more efficiently than an SFV-TR,12 we also constructed SINV-derived TR and STR. Interestingly, STRs of SFV and SINV resulted in greater expression than respective TRs of the same viruses, reflected by a statistically significant difference of GFP-mean fluorescence intensities (MFIs) between both TR formats (Figure S3B). As hypothesized, exchanging SFV- for SINV-(S)TR improved the expression of SFV-replicase-driven taRNA by significantly increasing the rate of transfected cells rather than the MFI of GFP-positive cells (Figures 1C and S3). Noteworthy, the benefit of SINV-STR for taRNA expression was not limited to K562 cells, which we used for the majority of experiments in this study. We made similar observations in BHK21 cells, which are widely used for alphaviral vector development. Also, absolute expression levels in both cell types were comparable (Figure S4A). Although statistically not significantly different, there was a trend toward higher viability of K562 cells upon transfection with the cytotoxic SFV replicase (Figure S4B). The greater expression achieved with the SINV-STR is remarkable because it indicates that SFV evolution did not result in RNA promotor elements supporting maximal RNA replication in trans by the SFV replicase.
Figure 1.

Transreplicon simplification and mechanism of trans-replication
(A) Design of simplified and shortened transreplicons. Conserved sequence elements (CSEs) serving as replicase recognition signals are located within the 5′ UTR of viral origin (5′ vUTR), at the beginning of nsP1, at the end of nsP4 (subgenomic promoter [SGP]), and at the end of the 3′ vUTR extending into the poly(A) tail (reviewed in Pietilä et al.11). In the first generation of transreplicons (TRs, top), the replicase gene was largely deleted, but all CSEs were conserved. Consequently, 221 nucleotides of nsP1 (1∗) and 984 nucleotides of nsP4 (4∗) comprising the 5′ CSE or the SGP, respectively, remained in the TR. To trim and simplify the TR, we removed the nsP4-SGP (indicated by the gray triangular background) and mutated all AUG triplets within the 5′ CSE/nsP1 (gray shaded 1∗), resulting in the start codon of the transgene becoming the most upstream one. Thus, the short TR (STR; bottom) closely resembles an mRNA and can be translated without replication if capped during in vitro transcription. (B) Mechanism of TR and STR trans-replication. Full-length immature replicase (imm. REPL, center) is translated from in-vitro-transcribed (IVT) nrRNA-REPL and then processed by intramolecular proteolysis mediated by nsP2. First, the negative-strand-specific replicase complex made of nsP123 and nsP4 is formed, followed by further cleavages and release of nsP1, nsP2, and nsP3, eventually forming the mature positive-strand-specific replicase complex ((+)REPL). Common replication steps of TR (left) and an STR (right) include the synthesis of a negatively sensed copy of the positively sensed transfected IVT RNA vectors in full length, followed by transcription of novel full-length positive-sensed copies ((+)TR-TG or (+)STR-TG, respectively). These steps mirror the replication of genomic RNA of alphaviruses. In the case of the TR-TG, the (+)REPL transcribes, in addition, a subgenomic RNA (sgRNA-TG) identical to that found in the parental saRNA, which is translated into the transgene (left, dashed box no. 1). Due to the presence of the original replicase start codon, a truncated nsP1 peptide (nsP1∗; 1∗) may be translated from the TR transgene (left, dashed box no. 2). The replication of the STR, however, no longer supports subgenomic transcription, and the AUG codon mutations prevent a putative nsP1-peptide translation (right). Thus, STR replication is limited to genomic-like replication, and all positive-stranded copies are translated exclusively to the transgene (right, dashed box no. 3). (C) STRs improve taRNA performance compared with TRs. K562 cells were co-electroporated with 1 μg nrRNA encoding either inactive or active SFV replicase and 0.2 μg TR or STRs of SFV or SINV. The GFP expression mediated by TR and STRs was measured by flow cytometry to investigate transfection rates and level of expression in individual cells. Data are shown as mean ± SD of three independent experiments.
The SINV-STR is the most efficiently amplified alphaviral STR, but it is outcompeted by a 5′-modified SFV-STR upon directed evolution
Recent literature suggests that heterologous substrate usage is rather common among alphaviral replicases.13 Nevertheless, we wondered whether it was exceptional that the SFV replicase amplified the SINV-(S)TR more efficiently than SFV-(S)TR, or whether this heightened efficiency would apply to STRs of other alphaviral origins as well. To answer this, we generated STRs from a number of alphaviral species (Fort Morgan virus [FMV], Aura virus [AURAV]; Highlands J virus [HJV], Madariaga virus [MADV], Chikungunya virus [CHIKV]) following the procedure applied to the 5′ end of SFV and SINV. To standardize the 3′ end, we used the 3′ terminal 100 nucleotides of each viral genome as negative strand promoter and 3′ UTR, which might not be optimal in each case. To prevent direct translation of the IVT STRs in the absence of replicase, we omitted capping during in vitro RNA production, which, in our previous study, did not affect trans-replication.6 Thereby STR expression indicates de novo synthesis of capped plus-stranded STR copies in cells co-transfected with SFV-replicase nrRNA. The SFV replicase amplified all alphaviral STRs tested (Figure S5A), thus confirming the described cross-utilization.13 With the exception of the CHIKV-STR, the heterologous STRs provided greater expression than the SFV-STR. Although co-transfecting STRs and SFV-saRNA instead of SFV-replicase nrRNA confirmed the findings, differences in expression were much more contrasted (Figure S5B). SINV-STR expression was 100-fold greater than SFV-STR expression when using SFV-saRNA compared with a 3.5-fold advantage using the SFV-replicase nrRNA. The SINV-STR was also a strong inhibitor of the SFV-saRNA (Figure S5C), indicating competition of both replication-competent RNAs for the saRNA-encoded replicase. Inhibition of the expression of SFV-saRNA by other strongly expressed STRs was less pronounced than that by the SINV-STR. Overall, the SINV-STR was the best expressed of all STRs tested.
To assess whether a SINV-STR can prevail against other STRs, we took a second approach. Pools of equimolar-mixed STRs expressing a GFP-SecNLuc fusion reporter originating from FMV, AURAV, HJV, MADV, CHIKV, and SFV were spiked with a 10- to 1,000-fold lower molar amount of SINV-STR expressing a GFP-iRFP fusion reporter, giving a final SINV-STR abundance of 1.6%–0.016%. Upon co-transfection of these pools with nrRNA-SFV replicase, flow cytometry revealed that the iRFP expression translated from the SINV-STR (double-positive cells) was much greater (more than 60%) than expected from its abundance at the time of transfection (1.6%) compared with other STRs (Figure 2A). This indicates that the SINV-STR outcompeted other co-replicating STRs in the cells. To assess whether the SINV-STR bears the optimal replication potential, or whether it could be overtopped, we subjected pools of STRs to a directed evolution, exploiting the inaccuracy of the SFV replicase as a natural source for mutations. Since they lack proofreading activity, alphaviral replicases have an error rate of about 10−4.14 Therefore, mutations within the CSE that would enhance replication should arise during continued replication, and mutated STRs should accumulate at the expense of parental STRs. To prolong replication and foster the accumulation of mutants, we resorted to a serial RNA transfer. We extracted cellular total RNA 1 day after transfection of replicase nrRNA and the STR mixture, added “fresh” IVT SFV-replicase nrRNA to the extracted RNA (which includes STR-RNA), and transfected new cells. In the initial pools, a GFP-iRFP-expressing SINV-STR was added since we expected it to accumulate. To provide an advantage to STRs other than SINV-STR, we generated STR mixtures with a low SINV-STR abundance of 1.6%–0.0016% and repeated the described serial RNA transfer four times and followed total STR expression (GFP) and SINV-STR expression (GFP and iRFP) by flow cytometry. Indeed, according to the iRFP expression, the SINV-STR abundance grew at each dilution within two serial transfer steps (P0 to P2), reflecting its replicative advantage in the starting pools (Figure 2B). However, upon continued serial transfer, the iRFP expression decreased (P3 and P4), except for the pool with the greatest starting amount of SINV-STR. Since the expression of pool-encoded GFP remained consistently high, we excluded a general loss of STRs due to ineffective serial RNA transfer. Hence, the loss of SINV-STR expression indicated that either the SINV-STR lost iRFP expression (for instance by a frameshift mutation) or that another STR became dominant.
Figure 2.

The SINV-STR is preferentially amplified by the SFV replicase, but it is outcompeted by a 5′-modified SFV-STR upon directed evolution
(A) Preferential expression of SINV-STR. GFP-SecNLuc-encoding uncapped STRs of 6 alphaviruses (FMV, AURAV, HJV, MADV, CHIKV, SFV) were mixed in equal amounts and spiked with a minor amount of an uncapped SINV-STR encoding a GFP-iRFP fusion reporter. SINV-STR abundance ranged from 1.6% to 0.016%. This blend of STRs was mixed with capped nrRNA-SFV replicase and electroporated into K562 cells. Dot plots show the expression of both fluorescent reporters of a representative experiment the day after electroporation (left). Data are also shown as mean and SD of 3 independent experiments (right). (B) Evolution of iRFP expression upon serial RNA transfer. K562 cells were co-electroporated with SINV-spiked uncapped STR mixtures and capped nrRNA-replicase, with SINV-STR abundance ranging from 1.6% to 0.0016%. After 16 h, GFP and iRFP expression was measured before the total RNA of the transfected cells was extracted, supplemented with IVT nrRNA-replicase, and used to electroporate K562 cells again. This serial RNA transfer was done four times, iRFP expression indicates the abundance of the SINV-STR after each passage (P0 = cells transfected with IVT RNA; P1 to Px = cells transfected with cellular RNA). (C) Competitive inhibition of a SINV-STR by cellular RNA of serially transfected cells. Total cellular RNA of mock electroporated K562 cells or total cellular RNA of passages P1 and P3 were mixed with very low amount (250 pg) of uncapped IVT SINV-STR coding for luciferase. Upon co-electroporation with SFV-replicase nrRNA into K562 cells, luciferase expression was measured after 24 h. Data are shown as the expression relative to the mock control. (D) Identification of a 5′-extended SFV-STR enriched upon serial transfer. Cellular RNA was extracted from cells that were serially transfected 4 times with the STR mixture containing 0.0016% SINV-STR. The RNA was reverse transcribed, and the resulting cDNA was used to amplify the 5′ end of STRs. 5′ end PCR products were subcloned and sequenced. SFV-STRs extended at the 5′ end by AUAAAA or AUAAAAA were found in 59% of the clones.
To test for the enrichment of a dominant STR, we supplemented the total cellular RNA of P1 and P3 cells with a low amount of an IVT luciferase encoding SINV-STR and co-transfected this mixture with SFV-replicase nrRNA. Compared with total RNA of mock-transfected cells, we observed almost a complete loss of SINV-STR expression with the P3 RNA (Figure 2C), which strongly suggested the emergence of a competing STR. Since the 3′ CSE of alphaviruses is short and strongly conserved, we assumed that a competing STR most likely acquired mutations within the longer and more complex 5′ CSE. Indeed, 5′ end sequencing revealed that the STR pool was dominated by a 5′-extended SFV-STR (eSTR), the extension being 5′-AUAAAA-3′ (32 of 66 clones) or 5′-AUAAAAA-3′ (7 of 66 clones) (Figure 2D). Similar extensions have been found in pseudorevertant chimeric SFV/SINV viruses upon serial transfer.15 Interestingly, these extensions were added upstream to the 5′ terminal G that is required for efficient IVT of the STRs but were not present in the viral genomic RNA.
eSTRs competitively inhibit co-transfected STRs and saRNA, replicate faster, and increase expression of taRNA
To characterize the evolved SFV-derived 5′-extended STRs (eSTRs), we cloned eSTRs with the more frequent 5′ extension 5′-AUAAAA-3′ (AUA4) into plasmids for IVT. Since the eSTR was identified in a pool of STRs outcompeting the formerly dominant SINV-STR, we first assessed whether an IVT eSTR inhibits the co-replication of a SINV-STR. To this aim, SFV-replicase nrRNA, a SINV-STR encoding firefly luciferase, and either the parental SFV-STR or the eSTR were co-transfected in different molar ratios. The next day, luciferase expression confirmed that eSTRs strongly inhibit co-replicating SINV-STRs (Figure 3A). The same experiment further confirmed that replication of both the parental SFV-STR (Figure 3B) as well as a full-length SFV-saRNA (Figure 3C) were inhibited by the eSTR. Furthermore, the eSTR was amplified much faster in response to SFV-replicase nrRNA. Within 3 h, the eSTR reached copy numbers comparable to the parental SFV-STR after 24 h and produced copies at an order of magnitude greater after 24 h (Figure 3D). This greatly enhanced replication of eSTRs was reflected by the transgene expression, which was kept at high levels with much lower amounts of transgene encoding eSTR. The lower the dose of eSTR, the greater was the copy benefit compared with the STR, culminating in a 20-fold increase of the transfection rate with the lowest dose tested (Figure 3E). Thus, directed evolution of the STR by serial RNA transfer improved the performance of taRNA in vitro, allowing a reduction in the resultant eSTR dose of at least 10-fold while maintaining high, consistent expression. Furthermore, the eSTR improved taRNA expression not only in K562 cells but also in antigen-presenting primary human immature dendritic cells (hiDCs), primary human foreskin fibroblasts (HFFs), and rat and mouse fibroblast cell lines (Rat-2, 3T3-L1) (Figure 3F). Altogether, we conclude from these findings that the SFV-derived eSTR is a promising candidate for improving SFV-taRNA vaccine performance.
Figure 3.

eSTRs competitively inhibit co-transfected STRs and saRNA, replicate faster, and increase expression of taRNA
(A and B) Expression of a SINV- or an SFV-STR in the presence of eSTRs. K562 cells were co-electroporated with 3.3 nM capped SFV-replicase nrRNA and 0.033 to 3.3 nM capped wild-type (WT) SFV-STRs or capped eSTRs (5′ extension AUAAAA) expressing GFP-SecNLuc. Capped WT SINV-STR (A) or capped SFV-STR (B) expressing firefly luciferase (0.33 nM) were added, and luciferase expression was measured 24 h after transfer and normalized to the expression of the uncompeted control. (C) Inhibition of SFV saRNA by eSTRs. K562 cells were co-electroporated with 3.3 nM capped SFV saRNA encoding firefly luciferase and 0.033 to 3.3 nM capped WT SFV-STR or capped eSTRs. Controls were electroporated with saRNA alone and used to normalize the luciferase expression data measured 24 h after transfection. Data are shown as mean and SD of 3 independent experiments (A–C). (D) Replication rate of eSTR compared with STR. BHK21 cells were co-electroporated with 3.3 nM capped SFV-replicase nrRNA and 0.33 nM capped WT SFV-STR or capped eSTR encoding firefly luciferase. RNA was extracted at indicated time points, and STR was quantified by qRT-PCR. Controls were co-electroporated with inactive replicase and levels of STRs found in these cells used to calculate the factor of amplification at each time point. Data of three independent experiments are shown as individual line graphs. (E) Expression of taRNA with STR and eSTR. K562 cells were co-electroporated with 3.3 nM capped SFV-replicase nrRNA and 0.033 to 3.3 nM capped WT SFV-STR or capped eSTR. GFP expression resulting from STRs was assessed by flow cytometry 24 h after transfer. Data from three independent experiments are shown (mean ± SD). Unpaired Student’s t test was performed (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). (F) Improvement of taRNA expression by eSTR in various cell types and species. Human immature dendritic cells (hiDCs), human foreskin fibroblast (HFFs), Rattus norvegicus fibroblasts (Rat2), and Mus musculus fibroblasts (3T3-L1) were seeded into 96-well plates lipofected with 20 ng taRNA encoding firefly luciferase per well (19.6 ng capped SFV-replicase nrRNA and 0.4 ng capped SFV-STR or eSTR), using 80 nL MessengerMax per well. 24 h after transfection, luciferase expression levels were measured and the fold increase in expression by eSTR over STR calculated for each cell type. Relative expression (eSTR/STR) of three independent experiments are shown (mean ± SD).
eSTRs improve vaccination with SFV-taRNA
Having achieved significantly improved taRNA performance in tissue culture, we investigated whether the eSTR improves taRNA vaccination outcomes in mice. We thus inserted influenza A virus hemagglutinin (HA) into the parental SFV-STR and eSTR and synthesized uncapped (e)STR-RNA. For intradermal (i.d.) co-injection, we mixed 20 μg of the capped nrRNA-SFV replicase and 50 ng of the uncapped STR-HA or eSTR-HA per animal in PBS. As a reference group, we used 20 μg of the same batch of nrRNA replicase and 50 ng uncapped SFV-TR-HA, which effectively induced immune responses in our previous study.6 After a prime-boost regimen, blood was collected to obtain sera. The endpoint titration of HA-specific antibodies revealed a significantly greater concentration of circulating HA-specific immunoglobulin G (IgG) in the peripheral blood of mice vaccinated with eSTR compared with the TR reference (Figure 4A). Importantly, increased antibody concentrations were reflected by significantly increased influenza virus neutralizing titers (VNTs) when comparing eSTR- with TR-vaccinated mice (Figure 4B). Although differences in both peripheral IgG titers and VNTs between STR and eSTR were not statistically significant, the immune responses using eSTR were nevertheless elevated compared with STR.
Figure 4.

eSTRs improve vaccination with SFV taRNA
BALB/c mice (group sizes n = 5) were immunized at days 1 and 29 by intradermal injection of 50 ng uncapped SFV-TR, SFV-STR, or eSTR encoding the influenza A virus hemagglutinin (HA) together with 20 μg capped nrRNA encoding SFV replicase. Negative controls received 20 μL buffer without RNA. Sera for the quantification of IgG titer were sampled on day 55. (A) Total anti-HA IgG antibody in sera of responding mice were determined by ELISA. Unpaired Student’s t test was performed for each dilution step (TR vs. eSTR: ∗p < 0.05, ∗∗p < 0.01; TR vs. STR: all non-significant). (B) Virus neutralization test (VNT) titer of each responding animal is displayed, with the limit of detection of 10. All animals receiving PBS alone were non-responding, and therefore data are not shown. For all other groups, the number of responding over total number of animals is given below the x axis. One-way ANOVA was performed (∗p < 0.05; ns p > 0.05).
In summary, we show that serial RNA transfer is an effective method to select for trans-replicating RNA with greatly enhanced amplification rates. Using this approach, we vastly reduced the required amount of trans-replicating RNA while conserving high expression levels of taRNA in vitro; moreover, immunization with the eSTR improved the immune response in vivo.
Discussion
In this study, we made alterations to the trans-replicating RNA part of the taRNA split-vector system with a view to optimize its replication efficiency. Our first measure was to shorten the TR and to delete the SGP, which simplified its structure and replication process. As a second measure, we used directed evolution to identify RNAs that trans-replicate faster. Thereby, we generated a taRNA with improved replication efficiency and speed, leading to enhanced immune responses in vivo.
First, we reworked the sequence design of the TR in two steps that resulted in a shorter TR (accordingly called STR) that structurally closely resembled an mRNA. We removed remnants of the nsP4 ORF including the SGP, thereby eliminating almost 1 kb of the RNA backbone. Furthermore, we mutated the original nsP1 start codon, as well as further AUG triplets within the 5′ CSE, to prevent translation initiation upstream of the transgene.12 Thus, we removed all potential start codons from the 5′ CSE, an essential modification for clinical application. In doing so, we created STRs that are translatable directly upon IVT and capping, thus, in essence, these STRs resemble conventional mRNA, which is efficiently amplified in trans. taRNA expression improved upon this TR redesign, and expression in the presence of inactive replicase showed that an STR may facultatively be used as standalone mRNA or replicating RNA.
Although cross-utilization of template RNA by alphavirus replicases has been observed since the early 2000s,12 this phenomenon was recently reported in greater detail in mammalian and insect cells13 and observed to be controlled by the nsP4 subunit.16 Inspired by prior publications, we found that the SFV replicase amplified SINV-derived (S)TRs more efficiently than SFV-(S)TRs. In our study, the heterologous alphaviral STRs were replicating more efficiently than the SFV-derived STR, except for the CHIKV-STR. We found it remarkable that so many heterologous substrates outcompeted the SFV-derived substrate, which is the only one shaped and adapted to the SFV replicase by viral evolution. Possibly, higher levels of genomic RNA replication in the case of SFV would be detrimental to viral fitness. However, we have to emphasize that we screened exclusively STRs lacking SGPs. A comparable collection of TRs with an SGP of the respective virus could alter the spectrum and magnitude of substrate RNA expression. Furthermore, we cannot exclude that the arbitrary 3′ UTR length of 100 nucleotides biased some results, nor can we exclude that the parental SFV strain of the replicase that we chose for this study differs from other SFV strains. Overall, the replication of a number of heterologous STRs in response to the SFV replicase demonstrates that taRNA is a very flexible platform. The replicating template and the replicase can originate from different viruses, and non-natural combinations can be freely made and may even improve the vector platform.
The flexibility of taRNA also allowed us to exploit a directed evolution based on serial transfer of the replicating template RNA, thereby selecting for templates with increased replication rates. In essence, the serial RNA transfer greatly extended the time for replication. Upon a single transfection, taRNA expression peaks within 24 h.6,16 Thus, extraction and retransfection after 24 h keep replication at high rates, facilitating the accumulation of faster-replicating mutant templates. In our experiment, SFV-derived STRs acquired accelerated replication through the addition of 5′ terminal nucleotides. Since these extensions were upstream of the 5′ terminal G of the IVT STR, we conclude that these nucleotides were added by the replicase in a template-independent manner, which has been observed before.15 Since the SFV genomic RNA lacks such extensions, this is another argument for evolutionarily balanced genomic RNA replication, which does not necessarily result in the fastest possible RNA amplification. An accelerated amplification of SFV-saRNA would also accelerate the accumulation of transcripts encoding the cytotoxic SFV replicase. In contrast to saRNA, the taRNA split vector system liberates RNA replication from replicase gene amplification. Therefore, template replication in trans possibly can reach higher rates, at least when the transgene is non-toxic.
The 5′ eSTR not only replicated faster compared with the parental STR but also proved to be a strong competitive inhibitor of the formerly best replicating SINV-STR, the SFV-STR, and of an SFV-saRNA. Competing with other STRs is probably the direct result of faster replication, but the inhibition of the SFV-saRNA by the eSTR indicates that the 5′ extension could also compete with cis preferential replication, possibly by increased affinity to replicase, which could also be the case for the SINV-STR. Moreover, it is reasonable to assume that accelerated replication of eSTRs increases the likelihood of initiating productive replication in individual cells. Accordingly, we observed greater transgene expression rates when the eSTR was subjected to limiting titrations. In accordance with in vitro improved taRNA expression in cell types that mount an effective innate immune response, the immune response of taRNA-vaccinated mice was increased by eSTR. However, the immunization with naked RNA was overall not very robust, leading to some non-responding animals, which reduces the reliability of statistical analysis. Nevertheless, we would conclude that the combined effects of the TR simplification together with the eSTR’s 5′ extensions provide a significantly improved version of taRNA. Screening for appropriate formulations will be a necessary next step to optimize taRNA-based vaccination, but this was beyond the scope of this study. Here, the size difference of the two RNAs that need to be co-transferred into individual cells for being effective challenges formulation development. Furthermore, future work to improve taRNA performance should also combat the inhibitory action of innate immunity, similar to approaches applied to saRNA.17,18 Nevertheless, mice receiving taRNA with STRs and eSTRs developed greater antibody titers compared with the parental first generation TR, indicating that the simplified structure of STRs was overall beneficial and an improvement of taRNA.
In summary, we demonstrate that taRNA is a very flexible modular vector system that allows assembly of components of heterologous viral origin. Furthermore, in vitro molecular evolution occurs rapidly in this system, promising that similar studies with appropriate selective pressures may further improve the vectors. We believe that the simplified, mRNA-like design of STRs developed in this study paves the way for future optimization of taRNA for use as a vaccine platform.
Materials and methods
Plasmids and RNA
Plasmids serving as templates for in vitro transcription of mRNA encoding the SFV replicase (accession number NCBI: KP699763), an inactive replicase variant, and first-generation SFV TRs were constructed and have been previously described.6 The coding sequences for transgenes and replicases were preceded by the Kozak consensus sequence 5′-GCCACC-3′. Template plasmids for in vitro synthesis of STRs were designed in silico and empty vectors ordered by custom gene synthesis; the sequences flanking the transgenes are provided as supplemental information (Table S1). Genes of interest were inserted using appropriate restriction sites.
Downstream of the alphaviral 3′ CSEs, all STR plasmids encoded a poly(A) tail followed by a type IIS restriction enzyme site SapI to generate an unmasked poly(A) tail.19
Synthesis and purification of RNA have been previously described.19,20 TR or STRs were either left uncapped or co-transcriptionally capped with the beta-S-ARCA cap.21,22 Replicase-coding nrRNA was co-transcriptionally capped using the beta-S-ARCA cap. Detailed RNA-capping information is given in each figure legend. RNA concentration and purity were assessed by spectrophotometry (NanoDrop 2000c, Thermo Fisher Scientific). Typical reaction yields are 5–10 μg RNA per μL reaction volume irrespective of the type or length of RNA. RNA integrity of synthetic RNA was assessed by capillary electrophoresis (Fragment Analyzer; Agilent).
Cell culture
All growth media, fetal calf serum (FCS; Sigma), and supplements were supplied by Life Technologies/Gibco or HiMedia Laboratories. BHK21 cells (ATCC; CCL10) were grown in minimum essential medium, and K562 cells (ATCC; CCL-243) were grown in RPMI medium. Media of both cell types were supplemented with 10% FCS. HFFs were obtained from System Bioscience (HFF, neonatal) cultivated in minimum essential media (MEMs) containing 15% FCS, 1% non-essential amino acids, and 1 mM sodium pyruvate. Rat2 (ATCC; CRL-1764) and 3T3-L1 fibroblasts (ATCC; CL-173) were cultivated in Dulbecco’s modified Eagle medium containing 10% FCS.
hiDCs were generated by differentiation of CD14-positive cells isolated from peripheral blood mononuclear cells by magnetic cell sorting as described previously19,23 and cultivated in RPMI medium supplemented with 5% plasma-derived pooled human serum (OneLambda), 1% non-essential amino acids, 1 mM sodium pyruvate, 1,000 U/mL human interleukin-4 (IL-4) premium grade (Miltenyi Biotec), and 1,000 U/mL human GM-CSF premium grade (Miltenyi Biotec). All cells were grown at 37°C in humidified atmosphere equilibrated to 5% CO2.
RNA transfection
Electroporation of RNA (nrRNA, saRNA, and taRNA) was carried out at room temperature using X-Vivo 15 serum-free medium (Lonza) as electroporation buffer and applying defined pulses with a square-wave electroporator (BTX ECM 830, Harvard Apparatus). BHK21 cells were electroporated at 750 V/cm with one pulse of 16 ms; K562 cells were electroporated at 650 V/cm with 3 pulses of 8 ms interrupted by 400 ms intervals. Molarities or amounts of RNAs used in the experiments are indicated in the figure legends. Lipofections were done using Lipofectamine MessengerMax (Thermo Fisher Scientific) following the manufacturer’s instructions. To this aim, cells were plated into 96-well plates at the following densities in 100 μL growth medium: K562 cells: 40,000 cells/well; hiDC, HFF, Rat2, and 3T3-L1: 10,000 cells/well. For each well, 0.4 μL MessengerMax diluted in 5 μL OptiMEM medium (Thermo Fisher Scientific) was mixed with 100 ng total RNA in 5 μL OptiMEM and added to plated cells. As indicated in the figure legends, some experiments were done using reduced RNA amounts (20 ng) and 80 nL MessengerMax per well.
Luciferase and viability assay
Firefly luciferase or secretable nano-luciferase expression was assessed using either the Bright-Glo Luciferase assay system or the NanoGlo assay system (both Promega) according to the manufacturer’s instructions. For more information about the NanoLuc reporter, please refer to literature.24 Viability of transfected cells was assessed using a luminescence-based method assaying the ATP concentration after 48 h (CellTiter-Glo assay; Promega) according to the instructions of the manufacturer. Relative viability was calculated by normalizing the value of each sample to the signal of a reference sample indicated in the respective figure legends. Bioluminescence (photons per second [p/s]) of all assays was measured using a microplate luminescence reader Infinite M200 (Tecan Group).
Flow cytometric analysis
A FACS Canto II flow cytometer (BD Bioscience) was used to determine fluorescent protein expression. To this aim, the cells were harvested, washed with PBS, and fixed with PBS containing 4% formaldehyde. Data were analyzed using the FACSDiva or FloJo v.10 software (both BD Bioscience).
Serial RNA transfer
IVT STRs of SFV, FMV, AURAV, HJV, MADV, and CHIKV encoding a GFP-SecNLuc fusion reporter were mixed in equal amounts (0.3 μg each). A SINV-STR encoding a GFP-iRFP double-fluorescent reporter was spiked into this mixture at a 1:100 to 1:100,000 lower amount. For the first electroporation, 4 μg nrRNA coding for the SFV replicase was mixed with 1.8 μg of the STR mixture and electroporated into 1.2 × 107 K562 cells. The cells were cultivated overnight, and reporter gene expression was assessed the next day with a small sample of the cells using flow cytometry. Total RNA of the cells was extracted using the RNeasy Mini Kit (Qiagen) and quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Ten μg total RNA was mixed with 4 μg IVT nrRNA-SFV replicase and used to electroporate K562 cells. This procedure was repeated 4 times.
Quantitative real-time reverse-transcriptase PCR (RT-PCR)
To assess RNA amplification rates, cells were washed once and then lysed directly in the plates. To this aim, 20,000 cells were lysed with 50 μL iScript RT-qPCR Sample Preparation Reagent (BioRad). One μL iScript lysate was reverse transcribed using Superscript IV reverse transcriptase (Invitrogen) and oligo-dT18. Quantitative real-time RT-PCR was performed using the ABI 7300 Real-time PCR System, the companion SDS analysis software (Applied Biosystems), and the QuantiTect SYBR Green PCR Kit (Qiagen). Protocol followed the manufacturer’s instruction with 15 min at 95°C and 40 cycles of 30 s at 95°C, 30 s at oligo specific annealing temperature stated below, and 30 s at 72°C. Analysis was performed using the 2-ΔΔCT method,25 normalized to the housekeeping gene HPRT1. The following specific primers and annealing temperatures were used. GFP, forward 5′-ATCCGCCACAACATCGAGGAC-3′, reverse 5′-CTCCAGCAGGACCATGTGATC-3′ (62°C); luciferase, forward: 5′-TACGTTAACAACCCCGAGGC-3′, reverse: 5′-CAGGATGCTCTCCAGTTCGG-3′ (60°C); HPRT1, forward: 5′-TGACACTGGCAAAACAATGCA-3′, reverse: 5′-GGTCCTTTTCACCAGCAAGCT-3′ (62°C).
RNA 5′ end sequencing
To sequence the 5′ CSE region of eSTR including the 5′ end, we applied SMART cDNA synthesis (Switching Mechanism at 5′ end of RNA transcript)26 using the Mint-2 cDNA synthesis kit (Evrogen), followed by a specific PCR to amplify the 5′ CSE region. The PCR product was inserted seamlessly into pST1 using ColdFusion cloning (System Biosciences). E. coli cells were transformed, and 96-clones were chosen arbitrarily for plasmid preparation. The inserts were identified by Sanger sequencing.
Animals
Female Balb/c_Rj mice were purchased at an age of 7 weeks (Janvier Labs). German laws and guidelines for animal welfare were respected, and experiments were approved by the local authorities (Landesuntersuchungsamt Koblenz/Rhineland-Palatinate, Germany).
Animal immunizations
Immunization of mice were done as described before.6 Briefly, mice were put under isoflurane anesthesia, and single i.d. injections at the shaved flank were performed on days 1 and 29. RNAs were mixed as described in the results and resolved in a total volume of 20 μL RNAse-free PBS. Controls received buffer alone.
Virus neutralization test
For antibody analysis, blood samples were taken via the vena facialis on day 55 after the first immunization. Virus neutralization test (VNT) to determine VN antibodies in the serum was performed based on the WHO’s Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza (WHO Global influenza Surveillance Network) and as described.6 Briefly, serum levels of HA-neutralizing antibodies were quantified by incubating serial dilutions of the mouse sera for 2 h with 100 tissue culture infection dose 50 (TCID50) of active Cf4/H1N1 influenza virus generated as described6 and then applied onto a confluent MDCK cell monolayer in 96-well plates. Upon incubation for 3 days with MDCK cell culture, supernatant was harvested and mixed 1:2 with 0.5% chicken red blood cells (RBCs; Lohmann Tierzucht, Cuxhaven, Germany). RBC agglutination was visually observed in round-bottom 96-well plates, and the VN serum titer was recorded as the inverse of the lowest dilution that inhibited agglutination (VNT titer/50 μL serum).
ELISA
For ELISA, recombinant, His-tagged influenza HA (A/California/04/2009 (Cf4/H1N1)) (Sino Biological) was biotinylated utilizing the EZ-Link Sulfo-NHS-LC-Biotinylation kit according to supplier’s protocol (Thermo Fisher Scientific). 96-well streptavidin plates (Thermo Fisher Scientific) were coated with the biotinylated HA protein at 4°C overnight. Upon washing and blocking, serum samples were screened for HA-specific antibodies by incubation on plates for 1 h at 37°C while shaking at 350 RPM. After incubation and including assay controls, plates were washed again and incubated with HRP-labeled secondary anti-mouse IgG antibody (Jackson Immuno Research) for another 45 min at 37°C before TMB one substrate (Kementec) was applied after another washing step. Upon stopping the reaction with 25% sulfuric acid (Merck), colorimetric detection was monitored using the EPOCH 2 Microplate Reader (BioTek) and optical density read at 450 nm calculated to a wavelength reference of 620 nm (Δ450–620nm). ELISA were performed in duplicates for each sample and normalized to background signals obtained from a non-coated control plate.
Statistics
The data of independent experiments were summarized and displayed as mean ± standard deviation. All statistical analyses were performed with GraphPad Prism 9. Tests applied to the experiments are mentioned in the respective figure legends. No statistical methods were applied to predetermine sample size for animal experiments.
Acknowledgments
The authors would like to gratefully thank all involved team members of TRON’s service units Cloning and Animal Models for excellent support in vector cloning and animal experiments. Further, we thank Janina Vogt and Klara Zwadlo for performing VNT assays and helping us to establish anti-HA ELISA and Lien Cao for supporting cell culture experiments. We would like to acknowledge Fulvia Vascotto for detailed proofreading of the manuscript and Karen Chu for copy editing.
Author contributions
Conceptualization, T.B. and M.P.; methodology, T.B., M.P., and S.G.; investigation, S.G., T.H., S.B., and E.N.; writing, reviewing, and editing, T.B., M.P., S.G., and U.S.; visualization, T.B., M.P., and E.N.; supervision, U.S. and T.B.
Declaration of interests
U.S., T.B., and M.P. are inventors on patents and patent applications, which cover parts of this article. U.S. is employee at BioNTech Corporation (Mainz, Germany), a privately owned company developing therapeutic RNA.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.01.019.
Contributor Information
Ugur Sahin, Email: ugur.sahin@biontech.de.
Tim Beissert, Email: tim.beissert@tron-mainz.de.
Supplemental information
Data availability
All data supporting the statements and conclusions made by the authors are included in the figures of the manuscript and in the supplemental information provided online at the journal’s homepage.
References
- 1.Kumar A., Blum J., Thanh Le T., Havelange N., Magini D., Yoon I.-K. The mRNA vaccine development landscape for infectious diseases. Nat. Rev. Drug Discov. 2022;21:333–334. doi: 10.1038/d41573-022-00035-z. [DOI] [PubMed] [Google Scholar]
- 2.Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Pérez Marc G., Moreira E.D., Zerbini C., et al. Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. N. Engl. J. Med. 2020;383:2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simon-Loriere E., Schwartz O. Towards SARS-CoV-2 serotypes? Nat. Rev. Microbiol. 2022;20:187–188. doi: 10.1038/s41579-022-00708-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloom K., van den Berg F., Arbuthnot P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021;28:117–129. doi: 10.1038/s41434-020-00204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kis Z., Kontoravdi C., Shattock R., Shah N. Resources, production scales and time required for producing RNA vaccines for the global pandemic demand. Vaccines. 2020;9:3. doi: 10.3390/vaccines9010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beissert T., Perkovic M., Vogel A., Erbar S., Walzer K.C., Hempel T., Brill S., Haefner E., Becker R., Türeci Ö., Sahin U. A trans-amplifying RNA vaccine strategy for induction of potent protective immunity. Mol. Ther. 2020;28:119–128. doi: 10.1016/j.ymthe.2019.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuller D.H., Berglund P. Amplifying RNA vaccine development. N. Engl. J. Med. 2020;382:2469–2471. doi: 10.1056/NEJMcibr2009737. [DOI] [PubMed] [Google Scholar]
- 8.Vogel A.B., Lambert L., Kinnear E., Busse D., Erbar S., Reuter K.C., Wicke L., Perkovic M., Beissert T., Haas H., et al. Self-amplifying RNA vaccines give equivalent protection against influenza to mRNA vaccines but at much lower doses. Mol. Ther. 2018;26:446–455. doi: 10.1016/j.ymthe.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blakney A.K., Ip S., Geall A.J. An update on self-amplifying mRNA vaccine development. Vaccines. 2021;9:97. doi: 10.3390/vaccines9020097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lundstrom K. Self-replicating RNA viruses for vaccine development against infectious diseases and cancer. Vaccines. 2021;9:1187. doi: 10.3390/vaccines9101187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pietilä M.K., Hellström K., Ahola T. Alphavirus polymerase and RNA replication. Virus. Res. 2017;234:44–57. doi: 10.1016/j.virusres.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Frolov I., Hardy R., Rice C.M. Cis-acting RNA elements at the 5' end of Sindbis virus genome RNA regulate minus- and plus-strand RNA synthesis. RNA. 2001;7:1638–1651. doi: 10.1017/s135583820101010x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lello L.S., Utt A., Bartholomeeusen K., Wang S., Rausalu K., Kendall C., Coppens S., Fragkoudis R., Tuplin A., Alphey L., et al. Cross-utilisation of template RNAs by alphavirus replicases. Plos Pathog. 2020;16 doi: 10.1371/journal.ppat.1008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rozen-Gagnon K., Stapleford K.A., Mongelli V., Blanc H., Failloux A.-B., Saleh M.-C., Vignuzzi M. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. Plos Pathog. 2014;10 doi: 10.1371/journal.ppat.1003877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorchakov R., Hardy R., Rice C.M., Frolov I. Selection of functional 5' cis-acting elements promoting efficient sindbis virus genome replication. J. Virol. 2004;78:61–75. doi: 10.1128/JVI.78.1.61-75.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lello L.S., Bartholomeeusen K., Wang S., Coppens S., Fragkoudis R., Alphey L., Ariën K.K., Merits A., Utt A. nsP4 is a major determinant of alphavirus replicase activity and template selectivity. J. Virol. 2021;95 doi: 10.1128/JVI.00355-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beissert T., Koste L., Perkovic M., Walzer K.C., Erbar S., Selmi A., Diken M., Kreiter S., Türeci Ö., Sahin U. Improvement of in vivo expression of genes delivered by self-amplifying RNA using vaccinia virus immune evasion proteins. Hum. Gene Ther. 2017;28:1138–1146. doi: 10.1089/hum.2017.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blakney A.K., McKay P.F., Bouton C.R., Hu K., Samnuan K., Shattock R.J. Innate inhibiting proteins enhance expression and immunogenicity of self-amplifying RNA. Mol. Ther. 2021;29:1174–1185. doi: 10.1016/j.ymthe.2020.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holtkamp S., Kreiter S., Selmi A., Simon P., Koslowski M., Huber C., Türeci O., Sahin U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108:4009–4017. doi: 10.1182/blood-2006-04-015024. [DOI] [PubMed] [Google Scholar]
- 20.Kreiter S., Konrad T., Sester M., Huber C., Türeci O., Sahin U. Simultaneous ex vivo quantification of antigen-specific CD4+ and CD8+ T cell responses using in vitro transcribed RNA. Cancer Immunol. Immunother. 2007;56:1577–1587. doi: 10.1007/s00262-007-0302-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grudzien-Nogalska E., Jemielity J., Kowalska J., Darzynkiewicz E., Rhoads R.E. Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells. RNA. 2007;13:1745–1755. doi: 10.1261/rna.701307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuhn A.N., Diken M., Kreiter S., Selmi A., Kowalska J., Jemielity J., Darzynkiewicz E., Huber C., Türeci O., Sahin U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010;17:961–971. doi: 10.1038/gt.2010.52. [DOI] [PubMed] [Google Scholar]
- 23.Orlandini von Niessen A.G., Poleganov M.A., Rechner C., Plaschke A., Kranz L.M., Fesser S., Diken M., Löwer M., Vallazza B., Beissert T., et al. Improving mRNA-based therapeutic gene delivery by expression-augmenting 3' UTRs identified by cellular library screening. Mol. Ther. 2019;27:824–836. doi: 10.1016/j.ymthe.2018.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall M.P., Unch J., Binkowski B.F., Valley M.P., Butler B.L., Wood M.G., Otto P., Zimmerman K., Vidugiris G., Machleidt T., et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012;7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Zhu Y.Y., Machleder E.M., Chenchik A., Li R., Siebert P.D. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. BioTechniques. 2001;30:892–897. doi: 10.2144/01304pf02. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the statements and conclusions made by the authors are included in the figures of the manuscript and in the supplemental information provided online at the journal’s homepage.