

Abstract
Emphasis was placed in this work on the assessment of biological features of 2,2,4-triaminooxazolone, a major one-electron and ·OH-mediated oxidation product of guanine. For this purpose, two oligonucleotides that contain a unique oxazolone residue were synthesized. Herein we report the mutagenic potential of oxazolone during in vitro DNA synthesis and its behavior towards DNA repair enzymes. Nucleotide insertion opposite oxazolone, catalyzed by Klenow fragment exo– and Taq polymerase indicates that the oxazolone lesion induces mainly dAMP insertion. This suggests that the formation of oxazolone in DNA may lead to G→T transversions. On the other hand, oxazolone represents a blocking lesion when DNA synthesis is performed with DNA polymerase β. Interestingly, DNA repair experiments carried out with formamidopyrimidine DNA N-glycosylase (Fpg) and endonuclease III (endo III) show that oxazolone is a substrate for both enzymes. Values of kcat/Km for the Fpg-mediated removal of oxidative guanine lesions revealed that 8-oxo-7,8-dihydroguanine is only a slightly better substrate than oxazolone. In the case of endo III-mediated cleavage of modified bases, the present results suggest that oxazolone is a better substrate than 5-OHC, an oxidized pyrimidine base. Finally, MALDI-TOF-MS analysis of the DNA fragments released upon digestion of an oxazolone-containing oligonucleotide by Fpg gave insights into the enzymatic mechanism of oligonucleotide cleavage.
INTRODUCTION
Oxidative damage to DNA is known to be mutagenic and is possibly involved in the aging process and human diseases, including cancer (1–4). Reactive oxygen species are probably the most important source of spontaneous damage to DNA and up to now more than 50 base modifications have been identified (5–8). The mutagenicity of damaged DNA is expressed during its replication by a DNA polymerase (9,10). However, oxidative DNA lesions may be removed in cells by a variety of repair enzymes (9,11,12). Most of the oxidized base lesions are excised by the base excision repair (BER) mechanism (13,14). Several DNA N-glycosylases which act by excising the modified base have been identified in bacterial, yeast and mammalian cells.
Among the four DNA bases, guanine is the most susceptible to one-electron oxidation due to its low redox potential (15). During the past decade, many studies were conducted in order to identify the main oxidative damage to guanine. One-electron oxidation of guanine gives rise to the related radical cation, which has been shown to mostly undergo deprotonation within nucleosides and single-stranded DNA. On the other hand, both deprotonation and hydration occur within duplex DNA (Fig. 1) (6,16). 8-Oxo-7,8-dihydroguanine (8-oxoGua) is the major oxidative lesion formed in DNA through the hydration pathway (5,8,17–19). However, 2,2-diamino-4-[(2-deoxy-β-d-erythro-pentofuranosyl)-amino]-5(2H)-oxazolone (oxazolone or Z) together with its precursor 2-amino-5-[(2-deoxy-β-d-erythro-pentofuranosyl)-amino]-4H-imidazol-4-one (imidazolone or Iz) were found to be the major oxidation compounds upon γ-irradiation or type I-mediated photosensitization of 2′-deoxyguanosine in aerated aqueous solution (20). The half-life for hydrolysis of the imidazolone nucleoside, in neutral aqueous solution at 37°C, is 2.5 h (21). Oxazolone has also been characterized in a short oligonucleotide as a major decomposition product upon one-electron oxidation of guanine (22). Evidence was provided for the formation of oxazolone in double-stranded DNA (23,24). It was also found that imidazolone, the precursor of oxazolone, is generated in duplex DNA under type I photooxidation conditions (25). More recently Vialas et al. (26) pointed out the importance of the imidazolone and oxazolone decomposition pathway in the two-electron oxidation of 2′-deoxyguanosine. Oxazolone was also identified as a major singlet oxygen oxidation product of 8-oxo-7,8-dihydro-2′-deoxyguanosine (27) and calf thymus DNA (28). In the latter case, 8-oxoGua was proposed to be an intermediate in the formation of oxazolone. Thus, it is reasonable to assume that oxazolone is a key compound in the oxidation of both guanine and 8-oxoGua.
Figure 1.

Fate of the guanine radical cation: formation of 8-oxoGua versus imidazolone and oxazolone.
Determination of the biological role of oxidative damage has been made possible by the synthesis of site-specific modified oligonucleotides. Availability of such probes facilitates the assessment of the mutagenic potential of the damage during DNA synthesis by a DNA polymerase. In addition, the lesion-containing oligonucleotides are suitable for evaluation of the substrate specificity of DNA repair enzymes (9). Up to now the biological features of 8-oxoGua have been extensively studied.
Primer extension experiments using an 8-oxoGua-modified template have shown that DNA polymerases are able to bypass the lesion and to selectively incorporate dAMP in addition to dCMP opposite the damage (29–31). A repair system for 8-oxoGua, which involves several enzymes, has been elucidated in Escherichia coli (32,33). It was shown that the Fpg protein, also known as MutM (34) or 8-oxoguanine glycosylase, removes 8-oxoGua from double-stranded DNA when it is paired with a cytosine base. It has been reported that Fpg exibits both N-glycosylase and AP lyase activities (34,35) that cleave the DNA backbone through a β-δ-elimination reaction, thus leading to the release of the sugar from the DNA strand (36,37). Although 8-oxoGua has received considerable attention, little is known about the mutagenicity and the repairability of other important oxidized purine bases such as oxazolone.
In this study we synthesized two oxazolone-containing oligonucleotides. This was achieved by specific riboflavin-mediated photosensitization of oligomers having a unique guanine residue. First, we examined the nucleotide insertion opposite oxazolone during in vitro DNA synthesis catalyzed by Klenow fragment exo– (Kf exo–), DNA polymerase β (pol β) and Taq polymerase (Taq). DNA repair experiments were performed in order to check whether oxazolone was a substrate for Fpg and endo III. Km and Vmax values were determined for a comparative study of the excision efficiencies of oxazolone and 8-oxoGua by Fpg or oxazolone and 5-hydroxy-2′-deoxycytidine (5-OHC) by endo III. The DNA fragments generated upon excision of oxazolone by Fpg were also analyzed by MALDI-TOF-MS in order to assess the mechanism of cleavage of the modified oligonucleotide by Fpg.
MATERIALS AND METHODS
Materials
T4 polynucleotide kinase, Kf exo–, [γ-32P]ATP, dNTPs, NAP-25 Sephadex and MicroSpin G-25 columns were obtained from Amersham Pharmacia Biotech (Uppsala, Sweden). Pol β and uracil N-glycosylase were from Trevigen-Interchim (Montluçon, France). Taq was purchased from Boehringer Mannheim (Mannheim, Germany). Fpg and endo III were kind gifts from Dr Serge Boiteux (CEA, Fontenay-aux-Roses, France).
Oligodeoxynucleotide synthesis
Oligonucleotides (Fig. 2) were synthesized by standard phosphoramidite chemistry using an Applied Biosystems 392 DNA synthesizer. The 8-oxoGua-containing oligonucleotide 5′-d(CTCCTCT[8-OG]TCACTCC) (6) was prepared using a commercially available phosphoramidite monomer of 8-oxo-deoxyguanosine (Glen Research, Sterling, VA). The 5-OHC-containing oligonucleotide (9) was synthesized using the 5-hydroxy-2′-deoxycytidine phosphoramidite as previously described (38). Unmodified oligonucleotides 1, 2, 4, 5, 8 and 10 were deprotected in a concentrated aqueous ammonia solution (32%) for 15 h at 55°C. The 5-OHC-containing oligomer 9 was deprotected in a concentrated ammonia solution (32%) for 4 h at 25°C. Oligonucleotide 6 was deprotected in a concentrated ammonia solution of 0.25 M β-mercaptoethanol for 15 h at 55°C to prevent further oxidation of 8-oxoGua during the deprotection step. Oligonucleotide 11, containing a natural abasic site (deoxyribose, Ab), was prepared using 10 in the presence of uracil N-glycosylase as described in the enzyme activity synopsis provided by the manufacturer. The oligonucleotides were purified by PAGE using a 20% polyacrylamide/7 M urea gel and then desalted using NAP-25 Sephadex columns. The integrity of the 8-oxoGua-containing oligonucleotide was assessed by electrospray ionization mass spectrometry (Platform 3000 model spectrophotometer; Micromass, Manchester, UK). The integrity of oligonucleotides 1, 2, 4, 5 and 8–11 was assessed by MALDI-TOF-MS (Voyager-DE; Perseptive Biosystems, Farmingham, MA). Oligonucleotides were 5′-end-labeled using T4 polynucleotide kinase and [γ-32P]ATP prior to purification on MicroSpin G-25 columns.
Figure 2.

Sequences of oligonucleotides.
Synthesis of oxazolone-containing oligonucleotides
The preparation of modified oligonucleotides containing the oxazolone motif has been recently described (22). Briefly, 50 µl of an aqueous saturated solution of riboflavin was added to 450 µl of a 100 µM aqueous solution of either 5′-d(CTCTGTCTCCACTCC) (1) or 5′-d(CTCCTCTGTCACTCC) (5). Then, the resulting solution was exposed for 30 min to 16 black- light lamps (λ = 350 nm) of a Rayonet photochemical reactor (New England UV Co., Hamden, CT). The irradiated mixture was kept at room temperature for 20 h in order to allow conversion of imidazolone to oxazolone. The corresponding oxazolone-containing oligonucleotides 5′-d(CTCT[Z]TCTCCACTCC) (3) and 5′-d(CTCCTCT[Z]TCACTCC) (7) were purified on a Hypersil (Interchim) ODS column (5 µm, 250 × 4.6 mm) with a gradient of acetonitrile (0–10% in 90 min) in 25 mM TEAA (pH 7) at a flow rate of 1 ml/min. Detection of the oligonucleotide was achieved using a UV-visible spectrophotometer set at 260 nm. The purity of 3 and 7 was controlled by denaturing PAGE analysis. The presence and the integrity of oxazolone in the oligonucleotides were assessed by MALDI-TOF-MS measurements.
Primer extension
Reactions catalyzed by Kf exo– were carried out in 10 µl of 50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 0.05 mg/ml bovine serum albumin (BSA) and 1 mM dithiothreitol (DTT). Primer extension reactions using pol β were performed in 10 µl solutions of 50 mM Tris–HCl (pH 8.8), 10 mM MgCl2, 10 mM KCl, 0.4 mg/ml BSA, 1 mM DTT and 1.5% glycerol. Reactions catalyzed by Taq were conducted in 10 µl solutions of 10 mM Tris–HCl (pH 8.3), 1.5 mM MgCl2 and 50 mM KCl. Buffered solutions containing the oligonucleotide template 1, 3 or 11 and the 5′-end-labeled primer 5′-d(GGAGTGGAGA) (2), in a template:primer nanomolar ratio 1:2, 3:2 or 11:2 = 45:15, were heated to 60°C for 5 min and then cooled to 4°C over a period of 2 h. DNA polymerization reactions were carried out with either 100 µM solutions of a single dNTP or a mixture of all four dNTPs. The solutions were maintained at 25°C for 10 min in the presence of 0.01 U of Kf exo–. The samples were incubated for 30 min at 25°C in the presence of 0.1 U of either pol β or Taq. Reactions were stopped by adding 5 µl of a solution containing 95% formamide, 0.1% bromophenol blue and 0.1% xylene cyanol (formamide dye). Then, the samples were heated at 70°C for 3 min prior to application to a 20% polyacrylamide/7 M urea gel. Subsequently, analysis of the radiolabeled bands was achieved by phosphorimaging (Molecular Dynamics Phosphorimager) using Image QuanT software.
Fpg and endonuclease III repair studies: assays for nicking activity
DNA repair experiments were carried out with Fpg and endo III on modified double-stranded DNA fragments that contained a unique oxazolone residue. Typically, 0.5 pmol of 32P-labeled modified oligonucleotide 3 were annealed to 0.75 pmol of the complementary strand (4A, 4C, 4G or 4T) by heating at 80°C for 5 min and subsequent slow cooling to room temperature over a period of 2 h. The integrity of the modified duplex was then assessed by MALDI-TOF-MS. The enzymatic reactions were performed in 10 µl solutions of 20 mM Tris–HCl (pH 7.5), 1 mM EDTA, 100 mM KCl at 37°C for 30 min with increasing concentrations of either Fpg (1–10 ng/µl) or endo III (2–20 ng/µl). For control experiments that require denatured Fpg or endo III, the enzyme was first dissolved in formamide and heated at 65°C for 20 min prior to incubation with the modified duplex DNA. Enzymatic reactions were stopped by adding 5 µl of formamide dye and the samples were then subjected to denaturing 20% PAGE. The gels were then analyzed as previously described by phosphorimaging.
Kinetic studies with determination of Vmax and Km
The concentration range of modified oligonucleotide 6, 7 or 9, in the presence of 1.5 equiv. of the complementary strand 8C (for 6 and 7) or 8G (for 9) was 0.1–4 µM. Substrate concentrations were chosen in order that the Michaelis–Menten curves reached a plateau. For each reaction, in 10 µl volume, the amount of 32P-labeled oligonucleotide was 1 pmol (0.1 µM). The concentrations of Fpg (30.2 kDa) were 2 and 0.8 ng/µl for the oxazolone- and 8-oxoGua-modified duplexes, respectively. The concentrations of endo III (23 kDa) were 1 and 3 ng/µl for the oxazolone- and 5-OHC-modified double strands, respectively. The enzymatic reactions were allowed to proceed at 37°C for 10 min in the presence of Fpg or 2 or 5 min in the presence of endo III for the oxazolone and 5-OHC duplexes, respectively. The reactions were then stopped by adding formamide dye. The samples were subjected to 20% denaturing PAGE and the resulting gel was analyzed by phosphorimaging. Bands corresponding to cleavage products and unreacted oligonucleotides were quantified using Image QuanT software. Vmax and Km were calculated by non-linear least squares fitting of the data points using Microcal Origin, on the basis of, at least, three separate experiments. Reaction velocity (V) was expressed in pmol substrate/min/ng enzyme while the substrate concentration was given as µmolarity (Figs 6 and 7).
Figure 6.

(A) Michaelis–Menten kinetics. Cleavage of oxazolone-containing oligonucleotide by Fpg (2 ng/µl). (B) Km and Vmax values for the removal of oxazolone and 8-oxoGua by Fpg (2 and 0.8 ng/µl, respectively). Numbers in parentheses indicate standard errors.
Figure 7.

(A) Michaelis–Menten kinetics. Cleavage of oxazolone-containing oligonucleotide by endo III (1 ng/µl). (B) Km and Vmax values for the removal of oxazolone and 5-OHC by endo III (1 and 3 ng/µl, respectively). Numbers in parentheses indicate standard errors.
MALDI-TOF-MS analysis of Fpg-mediated cleavage of oxazolone-containing oligonucleotide
Enzymatic reaction was carried out with 25 pmol of modified DNA duplex 3/4C in 10 µl solutions of 20 mM Tris–HCl (pH 7.5), 1 mM EDTA and 100 mM KCl. DNA repair protein Fpg was added at a final concentration of 5 ng/µl and the resulting solution was incubated at 37°C for 30 min. The reaction was quenched by freezing the solution in liquid nitrogen followed by lyophilization. The oligonucleotides were subsequently precipitated with 3 M ammonium acetate/ethanol (1/3 v/v) and then analyzed by MALDI-TOF-MS. Mass spectra were obtained with a commercial time-of-flight mass spectrometer (Voyager-DE; Perseptive Biosystems) equipped with a 337 nm nitrogen laser and pulsed delay source extraction. The spectra were recorded from 256 laser shots with an accelerating voltage of 25 kV in the linear and positive modes. For the matrix, a mixture of 3-hydroxypicolinic acid and picolinic acid in a 4:1 (w/w) ratio was dissolved in a 50% acetonitrile aqueous solution that contained 0.1% trifluoroacetic acid (TFA) and a small amount of Dowex-50W 50X8-200 (Sigma) cation exchange resin. Then, 1 µl of a 0.1% TFA aqueous solution of the sample was added to 1 µl of the matrix and the resulting solution was stirred. The sample was subsequently placed on top of the target plate and allowed to dry by itself. The spectra were calibrated with a 1 pmol/µl solution of myoglobin (m/z 16 952), using the same assay conditions as described for the oligonucleotides.
RESULTS
Insertion of dNTPs opposite oxazolone
In the present study, attempts were made to determine the mutagenic potential of oxazolone, a major oxidative product of 2′-deoxyguanosine in single-stranded DNA, during in vitro DNA synthesis. A 15mer oligonucleotide template 3 (Fig. 2) containing the oxazolone moiety at position 5 from the 5′-end was subjected to primer extension using a 10mer primer 2 (Fig. 2). The primer was extended using three different DNA polymerases in the presence of either a single dNTP or a mixture of all four dNTPs. Unmodified template 1 (Fig. 2) was used as a control for nucleotide incorporation. Results of primer extension reactions catalyzed by Kf exo– (lanes 1–5), pol β (lanes 6–10) and Taq (lanes 11–15) are shown in Figure 3. DNA synthesis using template 1 (Fig. 3A) led to the expected insertion of dCMP opposite dG (lanes 2, 7 and 12). In the presence of the four dNTPs (lanes 5, 10 and 15), extension of the oligonucleotide to full length was observed. The relative amounts of dCMP insertion opposite dG obtained for the three DNA polymerases are dependent on both the concentration of the enzymes and the polymerization time (data not shown). Different parameters, including the template:primer ratio, the amount of enzyme and the polymerization times (see Materials and Methods), were optimized to avoid misincorporation of dNTPs opposite dG in the control experiments. When modified oligonucleotide 3 was used as the template for Kf exo–-mediated polymerization (Fig. 3B), dAMP was exclusively incorporated opposite oxazolone (lane 1). Interestingly, the enzyme was able to extend the oxazolone/dA (dZ/dA) mispair and a second dAMP was incorporated opposite dT. When all four dNTPs were present, transient inhibition was observed opposite the damage and only a small amount of full-length product was obtained (lane 5). However, the integrity of the complement was not determined. Primer extension reactions catalyzed by Taq using template 3 (Fig. 3B, lanes 11–15) led mainly to dAMP insertion opposite the lesion (lane 11). In addition, small amounts of dCMP and dGMP were incorporated (lanes 12 and 13, respectively). In contrast to Kf exo–, Taq was not able, in the presence of dATP, to extend the dZ/dA mispair beyond the damage. In the presence of all four dNTPs, only a small amount of a nucleotide likely to be dAMP was inserted opposite oxazolone, with no fully extended primer (lane 15). The results suggest that the oxazolone lesion may represent a stop point for the enzyme. DNA synthesis catalyzed by pol β was also conducted with oligonucleotide 3, and the results are reported in Figure 3B (lanes 6–10). Interestingly, the oxazolone lesion inhibits primer extension mediated by pol β, with no dNTP insertion. This suggests that dZ may induce a change in the conformation of the template which is not suitable for DNA synthesis by pol β. In vitro DNA synthesis opposite a normal abasic site (2′-deoxyribose, template 11) was also performed using Kf exo–, pol β and Taq (data not shown). Similar results to those observed for oxazolone were obtained. The primer was not extended in the presence of pol β, while dAMP was exclusively incorporated by Kf exo– opposite the abasic site. When all four dNTPs were present, inhibition of DNA synthesis was observed opposite the damage. When DNA synthesis was performed with Taq, dAMP was incorporated in addition to dGMP. In the presence of the four dNTPs, transient inhibition was observed opposite the abasic site with no full extension.
Figure 3.
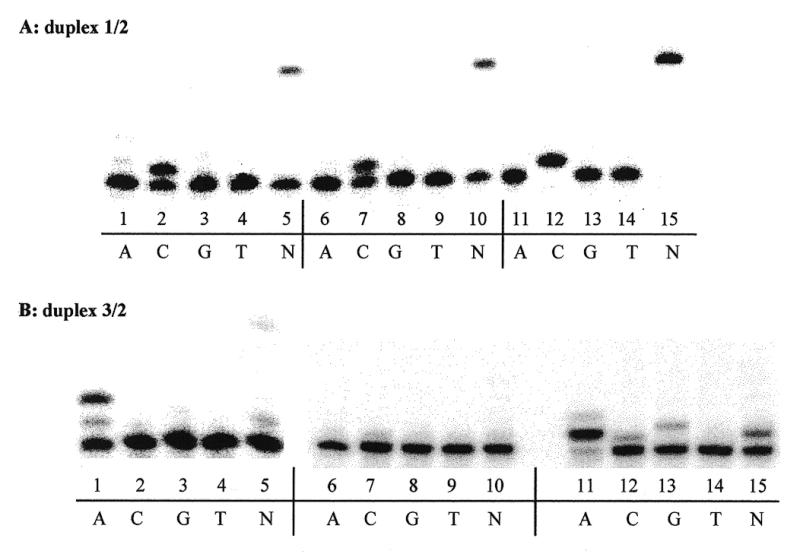
In vitro DNA synthesis. Primer extension reactions catalyzed by Kf exo– (lanes 1–5), pol β (lanes 6–10) and Taq (lanes 11–15) using a single dNTP (A, C, G or T) or a mixture of the four dNTPs (lane N). (A) Unmodified template. (B) Oxazolone-containing template.
Oxazolone excision by Fpg and endo III DNA repair proteins
Substrate specificity of Fpg and endo III towards the oxazolone damage was investigated using a 15mer DNA duplex containing a single oxazolone opposite any of the four DNA bases (Fig. 2, duplexes 3/4A, C, G and T). A control experiment to check for possible depurination of the modified base during the hybridization step is shown in Figure 4A. The modified duplex was analyzed by MALDI-TOF-MS. The peak observed at m/z = 4396.4 is accounted for by oligonucleotide 3 containing the oxazolone nucleoside (calculated M + H+ = 4395), while the peak at m/z = 4732.6 corresponds to the complementary strand 4C (calculated M + H+ = 4732). The mass spectrum did not show evidence for the presence of an abasic site resulting from the loss of oxazolone during the annealing step. Modified DNA oligonucleotides were then subjected to enzymatic reactions using increasing concentrations of Fpg and endo III. Interestingly, both DNA N-glycosylases were shown to incise oligonucleotide 3 at the oxazolone site. Figure 4B shows the cleavage efficiency of the two DNA repair enzymes towards the oxazolone lesion. In control experiments, where the 3/4C duplex was incubated in the presence of denatured Fpg (lane 2) or endo III (lane 3), no enzymatic cleavage was observed. The substrate remained intact by comparison with the starting oligonucleotide (lane 1). Lanes 4–6 show that the oxazolone lesion is recognized and efficiently cleaved by Fpg when it is paired with dC. Complete cleavage of 3 was observed at a concentration of 5 ng/µl of Fpg. Experiments conducted with endo III on duplex 3/4C showed that the oxazolone lesion is also a substrate for endo III (lanes 7–9). Significant cleavage of the duplex was observed at a concentration of 20 ng/µl. PAGE analysis of the fragments obtained upon Fpg- and endo III-mediated excision of the oxazolone and subsequent cleavage of the modified oligonucleotide was compared. This indicates different processing of the damage by the two proteins, providing different products. Cleavage of duplex 3/4C by endo III (lanes 7–9) led to two distinct bands on the gel, while Fpg generates a single band which migrates faster on the gel (lanes 4–6). Similar observations were made for cleavage of duplexes 3/4G and 3/4T by Fpg and endo III (lanes 16–27). This also applies to the processing of oxazolone by endo III when dZ is paired with dA (duplex 3/4A, lanes 13–15). Interestingly, cleavage of duplex 3/4A by Fpg led to a mixture of, at least, three fragments (lanes 10–12). On the other hand, only one cleaved oligonucleotide was observed for the three other duplexes. This is suggestive of a different enzymatic processing of the lesion. No cleavage was observed with low amounts of Fpg (lane 10), however, at higher concentrations of the enzyme three bands were observed on the gel.
Figure 4.

(A) MALDI-MS analysis of the modified duplex 3/4C. (B) PAGE analysis of oxazolone strand cleavage by Fpg (lanes 4–6, 10–12, 16–18 and 22–24) or endo III (lanes 7–9, 13–15, 19–21 and 25–27), using for each duplex 1, 5 or 10 ng/µl of Fpg or 2, 10 or 20 ng/µl of endo III.
MALDI-TOF-MS analysis of products resulting from the cleavage of dZ by Fpg
Figure 5 shows the mass spectra of the fragments generated by Fpg from duplex 3/4C. The oligonucleotides are present in the protonated form, together with K+ or Na+ counterions, leading to a distribution of peaks (unmarked peaks). The peak observed at m/z = 4731 corresponds to the complementary strand. Fragments at m/z = 1206 and 2980 arise from cleavage of 3 as follows: the fragment at m/z = 1206 is accounted for by the 4mer oligonucleotide released 5′ to the lesion (5′-CTCTp-3′, calculated M+H+ = 1205.8), while m/z = 2980 corresponds to the 10mer oligonucleotide released 3′ to the damage (5′-pTCT-CCACTCC, calculated M+H+ = 2980). Taken together, these results indicate that oxazolone is excised by Fpg, followed by enzymatic cleavage of both the 3′ and the 5′ phosphodiester bonds at the site of damage. These findings are in agreement with a β-δ-elimination mechanism as previously reported for Fpg (37).
Figure 5.

MALDI-MS analysis of oxazolone strand cleavage by Fpg
Comparative kinetics, determination of Vmax and Km values
In order to assess the relative efficiency of the excision of 8-oxoGua and oxazolone by Fpg, site-specifically modified oligonucleotides 6 and 7, containing the 8-oxoGua and oxazolone lesions, respectively, were annealed to the complementary strand 8C and subjected to Fpg cleavage. Experiments in which the concentration of the substrate was varied were carried out and values of Vmax and Km were determined for each lesion (Fig. 6A, oxazolone excision by Fpg). As shown in Figure 6B, Vmax was found to be twice as high for 8-oxoGua compared to that of oxazolone. Moreover, the Fpg protein had an ~2-fold lower Km for removing 8-oxoGua in comparison to oxazolone. Taken together, the results indicate that 8-oxoGua is repaired more efficiently than oxazolone. The relative efficiency of cleavage of oxazolone- and 5-OHC-containing oligonucleotides by endo III was also determined. Site-specifically modified duplexes 7/8C and 9/8G were subjected to endo III cleavage. Experiments in which the concentration of the substrate was varied were performed and values of Vmax and Km were determined for each modified duplex (Fig. 7A, oxazolone excision by endo III). As shown in Figure 7B, Vmax was 75 times higher for oxazolone compared to 5-OHC. Moreover, the endo III protein has a 15-fold lower Km for removing oxazolone in comparison to 5-OHC. This result suggests that oxazolone is a much better substrate than 5-OHC for endo III.
DISCUSSION
In the present study we examined the mutagenic potential of oxazolone during in vitro DNA synthesis, as well as the capacity of this lesion to be recognized and excised by Fpg and endo III. Polymerase insertion of nucleotides opposite oxazolone has been investigated using Kf exo –, pol β and Taq as DNA polymerases. In vitro DNA synthesis opposite the lesion and full extension of the primer were performed using either a single dNTP or a mixture of all four dNTPs. Replication of template 3 indicates that dAMP was selectively incorporated opposite the damage by Kf exo– and Taq. Moreover, small amounts of dGMP and dCMP were also inserted by Taq (Fig. 3B). Primer extension reactions catalyzed by pol β led to different results in comparison to Kf exo– and Taq. In fact, pol β was unable to insert a nucleotide opposite the lesion, leading to a stop at the site of damage. This result might suggest a change in the template conformation in such a way that the modified base is no longer a substrate for the enzyme. Alternatively, small amounts of an incorrectely inserted nucleotide may have been removed by the exonuclease activity of pol β. As previously reported for other oxidized purines or pyrimidines (9), nucleotide insertion opposite oxazolone appears to be dependent on the polymerase used. The present results suggest that oxazolone formation in DNA may lead to G→T and, to a much lesser extent, G→C transversions. Furthermore, oxazolone may also represent a block during DNA synthesis. Interestingly, nucleotide incorporation induced by oxazolone using Kf exo– and Taq is similar to that observed with a natural abasic site, where dAMP incorporation is favored. dAMP insertion opposite a non-coding lesion such as an abasic site fits the A rule (39). However, in the case of oxazolone, the heterocycle presents a variety of hydrogen bonding possibilities. On the other hand, it cannot be excluded that dAMP may be incorporated opposite oxazolone on the basis of size and shape as opposed to highly specific and complementary hydrogen bonding, as suggested by Moran et al. for non-hydrogen bonding nucleoside analogs (40,41).
In vitro repair experiments were performed in order to assess the capacity of Fpg and endo III, two E.coli repair enzymes, to remove the oxazolone lesion from modified duplexes. Endo III has already been shown to recognize a wide range of modified pyrimidine bases (34,42–46), including 5-hydroxy-2′-deoxycytidine, thymine glycol, 5,6-dihydrothymine, 5-hydroxy-5,6-dihydrothymine, 5,6-dihydrouracil and 5-hydroxy-5,6-dihydrouracil. Fpg protein acts mainly on alterated purines (47) and was initially identified for its activity on formamidopyrimidines (48). However, its main biological role is the removal of 8-oxo-7,8-dihydroguanine (32,36). Our results indicate that oxazolone is removed from DNA by both Fpg and endo III DNA repair proteins (Fig. 4B). Interestingly, both enzymes excised the oxazolone lesion with similar efficiencies regardless of the base on the opposite strand. However, in the case of a dZ/dA base pair, oxazolone is cleaved less efficiently by the Fpg protein, since significant amounts of the starting duplex are still observed (Fig. 4B, lanes 10–12). Furthermore, in contrast to what is observed for dZ/dC, dZ/dG or dZ/dT base pairs, Fpg appears to process the dZ/dA mispair by at least two distinct mechanisms. Interestingly, our results provide the first evidence for incision at an oxidized purine base by endo III. The relatively small size of the oxazolone residue, in comparison to other purine oxidized products, might explain its incision by endo III. On the other hand, oxazolone cleavage by Fpg confirms that the latter DNA N-glycosylase can also accommodate small substrates, in addition to modified purines, in its active site. This property of the Fpg protein has been previously reported since oxidized pyrimidines such as 5-hydroxycytosine, 5-hydroxyuracil and uracil glycol are cleaved by Fpg (43,49). The kinetic parameters for the excision of oxazolone and 8-oxoGua by Fpg are reported in Figure 6B. Based on values of kcat/Km, 8-oxoGua was found to be a slightly better substrate for Fpg than oxazolone. The removal of 8-oxoG by Fpg is favored only 4-fold. It should be noted that the value of kcat/Km for the removal of 8-oxoGua by Fpg obtained in this study is in good agreement with data previously reported (47). The kinetic parameters for the excision of oxazolone and 5-OHC by endo III are reported in Figure 7B. The kcat/Km value was ~100-fold higher for oxazolone compared to 5-OHC, thus suggesting that the former modified base is a better substrate for endo III. As previously reported, uracil glycol (42) and thymine glycol (50) were found to be the best substrates for endo III. From our results, based on the kcat/Km values, it appears that oxazolone is a better substrate for endo III than thymine glycol and that the removal of uracil glycol by endo III is favored only 1.5-fold. However, as shown in Figure 7B (footnotes b and c) kcat/Km values for excision of a defined modified base by a DNA repair enzyme may differ from one study to another. While oxazolone appears to be a much better substrate than 5-OHC for endo III in our study and for D’Ham et al. (50), Wang and Essigmann (42) reported a kcat/Km value for the excision of 5-OHC by endo III comparable to that assessed for oxazolone. The latter observations underline the importance of using the same batch of DNA repair protein when determining the relative efficiency of oligonucleotide cleavage. Moreover, when working with modified oligonucleotides different DNA sequences may also complicate comparison of the results obtained for the excision of modified DNA bases with a particular DNA repair enzyme.
As previously reported by our group for other DNA lesions (50,51), MALDI-MS analysis of the fragments generated upon enzymatic cleavage provides a straightforward method to gain insights into the mechanistic pathway of repair enzymes. As shown in Figure 5 for oxazolone, Fpg removes the modified base and cleaves the resulting abasic site, giving rise to fragments which are in agreement with a β-δ-elimination mechanism. In the case of endo III, it is generally assumed that excision of a base damage is followed by β-elimination of the resulting AP site (52,53). PAGE analysis of such an α-β unsaturated fragment leads to two distinct bands on the gel, as shown in Figure 4B (54,55). However, MALDI-MS analysis of the products obtained upon excision of oxazolone by endo III failed to detect a fragment bearing an alkenal residue (data not shown). More detailed studies, including the identification of a chemically generated alkenal residue by MALDI-MS, are currently in progress in order to assess the mechanism of endo III-mediated cleavage of modified bases.
Up to now the oxazolone base damage has not been detected at the cellular level and little is known about the relative yields of oxazolone in isolated DNA. In this study, the yield of oxazolone observed upon type I-mediated photosensitization of oligonucleotides 1 or 5 was ~20% (data not shown). However, indirect evidence for formation of oxazolone in double-stranded calf thymus DNA under oxidative conditions, such as γ-irradiation and type I-mediated photosensitization, has been obtained. This was gained from measurement of released guanidine, an alkali breakdown product of oxazolone.
Enzymatic base excision followed by strand scission or alkaline treatment, also leading to a strand break, is currently used to detect site-specific modifications in oxidized DNA (6). These methods rely upon either specific recognition of a modified base by DNA N-glycosylases or on the alkaline lability of the damaged nucleobase. In both cases the deglycosylation step is followed by phosphate elimination at the resulting abasic site, thus leading to strand scission. Interestingly, these methods are used to estimate and to identify oxidative damage at guanine sites within DNA. It is widely speculated that two main oxidative pathways are responsible for the decomposition of guanine. In one hand, the pathway leading to imidazolone and oxazolone in nucleosides or single-stranded DNA and, on the other, to the favored formation of 8-oxoGua in duplex DNA. Until now the two pathways and the resulting products have been tentatively identified and quantitated by PAGE through their different sensitivities to either alkaline treatment or Fpg cleavage. Both imidazolone and oxazolone are sensitive to piperidine treatment, while 8-oxoGua is poorly cleaved upon basic treatment and is released by Fpg (19). However, the present findings indicate that Fpg is able to efficiently recognize oxazolone. Thus, Fpg treatment of damaged DNA to estimate the amount of 8-oxoGua might generate strand breaks not only at 8-oxoGua sites but also at oxazolone lesions. The use of Fpg to detect 8-oxoGua is likely to overestimate the level of the latter lesion within DNA.
In conclusion, our results provide the first evidence that oxazolone, a major guanine oxidation product, induces misincorporation of dAMP and, to a much lesser extent, of dGMP during in vitro DNA synthesis. Alternatively, it may constitute a block for DNA elongation. It is therefore quite interesting that oxazolone is a substrate for both the Fpg and endo III DNA repair enzymes. It will now be important to determine if oxazolone is recognized and excised by other DNA repair enzymes, including Ogg1, Ntg1 and Ntg2. Such studies are currently in progress. Furthermore, given the importance of oxazolone in guanine oxidation, investigations are currently being designed to directly detect this lesion in isolated DNA and at the cellular level using a highly sensitive HPLC-tandem mass spectrometry assay.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Serge Boiteux for the gift of endo III and Fpg proteins. We thank Prof. A. P. Grollman for helpful discussions on this work. We are indebted to Dr J. Laval for discussions on the initial part of the project. We also thank Dr Jean Luc Ravanat and Anne Gaëlle Bourdat for fruitful discussions during the accomplishment of the work. We are grateful to the Comité de Radioprotection (Electricité de France) for financial support.
REFERENCES
- 1.Ames B.N., Shigenaga,M.K. and Hagen,T.M. (1993) Proc. Natl Acad. Sci. USA, 90, 7915–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ames B.N. (1983) Science, 221, 1256–1264. [DOI] [PubMed] [Google Scholar]
- 3.Floyd R.A. (1990) Carcinogenesis, 11, 1447–1450. [DOI] [PubMed] [Google Scholar]
- 4.Ames B.N. and Gold,L.S. (1991) Mutat. Res., 250, 3–16. [DOI] [PubMed] [Google Scholar]
- 5.Breen A.P. and Murphy,J.A. (1995) Free Radic. Biol. Med., 18, 1033–1077. [DOI] [PubMed] [Google Scholar]
- 6.Burrows C.J. and Muller,J.G. (1998) Chem. Rev., 98, 1109–1151. [DOI] [PubMed] [Google Scholar]
- 7.Cadet J., Berger,M., Douki,T. and Ravanat,J.L. (1997) Rev. Physiol. Biochem. Pharmacol., 131, 1–87. [DOI] [PubMed] [Google Scholar]
- 8.von Sonntag C. (1987) The Chemical Basis of Radiation Biology. Taylor and Francis, London, UK.
- 9.Wang D., Kreutzer,D.A. and Essigmann,J.M. (1998) Mutat. Res., 400, 99–115. [DOI] [PubMed] [Google Scholar]
- 10.Feig D.I. and Loeb,L.A. (1994) J. Mol. Biol., 235, 33–41. [DOI] [PubMed] [Google Scholar]
- 11.David S.S. and Williams,S.D. (1998) Chem. Rev., 98, 1221–1261. [DOI] [PubMed] [Google Scholar]
- 12.Laval J., Jurado,J., Saparbaev,M. and Sidorkina,O. (1998) Mutat. Res., 402, 93–102. [DOI] [PubMed] [Google Scholar]
- 13.Wallace S.S. (1994) Int. J. Radiat. Biol., 66, 579–589. [DOI] [PubMed] [Google Scholar]
- 14.Wallace S.S. (1998) Radiat. Res., 150, 60–79. [PubMed] [Google Scholar]
- 15.Steenken S. and Jovanovic,S.V. (1997) J. Am. Chem. Soc., 119, 617–618. [Google Scholar]
- 16.Steenken S. (1996) In Meunier,B. (ed.), DNA and RNA Cleavers and Chemotherapy ofCancer and Viral Diseases. Kluwer, Dordrecht, The Netherlands, pp. 225–247. [Google Scholar]
- 17.Kasai H., Yamaizumi,Z., Berger,M. and Cadet,J. (1992) J. Am. Chem. Soc., 114, 9692–9694. [Google Scholar]
- 18.Ravanat J.L. and Cadet,J. (1995) Chem. Res. Toxicol., 8, 379–388. [DOI] [PubMed] [Google Scholar]
- 19.Spassky A. and Angelov,D. (1997) Biochemistry, 36, 6571–6576. [DOI] [PubMed] [Google Scholar]
- 20.Cadet J., Berger,M., Buchko,G.W., Joshi,P.C., Raoul,S. and Ravanat,J.L. (1994) J. Am. Chem. Soc., 116, 7403–7404. [Google Scholar]
- 21.Raoul S., Berger,M., Buchko,G.W., Joshi,P.C., Morin,B., Weinfeld,M. and Cadet,J. (1996) J. Chem. Soc. Perkin Trans., 2, 371–378. [Google Scholar]
- 22.Gasparutto D., Ravanat,J.L., Gérot,O. and Cadet,J. (1998) J. Am. Chem. Soc., 120, 10283–10286. [Google Scholar]
- 23.Douki T. and Cadet,J. (1996) Free Radic. Res., 24, 369–380. [DOI] [PubMed] [Google Scholar]
- 24.Cadet J., Berger,M., Douki,T., Morin,B., Raoul,S., Ravanat,J.L. and Spinelli,S. (1997) Biol. Chem., 378, 1275–1286. [PubMed] [Google Scholar]
- 25.Kino K. and Saito,I. (1998) J. Am. Chem. Soc., 120, 7373–7374. [Google Scholar]
- 26.Vialas C., Pratviel,G., Claparols,C. and Meunier,B. (1998) J. Am. Chem. Soc., 120, 11548–11553. [Google Scholar]
- 27.Raoul S. and Cadet,J. (1996) J. Am. Chem. Soc., 118, 1892–1898. [Google Scholar]
- 28.Adam W., Saha-Möller,C.R. and Schönberger,A. (1996) J. Am. Chem. Soc., 118, 9233–9238. [Google Scholar]
- 29.Cheng K.C., Cahill,D.S., Kasai,H., Nishimura,S. and Loeb,L.A. (1992) J. Biol. Chem., 267, 166–172. [PubMed] [Google Scholar]
- 30.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 31.Lowe L.G. and Guenguerich,P. (1996) Biochemistry, 35, 9840–9849. [DOI] [PubMed] [Google Scholar]
- 32.Michaels M.L., Tchou,J., Grollman,A.P. and Miller,J.H. (1992) Biochemistry, 31, 10964–10968. [DOI] [PubMed] [Google Scholar]
- 33.Michaels M.L., Cruz,C., Grollman,A.P. and Miller,J.H. (1992) Proc. Natl Acad. Sci. USA, 89, 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boiteux S., O’Connor,T.R., Lederer,F., Gouyette,A. and Laval,J. (1990) J. Biol. Chem., 265, 3916–3922. [PubMed] [Google Scholar]
- 35.Boiteux S., O’Connor,T.R. and Laval,J. (1987) EMBO J., 6, 3177–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tchou J., Kasai,H., Shibutani,S., Chung,M.H., Laval,J., Grollman,A.P. and Nishimura,S. (1991) Proc. Natl Acad. Sci. USA, 88, 4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhagwat M. and Gerlt,J.A. (1996) Biochemistry, 35, 659–665. [DOI] [PubMed] [Google Scholar]
- 38.Romieu A., Gasparutto,D., Molko,D. and Cadet,J. (1997) Tetrahedron Lett., 38, 7531–7534. [Google Scholar]
- 39.Shibutani S., Takeshita,M. and Grollman,A.P. (1997) J. Biol. Chem., 272, 13916–13922. [DOI] [PubMed] [Google Scholar]
- 40.Moran S., Ren,R.X.F. and Kool,E.T. (1997) Proc. Natl Acad. Sci. USA, 94, 10506–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moran S., Ren,R.X.F., Rumney,S. and Kool,E.T. (1997) J. Am. Chem. Soc., 119, 2056–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D. and Essigmann,J.M. (1997) Biochemistry, 36, 8628–8633. [DOI] [PubMed] [Google Scholar]
- 43.Hatahet S., Kow,Y.W., Purmal,A.A., Cunningham,R.P. and Wallace,S.S. (1994) J. Biol. Chem., 269, 18814–18820. [PubMed] [Google Scholar]
- 44.Demple B. and Linn,S. (1980) Nature, 287, 203–208. [DOI] [PubMed] [Google Scholar]
- 45.Breimer L.H. and Lindahl,T. (1984) J. Biol. Chem., 259, 5543–5548. [PubMed] [Google Scholar]
- 46.Boorstein R.J., Hilbert,T.P., Cadet,J., Cunningham,R.P. and Teebor,G.W. (1989) Biochemistry, 28, 6164–6170. [DOI] [PubMed] [Google Scholar]
- 47.Tchou J., Bodepudi,V., Shibutani,S., Antoshechkin,I., Miller,J., Grollman,A.P. and Johnson,F. (1994) J. Biol. Chem., 269, 15318–15324. [PubMed] [Google Scholar]
- 48.Chetsanga C.J. and Lindahl,T. (1979) Nucleic Acids Res., 6, 2673–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purmal A.A., Lampman,G.W., Bond,J.P., Hatahet,J. and Wallace,S.S. (1998) J. Biol. Chem., 273, 10026–10035. [DOI] [PubMed] [Google Scholar]
- 50.D’Ham C., Romieu,A., Jaquinod,M., Gasparutto,D. and Cadet,J. (1999) Biochemistry, 38, 3335–3344. [DOI] [PubMed] [Google Scholar]
- 51.Bourdat A.G., Gasparutto,D. and Cadet,J. (1999) Nucleic Acids Res., 27, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim J. and Linn,S. (1988) Nucleic Acids Res., 16, 1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kow Y. and Wallace,S.S. (1987) Biochemistry, 26, 8200–8206. [DOI] [PubMed] [Google Scholar]
- 54.Bailly V. and Verly,G. (1987) Biochem. J., 242, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mazumder A., Gerlt,J.A., Absalon,M.J., Stubbe,J.A. and Cunningham,R.P. (1991) Biochemistry, 30, 1119–1126. [DOI] [PubMed] [Google Scholar]