

Abstract
As bacteria transition from exponential to stationary phase, they change substantially in size, morphology, growth and expression profiles. These responses also vary between individual cells, but it has proved difficult to track cell lineages along the growth curve to determine the progression of events or correlations between how individual cells enter and exit dormancy. Here, we developed a platform for tracking more than 105 parallel cell lineages in dense and changing cultures, independently validating that the imaged cells closely track batch populations. Initial applications show that for both Escherichia coli and Bacillus subtilis, growth changes from an ‘adder’ mode in exponential phase to mixed ‘adder–timers’ entering stationary phase, and then a near-perfect ‘sizer’ upon exit—creating broadly distributed cell sizes in stationary phase but rapidly returning to narrowly distributed sizes upon exit. Furthermore, cells that undergo more divisions when entering stationary phase suffer reduced survival after long periods of dormancy but are the only cells observed that persist following antibiotic treatment.
Bacteria in natural environments experience periods of starvation and stress, punctuated by the arrival of new nutrients that then are depleted again as cells grow and divide1,2. Many mechanisms have therefore evolved to help bacteria weather the busts and exploit the booms3–5. Traditionally, these have been studied by inoculating cultures of stationary phase cells into fresh medium and following them along the growth curve back into stationary phase6–8. However, batch assays consider average properties of cells, whereas single-cell studies have revealed great cell-to-cell heterogeneity during stress9–11. This could be a side effect of saturated pathways displaying less heterogeneity than sub-saturated pathways, making all cells ‘happy the same way but unhappy in different ways’, but could also be an adaptive response to maximize inclusive fitness in uncertain times12—for example, by making a small fraction of cells persistent to drugs13. Either way, because the heterogeneity is so substantial and so profoundly changes the fate of stressed cells14, effective studies of bacterial responses to stress and starvation should ideally monitor individual cells as they enter and exit stationary phase.
This is challenging for several reasons: first, it is straightforward to sample cultures at different times, manually15, using flow cytometry16 or with microfluidic automation17, but such snapshots provide little information about the progression of events or the connection between heterogeneity and growth18,19. The dynamics of fluctuations are also important; for example, since rapidly changing heterogeneity is easily time-averaged by affected processes, while slowly changing heterogeneity can establish effective cell states20. Second, studies of heterogeneity are difficult to interpret or reproduce unless local conditions are tightly controlled, which is particularly challenging to achieve when components become limiting and conditions keep changing. Third, many important outliers in stress response and stationary phase can be exceedingly rare, making it important to sample large numbers of cells. Some microfluidic devices boost throughput by accumulating data over time21,22, but experiments during changing conditions must instead rely on parallelism.
Here we present a microfluidic platform based on the ‘mother machine’ design23 that addresses these challenges. We monitor physiology and gene expression of cell lineages over multiple consecutive rounds in and out of stationary phase, while ensuring that the cells imaged behave quantitatively as the cells in a connected batch culture that can be simultaneously sampled. This provides a microcosm of bulk growth with exceptional resolution and control while enabling conventional bulk assays on the same culture. Our current throughput of more than 100,000 cell lineages, imaging 108 cells per day, each every few minutes, is high enough that we observe spontaneous persisters24 without using special mutants25. The setup also works at very high cell densities—for example, at an optical density of 10 at 600 nm—both for the Gram-negative E. coli and Gram-positive B. subtilis. In a first application, we probed the size-regulation principles during entry into and exit from stationary phase, and demonstrated trade-offs between the behaviours when entering and exiting stationary phase. All of the experiments described in this Article were performed in Neidhardt defined medium26 (rich, EZ-rich defined medium; poor, MOPS buffered medium; recipes for both media are provided in Supplementary Note 19), further optimized to achieve optimum osmolarity for E. coli27 and B. subtilis28.
Results
A platform for studying single cells along a growth curve.
Platforms such as the mother machine23, in which individual cells grow and divide in narrow channels that are fed diffusively by orthogonal flow channels, can achieve exceptional spatial and temporal uniformity. However, these consider balanced growth in fresh medium, in stark contrast to microbiology experiments more broadly8,29,30. The challenge is that fluid handling for non-viscous and unchanging liquids is very different from the handling of dense and changing cell cultures needed to study stationary phase cells or ecosystems. Also, it is easy to starve cells on any platform by simply letting the medium run out, but for reproducibility and interpretability, the imaged cells should be starved the same way as in corresponding bulk experiments. We therefore built a system to flow dense cultures from conventional batch experiments into a mother machine type of microfluidic device, (Fig. 1a,b), such that cells loaded in the mother machine experience the same environment as cells in the batch31. Since cells can be retained in the device for hundreds of generations, this enables us to observe how individual cell lineages pass through multiple rounds of entering and exiting stationary phase (Supplementary Video 1). The process is fully automated, has virtually zero dead volume and provides near-perfect timing in relation to media changes (Supplementary Note 1). The optical density is monitored continuously using a custom detector in series with the flow path, to align any part of the observed single-cell data to the bulk optical density (Supplementary Notes 2 and 7). This enables us to correlate specific states of nutrient depletion and bulk properties with their corresponding single-cell phenotypes at any time.
Fig. 1 |. Accurate high-throughput measurement of cell-growth physiology and gene expression along the growth curve.
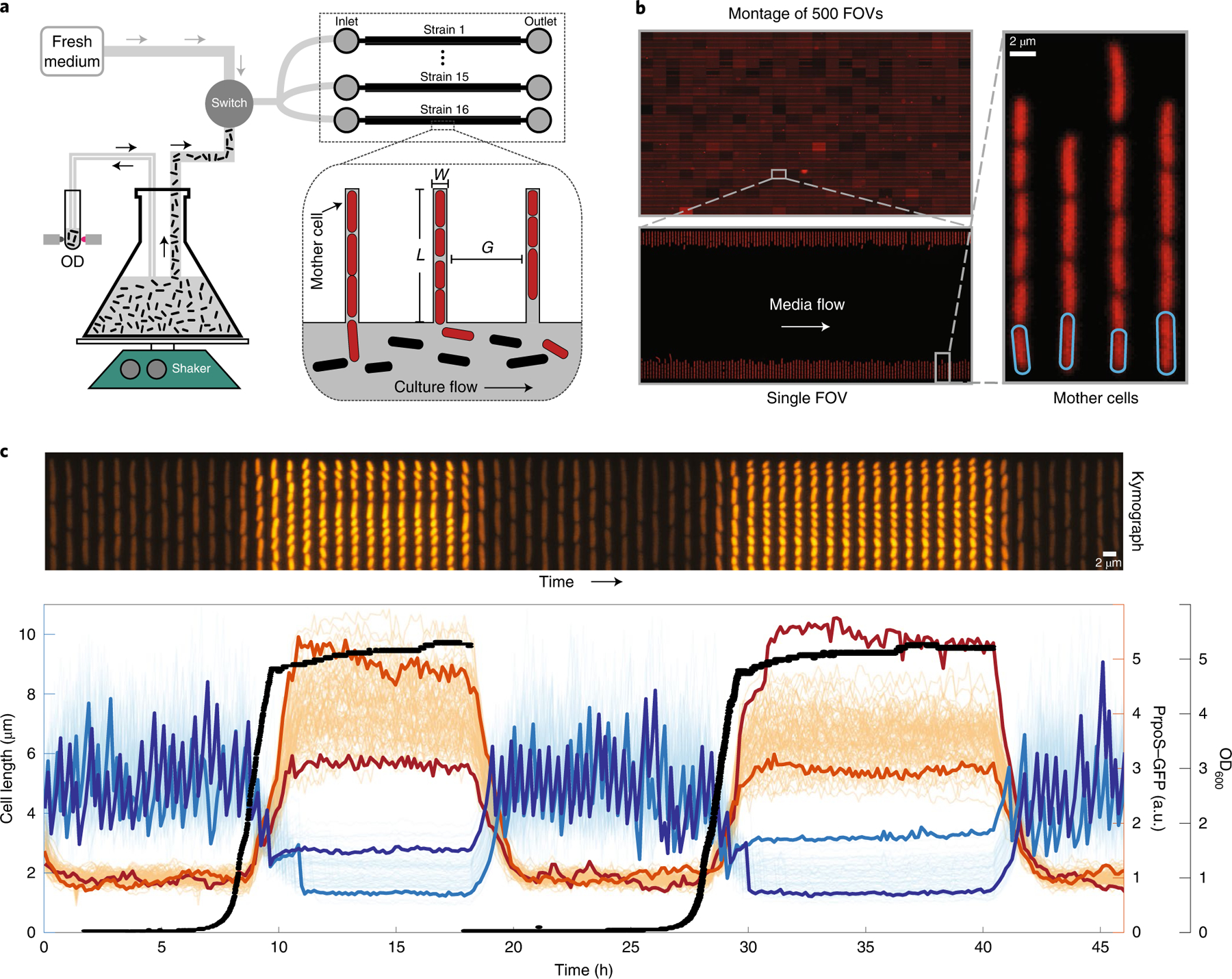
a, A simplified schematic depicting the growth-curve platform. The platform is based on the mother machine microfluidic device (right), in which cells under observation are grown in channels (shown in red) while liquid medium is pumped through an orthogonal flow channel. To observe growth dynamics, we flow actively growing bulk culture into the mother machine device while continuously observing its optical density (OD). As the bulk culture flows past (black cells) the red cells in the device (red) respond synchronously with the batch culture. At any point, the switch can enable flow of fresh medium, enabling observation of cells returning to optimal exponential growth. The dimensions (W, L and G in the inset) of the mother machine were highly optimized to meet the demanding requirements associated with flowing dense cultures through the flow channels. b, The optimizations made to the mother machine design have greatly improved the throughput. We can image up to 16 strains in parallel, with imaging of 705 FOVs, each containing 186 ± 1 (mean ± s.d.) lineages in under 5 min, giving a throughput of 131,072 (16 × 8,192) lineages imaged every 5 min, often for multiple days. We show a montage of 500 FOVs in the top left corner. The range of intensities present in the individual FOVs in the montage of FOVs has been increased for visualization purposes. c, Top: kymograph showing a single lineage of cells in a channel expressing a fluorescent RpoS transcriptional reporter as it goes through two consecutive rounds of growth curve (bottom). Bottom: 80 single-cell traces from a single FOV of RpoS expression (orange lines) and cell size (blue lines) as cells enter and exit from two consecutive rounds of stationary phase. Two cell-size traces and two expression traces are highlighted to illustrate high variability between the two rounds of stationary phase. The high-throughput measurements of each property enable us to measure accurate distributions of expression level and cell sizes at any time point along the growth curve. a.u., arbitrary units; OD600, optical density at 600 nm.
Several technical challenges must be overcome to ensure that cells in the mother machine are representative of the changing conditions in the flask: first, the shaking batch cultures must be separated from the microscope to prevent transmission of vibrations (Supplementary Note 3), and the flow path from flask to chip must be carefully designed to prevent biofilm formation, non-uniform temperatures, exposure of cells to surfaces other than inert tubing and cross-contamination between the fresh medium and culture paths. After extensive trial and error, our setup (Supplementary Note 1) addresses those challenges in a way that can be easily adapted for almost any setup. Second, shaking cultures contain bubbles that disrupt flow, requiring custom bubble traps (Supplementary Note 2). Third, maintaining proper diffusive feeding and retention of cells for several days is particularly difficult along the growth curve because cell volumes change markedly during entry and exit. We therefore evaluated 540 combinations of widths, heights and lengths of the narrow channels where cells grow and divide (L, W and G in Fig. 1a) to find near-optimal parameters for each growth condition (Supplementary Note 4). Fourth, since it is not possible to accumulate data over time, as for constant conditions, we systematically optimized the layout to maximize the number of cells imaged while maintaining effective feeding (Supplementary Note 6) and ensuring sufficient distances between channels to avoid overlapping point-spread functions of light between neighbouring lineages (Supplementary Note 5). The platform records cell size, morphology and changes in gene-expression levels as cells traverse the growth curve, while tracking either more than 8,000 lineages from each of 16 different strains or more than 120,000 lineages of a single strain (Supplementary Note 6), imaging every few minutes over the course of multiple days as cells experience multiple rounds of entry and exit from stationary phase (Supplementary Note 6). Because each channel contains multiple cells, this allows us to observe on the order of 100 million cell divisions per experiment—enough to observe many rare events and to accurately measure distributions of properties, while also correlating multiple events along the growth curve (Fig. 1).
Finally, our goal is not only to subject cells to some unknown level of stress and starvation, but also to observe cells along virtually the same growth curve as in the connected batch culture for quantitative comparisons. To confirm this, we first tracked the dynamics of cell size and a transcriptional reporter for the master regulator of stress response RpoS, PrpoS–green fluorescent protein (GFP), upon entry to stationary phase. As the density of the batch culture goes through a diauxic shift, the bulk growth rate drops momentarily, and both the cell length and PrpoS reporter fluorescence respond synchronously (Fig. 2a and Supplementary Video 2). Indeed, all observed bulk changes appear to affect the cells on the chip as expected and with minimal delay (Supplementary Note 8). To ensure quantitative similarity at the level of single cells, we compared cells on the chip to cells sampled from batch conditions for immediate microscopy. Cell length and PrpoS–GFP reporter activity at different time points along the growth curve (Fig. 2b) matched closely (Fig. 2c), to the point where the slight discrepancy of PrpoS dynamics reflected an imaging artefact in agar pads (Supplementary Notes 5 and 12), in which some of the light emitted by a cell is allocated to its neighbours—providing a further reason to use the microfluidic setup. Statistically representative sampling of cells exiting from stationary phase is difficult to achieve with traditional microscopy, since the cultures are so dilute. We therefore used a second microfluidic device, which makes it possible to take snapshots of cells from batch cultures with very high throughput32. The results again closely matched each other (Fig. 2d), showing that our platform quantitatively captures the single-cell dynamics of conventional batch culture.
Fig. 2 |. Tracking physiology and gene expression in single cells and bulk cultures.
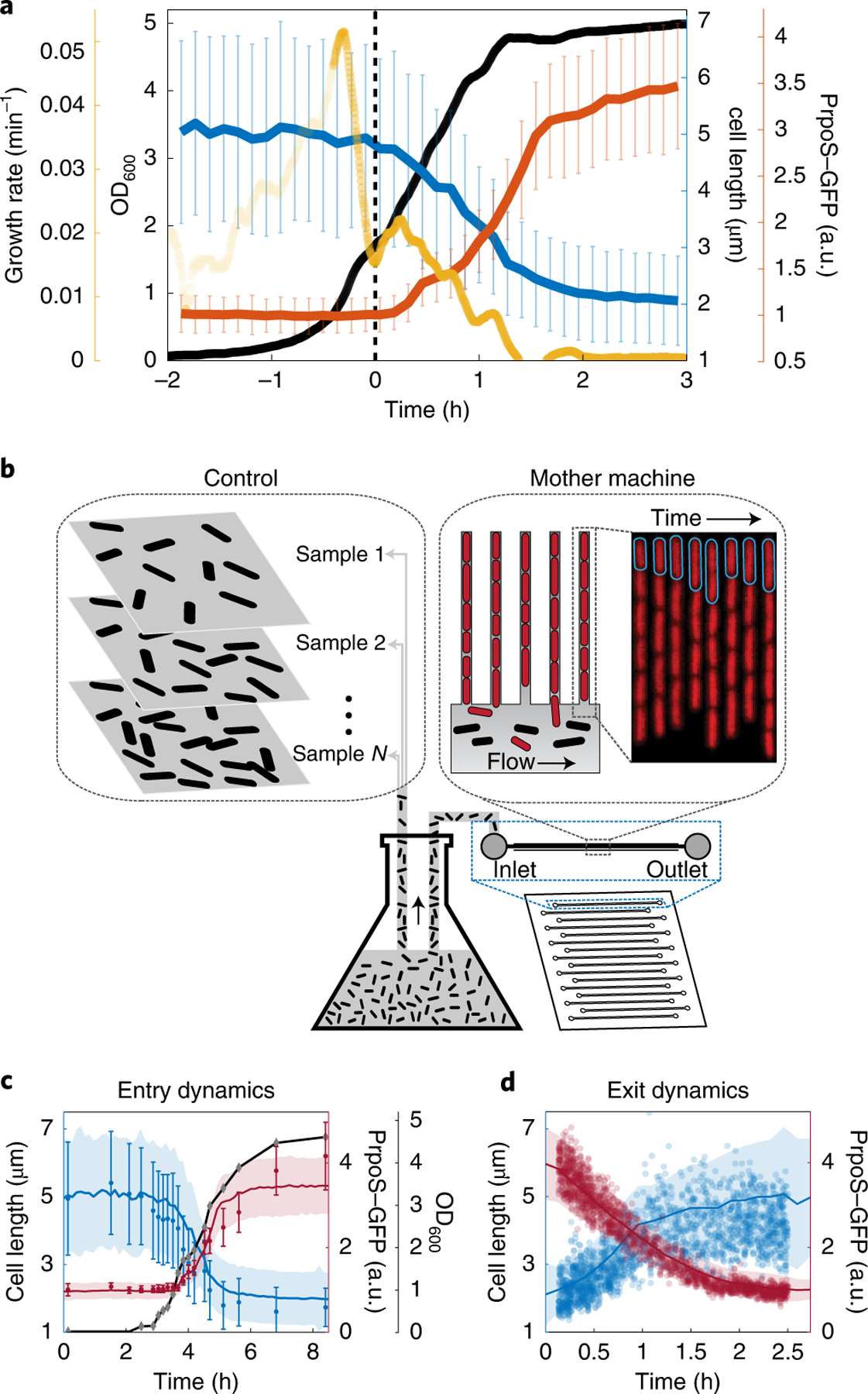
a, The bulk dynamics observed in the flask are synced with the dynamics of single cells in the mother machine. When the optical density (black) of the culture goes through an inflection point—such as the valley in bulk growth rate (yellow), represented by the rate of change in log(OD) per minute—there is a synchronous drop in the average cell length (blue) and an increase of RpoS transcriptional activity (red) of the cells in the mother machine device. To examine how well the cells in the mother machine channels mimic the cells grown in the bulk culture, we compared the average dynamics of cells in the mother machine with snapshots from cells grown in the flask. b, Schematic of the corresponding experimental setup. c, The average trend from the cells in the mother machine are plotted as solid lines (blue, cell length; red, RpoS transcriptional activity). Simple agar pad snapshots are compared with measurements in the mother machine during the entry to stationary phase. Black, OD600 of the culture during entry. d, During exit, snapshots were acquired with the microfluidics-assisted cell screening device28, which enables observation of very dilute cultures during exit from stationary phase. Values from individual cells are plotted as blue circles (length) and red circles (RpoS). In c and d, the shaded region (from the mother machine) and the error bars (from the agar pad) represent s.d. The average cell length from snapshots of bulk culture is generally shorter than average lengths in the mother machine, due to different age distribution, with more new cells in bulk culture compared with cells in the mother machine.
Size regulation during entry and exit from stationary phase.
The ability to quantitatively track growth and expression dynamics with single-cell resolution as bacteria enter and exit dormancy could provide important information about almost any process in the cell. We first applied it to study how cells modify their size along the growth curve, as they enter stationary phase from exponential phase and exit upon the introduction of fresh medium. Specifically, previous analyses have characterized where cell size regulation falls with respect to three simple phenotypes: adders, sizers and timers, which capture how individual cells in a population respond to being born smaller or larger than average. The adder mode is characterized by adding a constant cell volume each division (in the sense of being independent of birth size); the sizer mode is characterized by division at a fixed volume threshold, and timers are characterized by division after a fixed time interval33. To our knowledge, nearly all studies on size control have concluded that all bacteria, Archaea and eukaryotes closely follow adder-like phenotypes in a wide range of environments from fast growth to starvation34–37. However, growth phenotypes have—to our knowledge—not been tested as cells gradually enter into and exit from stationary phase.
We used a 100× apodized phase objective, imaging 2,215 lineages of E. coli MG1655 at 1 min intervals for more than 30 h to observe growth dynamics during balanced growth and then entry and exit from stationary phase. The use of the 100× apodized phase imaging with an oil-immersion objective and 1 min sampling rate reduces throughput but provides fine resolution and accuracy for the size-regulation study. We computed the specific growth rate (the rate of increasing cell mass) and the splitting rate (inverse of time between cell divisions) (Fig. 3a), as detailed in Supplementary Note 13. In balanced growth, the observed average specific growth rate (0.044 min−1) and splitting rate (0.043 min−1) are both similar to the estimated bulk growth rate (0.044 min−1) (Supplementary Notes 8 and 13). The coefficient of variance in specific growth rate (0.053) was approximately one-third the coefficient of variance in the splitting rate (0.16), in agreement with previous studies34. After the period of balanced growth, we observed an initial sharp drop in the specific growth rate followed by a brief increase (Fig. 3c) before it steadily declined to zero, as expected when cells go through a diauxic shift but then deplete all nutrients (Fig. 3b,c and Supplementary Note 8). By contrast, the splitting rate is refractory through the diauxic shift for several more divisions (Fig. 3c). This causes cells to produce daughters with ever decreasing sizes as cells enter stationary phase, causing the average cell size to decrease rapidly (Supplementary Note 14). A similar phenomenon has previously been observed in bulk experiments during nutrient downshifts and attributed to ‘rate maintenance’38, caused by the fact that cells can split into only two daughter cells once initiated rounds of replications are completed.
Fig. 3 |. Cell-size regulation during entry and exit from stationary phase reveals novel control principles.
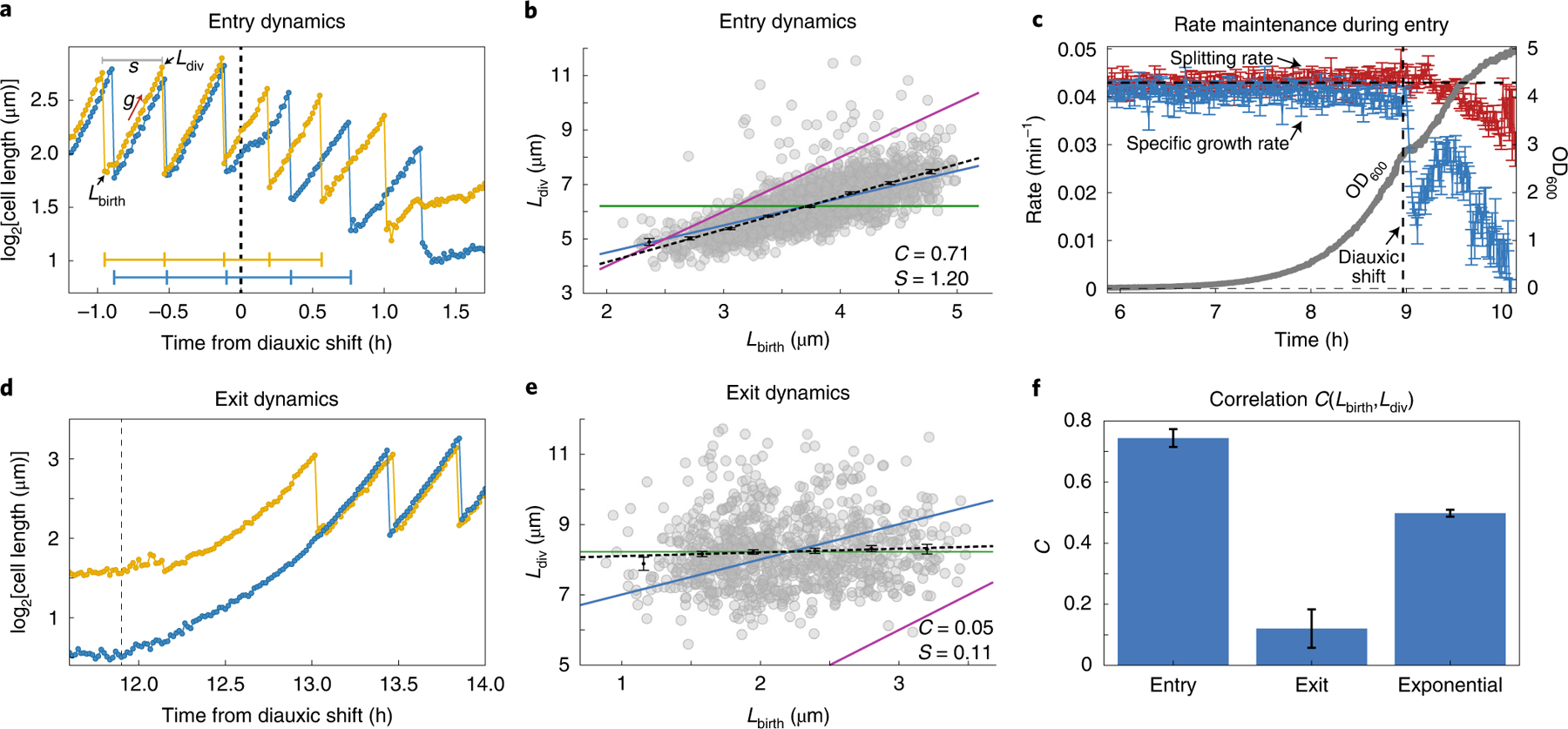
a, Cell lengths of two representative bacteria are shown (log scale) as they enter stationary phase after passing through diauxic shift. Individual cells slow their growth during the diauxic shift period. However, the interdivision times do not slow down, as shown by the horizontal blue and yellow lines that mark the division times for the two traces. Both the specific growth rate (g) and splitting rate (s) can be computed from a single trace (details are presented in Supplementary Note 13). Since the data are collected every 1 min, we get a high-resolution estimate of the size at birth (Lbirth) and size at division (Ldiv). b, Correlation between cell lengths at birth and at division are plotted during the entry to stationary phase (1.0 h from diauxic shift). The Pearson correlation coefficient (C) and the slope of the linear fit (black dotted line; S) are shown on the plot. Theoretical lines for the adder, timer and sizer models are shown as blue, purple and green lines, respectively. c, The average splitting rate and specific growth rate of the population are plotted in terms of number of doublings per minute at each time point of the experiment relative to the diauxic shift time point. During the entry to stationary phase, the cell splitting rate (red) changes gradually through the diauxic shift (dotted line), but the natural specific growth rate (blue) drops precipitously. d, Cell lengths of two bacteria during the exit from stationary phase. e, Correlation between cell length at birth and at division is shown during the exit from stationary phase. f, Summary of size-regulation analysis from 5 different growth-curve experiments. Cells behave as mixed adder–timers during entry to stationary phase (<Centry> = 0.74) and act as sizers during exit from stationary phase (<Cexit> = 0.12) and return to exponential phase where they behave as adders (<Cexponential> = 0.50). a,d, Time stamps are shifted with respect to diauxic shift (t = 0 at diauxic shift).
To investigate the size-regulation strategies used by cells in different growth phases, we report the Pearson correlation coefficient (C) between cell size33 at birth (Lbirth) and division (Ldiv). The correlation metric spans a continuum of phenotypes, in which the classic modes of timer, sizer and adder corresponds to three specific values (C = 1, C = 0 and C = 0.5). We first confirmed that the size regulation of our cells during balanced exponential growth was consistent with the adder mechanism (C = 0.54; Supplementary Fig. 9a). For non-balanced conditions, the analysis must be done carefully to avoid confounding effects from an average that changes systematically in time. We used the high throughput to avoid such effects by only comparing cells born in the same brief time interval, analysing their subsequent growth as a function of their varying initial size (Supplementary Note 10). Consistent with the rate-maintenance phenomenon, where cells maintained a constant division time even as they started to grow more slowly, the size-control phenotype appears to be a mix of an adder and timer (C = 0.71; Fig. 3b). During exit from stationarity, the cells started out with widely varying sizes but still underwent the first division at a uniform size; that is, with almost pure sizer dynamics (C = 0.05; Fig. 3e). We further calculated the slopes of the linear fit of division length versus birth length, observing slopes of 0.998 for exponential phase, 1.20 for entry to stationary phase and 0.11 for exit from stationary, consistent with the proposed adder, mixed adder–timer and sizer modes, respectively. We note that although the cells act as near-perfect sizers in the first division during exit, they then immediately switch back to adder mode from the second division onwards. The size-regulation strategies observed were also consistent with measurements at different temperatures (30 °C and 40 °C) (Supplementary Note 15).
To explore whether such size-control phenotypes are shared by other bacteria, we performed similar measurements on the Gram-positive model organism B. subtilis, which is separated from E. coli by a billion years of evolution. Experimental details are provided in Supplementary Note 17. Similar to that of E. coli, the size regulation of B. subtilis cells during exponential phase was also consistent with an adder mechanism (C = 0.52), as previously observed39, but a near-perfect sizer (C = 0.09) for exit from stationary phase (Supplementary Figs. 9 and 11). Analysis of the size regulation during entry to stationary phase for the B. subtilis strain is complicated by lysis and consequent growth resumption of cells in the same channel (Supplementary Video 3), a known ‘cannibalistic’ behaviour of starving Bacillus strains40. However, we found that B. subtilis cells also display rate maintenance during the entry to stationary phase, which suggests that they could also follow mixed adder–timer dynamics.
It has been challenging to identify the underlying mechanisms of growth control, partly because so many mechanisms are consistent with adder phenotypes39,41–43. The distinct pattern of adders, timers and sizers that we observe in different growth phases may provide more discriminatory tests. For example, many growth-control models invoke replication patterns, which are different in entry and exit from stationary phase39,41. Dividing more times without additional protein production also causes changes in the levels of division proteins, which have also been invoked in explanations of growth control39,42. Repeating the growth-curve experiments with various genetic perturbations may thus help pinpoint the pathways involved.
Consequences of cell size variation in stationary phase.
Sizers, adders and timers are also expected to create very different overall heterogeneity in cell volumes, where pure timers fail to compensate for deviations in size and accumulate great variation, whereas sizers could accurately correct size deviations and adders are intermediate between the two. Partly as a consequence of the mixed adder–timer growth dynamics during entry to stationary phase, the distribution of cell volumes in stationary phase is therefore much broader than during exponential growth (coefficient of variance of about 0.26 in stationary phase compared with 0.16 in exponential growth). The sizer mechanism at exit from stationary phase then causes a rapid return to narrowly distributed cell sizes (coefficient of variance of 0.15). However, some size variation in stationary phase also reflects the fact that some cells undergo more divisions than others (Fig. 4a). These cells could have an advantage, since simply being more numerous enables more offspring to explore the world independently. However, those smaller cells would also exit from stationary phase with fewer internal resources. We next turn to investigating the advantages and disadvantages of these options.
Fig. 4 |. Stationary phase length distribution and survival of deep stationary phase.
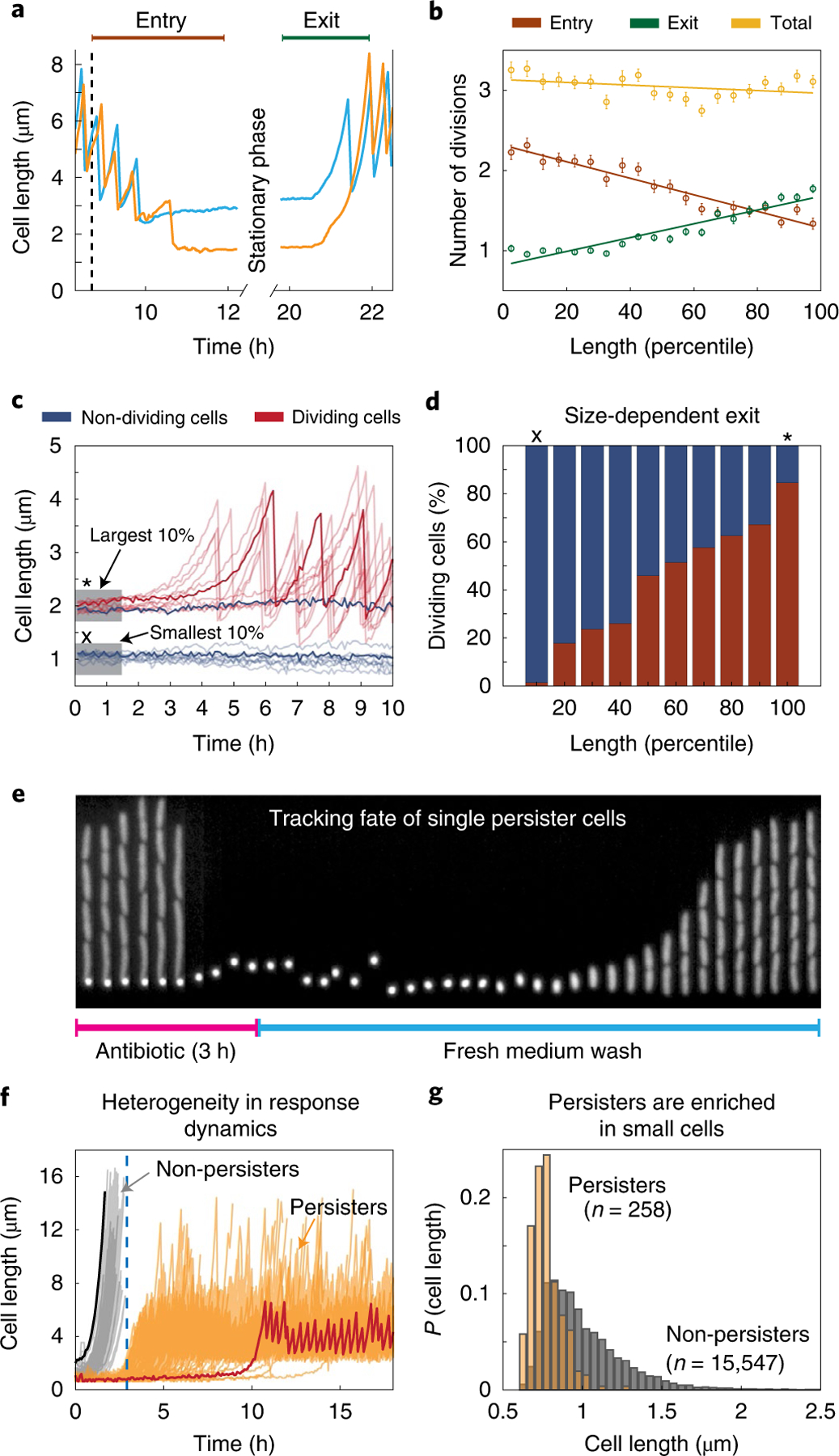
a, Two sample traces highlight the differing number of divisions performed during entry and exit for large (blue) and small (orange) stationary phase cells. b, The number of divisions performed during entry (brown), exit (green) and in total from entry through exit (yellow) are plotted against stationary phase cell size binned by percentile. The mean values are plotted with error bars (s.e.m.) and a linear regression line is fit to these points. c, Cells were subjected to a stationary phase lasting one week before being provided with fresh medium. Cell length time traces for ten example cells that exited from deep stationary phase are plotted in translucent red and a sample trace is overlaid in opaque red. Traces in blue show cell length of a sample of 10 cells that did not begin dividing within 10 h of receiving fresh medium. d, The percentage of cells of a given size that began dividing within 10 h (red) and the percentage that never began dividing (blue). In c and d, x represents the smallest quantile and * marks the largest quantile. e, Kymograph showing a channel containing persister mother cells during antibiotic treatment and recovery. f, Sample traces of 258 persister cells (orange) and non-persisters (cells that chain and die during ampicillin treatment; grey). The largest (black) and smallest (red) cells in the population are highlighted. Larger cells tend to exit more quickly and are therefore vulnerable to ampicillin treatment, whereas smaller cells exit stationary phase later and survive. The blue dashed line represents the timepoint where the media was switched to antibiotic-free rich growth medium. g, Distribution of cell sizes in stationary phase for cells that eventually become persisters in stationary phase (orange) compared with the overall population (grey). Each population has been normalized independently.
We found that in overnight stationary phase culture, cells display great heterogeneity in division patterns both when entering and exiting, but in a negatively correlated manner that has a net effect of cancelling out, thus creating virtually no variation in the total number of divisions (Fig. 4b). For example, during a time window around stationary phase, where every cell had at least one division before entry and after exit (Fig. 4a), the largest 10% of cells in stationary phase—which divided the least on entering stationary phase—on average go through 3.14 ± 0.07 (mean ± s.e.m.) doublings during that time window, whereas the smallest 10%—which divide the most as they enter stationary phase—similarly double 3.26 ± 0.05 times. The same principle was found under all conditions tested demonstrating the importance of tracking cells through stationary phase.
We then considered the same question for longer periods of stationary phase. After one week of starvation, we tracked exit from stationary phase upon addition of a poorer defined medium (MOPS buffered medium). This revealed a substantial net growth disadvantage for cells that divide more times as they entered stationary phase, where, for example, almost none (less than 2%) of the smallest 10% of cells began growth within 10 h of adding fresh medium, but 85% of the largest 10% of cells started to divide in that time window (Fig. 4c,d). Larger cells, which spend fewer resources on increasing their numbers in the entry to stationary phase, can thus reap a substantial net growth advantage after long periods of dormancy (Fig. 4d), perhaps explaining why not every cell divides maximally during the entry to starvation.
This raises the question of why all cells do not stop dividing earlier in stationary phase. One possibility is that this is due to the simple advantage of being more numerous when dispersing to a range of natural environments, but we also considered whether smaller cells could better persist when ‘waking up’ in antibiotic environments13. Specifically, a small fraction of cells trigger persistence after starvation24, but monitoring this has been difficult, partly because most platforms cannot ensure local uniformity under stress and partly because persistence is rare. Our combination of tightly controlled local conditions and extreme imaging throughput enables us to directly track large numbers of natural persisters, without mutants that may change both the frequency and properties25 of persisters. Because cells in our setup occupy dedicated growth channels, cells at the end of each channel are also shielded from competition, which enables long-term monitoring of slow growers that would otherwise be lost by dilution in bulk cultures. However, since the cells at the end of the channel have old poles and remained from the initial loading, it is possible that these conditions might influence the observations. To avoid this, we loaded cells that were grown into stationary phase in the bulk culture and treated them with medium containing antibiotics, and finally monitored the recovery of persister cells with freshly prepared growth medium.
Using this approach, we tracked more than 80,000 individual lineages that were grown in rich growth medium (EZRDM) and kept in stationary phase for 36 h. The cells were then treated for 3 h with fresh EZRDM containing a lethal dose of ampicillin (100 μg ml−1, 10× minimum inhibitory concentration, 99.7% mortality), an antibiotic known to specifically target actively dividing cells44. To determine which cells return to an actively growing state, antibiotic-free EZRDM was then flowed past the cells 3 h after antibiotic treatment for an additional 12 h. A majority of cells exiting stationary phase during this 3 h period was killed by the antibiotic treatment (grey traces in Fig. 4f), but some persister cells remained dormant during the antibiotic treatment and then switched into a non-persister growing state after the antibiotic was removed (Fig. 4e and Supplementary Video 4). We observed 258 such persister cells, at a frequency of 3 × 10−3 (orange in Fig. 4f), consistent with our bulk measurements (Supplementary Note 16) and previous reports13.
Examination of cells that turned into persisters showed that cells that were smaller as they entered stationary phase were much more likely to turn into persisters (Fig. 4g). Indeed, of the smallest 10% of cells, 3% turned into persisters, whereas none of the 45,000 cells that were larger than the average cell produced a single persister. Thus, we observe a clear tradeoff: prolonged stays in stationary phase confer great advantages to cells that stop dividing early if cells exit stationary phase without drugs, but confer a great disadvantage if cells exit stationary phase in the absence of drugs. This also provides what may be an important clue in the search for persister-creating mechanisms, by showing that the mechanism that triggers persistence is tied to the process of dividing more times during entry into stationary phase, which we suspect is triggered by the need to finish replication forks and place chromosomes in separate cells. This process could deplete resources, but persisters could also be created by the growth and division process itself—for example, due to fluctuations to low concentrations that arise when cells distribute some already low protein abundance into multiple small cells, which does not necessarily change the average concentration but should create more heterogeneity between cells. Indeed, we observe that only some of the small cells become persisters. That could constitute an elegantly simple mechanism for sensing that resources are running out, and using that to create relevant heterogeneity.
Discussion
The success of microorganisms depends on their ability to exploit booms and weather busts4,45, but sensing and adapting to environmental changes sufficiently quickly can be challenging. Cells may not survive the sudden arrival of drugs unless they are already in a drug-tolerant dormant state; conversely, they may be unable to take advantage of fresh resources before these are depleted unless they are immediately ready to grow and divide. Clonal populations could therefore benefit from hedging their bets and ensuring that individual cells are prepared for different future environments14,46. High phenotypic heterogeneity has indeed been observed both when cells enter and when they exit stationary phase18,47,48. However, analysing the temporal patterns of heterogeneity and their consequences for survival and propagation has been difficult owing to the limitations of existing methods. The platform that we present here enables us to track lineages entering and exiting stationary phase with sufficiently precise control of local environments to make the results straightforwardly interpretable, over a long enough time window, and with high enough throughput to observe key outliers. Because the cell culture can contain almost any mix of cells, our method could equally be used to study individual cells in ecosystems, such as in microbiome studies. We believe that the ability to track the growth, death and expression patterns of individual cells over time in dense, complex and changing cultures—potentially containing entire ecosystems—could be transformative in many areas of microbiology.
Methods
Strain construction.
Strain BO37 was used in the bulk culture flask. It was made by P1 transducing glmS::PRNAI-mCherry1–11-mKate-T1 terminator-FRT Kan FRT::pstS allele into MG1655 as described32. For all strains used in the mother machine, we used the MG1655 7740 background with ΔmotA. The strain SB8 was used for the phase-contrast imaging experiments depicted in Figs. 3b–g and 4a,b, and is the background strain for all the other strains containing transcription reporters used in this study. SB8 was constructed by a P1 transduction from the Keio collection strain CGSC#:9565, ΔmotA743::kan into MG1655 and flippase was used to remove the kanamycin resistance49. For fluorescent segmentation marker, we made a fluorescent version called SB7, by P1 transducing glmS::PRNAI-mCherry1–11-mKate-T1 terminator-FRT Kan FRT::pstS into this SB8 strain. We made 16 different strains containing transcription reporters for different processes, including the PrpoS reporter, by integrating the transcription reporters constructed by U. Alon’s group50 into the SB7 strain. To rigorously check if the ΔmotA was introducing any artefacts to our measurements, we compared our results with ΔfliC, which also renders the cell nonmotile, and finally compared with the MG1655 6300 strain, which is much less motile than 7740 version, and found that the results were identical (Supplementary Note 18).
Mother machine chip design and optimization.
The design of the mother machine also had to be highly optimized to handle the vast changes in cell size along the growth curve (for example, MG1655 cells are 15 times larger in exponential phase compared with stationary phase), and to maintain uniform nutrient access across the device. If cell channels are too wide, they cause double loading of cells in the same channel and affect data analysis. If channels are too narrow, then they affect loading efficiency and cell fitness due to mechanical stress on the cell wall and reduced flow of nutrient to the mother. Therefore, we first optimized the channel width. Once an optimum channel width was identified, we optimized for channel length and channel height. Channel length determines the nutrient availability and retention time. If the channel length is too short, then the cells do not stay in the channels for sufficient duration (due to occasional chain formation and being dragged by the flow in the medium flow channel). Conversely, if the channels are too long, then the medium does not reach the mother cell, as the cells below the mother consume it before it diffuses to the closed end, causing the mother cell to starve artefactually. The height of a channel determines whether cells are in focus or if they are pressed against the wall. Overall, we sampled 540 combinations of width, height and length of cell channels to find the optimum dimension for this experiment (channel length = 25 μm, channel width = 1.5 μm, channel height = 1.28 μm). Since E. coli and most other rod-shaped bacteria can change their size depending on the growth conditions, the exact choice of the parameters is expected to change. However, the sampler chip developed for this work and the analytic approach should help to quickly find the optimized parameters. The details of the results are provided in the Supplementary Note 4.
In addition to the dimensions of the cell channels, their layout was also substantially altered. We reduced the width of the flow channel to fit the channels on both sides within the same field-of-view. The height of the flow channel was increased to reduce the pressure and chances of biofilm formation. For experiments that require higher throughput to identify persister cells, we use a layout where we have combined 15 of these lanes in a snake-like formation. The design parameters of the snake were optimized to reduce the flow time through the entire path and to avoid clogs and blocks at the bends. This device enables us to load a single strain and get around 123,000 individual lineages imaged in a single experiment. Details of the layout optimization is provided in the Supplementary Note 6. To further improve the throughput, we reduced the channel gaps. In Supplementary Note 5, we discuss how the gap between channels determine the extent of bleed through of signal among cells from different channels and how we can optimize the channel gap to minimize such artefacts.
Different computer-aided designs for the sampler chip and the snake-like flow-channel layouts were done using the AutoCAD software. The layer containing the features for the cell channels is called channel layer and the one containing the flow-channel features is called the flow-lane layer. The designs were sent to Toppan for fabrication of custom chrome photomasks. The master moulds for the mother machine devices were prepared using these chrome photomasks. For a detailed protocol we refer to ref. 22. In brief, individual layers were sequentially deposited onto a silicon wafer using standard photolithography and aligned using included alignment marks on each chrome photomask. First, the cell-channel layer was created by applying a thin layer of SU-8 2001 photoresist (Microchem) on the silicon wafer using a spin coater and then the corresponding patterns were created by exposing the uncured SU-8 2001 with UV light through the chrome mask using a Suss MJB4 mask aligner. It is important to note here that the channel height is controlled only by the layer thickness, which depends on the photoresist and on the parameters of the spin coater, and not by the design in the chrome photomask. The wafer was then developed using SU-8 developer and rinsed with isopropyl alcohol. In the next step, a second layer was created for the flow channel using SU-8 2025 (Microchem) on top of the channel layer, and the features of the two layers were aligned using alignment marks. After development and rinse of the second layer, the silicon master was hard baked to be used for fabricating polydimethylsiloxane (PDMS) casts of the mother machine.
Mother machine chip preparation.
To prepare the PDMS casts for the mother machine chips, dimethyl siloxane monomer (Sylgard 184) was mixed in a 10:1 ratio with curing agent, defoamed and then poured onto the silicon master. The silicon master with the PDMS mix was then degassed for 1 h using vacuum, and then cured at 95 °C for 1 h. The cured PDMS was then carefully peeled off the silicon master. Individual mother machine chips were cut out of the PDMS mould and holes for the inlets and outlets were created using a biopsy puncher (0.75 mm diameter). The mother machine chips were then sonicated in isopropanol for 30 min, blow-dried with an air gun and baked at 95 °C for 30 min before bonding. Glass coverslips were cleaned by sonication in KOH followed by sonication in DI water, blow-dried and then baked for 30 min at 95 °C. The clean coverslips were then bonded to the feature side of the mother machine chips using oxygen plasma treatment (30 s at 50 W and O2 pressure set to 170 mTorr) followed by incubation at 95 °C for at least 1 h. The chips were generally bonded the day before being used in experiments.
Loading cells in the mother machine chips.
There are two types of experiments described in this Letter, which use two different types of mother machine devices (Supplementary Note 6). In general, 16 different strains can be loaded into the 16 different lanes of the type 1 device. We used 16 pairs of gel-loading tips to load those lanes and centrifuge the chips to load the cells into the channels that are orthogonal to the loading lanes. The high-throughput experiments involve a single strain to be loaded in a mother machine device in which 15 lanes are connected via a continuous snake-like feeding channel. In this case, the overnight culture is concentrated 10× before loading into the device. The chip is then spun at 4,500 rcf in a benchtop centrifuge for 2 min, using a custom-built holder. The centrifugal force rapidly loads the cells in the narrow channels. The dense culture from the feeding channel is washed quickly with fresh growth medium before starting the image acquisition.
Description of growth-curve platform.
To obtain reproducible profiles of growth curves and for relevance to conventional microbiology assays, we grew our cells in large batch cultures. For optimum growth, large batch cultures must be shaken well to enable thorough mixing and aeration. Cells were grown in a 500 ml baffled-bottom flask (ChemGlass) that is being shaken at 220 rpm on an orbital shaker with magnetic clamps (Benchmark Scientific Orbi-Shaker Jr). To isolate the imaging platform from the vibrations arising from the shaker system, we engineered a dual-incubator system. The culturing platform is housed in one incubator, and the microscope with the microfluidic platform is housed in another (Extended Data Fig. 1a). The incubator shown on the left of the Extended Data Fig. 1a was constructed using T-slot framing (cut at measure using a heavy-duty chop saw) and acrylic sheets (cut using a laser cutter). This incubator is heated with a resistive heater and fan, and the temperature of the incubator is constantly monitored in two places using a custom microcontroller solution and a data-logging thermometer. Shelves were created in this incubator to install multiple peristaltic pumps and solenoid pinch valves (Extended Data Fig. 1b, middle panel). A set of pumps takes the culture through the mother machine device loaded on the microscope stage. Another set of peristaltic pumps (Langer Instruments, T60-S2&WX10–14) circulates the culture to and back from a custom-designed inline optical density meter. The optical density meter uses an Arduino control device to control an LED to shine light on the cultures and reads the signal from the photodiode using an analogue-to-digital converter.
The second commercial incubator (Okolab) houses the microscope where the cells in the microfluidic device are imaged. The two incubators are connected via an insulating duct that maintains the temperature around the tubing that carries the cells from the culture flask to the microfluidic device. Hot air from the incubator is blown into the duct through fans at the junctions. To ensure that cells are maintained at 37 °C throughout the entire process, we used a handheld forward-looking infrared imaging system to monitor temperature of the tubing and different parts of the setup. The results are shown in Supplementary Note 3 and confirm that the temperature profile of the entire setup is uniform.
Description of a growth-curve experiment.
Before any experiment is run with the platform, an automated system is used to wash the entire medium and culture tubing path. The wash system consists of a four-way switch that can select liquid from three bottles containing bleach, ethanol and water, a peristaltic pump to flow the liquid to the culture and medium container (Extended Data Fig. 1b, left panel), and then two additional valves and a pair of pumps installed in the incubator to flow the liquid to the waste after each cycle. The entire system is controlled by a custom program installed on a Raspberry Pi computer. The wash cycle consists of 40 min wash with 20% bleach, followed by 40 min wash with 20% ethanol and finally a 40 min wash with distilled H2O. Then, just before running the experiment, a bottle containing EZRDM is hooked up to the platform and pumped through all the tubing to remove the water and replace it with medium. While this is taking place, the chip is loaded as described above. Once the fresh medium has washed out the water, the medium used for wash is removed and the flask containing the growth medium is attached to the setup.
At the beginning of an experiment, the 500 ml baffled flask containing 200 ml of pre-warmed medium is inoculated with 1:10,000 of liquid culture. It is then immediately placed into the incubator where it is both maintained at 37 °C and actively shaken at 220 rpm. The liquid from a flask containing either fresh medium or the actively growing culture is pumped through tubing then into the microscope incubator where it is connected to the mother machine via blunt-end needles. In this path, there is a small, sealed chamber that acts to remove bubbles and is also a vessel in which the optical density is continuously measured (Extended Data Fig. 1d). A different set of flask and pumps are used to flow fresh medium when we need to monitor the cells that exit from stationary phase.
The switch between the culture and the fresh medium is handled with an electrically controlled pinch-valves setup (Extended Data Fig. 1c), which minimizes any delay between the two conditions and avoids any possible contamination in the fresh medium from the culture (Supplementary Note 1). Extended Data Fig. 1c (bottom) shows the sequential steps of pinch-valve states to switch the flow towards the mother machine from the culture flask to the fresh medium. This setup also enables the culture setup to be kept isolated during the fresh medium flow and enables wash for restarting a second round of growth-curve experiment in the same device. The pinch valves were chosen since they avoid the medium getting into valve chambers and also keeps the tubing insulated from the temperature of the solenoid.
Microscopy and image acquisition.
We have custom-designed a microscope for the fast acquisition needed for the high-throughput imaging of mother machine. We replaced the turrets that house the dichroic mirror and emission filter to have a single quad band dichroic mirror for all the excitation and have a fast emission filter wheel for all the emission filters. This avoids the lags from the slow movement of the turret and improved our speed of acquisition tremendously. We then used an air objective with high numerical aperture (Apochromat ×40, 0.95 numerical aperture) to acquire the images, which made the entire time for stage movement comparable to exposure time, allowing us to scan the entire device in less than 5 min. Images were acquired with a sCMOS camera (ANDOR Zyla 4.2), which allowed us to have fast framerate with very high quantum efficiency (84%). We used a LED lamp as the white-light source for the phase-contrast imaging and the Spectra-X light engine LED excitation source (Lumencor) for fluorescence imaging. Using these settings, we acquired a total of 705 fields of view (FOVs) every 5 min, allowing us to track cells in 131,072 channels over time. For phase-contrast imaging, we switched to a high magnification apodized phase-contrast objective (×100 oil numerical aperture 1.3, PH3), that enabled us to get high-contrast images of cells in mother machine. We sampled 85 FOVs in 1 min using this oil objective allowing us to image a total of 3,500 channels. For long acquisition it is important to keep the entire sample in perfect focus, and so the focal drift was controlled via the Nikon Perfect Focus system (PFS). The entire setup of flow-switch and the Nikon Ti inverted microscope was housed in a temperature-controlled incubator (Okolab). We have used the Nikon Elements software for the acquisition control and to create position list using a grid structure. The fluorescent colours were ordered according to their order in the filter wheel, and the travel of the stage along the position list was optimized to get the fastest possible scanning of the entire chip. The PFS was always kept in ‘on’ option for fast scanning. The data were saved locally in ND2 format to improve writing and saving speed and eventually extracted for analysis into single TIFF files using a custom-made ND2extractor Python script.
Data processing for segmentation and single-cell tracking.
We developed an image-processing pipeline for processing the large volume of high-throughput data collected per experiment (approximately 1.5 TB per experiment). A multi-core extraction algorithm was used to extract single TIFFs from the ND2 file and organize them into folders for each FOV. We then proceeded to analyse the stack of single TIFFs for each FOV over time. The pipeline for the analysis involves four major sections: image segmentation, data extraction, single-cell tracking and trace cleaning. The image-segmentation step involves processing the entire FOV to identify single cells and find a contour for each cell from which the intensity data is to be extracted. One major challenge for single-cell segmentation for a data along the growth curve is that cells continuously change their size and shape along the growth curve. To address this, we developed a custom segmentation algorithm that is more flexible for variable cell sizes, which was roughly based on the adjustable watershed algorithm implemented in FIJI51,52. We first adjust contrast across all the frames of the time-lapse image stack to correct for changes in intensity of segmentation marker and phase contrast for every individual the TIFF files. Then we performed background subtraction intensity using a rolling ball algorithm. The background subtracted images were processed with a combination of global and local thresholding to create binary masks for cells from background and used a marker-based watershed algorithm to separate all the binary masks into masks of single cells in a reliable manner. The segmentation algorithms are different for the fluorescent and phase-contrast images. After the segmentation, we extracted the fluorescence intensity properties and shape properties for each connected component from the list of masks.
The next step was to track single masks over time to obtain time-lapse data. An individual FOV has about 230 channels with approximately 8 cells in each. Therefore, to track single cells over time, we have to assign the approximately 1,300 binary masks to their corresponding masks at different time points. This is a massive task if done by the traditional frame-by-frame nearest-neighbour assignment. To address this challenge, we developed a novel clustering algorithm that clusters all the cells in a single channel over the entire duration of the experiment and then assigns the top one in each frame as the mother cell. Specifically, we use DBSCAN53 clustering implemented in MATLAB54 to cluster the data into lineages from individual channels and then sort the data to get the top cell in each channel and assign all of its properties to mother-cell properties. The segmentation algorithm is not perfect, so we define criteria to help filter out data that is non-physical. The ratio of the division length and birth length should be 0.5, so we filter out any division interval where the length at birth over the length at the preceding division differs from 0.5 by more than 20%. The single-cell traces of the different properties of interest were further cleaned to remove unnatural trends using the reference marker intensity traces.
Extended Data
Extended Data Fig. 1 |. Operating principle of the single-cell growth-curve setup.
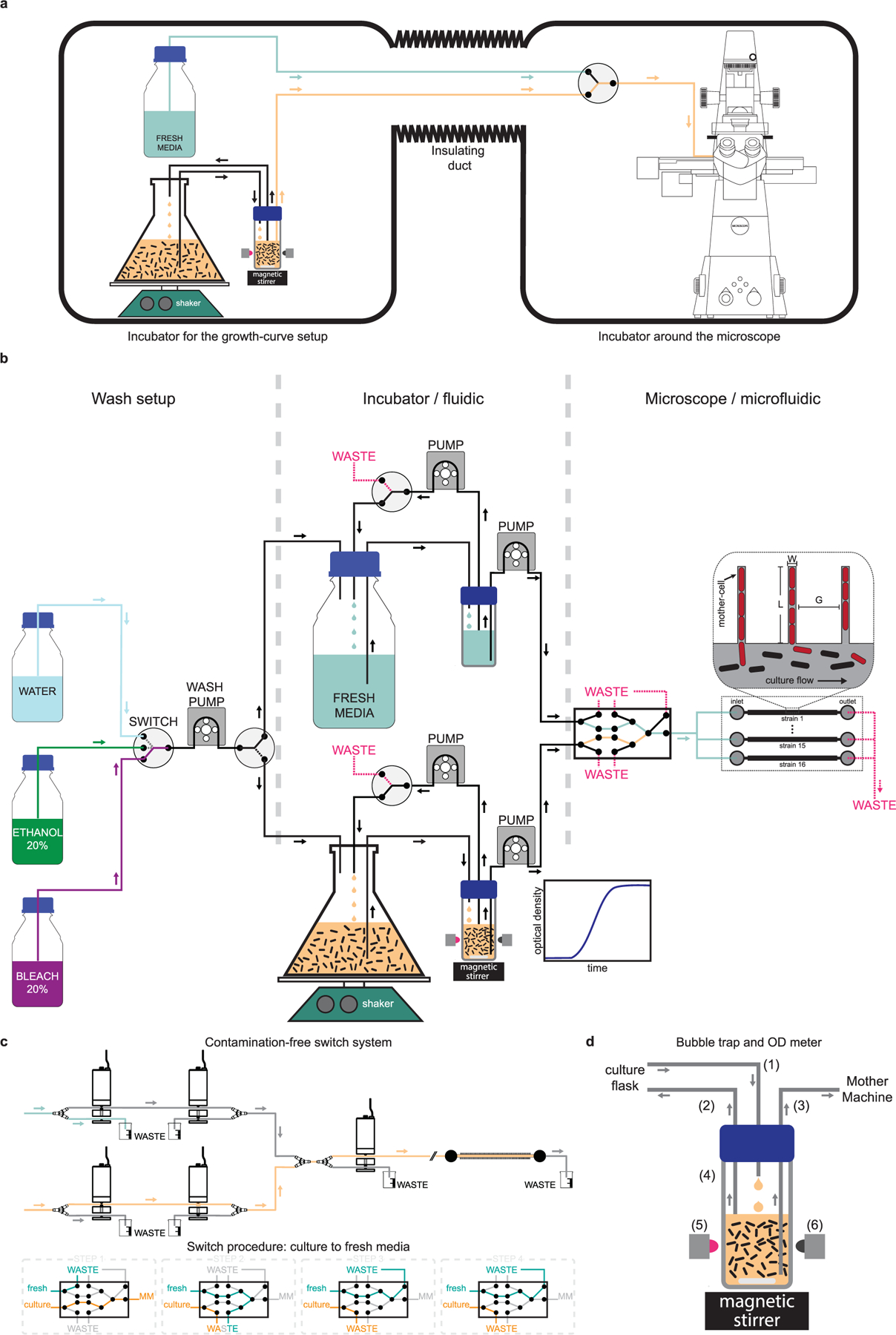
a, Schematic of the dual incubator setup is shown. The incubator in the left houses the growth curve setup (culture flask, shaker, and automated optical density measurement system), and the fresh media reservoir. The commercial incubator on the right houses the microscope and the microfluidic device in which cells are loaded. These two incubators are connected via an insulated duct through which the tubing that carry cells and media go from the flask to the microfluidic device. b, Detailed schematic of the flow-setup – showing the pumps, switches and liquid reservoirs. The box on the left shows the setup used for automatically washing the system before and after every run. A peristaltic pump and a 4-way switch are used to sequentially pump liquid from the reservoirs containing bleach, ethanol, and water to the culture flask or the media reservoir. The box in the center shows components that are housed in the incubator shown on the left of (a). Cells are grown in a baffled-bottom culture flask, which is placed on an orbital shaker. An inline automated OD-meter is used for continuously monitoring the optical density of the culture using a LED-photodiode pair. A set of pumps flow the culture to the OD meter and back, while another one flows it to the microfluidic device. A similar set of pumps are used on the path for fresh media, and both paths can be used interchangeably. The box on the right shows the components housed in the second incubator. These include a set of pinch valves that enable switch from culture to fresh media, and vice versa. We use five 3-way pinch-valves to make sure there is minimal delay in switch, the wash is clean and devoid of any residual bacteria from the culture, and the path is free of bubbles. This design also keeps individual paths isolated and enable wash with bleach, ethanol, and water, while the other path is in use. The media coming out of the final (rightmost) valve can be split using a manifold to flow through different flow-channels in the microfluidic device. d, A detailed schematic of the pinch valve assembly and sequences of steps involved in the media switch is shown. e, Schematic of the bubble-trap is shown to illustrate the principle of operation. Media from the culture flask is dripped into the reservoir (1) and a taken out using another tubing (2) that is placed at the liquid interface to remove bubbles and extra liquid. Due to this design, the media at the bottom of the reservoir stays essentially free of bubbles and is delivered to the mother-machine using a tubing (3) whose inlet is all the way at the bottom of the reservoir. The reservoir has transparent glass-walls (4) and the magnetic stirrer is used to prevent the culture from settling down and to keep it uniform. We use a LED (5) to shine near-IR light through the culture and monitor the transmittance using a photodiode (6) placed in the opposite direction to quantify the optical density of the culture.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (NIH) grant R01 AI141966 and DARPA agreement HR0011–16-2–0049. We thank S. Wan for her Python code for extracting TIFF files from the ND2 files; N. Lord for the master of the first mother machine we used; P.-Y. Ho and A. Amir for the discussion of the size-regulation analysis; and P. Gorelik and O. Mazor at the HMS Research Instrumentation Core Facility for assistance in instrument design and fabrication. R. Losick’s and D. Rudner’s laboratories provided the strains and necessary protocols for the B. subtilis experiments. The microfabrication involved in this work was performed in part at the Harvard Medical School Microfluidics Facility and Center for Nanoscale Systems, a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under National Science Foundation award no. 1541959. Center for Nanoscale Systems is part of Harvard University.
Footnotes
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
The code required to process the images (cell segmentation and measurements) are available at https://github.com/manukke/MMsegmenter.
Competing interests
The authors declare no competing interests.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41564-021-00900-4.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41564-021-00900-4.
Peer review information Nature Microbiology thanks the anonymous reviewers for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
Source data are provided with this paper.
References
- 1.Koch AL The adaptive responses of Escherichia coli to a feast and famine existence. Adv. Microb. Physiol 6, 147–217 (1971). [DOI] [PubMed] [Google Scholar]
- 2.Morita RY The starvation-survival state of microorganisms in nature and its relationship to the bioavailable energy. Experientia 46, 813–817 (1990). [Google Scholar]
- 3.Novitsky JA & Morita RY Possible strategy for the survival of marine bacteria under starvation conditions. Mar. Biol 48, 289–295 (1978). [Google Scholar]
- 4.Roszak DB & Colwell RR Survival strategies of bacteria in the natural environment. Microbiol. Mol. Biol. Rev 51, 365–379 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lennon JT & Jones SE Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol 9, 119–130 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Navarro Llorens JM, Tormo A & Martínez-García E Stationary phase in Gram-negative bacteria. FEMS Microbiol. Rev 34, 476–495 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Buchanan RE Life phases in a bacterial culture. J. Infect. Dis 23, 109–125 (1918). [Google Scholar]
- 8.Monod J The growth of bacterial cultures. Annu. Rev. Microbiol 3, 371–394 (1949). [Google Scholar]
- 9.Manina G, Dhar N & McKinney JD Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17, 32–46 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Grimbergen AJ, Siebring J, Solopova A & Kuipers OP Microbial bet-hedging: the power of being different. Curr. Opin. Microbiol 25, 67–72 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Casadesús J & Low DA Programmed heterogeneity: epigenetic mechanisms in bacteria. J. Biol. Chem 288, 13929–13935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bódi Z et al. Phenotypic heterogeneity promotes adaptive evolution. PLoS Biol 15, e2000644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balaban NQ, Merrin J, Chait R, Kowalik L & Leibler S Bacterial persistence as a phenotypic switch. Science 305, 1622–1625 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Ackermann M A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol 13, 497–508 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Givskov M, Eberl L, Møller S, Poulsen LK & Molin S Responses to nutrient starvation in Pseudomonas putida KT2442: analysis of general cross-protection, cell shape, and macromolecular content. J. Bacteriol 176, 7–14 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davey HM & Winson MK Using flow cytometry to quantify microbial heterogeneity. Curr. Issues Mol. Biol 5, 9–15 (2003). [PubMed] [Google Scholar]
- 17.Okumus B et al. Single-cell microscopy of suspension cultures using a microfluidics-assisted cell screening platform. Nat. Protoc 13, 170–194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sturm A & Dworkin J Phenotypic diversity as a mechanism to exit cellular dormancy. Curr. Biol 25, 2272–2277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mutlu A et al. Phenotypic memory in Bacillus subtilis links dormancy entry and exit by a spore quantity-quality tradeoff. Nat. Commun 9, 69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eldar A & Elowitz MB Functional roles for noise in genetic circuits. Nature 467, 167–173 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ullman G et al. High-throughput gene expression analysis at the level of single proteins using a microfluidic turbidostat and automated cell tracking. Philos. Trans. R. Soc. Lond. B 368, 20120025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norman TM, Lord ND, Paulsson J & Losick R Memory and modularity in cell-fate decision making. Nature 503, 481–486 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang P et al. Robust growth of Escherichia coli. Curr. Biol 20, 1099–1103 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balaban NQ et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol 17, 441–448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moyed HS & Bertrand KP hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol 155, 768–775 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neidhardt FC, Bloch PL & Smith DF Culture medium for enterobacteria. J. Bacteriol 119, 736–747 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konopka MC et al. Cytoplasmic protein mobility in osmotically stressed Escherichia coli. J. Bacteriol 91, 231–237 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barns KJ & Weisshaar JC Real-time attack of LL-37 on single Bacillus subtilis cells. Biochim. Biophys. Acta 1828, 1511–1520 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolter R Growth in studying the cessation of growth. J. Bacteriol 181, 697–699 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neidhardt FC Bacterial growth: constant obsession with dN/dt. J. Bacteriol 181, 7405–7408 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gefen O, Fridman O, Ronin I & Balaban NQ Direct observation of single stationary-phase bacteria reveals a surprisingly long period of constant protein production activity. Proc. Natl Acad. Sci. USA 111, 556–561 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okumus B et al. Mechanical slowing-down of cytoplasmic diffusion allows in vivo counting of proteins in individual cells. Nat. Commun 7, 11641 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amir A Cell size regulation in bacteria. Phys. Rev. Lett 112, 208102 (2014). [Google Scholar]
- 34.Taheri-Araghi S et al. Cell-size control and homeostasis in bacteria. Curr. Biol 25, 385–391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cadart C et al. Size control in mammalian cells involves modulation of both growth rate and cell cycle duration. Nat. Commun 9, 3275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandler-Brown D, Schmoller KM, Winetraub Y & Skotheim JM The adder phenomenon emerges from independent control of Pre- and post-start phases of the budding yeast cell cycle. Curr. Biol 27, 2774–2783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eun Y-J et al. Archaeal cells share common size control with bacteria despite noisier growth and division. Nat. Microbiol 3, 148–154 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Zaritsky A & Helmstetter CE Rate maintenance of cell division in Escherichia coli B/r: analysis of a simple nutritional shift-down. J. Bacteriol 174, 8152–8155 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Si F et al. Mechanistic origin of cell-size control and homeostasis in Bacteria. Curr. Biol 29, 1760–1770 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalez-Pastor JE, Hobbs EC & Losick R Cannibalism by sporulating Bacteria. Science 301, 510–513 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Amodeo AA & Skotheim JM Cell-size control. Cold Spring Harb. Perspect. Biol 8, a019083 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sekar K et al. Synthesis and degradation of FtsZ quantitatively predict the first cell division in starved bacteria. Mol. Syst. Biol 14, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chien A-C, Hill NS & Levin PA Cell size control in bacteria. Curr. Biol 22, R340–R349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohanski MA, Dwyer DJ & Collins JJ How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol 8, 423–435 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jørgensen BB & Boetius A Feast and famine—microbial life in the deep-sea bed. Nat. Rev. Microbiol 5, 770–781 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Veening J-W, Smits WK & Kuipers OP Bistability, epigenetics, and bet-hedging in Bacteria. Annu. Rev. Microbiol 62, 193–210 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Dworkin J & Shah IM Exit from dormancy in microbial organisms. Nat. Rev. Microbiol 8, 890–896 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Jõers A & Tenson T Growth resumption from stationary phase reveals memory in Escherichia coli cultures. Sci. Rep 6, 24055 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baba T et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol 2, 2006.0008 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaslaver A et al. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods 3, 623–628 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Schindelin J, Rueden CT, Hiner MC & Eliceiri KW The ImageJ ecosystem: an open platform for biomedical image analysis. Mol. Reprod. Dev 82, 518–529 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schindelin J et al. Fiji: an open-source platform for biological image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daszykowski M & Walczak B in Comprehensive Chemometrics 635–654 (Elsevier, 2009); 10.1016/B978-044452701-1.00067-3 [DOI] [Google Scholar]
- 54.MATLAB and Statistics and Machine Learning Toolbox Release 2018b (The MathWorks; Inc., 2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data are provided with this paper.