

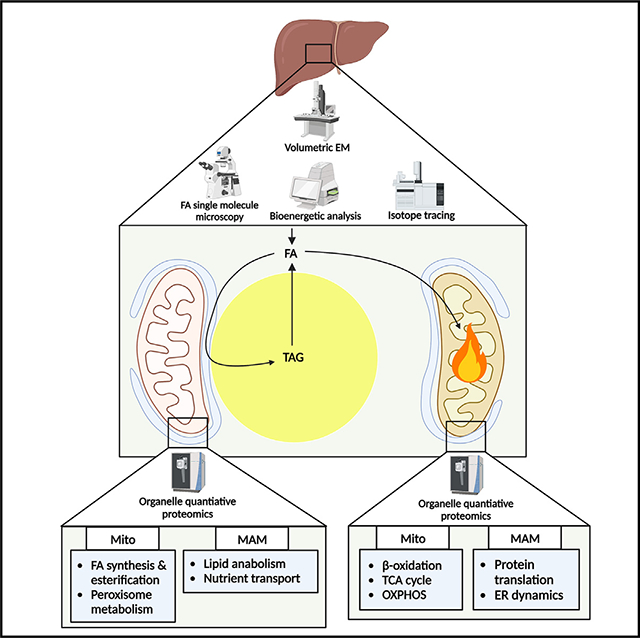
SUMMARY
Organelle interactions play a significant role in compartmentalizing metabolism and signaling. Lipid droplets (LDs) interact with numerous organelles, including mitochondria, which is largely assumed to facilitate lipid transfer and catabolism. However, quantitative proteomics of hepatic peridroplet mitochondria (PDM) and cytosolic mitochondria (CM) reveals that CM are enriched in proteins comprising various oxidative metabolism pathways, whereas PDM are enriched in proteins involved in lipid anabolism. Isotope tracing and super-resolution imaging confirms that fatty acids (FAs) are selectively trafficked to and oxidized in CM during fasting. In contrast, PDM facilitate FA esterification and LD expansion in nutrient-replete medium. Additionally, mitochondrion-associated membranes (MAM) around PDM and CM differ in their proteomes and ability to support distinct lipid metabolic pathways. We conclude that CM and CM-MAM support lipid catabolic pathways, whereas PDM and PDM-MAM allow hepatocytes to efficiently store excess lipids in LDs to prevent lipotoxicity.
In brief
Najt et al. reveal that the proteomes and functions of subpopulations of hepatic mitochondria and MAM differ based on their interactions with LDs. FAs are selectively trafficked to and oxidized in CM, especially during fasting, whereas PDM and associated MAM facilitate FA esterification and LD expansion.
Graphical abstract
INTRODUCTION
Lipid droplets (LDs) represent the primary energy storage depot in most cell types and act as hubs for lipid metabolism and signaling. One way in which LDs influence cellular function is through their interactions with other organelles.1 As an example, LDs are observed to frequently interact with mitochondria, an interaction that is enhanced by nutrient deprivation and/or fasting.2,3 While it would intuitively seem that this interaction would allow transfer of lipolysis-derived fatty acids (FAs) at the LD surface to mitochondria for their subsequent oxidation and energy production, studies demonstrating this accepted dogma are scant4 and typically infer changes in FA trafficking without distinguishing mitochondrial subpopulations.5,6 Additional studies in the liver suggest that lipophagy is the major route of LD degradation,7–10 arguing against direct transfer between LDs and mitochondria. Thus, our understanding of how FAs are trafficked and how lipid metabolism in general is compartmentalized in hepatocytes under different metabolic states is limited.
Studies in brown adipocytes show that subpopulations of mitochondria exist based on their interactions with LDs and suggest that these interactions may facilitate lipid synthetic pathways.11 Indeed, in addition to their capacity for oxidation, mitochondria also have resident proteins involved in lipid anabolism, including FA desaturation and elongation, select pathways of phospholipid (PL) synthesis, and proximal reactions of triacylglycerol (TAG) synthesis.12 Perilipin 5 (PLIN5) is a resident LD protein that bridges LDs and mitochondria.11,13 While PLIN5 is speculated to tether LDs and mitochondria for FA transfer for oxidation,14 this pleiotropic protein has many other functions that complicate its role in lipid metabolism. For example, PLIN5 has been implicated in regulating lipolysis15,16 by inhibiting adipose triglyceride lipase (ATGL) under nutrient-replete conditions,17–20 in facilitating FA esterification,11,21–24 and in nuclear signaling to control mitochondrial biogenesis and oxidative metabolism.25–28 Additional studies in white adipocytes show that interactions between mitochondria, LDs, and the endoplasmic reticulum (ER), which are driven by mitoguardin 2 (MIGA2), facilitate LD expansion, suggesting a complex triorganelle interaction to facilitate FA metabolic channeling.29 These data are supported by studies showing that mitochondrion-associated membranes (MAM), ER domains that are tethered to mitochondria, are enriched in enzymes involved in lipid synthetic pathways.30
Given the limited characterization of mitochondrial subpopulations and lack of understanding of subcellular compartmentalization of lipid metabolism in hepatocytes, we conducted studies to comprehensively define the proteome of these unique mitochondrial populations and characterize how LD interactions impact mitochondria, the ER, and FA trafficking and metabolism. Here, we find that the proteomes of cytosolic mitochondria (CM) are enriched for pathways of oxidative metabolism and are the primary site of FA trafficking and oxidation under fasting conditions. In contrast, peridroplet mitochondria (PDM) and MAM are enriched in proteins involved in lipid synthetic reactions and are the major site of FA trafficking and esterification under fed conditions or during FA exposure. Thus, LD-organelle interactions play a major role in coordinating FA trafficking and compartmentalizing hepatic energy metabolism.
RESULTS
CM and PDM have distinct proteomes
Recent studies have hypothesized that PDM facilitate FA trafficking toward mitochondrial β-oxidation,4,5,31 while others have suggested that mitochondrion-LD association enhances LD biogenesis and thereby protects mitochondria from lipotoxicity in adipose tissue.11,29 Given the conflicting literature, we sought to understand the role of PDM and CM in hepatic FA metabolism. In this study, we adapted an approach to isolate intact respiring mitochondria from the LD surface in addition to those free in the cytosol from the hepatic tissue (Figure S1A).11 Analysis of PDM and CM from livers of overnight-fasted mice revealed MitoTracker-positive particles and little to no BODPIY (a LD marker) staining (Figure S1B). Western blot analysis confirmed the lack of LD contamination in the PDM and CM fractions because no detection of the LD marker protein PLIN2 was detected (Figure S1C), whereas similar levels of COXIV (a mitochondrial marker) were present in the two mitochondrial subpopulations (Figure S1C). While the mitochondrial fractions were free of LD-contaminating proteins, the standard differential centrifugation method used did not remove tightly adhered ER/MAM from the mitochondria (Figure S1C). To comprehensively characterize the differences between PDM and CM, we subjected them to quantitative discovery-based proteomics (Figure S1D).
Following liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, we identified 3,190 proteins from a total of 62,793 unique peptides at a 2-peptide and a 1.1% false discovery rate (FDR) minimum. Of the 3,190 proteins, 794 mapped to the 1,140 (69.6%) validated mitochondrial proteins32 (Figure 1A). Analysis of previously published MAM proteomics data revealed that, of the remaining 2,396 proteins, 1,701 (71%) overlapped with reported MAM proteins (Figure 1A), while the remaining 695 proteins mapped to peroxisomes, lipid transfer proteins, and microbodies associated with the Golgi apparatus, among other cellular compartments (Figure 1A). These results suggest that mitochondrial protein enrichment in CM and PDM fractions was comparable and within the range of previously published methods11,33 and indicate tight organelle-organelle interactions outside of the LD-mitochondrion interactions that were not disrupted with the current isolation methodology. CM showed enrichment of 390 proteins over PDM (Figure 1B). Pathway analysis mapped these CM-enriched proteins to mitochondrial metabolic pathways, such as acetyl-coenzyme A (CoA) biosynthesis, FA β-oxidation, oxoglutarate oxidation, and NADH metabolism, in addition to the TCA cycle and mitochondrial complex protein formation (Figure 1C). In contrast, the PDM were enriched in 676 proteins that primarily mapped to pathways of lipid metabolism, peroxisome α-oxidation, and mitochondrion-ER targeting (Figures 1B and 1D). Hierarchical clustering highlighted the specific sets of proteins that were differentially enriched in CM or PDM (Figure 1E). Using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, statistically significant proteins that clustered together and were increased in CM mapped to mitochondrial electron transport, mitochondrial acetyl-CoA biosynthetic processing from pyruvate, mitochondrial β-oxidation, and TCA cycle proteins (Figure 1E). PDM clustered proteins indicate enrichment in glycerol lipid processing, FA α-oxidation, organelle-organelle binding, peroxisome fission, translation, and oxidation/reduction proteins. These processes were further broken down into the specific pathways mapped out in Figure 1F. CM-enriched pathways had between 10% and 60% of the genes for oxidative phosphorylation, FA β-oxidation, α-keto acid metabolism, valine degradation, acetyl-CoA biosynthetic processes, and the TCA cycle, while PDM were enriched in oleate biosynthesis, LXR/RXR signaling, TAG metabolism, FA α-oxidation, NOX/reactive oxygen species (ROS) signaling, and NAD biosynthesis. Given the relative increase in TCA cycle, oxidative phosphorylation (OXPHOS), and FA β-oxidation genes in our CM proteomes, we mapped the proteins detected in these pathways (Figures 1G and 1H). Western blot analysis of proteins involved in OXPHOS and FA β-oxidation confirmed the proteomics data (Figures 1I and S1E). Together, these results suggest that CM had higher expression of proteins involved in oxidative metabolism, while PDM were enriched in proteins supporting lipid anabolic pathways, supporting the idea that PDM exist to support LD expansion.
Figure 1. CM and PDM have unique proteomes.
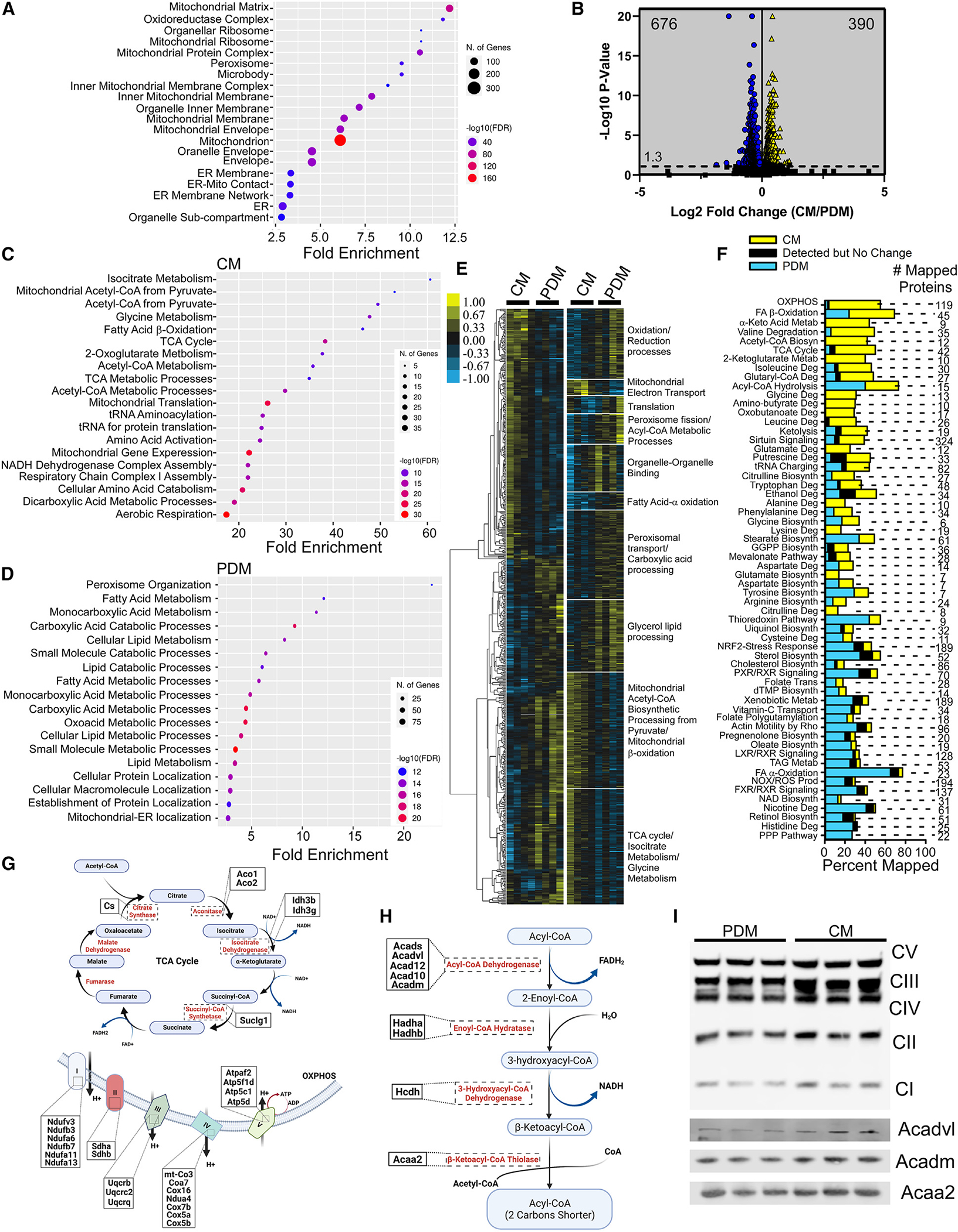
(A) Cellular compartment annotation of the entire proteomics dataset as determined by Gene Ontology (GO) analysis: cellular component.
(B) Volcano plot summarizing the changes in the proteomes of CM and PDM isolated from livers of overnight-fasted mice; n = 4. Blue indicates proteins that were enriched in the PDM fractions, and yellow indicates CM-enriched proteins. Significance was determined using non-parametric quantitative analysis in Scaffold; permutation tests between groups with Benjamini-Hochberg correction (p < 0.00968).
(C and D) Pathway analysis for the 390 and 676 statistically significant proteins in CM and PDM, respectively.
(E) Left: hierarchical clustering of proteins significantly different between PDM and CM across replicate fractions. The threshold for clustered proteins was determined by significance between groups. Right: proteins were further grouped using k-means statistics, breaking the PDM and CM significant genes into 10 protein clusters. The clustered proteins were mapped to specific metabolic pathways using Panther classification overrepresentation testing.
(F) GO terms from the pathway analysis of all significant proteins were expressed as a percentage of proteins enriched in PDM (blue), CM (yellow), or unchanged (black).
(G) Schematic of the TCA cycle and oxidative phosphorylation (OXPHOS) pathways, highlighting proteins significantly upregulated in CM.
(H) Schematic of the FA β-oxidation pathway, with proteins significantly upregulated in CM highlighted.
(I) Mitochondrial fractions were analyzed by SDS-PAGE and immunoblotting for the OXPHOS complex proteins Acadvl, Acadm, and Acaa2. Samples normalized to Ponceau S are shown in Figure S1E.
PLIN5 impacts cytosolic and peridroplet proteomes
PLIN5 is an LD-coating protein that is capable of recruiting mitochondria to LDs through its C-terminal region.11,13 We and others have also shown that PLIN5 promotes mitochondrial biogenesis.25,27 Given the link between mitochondrial function and localization to PLIN5, we assessed the proteomes of PDM and CM from mice treated with antisense oligonucleotides (ASOs) to knock down PLIN5 (Figure S2A). Of the 1,348 statically significant proteins across all four groups (CM, PDM, PLIN5 knockdown [KD]-CM, PLIN5 KD-PDM), only 21.4% were linked to KD of PLIN5 (Figure S2B). Downregulation of PLIN5 impacted the proteome of the CM to a greater extent, altering 317 proteins compared with PDM, where PLIN5 KD only altered 232 proteins. Pathway analysis of CM proteins impacted by PLIN5 were associated with mitochondrial gene expression and translation, carboxylic acid catabolism, amino acid metabolism, FA metabolism, and translation, in addition to other catabolic processes (Figure S2C). Proteins altered in PDM of PLIN5 KD livers were largely involved in pathways of FA oxidation and catabolism (Figure S2D). Hierarchical clustering of these genes highlighted specific protein clusters that PLIN5 KD impacted, specifically PPAR/PGC1α signaling, mitochondrial translation, lipid catabolism, and FA β-oxidation in the CM fractions (Figure S2E). More specifically, PLIN5 KD decreased PPAR/PGC1α-dependent protein expression and FA β-oxidation and increased peroxisome organization and peroxisomal import proteins, in addition to mitochondrial mRNA processing, RNA catabolism, and apoptotic mitochondrial changes (Figure S2E). PDM clustered proteins indicate that PLIN5 KD downregulates proteins associated with lipid metabolic processes, specifically lipid incorporation, FA desaturation, and de novo lipogenesis proteins, in addition to mitochondrion organization, transmembrane transport, and acetyl-CoA metabolism. Downregulation of these proteins in the PDM by PLIN5 KD was accompanied by increases in proteins linked to protein N-linked glycosylation, cholesterol biosynthetic processes, and peroxisome organization/FA α-oxidation (Figure S2F). We confirmed changes in select OXPHOS and FA oxidation enzymes, which also highlighted the effects of PLIN5 on normalizing protein levels (Figures S2G and S2H). Taken together, these results suggest that PLIN5 signaling plays a critical role in programming CM and PDM metabolic function and that loss of PLIN5 not only disrupts key protein expression via PPAR/PGC1α signaling, as expected, but also causes the differences between the CM and PDM to become less distinct.
CM have increased supercomplex I, II, and IV activity
Based on our proteomics analysis, we reasoned that increased OXPHOS and TCA cycle proteins would lead to increased oxidative capacity in CM. To test this, we utilized the Seahorse XF96 extracellular flux analyzer with PDM and CM fed different substrates to determine complex-specific oxygen consumption. Prior to the assay, we confirmed that equal levels of mitochondrial protein and functional mitochondria were present in CM and PDM, although PDM exhibited a slightly lower membrane potential (Figures 2A and 2B). CM fed pyruvate and malate as a fuel source showed increased basal and maximal ATP production in response to either ADP or FCCP, indicating elevated complex I activity (Figures S3A and S3B). CM assayed using succinate as a fuel showed increased basal, ADP-stimulated, and maximal ATP production, indicating increased complex II activity (Figures S3C and S3D). Next, we determined complex III and IV respiratory capacity using duroquinol and TMPD/ascorbate, respectively (Figures S3E–S3H). Duroquinol-driven respiration was not statistically significant between PDM and CM even though basal activity was increased in CM (Figure S3E). TMPD/ascorbate-driven respiration was 2-fold higher in CM, indicating increased complex IV activity (Figures S3G and S3H). Taken together, these results suggest that CM have enhanced OXPHOS capacity largely driven by increases in complex I, II, and IV, which parallels changes in the abundance of OXPHOS proteins (Figure 1).
Figure 2. CM have enhanced bioenergetic capacity.
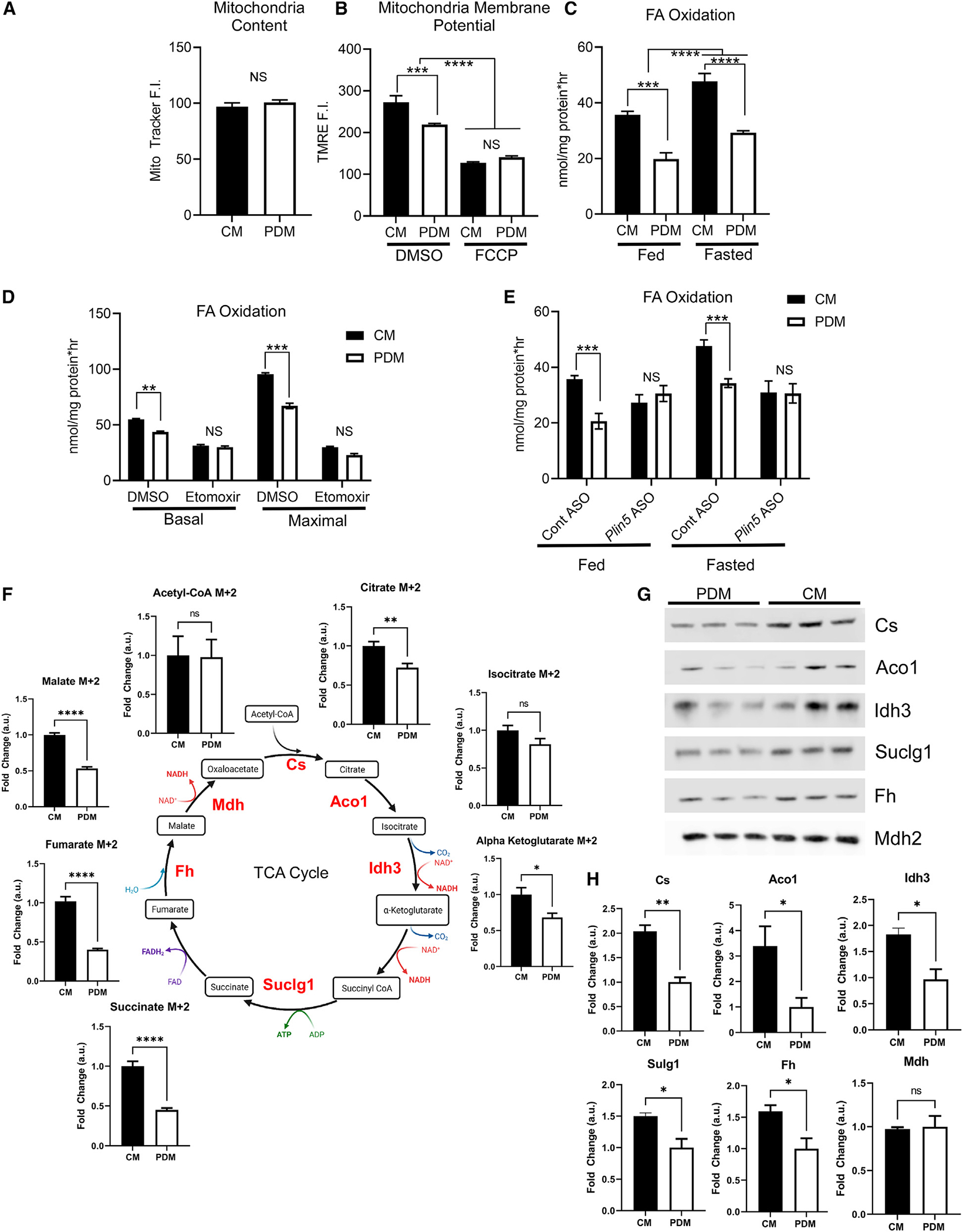
(A) Quantification of MitoTracker Deep Red fluorescence intensity (F.I.) of isolated CM or PDM to ensure equal mitochondrial content. 12–16 wells were quantified per condition and repeated on three independent isolations.
(B) Quantification of TMRE F.I. in control and FCCP-treated mitochondrial fractions; 12–16 wells, repeated on three independent isolations.
(C) Quantification of mitochondrial FA oxidation using [14C]oleate in mitochondrial fractions isolated from mice in the fed or fasted metabolic state. n = 3.
(D) The FA uptake inhibitor etomoxir was used to ensure the oxidation rates determined from our experimental design were due to mitochondrial FA β-oxidation. Shown is quantification of FA oxidation from PDM and CM fractions treated with DMSO or etomoxir in basal or ADP-stimulated maximal respiration. n = 6 from2 different mitochondrial isolations.
(E) Quantification of mitochondrial FA oxidation for CM and PDM isolated from control or PLIN5 KD animals in the fed or fasted metabolic state. n = 4 mitochondrial fractions isolated from independent mice.
(F) TCA cycle intermediates were analyzed by MS from PDM and CM treated with [13C]palmitate as the fuel source. Metabolite intermediates enriched in [13C] were quantified and expressed relative to CM. Shown is a schematic of the TCA cycle with the detected metabolites and the fold change between PDM and CM.
(G) Mitochondrial fractions were analyzed by SDS-PAGE and immunoblotting to Cs, Aco1, Idh3, Suclg1, Fh, and Mdh2. Samples normalized to Ponceau S are shown in Figure S1F.
(H) Quantification of immunoblots depicted in (H) relative to total protein loading.
ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
CM have increased FA oxidation and TCA cycle activity
Our results show that hepatic PDM are not the mitochondria population to support oxidation; instead, CM are the primary drivers of oxidation during fasting. We next measured FA catabolism in PDM and CM, given the differences in proteins involved in β-oxidation and the TCA cycle we observed in the PDM and CM proteomes. To test this, we measured FA-driven respiration and TCA cycle flux in isolated mitochondria using [14C]oleate or [13C]palmitate (Figure 2). FA oxidation was increased in CM compared with PDM and in response to fasting in CM and PDM (Figure 2C). Maximal FA oxidation stimulated by ADP mirrored the Seahorse data showing that CM had greater oxidative capacity and FA oxidative capacity (Figure 2D). We confirmed that CM increased FA oxidation capacity by inhibiting CM and PDM with etomoxir, which decreased fat oxidation rates to a similar level (Figure 2D).
Given the known positive effects of PLIN5 on mitochondrial oxidative capacity, we isolated CM and PDM from control and PLIN5 KD mice and repeated the FA oxidation assays. In control ASO-treated mice, CM maintained a greater oxidization rate compared with PDM in the fed and fasted state (Figure 2E). In contrast, treatment with the PLIN5 ASO negated the differences between CM and PDM, decreasing CM FA oxidation in the fed and fasted states and eliminating the increase in FA oxidation observed with fasting (Figure 2E). We furthered these data by evaluating [13C]palmitate incorporation into TCA cycle intermediates (Figure 2F). Although enrichment of acetyl-CoA was unchanged, almost all other TCA cycle intermediates showed increased enrichment of the label [M+2] in CM relative to PDM (Figure 2F). Consistent with this, the protein abundance of TCA cycle enzymes was consistently elevated in CM compared with PDM (Figures 2G, 2H, and S1F). Collectively, this work shows that CM possess increased FA oxidation and TCA cycle activity relative to PDM, again supporting a role of CM as drivers of OXPHOS in hepatic tissue.
FAs are differentially trafficked to mitochondrial subpopulations based on the metabolic state
We next sought to determine whether the differences in the proteomes of and flux through FA β-oxidation and OXPHOS pathways in isolated PDM and CM corresponded to alterations in FA trafficking in live cells. To gain high-resolution insights into the dynamics and nanoscopic localization of single FAs as they pertain to mitochondrion utilization or storage in LDs, we applied single-molecule localization microscopy (SMLM)34 with a recently developed approach using the fluorescently labeled FA analog BODIPY-C12 in living cells.35,36 SMLM offers the unique ability to determine the localization of single FAs and mitochondrion molecules in living cells with ~30-nm precision, enabling quantitative analysis of the localization density of one37 or two38 molecule species in a subcellular compartment. These SMLM experiments, therefore, fill the knowledge gap between static high-resolution electron microscopy (EM) studies and diffraction-limited conventional fluorescence microscopy and can quantify the density of FAs in different mitochondrial fractions with high precision. AML12 cells transfected with TOM20-HaloTag were placed in fed or fasting medium containing 150 nM BOPDIY-C12 complexed to BSA for 4 h (Figures 3A and 3B). Post counter staining for LDs with AutoDOT dye and labeling TOM20-HaloTag with the dye ligand JF646, we performed SMLM experiments to quantified the density of BODIPY-FA in CM and PDM under the different medium conditions (Figures 3C and 3D). In complete medium, PDM displayed a 5-fold increased density of FAs relative to CM (Figures 3E and 3F). In contrast, the FA density in CM was 2-fold higher compared with PDM under fasting medium conditions (Figures 3E and 3F). In addition to exogenous uptake, catabolism of LDs is also a major source of FAs, especially during fasting conditions. Thus, we conducted experiments with cells preloaded with the fluorescent BODPIY-FAs to determine whether endogenous FAs were differently trafficked between mitochondrial subpopulations (Figures 3G and 3H). Similar to what was observed with exogenous FAs, endogenously derived FAs were preferentially trafficked to PDM under fed conditions (Figure 3G) and to CM under fasting conditions (Figure 3E). While changes in the density of either exogenous or endogenous FAs in PDM were subtle between fed and fasting states, FA density was robustly increased in CM during fasting compared with the fed state (Figure 3H). For the experiments with endogenous FAs, we observed a similar, ~80% enrichment in the PDM FA density in the fed state but more than a 260% increase in the CM FA density during fasting (Figure 3H). These data indicate that there is a major increase in FA trafficking to CM during fasting states to fuel FA β-oxidation, which is supported by the increased capacity of the CM for FA utilization (Figure 2). The increased density of FAs in PDM during the fed state, when the need for FA oxidation is low, suggests that PDM are supporting lipid anabolic pathways.
Figure 3. SMLM quantifies differential FA trafficking to CM and PDM dependent on metabolic state.
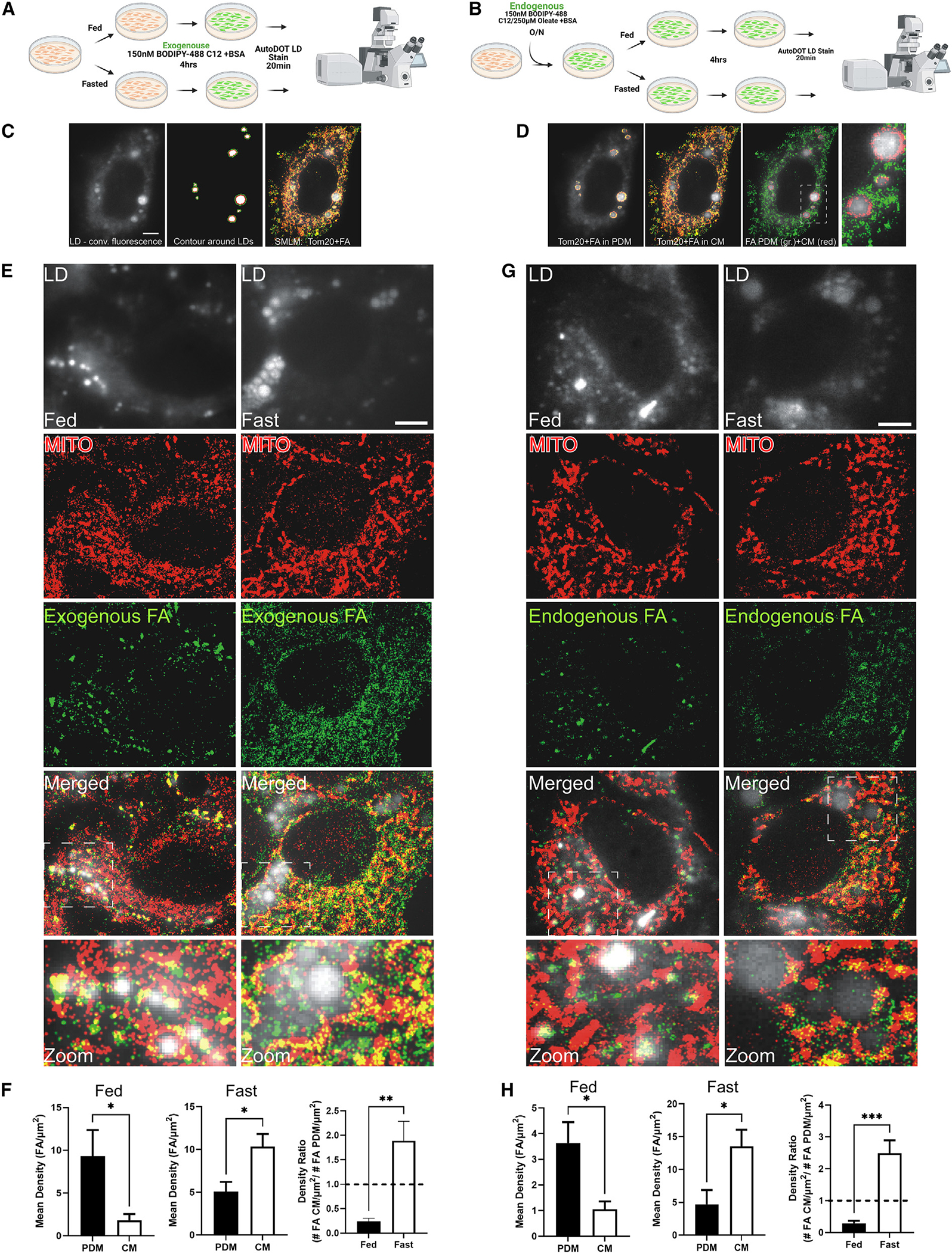
(A and B) Schematic of the experimental design for exogenous (A) or endogenous (B) FA tracing for SMLM experiments.
(C) Left: conventional fluorescence image of LDs in mammalian cells stained with the AutoDOT dye. Center: to discriminate CM from PDM, binary masks are created by thresholding the conventional fluorescence image of LDs. The depicted scaled contour of the binary LD signal is used to identify the mitochondrial fluorescence signal in proximity to LDs as PDM (red line, LD boundary; green line, PDM boundary). Right: two-color SMLM image of BODIPY-C12 (green) and Tom20-HaloTag JF646 localizations (red) superimposed on the conventional fluorescence LD signal.
(D) Left: BODIPY-C12 and Tom20-HaloTag JF646 localizations in PDM as identified by the scaled LD contour. Center: BODIPY-C12 and Tom20-HaloTag JF646 localizations in CM. Right: super-imposed SMLM image of BODIPY-C12 localizations in PDM (red) and BODIPY-C12 localizations in CM (green). Magnification: BODIPY-C12 in PDM and CM around an LD cluster. Scale bar, 5 μm.
(E) Exogenous FA tracing experiments; top to bottom: conventional fluorescence image of LDs stained with AutoDOT dye under fed (left) and fasted (right) conditions. SMLM image of mitochondria labeled with Tom20-HaloTag JF646. Shown are super-resolution images of the FA analog BODIPY-C12 under fed (left) and fasted (right) conditions and super-imposed SMLM images of Tom20-HaloTag JF646 and BODIPY-C12 localizations on top of conventional LD fluorescence images under fed (left) and fasted condition (right). Magnifications depict Tom20 and FA distribution around clustered LD regions. Scale bar, 5 μm.
(F) Quantification of the density of exogenous BODIPY-C12 FAs in PDM and CM in fed (left) and fasted (center) cells and the FA density ratio of CM/PDM (right).
(G) Endogenous FA tracing experiments as in (C) with the BODIPY-C12 (150 nM) preloaded in LDs overnight with non-fluorescent oleate (250 mM) and switched to fed (left) and fasting medium (right).
(H) Quantification of the density of endogenous BODIPY-C12 FA densities in PDM and CM in the fed (left) and fasted state (center) and FA density ratios of CM/PDM (right).
ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001.
PDM promote FA esterification and de novo lipogenesis
The increase in FA trafficking during the fed state along with increased proteins involved in lipid anabolic pathways suggest that PDM may support LD biogenesis or expansion. To determine whether PDM have increased capacity for FA esterification, we conducted [14C]oleate labeling experiments with PDM and CM along with isolated LDs (Figure 4A). Cell homogenates and isolated LDs exhibited the capacity to incorporate [14C]FA into neutral lipids, as observed previously.39 Co-incubation of LDs with PDM increased FA incorporation into TAG, diacylglycerol (DAG), PL, and cholesterol esters (CEs) compared with LDs alone or homogenates, while isolated PDM and CM alone had limited incorporation capacity (Figures 4B and 4C). In contrast, addition of CM to LDs failed to enhance FA esterification to PL, DAG, and CE but did marginally (~25%) increase TAG synthesis above LDs or liver homogenate alone (Figure 4C). These results match our FA trafficking and proteomics data, where PDM have increased levels of anabolic proteins and trafficked FAs to a higher degree in the fed state, when FA oxidation is low. To determine whether PDM only facilitated FA esterification into complex lipid species or whether PDM could also support de novo lipogenesis, we repeated the experiment using [14C]acetate (Figures 4A and 4D). Similar to the FA esterification assays, co-incubation of PDM with LDs enhanced incorporation of de novo-synthesized FAs into PL (2.25-fold) and TAG (2.1-fold) compared with co-incubation with CM (Figure 4D). Thus, the proteomics, energetics, and lipid incorporation assays contrast the existing dogma of PDM direct transfer of FAs for oxidation and instead show that PDM support the synthesis and esterification of FAs into complex lipids for storage in LDs.
Figure 4. PDM-LD association promotes lipid synthesis.
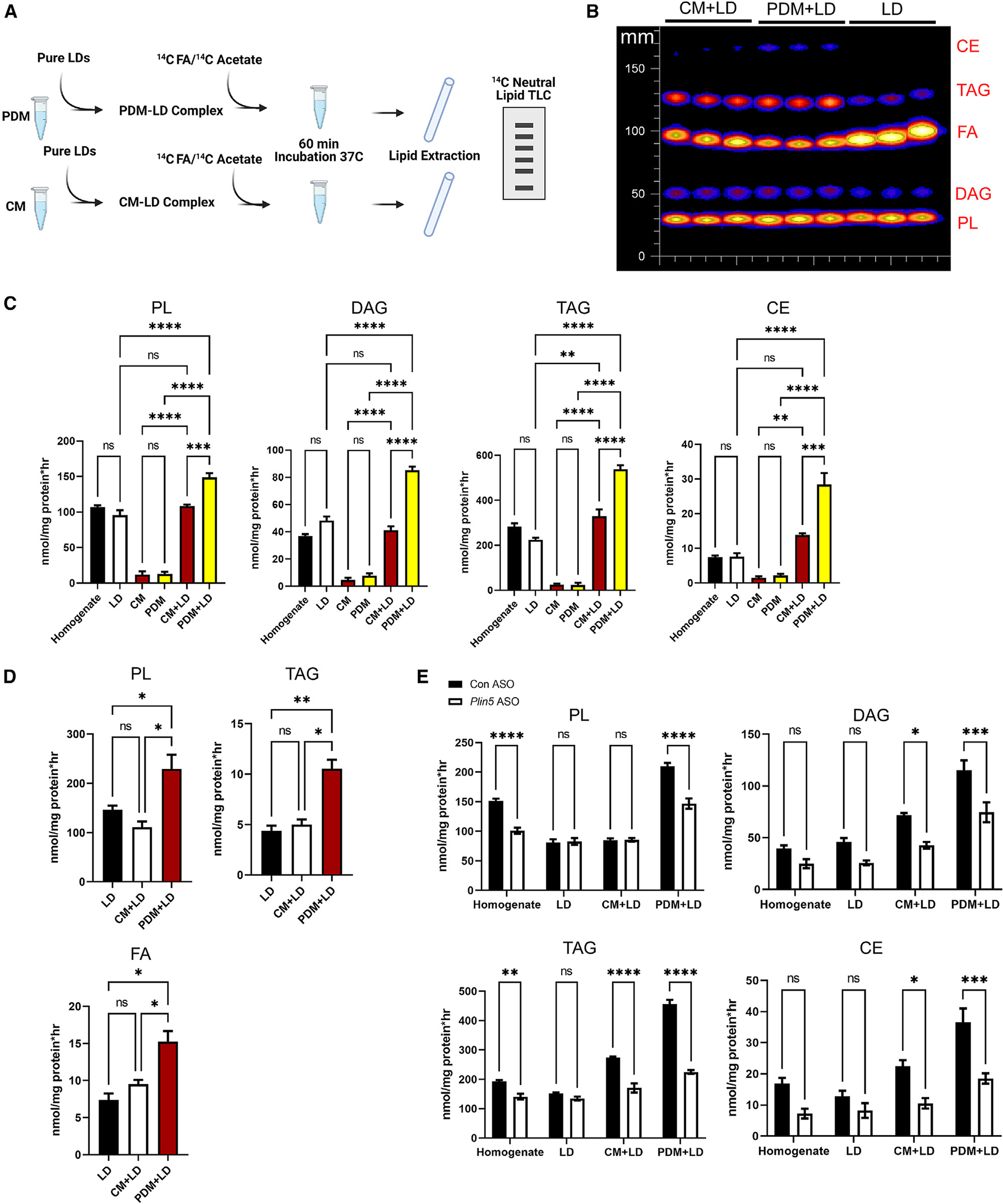
(A) Schematic of the experimental design using [1-14C]oleate or acetate as the substrate to assess FA incorporation into complex lipids from samples isolated from fasted mice.
(B) Representative thin-layer chromatography (TLC) of lipids extracted from liver homogenates, LDs, and mitochondrion-LD incubations.
(C) [1-14C]oleate incorporation into PL, DAG, TAG, and CE. n = 3 independent isolations.
(D) [1-14C]acetate incorporation into PL, TAG, and FA. n = 3.
(E) [1-14C]oleate incorporation into LDs co-incubated with CM and PDM isolated from control or PLIN5 KD fasted animals. n = 4.
ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Given the link between PLIN5 and mitochondrial recruitment to the LD surface, we also tested the impact of PLIN5 on the ability of PDM to support lipid anabolic pathways. Organelles isolated from mice treated with control or PLIN5 ASO were subjected to the same [14C]FA co-incubation as above (Figure 4A). PLIN5 KD significantly blunted PDM-mediated FA incorporation into all complex lipid species measured (Figure 4E). In addition, PLIN5 KD also blunted FA esterification into TAG, DAG, and CE in LDs co-incubated with CM as well esterification into PL and TAG in cell homogenates but not LDs (Figure 4E). Because FA incorporation was blunted with PLIN5 KD in PDM and CM, these data suggest that perhaps PLIN5 has a broader role in reprogramming mitochondria to support lipid anabolic pathways in addition to regulating FA catabolic pathways.
Metabolic states regulate mitochondrion, ER, and LD contacts
Given the role of PDM in supporting lipid anabolic pathways, we sought to characterize the underlying mechanism. The proteomics analysis of PDM and CM revealed that ER-mitochondria interactions were intact in our isolation. Given the tight association between the two organelles, we sought to compare the architecture of PDM- and CM-ER interactions. To do so, we employed serial block face scanning EM, which provides volumetric EM data, in liver samples (Figure 5A). Segmentation and analysis of the micrographs revealed that nearly every mitochondrion (CM or PDM) in hepatocytes was extensively associated with the ER (Figure 5A), which agrees with recent work describing this as “wrapped ER.”40 While direct interactions between LDs and mitochondria were observed, we found that contacts between mitochondria and the ER also play a role in mitochondrion-LD interactions (Figure 5A). We separated these organelle interactions into 3 classifications: mitochondria directly interacting with the LD surface and the ER wrapped around portions of the mitochondria (ER-MITO-LD; orange, Figures 5A and 5B), ER directly interacting with the LD in the absence of mitochondria (ER-LD; green Figures 5A and 5B), and ER sandwiched between the mitochondria and the LD (MITO-ER-LD; white Figures 5A and 5B). Quantification of these types of interactions revealed that direct contact of the mitochondria to the LD surface was the most common, followed by the ER sandwiched between the mitochondria and LDs and ER-LD contacts in the absence of mitochondria (Figure 5C).
Figure 5. Fasting-to-feeding transitions regulate mitochondrion-LD and mitochondrion-ER-LD contacts.
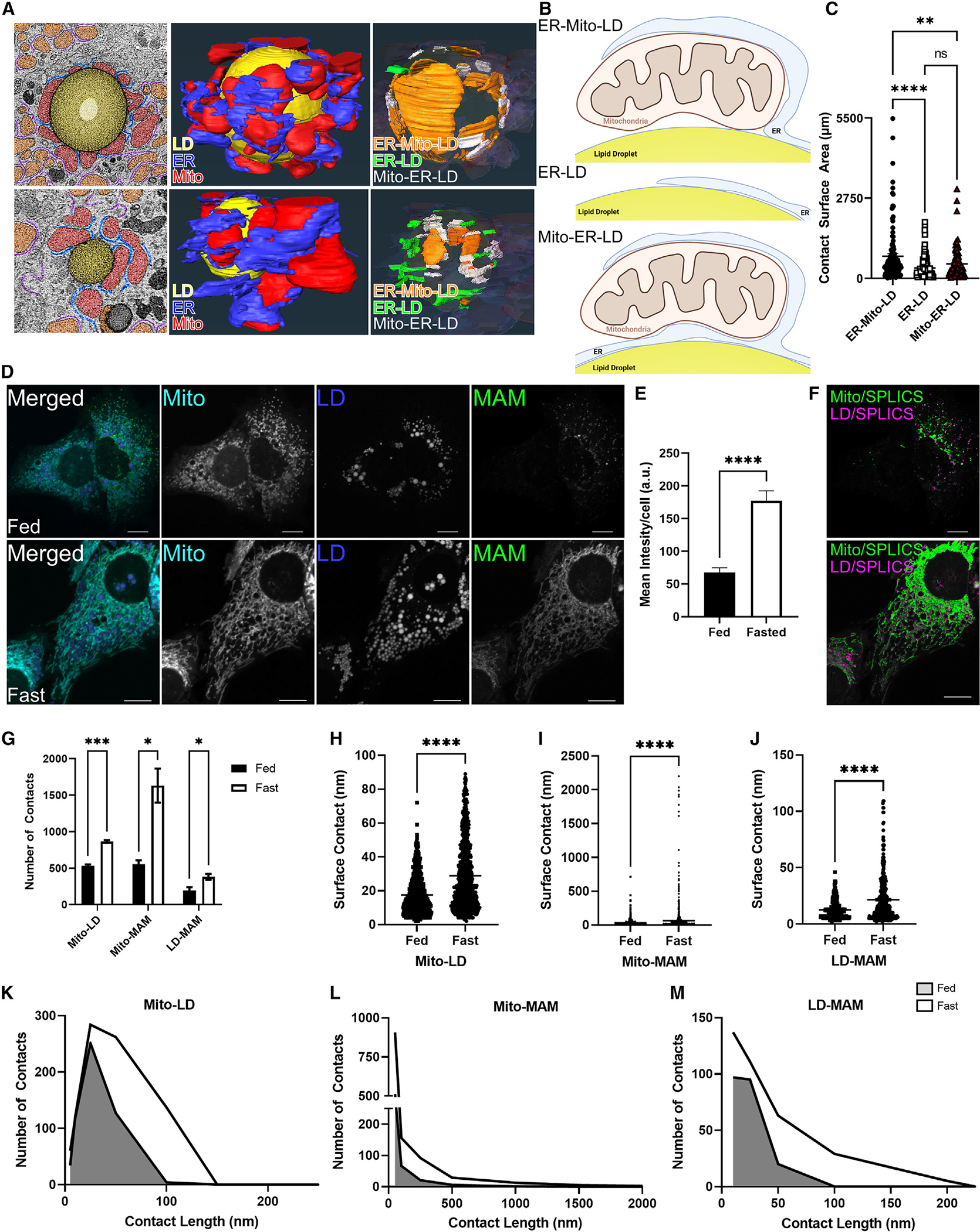
(A) Micrographs of LDs surrounded by PDM and CM from control ASO mice fasted overnight in addition to 3D reconstruction of serial tomograms compiled as a single image. Blue, ER; red, mitochondria; yellow, LDs. ER-MITO-LD (orange), ER-LD (green), and MITO-ER-LD (white) interactions were identified and segmented to render interaction maps in 3D and are shown in the far-right image.
(B) Schematic of the different interactions.
(C) Quantitative analysis of the ER-MITO-LD, ER-LD, and MITO-ER-LD contacts. Quantification was determined from 2 samples and 4 regions of interest.
(D) Representative confocal microscopy images of mitochondria, LDs, and the MAM sensor (SPLICSs) in AML12 cells in fed and fasting medium.
(E) Quantification of SPLICS by total mean intensity per cell. n = 6–8 images with 12–16 cells per condition.
(F) Colocalization analysis burned onto binary images, identifying the region of interaction between the SPLICS sensor and the mitochondrial signal (green) or the SPLIC sensor and the LDs (purple).
(G) DiAna plug-in colocalization was determined from images in (D), quantifying the number of MITO-LD, MAM-MITO, and MAM-LD contacts as determined by the DiAna plug-in.
(H) Surface contact length between mitochondria and LDs in the fed and fasted state.
(I) Surface contact length between SPLICS and mitochondria in the fed and fasted state as determined by the DiAna plug-in in FIJI.
(J) Quantification of surface contact length between SPLICS and LDs in fed and fasted states
(K–M) Histogram outlining the change in contact length determined in (H)–(I).
Data are expressed as means ± SEM. ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
The static high-resolution EM-tomography studies allow identification of mitochondrial-ER-LD structures in a given metabolic state with high precision but do not tell us how the architecture of the inter-organelle contacts changes because of the transition between feeding and fasting. To observe the dynamic changes between organelles under different conditions, we implemented a MAM reporter known as SPLICS.41 This sensor comprises an ER targeting sequence fused with the β−11 barrel of GFP and an outer mitochondrial membrane targeting sequence fused to GFP β barrels 1–10.41 When mitochondria interact with the ER to form MAM, GFP is complete and fluoresces (note: there are two SPLICS sensors; for these studies we used the short version, which measures tight ER-mitochondrion interactions less than 10.5 nm in distance). AML12 cells transduced with the SPLICS sensor and counterstained for mitochondria and LDs showed GFP puncta at or near the mitochondrion-LD interface in complete fed medium (Figure 5D). When the cells were placed in fasting medium, the SPLICS signal tripled relative to the fed state (Figures 5D and 5E). Moreover, the pattern of the SPLICS signal went from puncta in the fed state to a network mirroring the mitochondrial signal under fasting conditions (Figures 5D and 5F). The increase in MAM formation because of fasting was expected,41,42 but the rearrangement of the SPLICS from puncta to a network was not.
To quantify the change in the type of interactions we were observing with the mitochondria, LD, and MAM, we subjected the images to DiAna distance analysis.43 Using this tool, we were able to determine the number and length of interactions between mitochondria, LDs, and MAM (Figures 5G–5M). The number of mitochondrion-LD interactions increased because of fasting (Figure 5G), consistent with previous results.2,3,23 This ~30% increase also corresponded to an increase in the surface contact length of the mitochondrion-LD interactions (Figures 5H and 5K). Upon fasting, we observed a large increase (~3-fold) in the number (Figure 5G) and length (Figures 5I and 5L) of mitochondrion-MAM contacts. This dramatic increase in length (up to 2,200 nm) of the MAM underlies the observed changes in morphology, showing the change from puncta to a network as cells transitioned from fed to fasted states (Figures 5E, 5J, and 5K). When we conducted the DiAna analysis on LDs and MAM, we observed a 115% increase in the number and a 69% increase in the length of contacts (Figures 5G, 5J, and 5M). Thus, these data show that, in addition to the well-established increases in fasting-induced LD-mitochondrion interactions and MAM formation, there is also a simultaneous increase in the number and length of LD-MAM interactions.
FAs regulate mitochondrion-LD and mitochondrion-ER-LD contacts regardless of metabolic state
The mechanisms by which LD and mitochondria come together remain largely unknown; however, in hepatic tissue, fasting induces LD formation and FA oxidation in parallel with increased LD-mitochondrion interactions.23,44,45 These data could also be interpreted to indicate that the presence of excess lipids, rather than the need for energy production, could drive mitochondrion-LD interactions. To test this, we employed confocal microscopy along with the DiAna analysis in cells cultured under fed or fasting medium conditions with or without exogenous FAs. Addition of exogenous FAs during the fed state did not increase the number of LD and mitochondrion interactions, as observed with fasting alone; however, they did increase the length of LD-mitochondrion contacts, indicative of a wrapping morphology (Figures 6A–6D). These data, showing that FAs alone are sufficient to increase the area of mitochondrion and LD interactions under nutrient-replete conditions, suggest that FA availability may drive these organelles together.
Figure 6. FAs regulate mitochondrion-LD and mitochondrion-ER-LD contacts regardless of metabolic state.
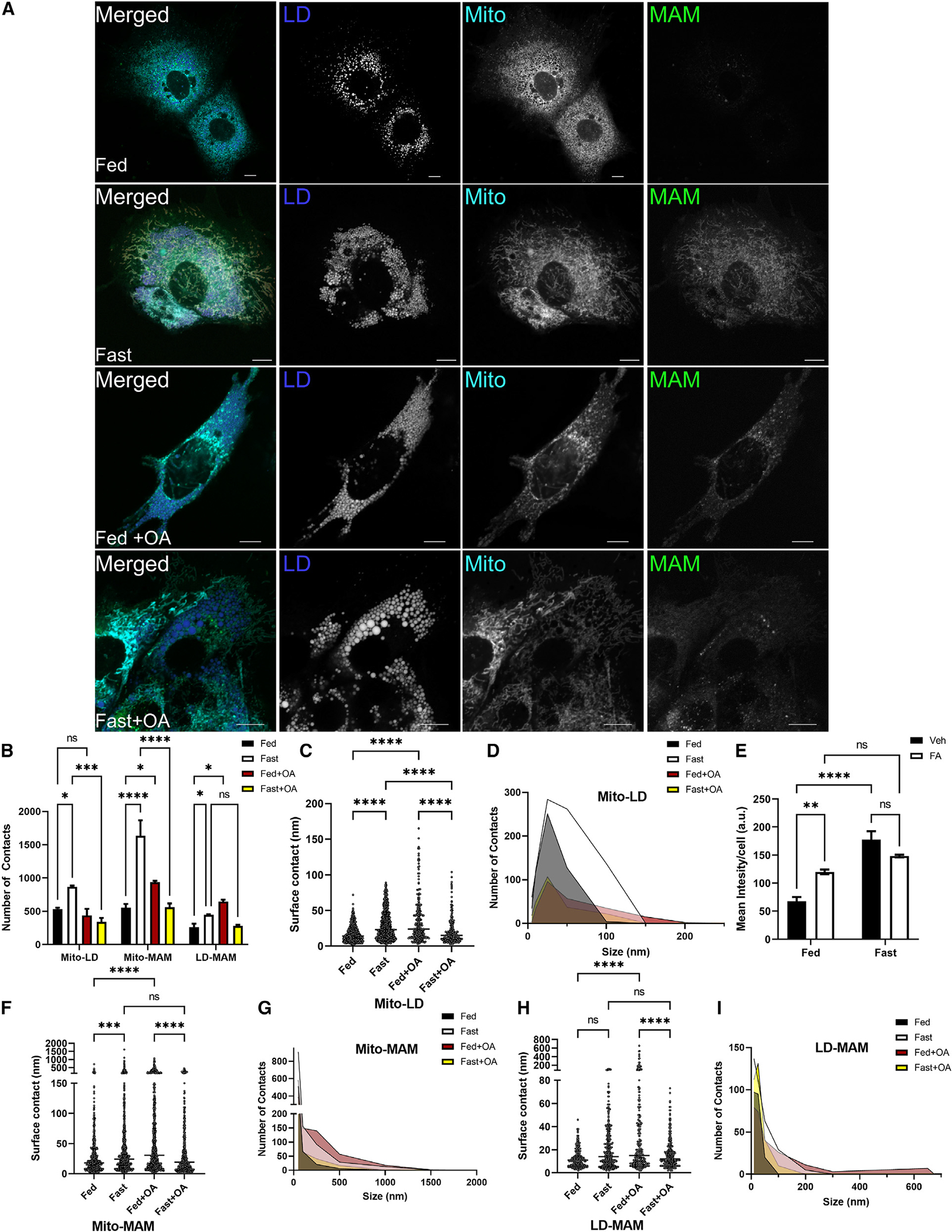
(A) Representative confocal microscopy images of mitochondria and LDs in AML12 cells in fed, fasting, fed plus 250 mM oleate, or fasting plus 250 mM oleate in the medium.
(B) DiAna plug-in colocalization determined from images in (A), quantifying the number of MITO-LD, MAM-MITO, and MAM-LD contacts as determined by the DiAna plug-in.
(C) Surface contact length between mitochondria and LDs in the fed and fasted state.
(D) Histogram outlining the change in contact length determined in (C).
(E) Quantification of SPLICS by total mean intensity per cell. n = 6–8 images with 12–16 cells per condition.
(F) Surface contact length between SPLICS and mitochondria in the fed and fasted state as determined by the DiAna plug-in in FUJI.
(G) Histogram outlining the change in contact length determined in (F).
(H) Quantification of surface contact length between SPLICS and LDs in fed and fasted states.
(I) Histogram outlining the change in contact length determined in (H).
ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
While mitochondria contain many enzymes involved in proximal pathways of glycerolipid synthesis, they lack many enzymes needed for complete synthesis of downstream products (TAG, DAG, and PL) in the Kennedy pathway. MAM are enriched in proteins involved in FA esterification and LD biogenesis and are known to complement mitochondrial pathways for neutral lipid synthesis. To determine whether exogenous FAs can also influence MAM dynamics, we utilized the SPLICS sensor. We observed similar interactions in the fed and fasted state as described above (Figure 5), where fasting greatly increased MAM formation (Figures 6A and 6E) along with increased number and length of contacts between MAM with mitochondria and LDs (Figures 6A, 6B, and 6F–6I). When excess FAs were introduced to fed medium, the SPLICS sensor increased in total intensity (Figures 6A and 6E). Mapping the interactions, we observed that addition of excess FAs to the fed medium increased the contact number and length of MAM-mitochondrion (Figures 6F and 6G) and MAM-LD interactions (Figures 6H and 6I). Surprisingly, addition of exogenous FAs under fasting conditions reduced the number, but not the length, of mitochondrion-MAM contacts, whereas LD-MAM contacts were unchanged (Figures 6H and 6I). Collectively, these data indicate that FAs are sufficient to drive MAM formation as well as MAM and mitochondrion interactions with LDs under fed conditions, suggesting that these organelles interact to facilitate lipid anabolic pathways rather than FA oxidation.
PLIN5 alters mitochondrion-LD interactions but does not impact MAM formation
We also tested the impact of PLIN5 KD on altering the architecture of PDM- and CM-ER interactions. Segmentation and analysis of the micrographs revealed that PLIN5 KD altered mitochondrion attachment to the LD surface (Figure S4A). While direct interactions between LDs and mitochondria were still observed, we found that contacts between mitochondria and the LD were shorter in overall contact surface area; a decrease of ~300 μm (Figure S4B). The shortening of mitochondrion-LD interactions was accompanied by an increase in the number of mitochondrion-LD contacts per LD (Figure S4C). These results indicate that PLIN5 may be responsible for stabilization of direct mitochondrion-LD contacts rather than their initiation. PLIN5 KD did not significantly impact the mitochondrion-ER-LD area of contact but did increase the number of contacts (Figures S4A–S4C). ER-LD contacts increased in overall length without changes in the total number of contacts in response to PLIN5 KD (Figures S4A–S4C). PLIN5 KD did not significantly impact the mitochondrion-ER-LD area of contact but did increase the number of contacts (Figures S4A–S4C). No differences in MAM formation were observed with PLIN5 KD or overexpression (Figures S4A–S4H). Given our results, PLIN5 impacts the stabilization of mitochondrion-LD interactions, increasing the surface area of these interactions, but does not impact mitochondrion-ER tethering.
Cytosolic and peridroplet MAM have unique proteomes
Given the presence of ER/MAM proteins in our mitochondrial preparations and the extensive presence of ER/MAM with LDs and mitochondria, we further sought to characterize the difference in MAM in PDM and CM subpopulations. To test this, we modified the protocol described by Wieckowski et al.46 to isolate MAM from CM and PDM (Figures S5A and S5B). The purity of the MAM fractions was established by western blotting (Figure S5C) by MAM markers outlined previously.40,46,47 Following LC-MS/MS analysis of the CM- and PDM-associated MAM (Figure S5D), we identified 3,790 proteins from a total of 121,196 unique peptide spectra at a 2-peptide and a 0.8% FDR minimum. These 3,790 proteins belonged to 3,253 protein clusters that map to the mitochondrion, ribosomes, and organelle-organelle contact points, as determined by Gene Ontology (GO) cellular compartment analysis (Figure 7A). Mapping the dataset to published work,40,48,49 we observed proteins known to be in the mitochondria, ER/MAM, and wrapped ER as well as the Golgi apparatus and peroxisomes (Figure 7B). Of these proteins, we found that 476 were significantly enriched in CM-MAM and 607 were enriched in PDM-MAM (Figure 7C). Heatmaps of hierarchical and K-means clustering showed distinct differences between MAM subpopulations, with the most notable changes in the processes involved in lipid metabolism, ER organization, RNA processing, and protein translation (Figure 7D). CM-MAM were enriched in many proteins and pathways involved in MAM protein translation and ER organization/MAM formation (Figures 7D and 7F). In contrast, PDM-MAM was enriched in pathways involved in lipid metabolism, long-chain acyl-CoA metabolism, as well as processing metabolites from pyruvate (Figures 7D and 7G). Given recent literature highlighting lipid-processing enzymes associated with ER-LD-mitochondrion interactions in adipocytes29 and the observation that PDM-MAM are highly enriched in lipid anabolic pathways, we mapped key enzymes associated with mitochondrion transport, FA synthesis, and elongation as well as complex lipid assembly (Figures 7E–7G). At every step in the lipid synthetic pathways, key enzymes were enriched in PDM-MAM, further supporting the role of PDM in lipid storage (Figure 7G). Immunoblotting of select proteins identified in the proteomics dataset confirmed the fold changes observed via MS (Figures S5E and S5F).
Figure 7. CM and PDM-MAM have unique proteomes that impact mitochondrial function.
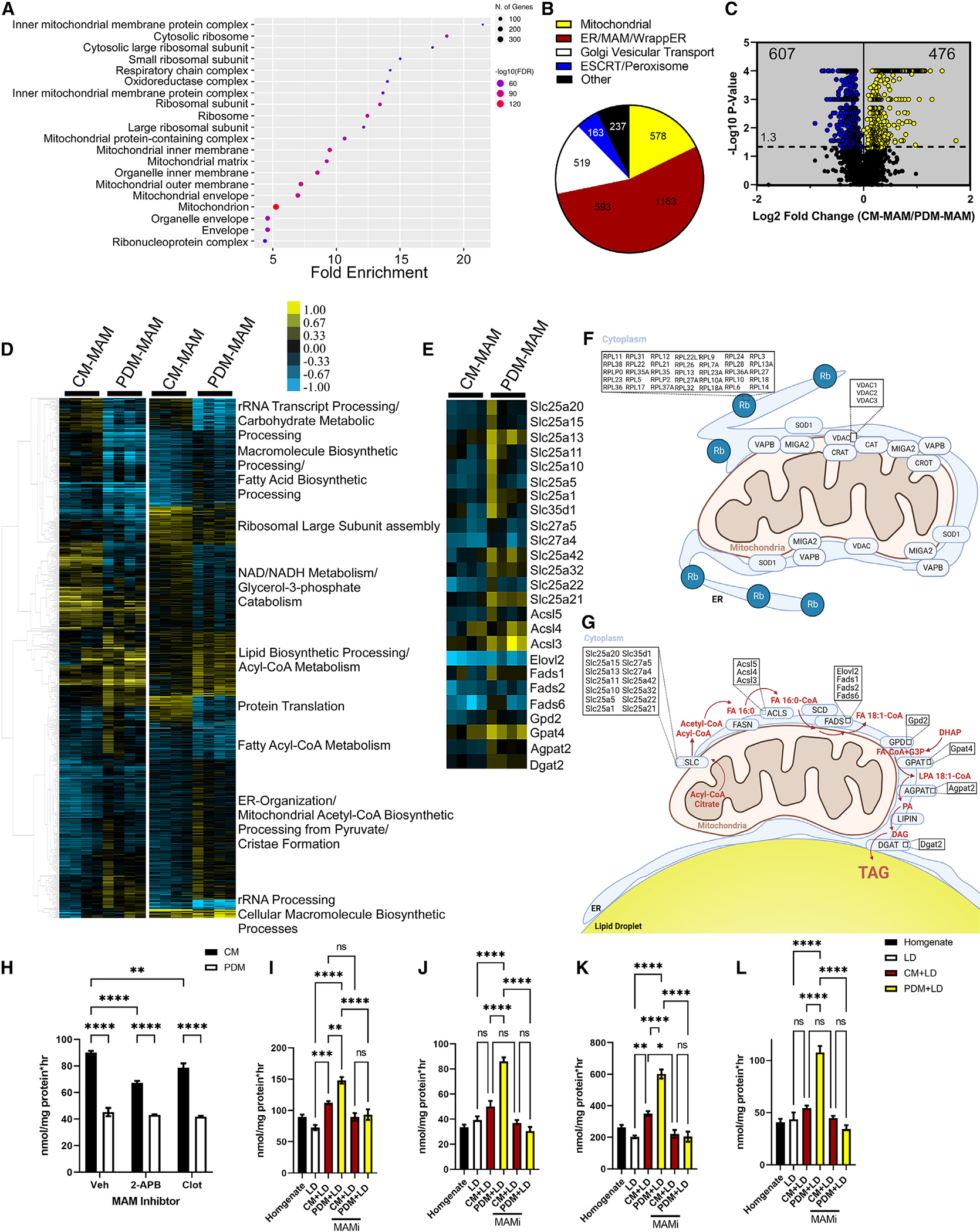
(A) Cellular compartment annotation of the entire proteomics dataset, both MAM subpopulations, as determined by GO analysis: cellular component.
(B) Pie charts summarizing the proteomics dataset in bona fide mitochondria, ER proteins, mitochondrion-associated proteins, wrapped ER mitochondrial (WAM) proteins, Golgi apparatus vesicular transport, and ESCRT/peroxisomes.
(C) Volcano plot summarizing the changes in the proteome of CM-MAM and PDM-MAM isolated from livers of overnight-fasted mice (n = 4 MAM fractions isolated from independent mice). Blue indicates proteins that were enriched in the PDM-MAM fractions, while yellow indicates CM-MAM-enriched proteins. Significance was determined using non-parametric quantitative analysis in Scaffold; permutation tests between groups with Benjamini-Hochberg correction (p < 0.006).
(D) Left: hierarchical clustering of proteins significantly different between PDM-MAM and CM-MAM across replicate fractions (threshold for clustered proteins was determined by significance between groups). Right: proteins were further grouped using k-means statistics, breaking the PDM-MAM and CM-MAM significant genes into 10 protein clusters. The clustered proteins were mapped to specific metabolic pathways using Panther classification overrepresentation testing.
(E) Heatmap of lipid incorporation proteins clustered around the GO term with the highest abundance of proteins increased in the PDM-MAM fraction.
(F) Schematic of CM and CM-MAM that depicts pathways with proteins significantly upregulated.
(G) Schematic of PDM and PDM-MAM that depicts pathways with proteins significantly upregulated.
(H) Quantification of FA oxidation from PDM and CM fractions treated with DMSO or 2-APB or clotrimazole (MAM decoupling compounds). n = 5 from 2 different mitochondrial isolations.
(I–L) Quantification of FA incorporation from PDM and CM promoted storage. Fractions were treated with DMSO, 2-APB, or clotrimazole.
Data are expressed as means ± SEM. ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
We also tested the impact of PLIN5 KD on altering the proteomes of PDM and CM-MAM. Unlike our mitochondrial proteomics dataset, KD of PLIN5 had dramatic effects on the PDM and CM-associated MAM proteomes (Figure S6A). PLIN5 KD impacted 52.4% of all detected proteins in the MAM data compared with 21.4% in the mitochondrial dataset (Figure S6A). Proteins involved in peroxisomal organization and lipid metabolism were altered in CM-MAM (Figures S6B and S6C). We also observed major changes in response to oxidative stress, mitochondrial respiratory chain complex assembly, as well as ER stress. Given the known effects of PLIN5 on mitochondrial biogenesis, these results were expected. Similar trends in reduced expression of proteins involved in oxidative metabolism pathways were observed in PDM-MAM (Figures S6D and S6E). PDM-MAM showed increased FA oxidation through α-oxidation in the peroxisome, reactive oxygen metabolic processes, and lipoprotein particle assembly and clearance (Figure S6E). Surprisingly, PLIN5 KD highlighted the compensation of mitochondrial function by the peroxisome (Figures S6D and S6E). These data indicate that the loss of PLIN5 signaling disrupts mitochondrial biogenesis and maintenance, causing an increase in peroxisome dynamics as a possible mechanism to compensate for the mitochondrial functional loss.
MAM differentially influence lipid metabolism in PDMs and CM
Given the presence of MAM on nearly all mitochondria, we sought to examine the impact of MAM on FA catabolic and anabolic pathways. To do this, CM and PDM isolated from hepatic tissue were subjected to FA oxidation or esterification assays with and without the small-molecule MAM disrupters 2-aminoethoxydiphenyl borate (2-APB) and clotrimazole (Figures 7H–7L). These disruptors impair ER-mitochondrion calcium signaling and mitochondrial function by targeting VDAC and GRP78/BiP but do not release the ER from the attached mitochondrion.50–53 In line with our results described above, CM have twice the oxidative capacity of PDM (Figure 7H). Administration of the MAM disruptor 2-APB or clotrimazole decreased FA oxidation in CM, but oxidation in PDM was not affected (Figure 7H), indicating CM are more dependent on MAM interactions for FA oxidation than PDM. In contrast, co-incubation assays with LDs and PDM or CM revealed that PDM-MAM-LD incubation had the highest rate of FA incorporation across all lipid species (Figures 7I–7L). MAM uncouplers dramatically decreased the ability of PDM to esterify FAs (Figures 7I–7L, yellow bars), decreasing PL, DAG, TAG, and CE by ~50%, 60%, 64%, and 56%, respectively. In contrast, MAM uncouplers only had subtle effects on reducing FA esterification to TAG in CM-LD incubation, while all other lipid species incorporation rates remained low and not significantly different (Figures 7I–7L, maroon bars). These data indicate that MAM play a major role in regulating the distinct lipid metabolic pathways in CM (oxidative) and PDM (anabolic).
DISCUSSION
Recent studies have hypothesized that PDM facilitate FA trafficking toward mitochondrial β-oxidation,4,5,31 while others have suggested that mitochondrion-LD association enhances LD biogenesis and thereby protects mitochondria from lipotoxicity.11,29,54,55 In this study, we adapted an approach to isolate intact respiring mitochondria from the LD surface in addition to those free in the cytosol from hepatic tissue. We determined the proteome, bioenergetic function, and lipid metabolic capabilities of CM and PDM in the liver. While PDM were metabolically active, they did not support high rates of FA oxidation or TCA cycle flux but instead facilitated lipid anabolic pathways, which mirror their proteomes. In contrast, CM were highly metabolically active, exhibiting increased rates of β-oxidation, TCA cycle, and OXPHOS activity, which was also supported by the proteomics analysis of CM showing enrichment of proteins in these pathways. By employing SMLM, we further discriminate PDM and CM fractions in living cells with ~30-nm resolution and demonstrated FA analog accumulation in PDM in fed cells and their re-distribution to CM upon fasting to facilitate their metabolism. Our data suggest that the transfer of FAs from LDs to adjacent mitochondria is not the major route of trafficking for FA oxidation and are supported by a small and growing group of studies in white and brown adipose tissue.11,24,29 In addition, studies in the liver suggest that lipophagy is a major route of LD degradation, arguing against the direct transfer of FAs from LDs to mitochondria as a major route of hepatic FA catabolism.7–10 These results show that hepatic PDM are not the mitochondrion population to support FA oxidation; instead, CM are the primary drivers of FA oxidation during fasting.
MAM were identified and first characterized in rat liver as being enriched in proteins that promote numerous lipid metabolic pathways, including synthesis of TAG, PL, cholesterol, CEs, and lipoproteins.56 More recent studies have detailed the extensive nature of ER and mitochondrion interactions in the liver and show that nearly all mitochondria are at least partially wrapped in ER.40 Moreover, the authors show that MAM are a subdomain of the more continuous “wrapped ER,” the latter of which has variable distances to the adjacent mitochondrial and variable density of ribosomes.40 While the role of the wrapped ER in disease is not known, alterations in MAM have been well defined to contribute to liver function and disease.57–60 Here, we report the characterization of subpopulations of MAM based on their associations with PDM or CM. These data reveal that MAM are heterogeneous and that key proteins that support the lipid anabolic pathways known to exist in MAM are more abundant in PDM-MAM to support LD biogenesis and/or expansion. In addition, disruption of MAM antagonizes the lipid anabolic pathways of PDM and the oxidative pathways of CM supporting the known role of MAM in regulating mitochondrial function.30,47 Collectively, these data suggest that differences in MAM subpopulations are highly intertwined with the unique functions of PDM and CM and further highlight integral roles of MAMs in coordinating hepatic lipid and energy metabolism.
The increase in mitochondria and LD interactions during fasting has contributed to the hypothesis that FAs flux from LDs, upon lipolysis, to mitochondria for oxidation.61 This model would predict that the demand for FA utilization and ATP production during nutrient deprivation would drive LD and mitochondria together to facilitate FA trafficking between the two organelles. However, our data show that FAs themselves can facilitate LD and mitochondrion interactions even in full medium, where the drive for FA oxidation and demand for ATP are low. These data are supported by another study showing that exogenous oleate, which is readily incorporated in LDs, can drive LD and mitochondrial colocalization more than palmitate, which is less readily esterified into the neutral lipids comprising LDs.62 Moreover, our data further extend this work and show that FAs can drive MAM and LD interactions (i.e., 3 organelle complexes) to facilitate FA channeling into LDs rather than their catabolism in CM. Thus, these data suggest that the availability of FAs, which are also increased with fasting, is likely the major driver of LD interactions with MAM and mitochondria instead of energetic demands stemming from nutrient deprivation.
These findings are supported by studies examining LD-mitochondrion interactions in adipose tissue through manipulation of MIGA2 and PLIN5.11,29 In these studies, ablation of MIGA2 in adipocytes disrupted mitochondrion-LD interactions as well as mitochondrion-LD-ER interactions preventing de novo TAG synthesis and LD expansion.29 Overexpression of full-length PLIN5 enhanced lipid storage in LDs, while the truncated version missing the mitochondrion-interacting domain had no effect.11 Taken together, our data and those of others emphasize that mitochondrion-LD interactions exist to facilitate lipid storage and LD expansion over lipid catabolism.
Numerous studies have linked lipolysis to changes in PGC-1α/PPARα signaling and oxidative gene expression.8,25,26,63–66 This signaling is thought to play a key role in increasing the oxidative capacity of the cell to match the supply of lipolytically supplied FAs. PLIN5 has been widely studied as a key LD protein that promotes oxidative metabolism and uncouples LD accumulation from lipotoxicity and insulin resistance.16,21,67–69 Our recent publication identified a role for PLIN5 as an FA binding protein that links LDs to nuclear control of mitochondrial biogenesis, coupling FA release to metabolic oxidative capacity.25 While loss of PLIN5 greatly reduces oxidative capacity and mitochondrial biogenesis, removal of PLIN5 mitochondrial tethering to LDs via truncation exerts a minor, if any, impact on mitochondrial FA oxidation.70 Other studies have also shown that PLIN5 at the LD surface couples mitochondria to the LD surface11 and that loss of PLIN5 decreases lipid storage capacity and LD expansion,23 while overexpression increases the number of LDs and incorporation of radiolabeled lipids into TAG in brown adipose tissue and cultured liver cells.11,14,21,23,71 Given the multi-faceted roles of PLIN5, we hypothesized that loss of PLIN5 would dramatically impact CM oxidative capacity through loss of nucleus-LD signaling and PDM function through loss of mitochondrion-LD interactions. Our results indicate that loss of PLIN5 decreased PDM LD expansion and lipid storage capacity. Conversely, PLIN5 KD decreased PGC-1α/PPARα signaling and impacted the CM proteome to decrease the oxidative capacity of the CM pool. Given these results, LD-bound PLIN5 appears to aid in promoting lipid storage and LD expansion, while nuclear signaling impacts CM functionality in terms of oxidative capacity. Taken together, our data indicate that a population of PLIN5 signals to the nucleus through our published mechanism, increasing mitochondrion biogenesis while the remaining LD-bound PLIN5 promotes LD-mitochondrion contacts to promote lipid storage and prevent lipotoxicity (Figure S7). Given our results and the known role that LDs, the ER, and mitochondria play in regulating cellular function, future studies should characterize the dynamics and these organelles in metabolic and aging-related diseases.
Tissues and cell types have distinct metabolic capabilities that contribute to overall organismal homeostasis. In addition, metabolism compartmentalization within cells exists to complete or link metabolic pathways and cellular processes that impact cell function. Here, we detail the complexities of organelle interactions with LDs to coordinate safe and efficient FA handling. We show that hepatic mitochondria and MAM are heterogeneous based on their interactions with LDs. Specifically, PDM and PDM-MAM possess the proteome and capacity to facilitate FA incorporation into LDs, which is supported by increased LD-MAM-mitochondrion interactions in response to FA availability. In contrast, CM and CM-MAM drive FA oxidation, and the increase in CM underlies the increase in FA oxidation during fasting/nutrient deprivation. Collectively, these studies provide an in-depth characterization of organelle subpopulations that impact lipid metabolism, which may provide insights into the many metabolic and aging-related diseases characterized by alterations in lipid metabolism.
Limitations of the study
We were unable to conduct comparable studies in female mice to determine sexual dimorphic aspects of hepatic FA trafficking. While these studies provide mechanistic insights into the compartmentalization of FA metabolism, we did not characterize how these dynamics change in the context of metabolic or aging-related diseases, which will be a major emphasis moving forward. Finally, these studies were conducted in mice or murine cell lines, which likely exhibit various degrees of difference in lipid metabolism relative to humans.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Douglas Mashek (dmashek@umn.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The two-color single molecule cross-correlation-colocalization filtering code has been up loaded to GitHub (PuchnerLab GitHub).
The proteomics raw data and datasets have been deposited on Massive: https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal experimentation
Mice and anti-sense oligonucleotide administration
All animal protocols were approved by the University of Minnesota Institutional Animal Care and Use Committee. Male 8–12-week-old C57BL6/J mice were obtained from Harlan Laboratories and housed under controlled temperature and lighting (20–22°C; 14:10-h light-dark cycle). The mice were fed a purified control diet (TD 94045; Harlan Teklad Premier Laboratory) and acclimatized for 1 week before any experimental procedure. Control and Plin5 antisense oligonucleotides (ASOs; Ionis Pharmaceuticals) were given via intraperitoneal injection twice per week at a dose of 40 mg/kg. ASO injections were carried out for three weeks, all mice were euthanized for liver tissue and serum collection. Knockdown was confirmed through protein (Western blot) analysis.
METHOD DETAILS
Peridroplet and cytosolic mitochondrial isolation
All procedures were performed using pre-chilled equipment and solutions. The following method was adapted from Benador et al.11 Livers from 12–16-week-old mice were harvested, rinsed in PBS, minced, and suspended in 6 mL Sucrose-HEPES-EGTA supplemented with FA-free BSA (SHE+BSA; 250 mM sucrose, 5 mM HEPES, 2 mM EGTA, 2% FA-free BSA, pH 7.4). The resuspended hepatic tissue was mechanically disrupted with 15 strokes in a glass Teflon Dounce homogenizer. The homogenate was then transferred to a 50 mL falcon tube and diluted up to 10 mL in ice-cold SHE+BSA. The lysates were centrifuged in an Avanti J-15R (Beckman Coulter) swinging bucket rotor at 900×g for 10 min at 4°C. The resulting sample was examined to ensure that a fat cake (containing PDM and LD) was floating on the top of the supernatant (containing CM) and cell debris was pelleted. Using a glass pasture pipette, the fat cake was removed from the supernatant. The fat cake was resuspended in SHE+BSA. The supernatant was transferred to a fresh pre-chilled 50 mL falcon tube and both the fat cake and supernatant were re-centrifuged at 900×g 10 min 4°C. Post centrifugation, the original fat cake and the remaining fat cake on the supernatant were combined and transferred into 2 mL Eppendorf tubes, being careful to remove only the fat cake and as little of the supernatant as possible. The supernatant containing the CM was also transferred to 2 mL Eppendorf tubes and the two fractions were centrifuged at 9,000×g for 10 min at 4°C. The mitochondrial pellets were re-suspended in SHE+BSA and centrifuged with the same settings twice more. Mitochondria were then re-suspended in SHE without BSA and protein concertation was determined by BCA (Thermo Scientific). In experimental designs where LDs were also needed, the residual fat cake stripped of PDM was kept. The fat cake was further purified to obtain LDs by transferring the fat cake to a 2mL Eppendorf tube, suspending them in SHE+BSA, and then centrifuged at 21,000×g for 20 min. Buoyant LDs were removed and placed into a fresh 2 mL tube using a glass pasture pipette and suspended in SHE without BSA. The LDs were again centrifuged at 21,000×g for 20 min before protein concentration was determined with BCA.
Purification of CM and PDM for quantitative proteomics
Samples for quantitative proteomic analysis were collected from four-control ASO and four PLIN5 ASO-treated mice. CM and PDM from these mice were harvested as described above. After the final pelleting of CM and PDM, mitochondrial fractions were resuspended in homogenate buffer [10 mM Tris-base pH 7.0 with protease (Complete protease inhibitor cocktail, Roche, Basel Switzerland), phosphatase (PhosSTOP, Sigma-Aldrich, St. Louis, MO), and deacetylase inhibitors (De-acetylase Cocktail, MedChem Express, Monmouth Junction, NJ)], in place of the SHE without BSA. The resuspended mitochondrial pellets were loaded onto a sucrose step gradient (4 mL 35% sucrose, 4 mL 25% sucrose) and centrifuged at 36,000 rpm in a Beckman SW41 swinging bucket rotor for 4 h. The mitochondria from the CM and PDM fractions appeared as a dark-brown pellet at the bottom of the tube. At this stage, the samples can be snap-frozen. This method is adapted from the full cellular fractionation described in.73
For proteomic analysis, mitochondria samples were extracted with proteomic lysis buffer [PLB1; 7 M urea, 2 M thiourea, 0.4 triethylammonium bicarbonate, pH 8.5, 20% acetonitrile, 10 mM tris (2-carboxyethyl) phosphine (TCEP), 40 mM chloroacetamide]. The samples were pipetted up and down several times and transferred to a PCT tube for the Barocycler NEP2320 (Pressure Biosciences, Inc., South Easton, MA) and cycled between 35 kPSI for 20 s and 0 kPSI for 10 s for 60 cycles at 37°C. Samples were centrifuged at 13000×g for 10 min. The supernatants were transferred to new 1.5 mL microfuge Eppendorf Protein LoBind tubes. Aliquots for each sample were taken for protein concentration determination by Bradford assay. A 25 μg aliquot of each sample was transferred to a new 1.5 mL Eppendorf Protein LoBind tube and brought to the same volume with PLB1. In addition, a pooled sample was created by combining equal amounts of each sample to use as a bridge sample across multiple TMT experiments. All samples were diluted five-fold with water, and then trypsin (Promega, Madison, WI) was added in a 1:40 ratio of trypsin to total protein. Samples were incubated overnight for 16 h at 37°C. After incubation, the samples were frozen at −80°C and dried in a vacuum concentrator. Each sample was cleaned with a 1 CC Waters Oasis MCX cartridge (Waters Corporation, Milford, MA), and eluates were vacuum dried and resuspended in 100 mM triethylammonium bicarbonate, pH 8.5 to a final concentration of 1 μg/μL. For each channel within the TMT10plex a 20 μg aliquot of the appropriate sample was made, and the samples were labeled with the corresponding TMT10plex Isobaric Label Reagent (Thermo Scientific, Waltham, MA) per the manufacturer’s protocol. After TMT labeling, all the samples were multiplexed together into a new 1.5 mL microfuge tube. The TMT sample was dried down in vacuo. The sample was cleaned with a 1 CC Waters Oasis MCX solid phase extraction cartridge (Waters Corporation, Milford, MA), and the eluate was dried in vacuo. The samples were offline fractionated as described previously.74 Each fraction was desalted on a C18 stage tip75 and reconstituted for analysis. Samples were separated on an Easy nLC-1000 UHPLC and analyzed by tandem mass spectra in a data-dependent manner with a Thermo Fisher Orbitrap Fusion mass spectrometer. In brief, we processed the MS peptide spectra using Sequest (Thermo Fisher Scientific, San Jose, CA, in Proteome Discoverer 2.2). The mouse (taxonID 10090) Universal Proteome (UP000000589) target protein sequence database was downloaded from UniProt (www.uniprot.org/) on Nov 19, 2019, and was merged with a common lab contaminant protein database (http://www.thegpm.org/cRAP/index.html); the number of protein sequences was 55,180. The digestion enzyme was trypsin, the fragment ion mass tolerance was 0.08 Da and the precursor tolerance was 15 ppm. We set the variable modifications for oxidation of methionine (+15.9949), pyroglutamic acid conversion from glutamine (−17.0265), deamidation of asparagine (+0.9840), protein N-terminal acetylation (+42.0106) and TMT 10plex (+229.1629) modification of lysine and peptide N-terminus. We specified carbamidomethyl of cysteine as a fixed modification. We used Scaffold Q+ (version 4.8.9, Proteome Software Inc., Portland, OR) for validation of tandem MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 94.0% probability to achieve an FDR less than 1.0% [mitochondrial proteomics] and greater than 91.0% probability to achieve an FDR less than 1.0% [MAM proteomics] by the Percolator posterior error probability calculation.76 Protein identifications were accepted if they could be established at greater than 5.0% probability to achieve an FDR less than 1.0% and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm.77 Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Proteins sharing significant peptide evidence were grouped into clusters. We used Scaffold Q+ (version 5.0.1, Proteome Software Inc., Portland, OR) for TMT-based peptide and protein quantification. Reporter ion intensities were adjusted by correction factors in all samples according to the algorithm described in i-Tracker78 according to the TMTpro 16plex Lot Number WF324548 product data sheet from ThermoFisher Scientific. Normalization was performed iteratively (across samples and spectra) on intensities, as described in statistical analysis of Relative Labeled Mass Spectrometry Data from Complex Samples Using ANOVA.79 Pooled protein samples from all samples in the two TMT experiments were used for normalization, using two TMT channels for each experiment. Medians were used for averaging. Spectra data were log-transformed, pruned of those matched to multiple proteins, and weighted by an adaptive intensity weighting algorithm. Differentially expressed proteins were determined by applying a permutation test with an unadjusted significance level p < 0.05 corrected by Benjamini-Hochberg.80
Heatmap and pathway analysis of proteomics data
Pathway analysis, hierarchical clustering, and heatmap production were carried out as previously described.81–87 The hierarchical clustering and heat maps were generated using Cluster 2.0 from the Eisen Laboratory modified by Michiel de Hoon (http://bonsai.hgc.jp/~mdehoon/software/cluster/). Java Tree Viewer was used to view and color the heatmap.
Isolated mitochondria respirometry using seahorse
The protein content of CM and PDM fractions were measured using BCA reagent, diluted to 0.1–0.4 μg/μL protein, and 2.5–10 μg protein (25–50 μL suspension) was added to each well in the Seahorse plate. The plate was immediately spun at 2,000×g for 20 min at 4°C to pellet mitochondria. Next, 130–150 μL of the appropriate mitochondrial assay buffer was added to the wells to bring up the volume to 180 μL before running the Seahorse assay. Complex I mediated respiration was measured by adding mito assay buffer containing 500 μm malate 5 mM pyruvate to the mitochondria which were followed by the sequential injections of 1) ADP (2 mM), 2) oligomycin (1.5 μM), 3) FCCP (1 μM), and 4) rotenone (0.5 μM)/antimycin A (0.5 μM). Complex II mediated respiration was measured by adding mito assay buffer containing 10 mM succinate and 5 μM rotenone to the mitochondria which was followed by the sequential additions of 1) ADP (2 mM), 2) oligomycin (1.5 μM), 3) FCCP (1 μM), and 4) malonate (mM). Complex III mediated respiration was measured by adding mito assay buffer without and substrates to the mitochondria which was followed by the sequential additions of 1) rotenone (5 μM)/malonate (5 mM), 2) duroquinol (2 μM), and 3) antimycin A (0.5 μM). Complex IV mediated respiration was measured by adding mito assay buffer without substrates to the mitochondria followed by the sequential additions of 1) rotenone (0.5 μM), malonate (5 mM), antimycin A (0.5 μM), 2) TMPD (1 mM), ascorbate (20 mM), and 3) sodium azide (20 mM).
Isolated mitochondria FA oxidation assay using [14C]oleate
Methods for measuring FA oxidation were adapted from Huynh et al.88,89 In brief, mitochondria pellets are resuspended in base mitochondria respiration buffer (MRB; 100 mM Sucrose, 10 mM Tris-HCL pH 7.4, 5 mM potassium phosphate, 0.2 mM EDTA, pH 8.0, 80 mM potassium chloride, 1 mM magnesium chloride, 2 mM carnitine, 0.1 mM malate, 0.05 mM coenzyme A, 2 mM ATP, 1 mM DTT pH 8.0). To ensure that the isolated mitochondria contents could be normalized and maintained respiratory function, we plated 50 μg total mitochondria in a 96-well plate, stained the mitochondria with MitoTracker Deep Red, and determined fluorescent intensity of mitochondria in CM and PDM (Figure 2A). We also plated 50 μg total mitochondria and stained them with the membrane potential sensitive dye TMRE (Abcam). The MitoTracker staining confirmed that protein abundance correlated with total mitochondrial content, and the TMRE indicated that both CM and PDM maintained mitochondrial membrane potential through the isolation procedures. The protein content of CM and PDM mitochondrial fractions were then measured using BCA reagent, diluted to 2 μg/μL protein, and 100 μg protein (50 μL suspension) was added to 350 μL MRB supplemented with 0.7% FA free BSA/500 μM oleate/0.5 μCi [14C]oleate. Samples were then incubated at 37°C for 1 h and quenched by the addition of 200 μL 1M perchloric acid, vortexed, and then centrifuged at >14,000×g for 15 min at room temperature. 400 μL of the supernatant was transferred to a scintillation vial and the radioactive acid-soluble products were counted on a scintillation counter. For maximal respiration measures, 2 mM ADP was introduced to the reaction mixture. Inhibitors, 60 μM etomoxir, 100 μM 2-APB, and 25 μM clotrimazole were also introduced to inhibit FA uptake into the mitochondria, and MAM, respectively.
Isolated mitochondria TCA metabolite measurements using [13C]palmitate
Similar to [14C]oleate studies, CM and PDM were resuspended in MRB and normalized to 10 μg/μL protein, and 500 μg protein (50 μL suspension) was added to 350 μL MRB supplemented with 0.7% FA free BSA/250 μM oleate/50 μM [U13C]palmitate. Samples were then incubated at 37°C for 1.5 h and quenched by snap freezing the samples in liquid N2 and lyophilized. For sample processing, 400 μL of ice-cold 2:2:1 acetonitrile:methanol:water containing the internal standard D8-valine was added to the lyophilized samples, and these mixtures were vortexed for 10 min, rotated at −20°C for 1 h, and centrifuged at 21,000×g at 4°C for 10 min. The supernatants were transferred to new microcentrifuge tubes and dried to completeness using a SpeedVac vacuum concentrator. Dried samples were resuspended in 30 μL of 1:1 acetonitrile:water, vortexed for 10 min, and stored at −20°C overnight. Resuspended samples were centrifuged at 21,000×g at 4°C for 10 min and supernatants were transferred to autosampler vials for analysis. 2 μL of the prepared samples were separated using a Thermo Vanquish Flex UHPLC and data were acquired using a Thermo Q Exactive MS as previously described.90 LC–MS data were processed using the Thermo Scientific TraceFinder (5.1) software. Targeted metabolites were identified using the University of Iowa Metabolomics Core facility standard-confirmed, in-house library defining accurate mass, retention time, and MS/MS fragmentation pattern when available.
Western blotting
Cell lysates (30–50 μg protein) were separated on 10–12% tricine gels using a Mini-Protean II cell (Bio-Rad lab, Hercules, CA) system at constant amperage (30 mA per gel) for about 3 h. Proteins were then transferred onto PVDF membranes at constant voltage (90 V) for 2 h. Blots were stained with Ponceau S to confirm uniform protein loading91,92 before blocking in 5% BSA or 7% non-fat milk in TBST (10 mM Tris-HCl, pH 8, 100 mM NaCl, 0.05% Tween 20) for 1 h. Blots were incubated with specific poly- or monoclonal antibodies overnight and were developed with IRDye 800CW (LI-COR) or IRDye 680RD (LI-COR) secondary antibodies. To visualize the bands of interest, blots were scanned using the LI-COR Odyssey imaging system (Lincoln, NE). Protein bands were quantitated by densitometric analysis after image acquisition using NIH Scion Image to obtain relative protein levels expressed as integrated density. All values were normalized to Ponceau S staining.93–96 Antibodies were purchased or obtained from the following sources; Total-Plin5 (Progen; Heudelberg, Germany), OXPHOS ACADVL, ACADM, ACAA2, CS, ACO1, IDH3, SUCLG1, FH, MDH2, Catalase, COXIV, Calreticulin, VAP-B, MIGA2, VAPB, SLC25a1, FAS, ACSL1, ACSL3, ACSL 5, SCD1, FADS6, AGPAT2, CPT1α, BIP, Calreticulin, and PLIN2 [Barbara Atshaves developed in72].
Cell culture
All cells were maintained in a humidified incubator at 37°C under 5% CO2. AML12 cells were obtained from the ATCC (Manassas, VA). Cells were grown in DMEM:F12 (1:1) supplemented with 10% FBS, ITS (10 mg/mL insulin, 5.5 mg/mL transferrin, 5 ng/mL selenium), and 40 ng/mL dexamethasone. For imaging experiments, cells were treated under one of the following conditions: fed media [DMEM:F12 (1:1) supplemented with 10% FBS, ITS (10 mg/mL insulin, 5.5 mg/mL transferrin, 5 ng/mL selenium, and 40 ng/mL dexamethasone], fasting media (DMEM phenol free basic media supplemented with 0.5 g/L D-glucose, 5.5 g/L HEPES, 2.2 g/L Sodium bicarbonate, pH 7.4), and either fed or fasting media +250 μM oleate complexed to FA-free BSA at a 3:1 ratio (FA:BSA). For experiments with the MAM sensor SPLICSs (SPLICS short measuring 10.4 nm mitochondria-ER interactions), AML12 cells were transduced with pLV-Bsd-TRE-SplitGFP (SPLICSs-MT-ER)-CMV-rtTA and screened for clones using blasticidin. The SPLICSs construct was induced with 500 nM tetracycline the night before. Cells were countered stained for mitochondria using MitoTracker Deep Red (Thermo) and AutoDOT neutral lipid stain (mondansylpentane [MDH] Lipid Droplet Staining Tool; Absepta) 20 min prior to imaging. For the SMLM studies, AML12 cells were transfected with pSEMS-TOM20-HaloTag (Addgene#111135) using Qiagen’s Effectine transfection reagent. Two types of experiments were carried out, one where BODIPY-C12 FL (BODIPY-C12 488nm) was added exogenously while the cells were in fed or fasting media and the second where cells were loaded overnight with BODIPY-C12 FL and non-fluorescent oleate so that the FA was in endogenous lipid stores. In both experimental set-ups, 150 nM BODIPY-C12 FL was complexed to FA-free BSA. For the overnight loading cells were treated with 150 nM BODIPY-C12 FL/250 μM oleate complexed to FA-free BSA. Cells were placed in the corresponding fed or fasting media for 4 hrs, stained with AutoDOT LD stain and HaloTag reagent JF646 for 30 min, washed, then placed in phenol-free media for imaging.
Microscope setup and imaging for SMLM studies
A Nikon inverted microscope (Eclipse Ti-E) equipped with a perfect focus system and a motorized sample stage (Pecon) maintained at 37°C and 5% CO2 was used for all experiments. All movies were recorded on an Andor iXon 897 Ultra DU-897U electron-multiplying charge-coupled detector (EMCCD) camera, which was cooled to −70°C and set to a prem-amp gain of 5.1 and an EMCCD gain of 30. The camera was calibrated as described in our previous paper with camera calibration35 and the calculation of photons/count.35–37
The 3 excitation lasers (405 nm, 561 nm, 640 nm OBIS-CW, Coherent Optics) were aligned, expanded, and focused into the back focal plane of the objective (Nikon CFI 100× 1.49 NA oil immersion) using a variety of dichroic mirrors, beam expanders, and lenses. The laser intensity was controlled digitally by a computer. A quad band dichoric mirror (ZT405/488/561/640rpc; Chroma) was used to separate fluorescence emission from excitation light. The fluorescence emission was further split into red and far-red channels by a dichroic longpass beamsplitter (FF652-Di01; Semrock) and further filtered by bandpass filters: ET610/75 (Chroma) in the red and FF731/137 (Semrock) in the far-red channel.
Programmable shutter sequences were loaded on an NI-DAQ board to synchronize the laser outputs with camera frame duration. The HAL4000 software (Zhuang lab Github: https://github.com/ZhuangLab/storm-control) was used for adjusting laser powers, programmable shutter sequence output, programming camera settings, and image acquisition as described.35–37 In brief, for three-color imaging of AutoDOT, BODIPY-C12, and Tom20-Haloag (JF646), an 11-frame shutter sequence at 20 Hz was employed with one 405 nm excitation frame (~0.5 W/cm2) for conventional imaging of LDs followed by five frames of 561 nm excitation (~1 kW/cm2) for D∥ states imaging of BODIPY-C12 and five frames of 640 nm excitation (~2 kW/cm2) for JF646 imaging. Single-molecule signals of D∥ states of BODIPY-C12 dyes and JF646 dyes were detected in the red and far-red channels, respectively. Emission of the AutoDOT dye localized to LDs was detected in the red channel with 405 nm excitation due to its broad emission spectrum and was used to identify individual LDs (Figure 3). The shutter sequence was repeated for ~7000 frames to collect enough BODIPY-C12 and Tom20-HaloTag-JF646 localizations to reconstruct a well-sampled two color SMLM image.
Bead calibration for two-channel alignment of single molecule localizations for SMLM co-localization quantification
To accurately superimpose localizations from the far-red and to the red microscope channel, a bead calibration experiment was performed on each imaging day with fluorescent microsphere beads (TetraSpeck microspheres, Invitrogen T7279) that were excited at 561 nm and 640 nm and detected in both channels. Fluorescence puncta of individual beads were sparsely distributed throughout the field of view in each channel and fitted with Gaussians as in SMLM experiments. From the coordinates of each bead in far-red and red channels, a third-degree polynomial transformation was determined that maps an arbitrary coordinate from the far red to the red channel. This transformation was verified by transforming the localizations from the different sets of beads to have a mean accuracy of 18.23 nm.97 The obtained transformation was used to superimpose the Tom20-HaloTag-JF646 localizations from the far-red channel on the BODIPY-C12 localizations and LD signal in the red channel for further co-localization analysis.
Single-molecule localization analysis
The Insight3 software from Xiaowei Zhuang’s group (https://doi.org/10.5281/zenodo.3528331) was used to identify single molecules and to fit them with 2D Gaussians with the following parameters: 7 × 7 pixel ROI, widths between 250 and 700 nm, and a minimum of 100 photons. The x- and y coordinates of the localizations, along with the intensity, width, background, frame number, and other parameters were saved in a single molecule localization list. Three-color images are obtained by transforming the JF646 localizations from the far-red channel to the red channel and by rendering JF646 and BODIPY-C12 localizations as 2D-Gaussians on the top of averaged conventional fluorescence image of LDs.
Co-localization analysis to determine the density of BODIPY-C12 in PDM and CM
To measure the degree of co-localization of BODIPY-C12 in PDM and CM under each condition, we assigned BODIPY-C12 and JF646 localizations to each mitochondrial fraction in the following way. First, the conventional fluorescence image of LDs was averaged and thresholded using a locally low-rank soft thresholding approach to remove background fluorescence and identify individual LDs. Thresholded images were then converted into a binary mask using the imbinarize function in MATLAB to identify boundaries of LDs. Next, a contour was drawn around identified LDs using the bwboundaries function in MATLAB and scaled by a factor of 1.2 to include mitochondria that are wrapped around or in contact with LDs. This scaling was confirmed to include PDM fractions across all datasets. TOM-20-HaloTag JF646 localizations within the scaled contour but outside the LD boundaries were considered PDM and all other localizations outside the scaled contour were considered CM. BODIPY-C12 localizations, within a 120 nm distance from a PDM JF646 localization were considered to be associated with PDMs and BODIPY-C12 localizations within a 120 nm from a CM JF646 localization were considered to be associated with CMs. This distance threshold, determined by cross-correlation analysis accounts for the localization precision, limited sampling of the mitochondrial surface and potential motion of mitochondria during the data acquisition time. Details for this analysis can be found in ref. 38 Using the area of the PDM contour surrounding the LDs and the number of co-localized BODIPY-C12 localizations, the density of BODIPY-C12 in PDM was determined. For estimating the CM density of BODIPY-C12, the number of co-localized BODIPY-C12 outside the scaled contour around LDs is divided by the cytoplasmic area (cell area minus the area of the nucleus and scaled LD area). The density of BODIPY-C12 across different movies was normalized by the number of data acquisition frames for accurate comparison (see37). To account for any bias and systemic error in determining BODIPY-C12 densities in the PDM and CM areas, the ratio of the CM and PDM BODIPY-C12 density was calculated for the relative measure of FA redistribution under different imaging conditions.
CM and PDM lipid incorporation assays using [14C]oleate and [14C]acetate
Lipid incorporation assays were adapted from.98 In summary, PDM and CM isolated as described above were normalized to 2 μg/μL in MRB. LDs kept from the isolation and purified as described above were also suspended in MRB at 5 μg/μL. PDM and CM were combined with the purified LDs in a 1:1 ratio (25 μL mitochondria/25 μL LDs; 50 μg/250 μg). Mitochondria and LDs were allowed to incubate together for 20 min before the addition of 350 μL lipid incorporation buffer (LIB; 100 mM Sucrose, 10 mM Tris-HCL pH 7.4, 5 mM potassium phosphate, 0.2 mM EDTA pH 8.0, 80 mM potassium chloride, 1 mM magnesium chloride, 2 mM carnitine, 0.1 mM malate, 0.05 mM coenzyme A, 1 mM ATP, 1 mM ADP, 1 mM DTT, 0.7% FA free BSA/500 μM oleate/1 μCi [14C]oleate, pH 8.0). Mitochondria-LDs were incubated at 37°C for 90 min in the reaction mixture. Chloroform-methanol (400 μL; 1:2 v/v) was added to terminate the reaction. Samples were vortexed and allowed to extract at −20°C for 1 h. An additional 700 μL chloroform was added to the reaction mixture, vortexed again, then centrifuged at 1,500×g for 10 min. The hydrophobic-chloroform phase was transferred to a glass tube and dried under nitrogen. The dried lipid extracts are then resuspended in 30 μL chloroform and 15 μL were then spotted on Analtech Preadsorbent Silica gel HL thin-layer chromatography (TLC) plates and 5 μL of the resuspension was added to scintillation fluid and counted. TLC plates were run to separate major lipid species PL, cholesterol, DAG, FFA, TAG, and CE using hexane, diethyl ether, and acetic acid (4/1/0.1 v/v/v) mobile phase. Radioactivity of the TLC plates was counted on a Bioscan AR-2000 TLC plate scanner and analyzed using WinScanV3 software (Figure 4B). Experiments were repeated using LIB with 5 μCi [14C]acetate in place of oleate to determine de novo lipogenesis in place of FA esterification. Incubations for the [14C]acetate de novo lipogenesis were still carried out at 37°C however the time was extended to 2 hrs to allow for greater acetate incorporation into complex lipid species.
Serial block-face electron microscopy
Methods were adapted from99 and were performed in collaboration with the Mayo Clinic Microscopy and Cell Analysis Core (MACA) and the University of Minnesota Imaging Center (UMIC). Fixation of 1 mm by 1 mm sections of hepatic tissue was achieved by SBFSEM fixative (2% glutaraldehyde, 2% formaldehyde, in 0.15 M cacodylate buffer with 2 mM calcium chloride). Submerged tissues in fixative were stored in glass vials at 4°C for a minimum of 1 h. The tissue was rinsed with 0.15 M cacodylate buffer containing 2 mM calcium chloride for 3 min while rotating. The wash step was repeated a minimum of four times. The samples were then incubated in fresh 2% osmium tetroxide in 0.15 M cacodylate buffer, pH 7.4 for 1.5 h at room temperature with rotation. Rinsed in ultra-pure water for 3 min a minimum of four times. Tissues were again incubated in 2% osmium tetroxide without cacodylate for 1.5 h at room temperature followed by an additional four washes in ultra-pure water. After the last water wash, tissue samples were incubated in 1% aqueous uranyl acetate at 4°C overnight. The following day, samples were baked at 50°C for 1 h, rinsed in ultra-pure water, and incubated in Walton’s Lead aspartate for 1 h at 50°C. A final four water rinses were carried out before dehydrating the samples and infiltrating them in 812 resin. The mounting, trimming, and imaging was performed by the Mayo Clinic Microscopy and Cell Analysis Core Facility.
Three-dimensional reconstruction & analysis
For each electron tomography analysis, serial tomograms were compiled as a single TIFF file and analyzed using Avizo 3D Software (v. 2021.1.; Thermo Fisher Scientific). The structure of interest (e.g., LD, mitochondria, ER.) was carefully manually traced on each virtual slice using a Wacom graphic tablet and then reconstructed in three dimensions, with rendering generated using unconstrained smoothing parameters. The surfaces of adhesion/contact sites and mitochondria were measured using the Surface Area Volume module. Movies were generated using Avizo 3D Software (v. 2021.3, Thermo Fisher Scientific) and then edited with the Microsoft Photos application (v. 2022.30060.30007.0; Microsoft).
Live cell imaging
Fluorescence imaging experiments were performed with cells seeded at a density of 50,000 cells/plate on Mat-Tek cover-glass plates (Ashland, MA). The plasmid expressing SPLICSs (10.4 nm) was purchased from Addgene (#164108) and cloned into the tetracyclin inducible lentivirus system pLV-Bsd-TRE-CMV-rtTA. AML12 cells were transduced and put under blasticidin selection before being screened for positive clones. Digital images were acquired using a Nikon A1Rsi Laser Scanning Confocal Imaging System (LSCIS; Nikon, Melville, NY) equipped with 405 nm, 488 nm, 561 nm, and 640 nm laser, four-channel GaSP detectors, and a 60x water immersion objective. To determine the subcellular localization of LDs, MAM, and mitochondria under fed and fasted conditions, cells were treated with AutoDOT neutral lipid stand and MitoTracker Deep Red. Media conditions are described above for fed and fasting media. For probe excitation, the A1Rsi LSCIS utilized the 640 nm diode laser (MitoTracker Deep Red), the 405 nm laser line (AutDOT Neutral lipid stain), and the 488 nm laser line (SPLICSs) to acquire images of the cells by sequential excitation. Image files were analyzed using NIS-Elements software or FIJI-Image software. z-stack or multiple focal planes were imaged to ensure compartmentalization and localization.
Diana surface contact analysis
Mitochondria-LD, LD-MAM, and MAM-mitochondria interactions were characterized using the DiAna ImageJ tool.43 In brief, images were individually segmented and analyzed using the DiAna plugin in FIJI. Lipid droplets were segmented using the iterative segmentation method with a minimum volume of 3 pixels, maximum volume of 2000 pixels, minimum threshold of 100, and STEP value of 100. The mitochondria and MAM (SPLICS) were segmented using the classic segmentation method, with either no filter (mitochondria) or a Gaussian filter (MAM) applied during thresholding, an XY radius of 0.5, minimum object size of 3 pixels, and maximum object size of 20,000 or 50,000 pixels. The maximum object size was determined based on thresholding for each image. DiAna colocalization analysis was performed to calculate the surface in contact length between lipid droplets and mitochondria, lipid droplets and MAM (SPLICS), and mitochondria and MAM (SPLICS) for each image. The maximum distance to establish contact between objects was set at 50 nm.
Isolation of MAM from CM and PDM
Isolation of MAM from CM and PDM was achieved by adapting the following protocol46 (Figure S5A). In brief, CM and PDM were isolated as described above and in Figure S1. CM and PDM pellets were resuspended in mitochondria resuspending buffer (MRSB; 250 mM mannitol, 5 mM HEPES, 0.5 mM EGTA, pH 7.4) and layered on a Percoll medium separation gradient (PM; 225 mM mannitol, 25 mM HEPES, 1 mM EGTA, 30% Percoll (v/v), pH 7.4) in a Polyallomer ultracentrifuge tube. The remainder of the tube volume above the sample was topped off with MRSB. Samples were then centrifuged at 95,000×g for 30 min at 4°C in an SW-41Ti swing bucket rotor. The MAM appeared as an off-white band in the middle of the ultracentrifuge tube while the mitochondria were a dark brown band at the bottom just above the Percoll pellet (Figure S5B). Using a glass pasture pipette both the MAM and mitochondrial fractions were collected. MAM and mitochondria were transferred to a clean falcon tube and diluted ten times with MRSB then centrifuged at 6,300×g for 10 min at 4°C. The MAM supernatant was transferred to a polycarbonate tube with cap assembly (the mitochondrial pellet was discarded) and centrifuged at 100,000×g for 1 h in a 70-Ti rotor at 4°C. The mitochondrial supernatant was discarded from the pure mitochondrial fraction. The pelleted mitochondria were again resuspended in MRSB and centrifuged a second time at 6,300×g for 10 min at 4°C. The supernatant was discarded, and mitochondria were resuspended in a small volume of MRSB and flash frozen. The MAM samples post 100,000×g spin are pelleted at the bottom of the ultracentrifuge tube. The MAM supernatant was discarded and the pelleted MAM was resuspended in a small volume of MRSB and snap-frozen for later analysis. The purity of the fractions was determined via immunoblotting (Figure S5C).
Proteomic analysis of MAM from CM and PDM
For proteomic analysis of CM-MAM, PDM-MAM isolated from control and PLIN5 knockdown samples, mitochondria samples were extracted with proteomic lysis buffer [PLB2; 7 M urea, 2 M thiourea, 0.4 M tris pH 8, 20% acetonitrile, 10 mM tris (2-carboxyethyl) phosphine (TCEP), and 40 mM chloroacetamide]. The samples were pipetted up and down several times and transferred to a PCT tube for the Barocycler NEP2320 (Pressure Biosciences, Inc., South Easton, MA) and cycled between 35 kPSI for 20 s and 0 kPSI for 10 s for 60 cycles at 37°C. Samples were centrifuged at 13000×g for 10 min. The supernatants were transferred to new 1.5 mL microfuge Eppendorf Protein LoBind tubes. Aliquots for each sample were taken for protein concentration determination by Bradford assay. A 25 μg aliquot of each sample was transferred to a new 1.5 mL Eppendorf Protein LoBind tube and brought to the same volume with PLB2. In addition, a pooled sample was created by combining equal amounts of each sample to use as a bridge sample across multiple TMT experiments. All samples were diluted fivefold with water, and then trypsin (Promega, Madison, WI) was added in a 1:40 ratio. Samples were incubated overnight for 16 h at 37°C. After incubation, the samples were frozen at −80°C and dried in a vacuum concentrator. Each sample was cleaned with a 1 CC Waters Oasis MCX cartridge (Waters Corporation, Milford, MA), and eluates were vacuum dried and resuspended in 100 mM triethylammonium bicarbonate, pH 8.5 to a final concentration of 1 μg/μL. For each channel within the TMT10plex a 20 μg aliquot of the appropriate sample was made, and the samples were labeled with the corresponding TMT10plex Isobaric Label Reagent (Thermo Scientific, Waltham, MA) per the manufacturer’s protocol. After TMT labeling, all the samples were multiplexed together into a new 1.5 mL microfuge tube. The TMT sample was dried down in vacuo. The sample was cleaned with a 1 CC Waters Oasis MCX solid phase extraction cartridge (Waters Corporation, Milford, MA), and the eluate was dried in vacuo. The samples were offline fractionated as described previously.74 Each fraction was desalted on a C18 stage tip75 and reconstituted for analysis. Samples were separated on a Thermo Fisher Scientific, Inc (Waltham, MA) Dionex UltiMate 3000 RSLCnano system and analyzed by tandem mass spectra in data-dependent acquisition (DDA) mode on a Thermo Fisher Orbitrap Fusion mass spectrometer. Analysis of peptides was monitored according to previously established methods74 with the following modifications: peptides were separated on a 40 cm column with the following elution profile: 5–35% B solvent from 0 to 70 min, 35–90% B solvent from 70 to 80 min and held at 90% B from 80 to 86 min at a flowrate of 325 nL/min, where solvent A was 0.1% formic acid in water and solvent B was 0.1% formic acid in ACN; the Orbitrap MS1 injection time was 100 msec with 4 × 105 AGC (100%); DDA was performed on the top 12 most intense ions; HCD (higher energy collision dissociation) energy was 40%; MS2 injection time was 150 msec with 4 × 105 AGC (200%).
Statistical analysis
Values were expressed as the means ± SEM. In comparisons made between two groups, such as CM versus PDM, Student t-tests were performed using GraphPad Prism (San Diego, CA). When more than two groups were compared, such as in our analysis across control and PLIN5 knockdown samples, analysis of variance (ANOVA) with a Newman Keuls posthoc test was performed. Values with p < 0.05 were considered statistically significant. For proteomics quantification, we used Scaffold 5 Label Based Quantitation plug in to determine significant changes. Statistical significance between samples and groups was determined using a Permutation Test with a Benjamini-Hochberg posthoc test (Proteome Software: https://www.proteomesoftware.com/products/scaffold-5).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Guinea pig polyclonal anti-PLIN5 | Progen | GP44 |
| Rabbit polyclonal anti-PLIN2 | (Atshaves BP et al., 1999)72 | N/A |
| Mouse polyclonal anti-OXPHOS cocktail | Abcam | ab110413 |
| Rabbit polyclonal anti-Acadvl | Abcam | ab188872 |
| Rabbit monoclonal anti-Acadm | Abcam | ab92461 |
| Rabbit polyclonal anti-Acaa2 | Abcam | Ab128911 |
| Mouse monoclonal anti-Cs | Cell Signaling | 14309 |
| Rabbit monoclonal anti-Aco1 | Abcam | Ab183721 |
| Mouse Monoclonal anti-Idh3 | Abcam | ab154886 |
| Mouse Monoclonal anti-Suclg1 | Abcam | ab97867 |
| Mouse Monoclonal anti-Fh | Abcam | Ab233394 |
| Mouse Monoclonal anti-Mdh2 | Abcam | Ab181873 |
| Rabbit Polyclonal anti-Catalase | Cell Signaling | 14097S |
| Rabbit Polyclonal anti-COXIV | Cell Signaling | 4850S |
| Rabbit Polyclonal anti-Calreticulin | Cell Signaling | 12238S |
| Mouse Polyclonal anti-VAPB | ThermoFisher | MA5–24318 |
| Rabbit Polyclonal anti-Cpt1-a | Protein Tech | 15184–1-AP |
| Rabbit Polyclonal anti-BiP | Cell Signaling | C50B12 |
| Rabbit Polyclonal anti-MIGA2 | ThermoFisher | PA-5–71096 |
| Rabbit Polyclonal anti-SLC25a1 | Thermo | 15235–1-AP |
| Rabbit Polyclonal anti-FAS | Cell Signaling | 3180S |
| Rabbit Polyclonal anti-ACSL1 | Abcam | Ab189939 |
| Rabbit Polyclonal anti-ACSL5 | Santa Cruz | SC-47999 |
| Rabbit Polyclonal anti-FADS6 | Novus | NBP2–83031 |
| Rabbit Polyclonal anti-AGPAT2 | Cell Signaling | D8W9B |
| Donkey anti-Guinea pig IRDye 800CW | LI-COR | Cat No. 926–32411 |
| Donkey anti-Guinea pig IRDye 680RD | LI-COR | Cat No. 926–68030 |
| Donkey anti-Rabbit pig IRDye 800CW | LI-COR | Cat No. 926–32213 |
| Donkey anti-Rabbit pig IRDye 680RD | LI-COR | Cat No. 926–68022 |
| Donkey anti-Mouse pig IRDye 800CW | LI-COR | Cat No. 925–32212 |
| Donkey anti-Mouse pig IRDye 680RD | LI-COR | Cat No. 926–68023 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| DH5a | Invitrogen | 18258–012 |
| XL1-Blue | Agilent | Cat No. 200150 |
| Stable | NEB | C3040H |
| pLV-Bsd-TRE-SplitGFP (SPLICSs- MT-ER)-CMV-rtTA | This paper | N/A |
|
| ||
| Chemicals | ||
|
| ||
| Trypsin Gold, Mass Spec Grade | Promega | V5280 |
| MitoTracker Deep Red | Invitrogen | M22426 |
| [14C]oleate | Perkin Elmer | NEC317250UC |
| FCCP | Abcam | Ab120081 |
| TMRE | Abcam | Ab113852 |
| Etomoxir | Abcam | Ab254445 |
| [13C]palmitate | Sigma-Aldrich | 687871–3.80G |
| Ponceau S | Fisher BioReagents | BP103–10 |
| AutoDOT-Neutral lipid Stain | Abcepta | SM1000a |
| BODIPY-C12FL | Invitrogen | D3821 |
| Halotag reagent JF646 | Promega | GA1120 |
| [14C]acetate | American Radiolabeled Chemicals, Inc. | ARC0173 |
| Analtech Preadsorbent Silica gel HL Plates | Miles Scientific | P43911 |
| Percoll | Sigma | P4937–100ML |
| 2-APB | Tocris | 1224 |
| Clotrimazole | Tocris | 4096 |
| ADP | Sigma-Aldrich | A5285 |
| Oligomycin A | Sigma-Aldrich | 75351 |
| Rotanone | Sigma-Aldrich | R8875 |
| Antimycin A | Sigma-Aldrich | A8674 |
| Malonate | Sigma-Aldrich | 63409 |
| Duroquinol | Sigma-Aldrich | D223204 |
| TMPD | Sigma-Aldrich | T7394 |
| Ascorbate | Sigma-Aldrich | A4034 |
| Sodium Azide | Sigma-Aldrich | S8032 |
|
| ||
| Critical commercial assays | ||
|
| ||
| TMT-10plex | ThermoFisher | 90110 |
| Seahorse kit | Agilent | 103015–100 |
| Effectene | QIAGEN | 301425 |
|
| ||
| Deposited data | ||
|
| ||
| CM-PDM Control-PLIN5 knockdown proteomics | MassIVE:https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp | ftp://massive.ucsd.edu/MSV000091575/ |
| CM-MAM-PDM-MAM Control-PLIN5 knockdown proteomics | MassIVE:https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp | ftp://massive.ucsd.edu/MSV000091576/ |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| AML12 | ATCC | CRL-2254 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Male C57BL6/J mice | Envigo | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Control Anti-Sense Oligonucleotide | Ionis Pharmaceuticals; Mark Graham | N/A |
| Perilipin5 Anti-Sense Oligonucleotide | Ionis Pharmaceuticals; Mark Graham | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSEMS-TOM20-Hal7Tag | Addgene | 111135 |
| pLV-Bsd-TRE-SplitGFP(SPLICSs- MT-ER)-CMV-rtTA |
This paper | N/A |
| pEZ-M29-mCherry-mPLIN5 | GeneCopoeia | EX-mm27089-m29 |
| p-SPLICSs-(Split-GFP ER-Mito Sensor) | CieriDetal. (2017)41 | N/A |
| pLV-Bsd-TRE-CMV-rtTA | Addgene | 128061 |
|
| ||
| Software and algorithms | ||
|
| ||
| SigmaPlot 11 | Systat Software, Inc | N/A |
| Prism8 | GraphPad | N/A |
| Canvas 11 | Canvas | N/A |
| Avizo 3D Software | Thermo Fisher Scientific | N/A |
| Image Studio v5 | LI-COR Biosciences | N/A |
| NIS-Elements 4 | Nikon | N/A |
| Microsoft Photos Application | Microsoft | N/A |
| HAL4000 | Zhuang lab | Github: https://github.com/ZhuangLab/storm-control |
| Insight3 | Xiaowei Zhuang lab | Github: https://github.com/BoHuanglab |
| Cross-correlation-filtering | Puchner Lab | Github: https://github.com/PuchnerLab |
| MATLAB | MathWorks | MATLAB |
| Diana-FIJI | Gilles JF et al. (2017)43 | N/A |
| Scaffold 5 | Proteome Software | Scaffold 5 |
Highlights.
CM are enriched in oxidative proteins and utilize FAs during fasting
PDM are enriched in lipid anabolism proteins and facilitate FA esterification
MAM around PDM and CM are unique and differentially regulate lipid metabolism
PLIN5 regulates the proteomes and functions of mitochondrial and MAM subpopulations
ACKNOWLEDGMENTS
We thank the University of Minnesota Imaging Center (Marker Sanders, Gail Celio, Guillermo Marques, and Thomas Pengo), the Center for Mass Spectrometry and Proteomics (Candace Guerrero), the Mayo Clinic Microscopy and Cell Analysis Core Facility (Trace Christensen and Jeffrey Salisbury), and the University of Iowa Metabolomics Core Facility for providing instrumentation and expertise. We thank Mahima Devarajan, Ann Hertzel, and David Berhnlor for scientific discussions and Margaret Harter for technical assistance. Funding was provided for C.N. (NIH: T32AG029796, K99AG070104, AHA: 20POST35180115), T.D.H. (NIH: K01DK125258), E.W.K. (NIH: T32DK007203), E.B.T. (NIH: R01DK104998 and University of Iowa Healthcare Distinguished Scholars Award), E.M.P. (NIH: R21GM127965), and D.G.M. (NIH: R01DK132849 and R01DK114401 and the University of Minnesota Institute on Diabetes and Obesity and the University of Iowa Fraternal Order of Eagles Diabetes Research Center Pilot and Feasibility grant).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112435.
REFERENCES
- 1.Henne M (2019). And three’s a party: lysosomes, lipid droplets, and the ER in lipid trafficking and cell homeostasis. Curr. Opin. Cell Biol. 59, 40–49. 10.1016/j.ceb.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Kramer DA, Quiroga AD, Lian J, Fahlman RP, and Lehner R (2018). Fasting and refeeding induces changes in the mouse hepatic lipid droplet proteome. J. Proteomics 181, 213–224. 10.1016/j.jprot.2018.04.024. [DOI] [PubMed] [Google Scholar]
- 3.Sadh K, Rai P, and Mallik R (2017). Feeding-fasting dependent recruitment of membrane microdomain proteins to lipid droplets purified from the liver. PLoS One 12, e0183022. 10.1371/journal.pone.0183022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J, Fang N, Xiong J, Du Y, Cao Y, and Ji WK (2021). An ESCRT-dependent step in fatty acid transfer from lipid droplets to mitochondria through VPS13D-TSG101 interactions. Nat. Commun. 12, 1252. 10.1038/s41467-021-21525-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rambold AS, Cohen S, and Lippincott-Schwartz J (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692. 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herms A, Bosch M, Reddy BJN, Schieber NL, Fajardo A, Rupérez C, Fernández-Vidal A, Ferguson C, Rentero C, Tebar F, et al. (2015). AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 6, 7176. 10.1038/ncomms8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui W, Sathyanarayan A, Lopresti M, Aghajan M, Chen C, and Mashek DG (2021). Lipophagy-derived fatty acids undergo extracellular efflux via lysosomal exocytosis. Autophagy 17, 690–705. 10.1080/15548627.2020.1728097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sathyanarayan A, Mashek MT, and Mashek DG (2017). ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep. 19, 1–9. 10.1016/j.celrep.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, and McNiven MA (2019). Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 218, 3320–3335. 10.1083/jcb.201803153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA, Acín-Pérez R, Shum M, Oliveira MF, Cinti S, et al. (2018). Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 27, 869–885.e6. 10.1016/j.cmet.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coleman RA, and Mashek DG (2011). Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem. Rev. 111, 6359–6386. 10.1021/cr100404w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong D. w., Stanley WC, and Sztalryd C (2011). Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 52, 2159–2168. 10.1194/jlr.M017939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong DW, Stanley WC, and Sztalryd C (2011). Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 52, 2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Granneman JG, Moore HPH, Mottillo EP, Zhu Z, and Zhou L (2011). Interactions of perilipin-5 (plin5) with adipose triglyceride lipase. J. Biol. Chem. 286, 5126–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pollak NM, Jaeger D, Kolleritsch S, Zimmermann R, Zechner R, Lass A, and Haemmerle G (2015). The interplay of protein kinase A and perilipin 5 regulates cardiac lipolysis. J. Biol. Chem. 290, 1295–1306. 10.1074/jbc.M114.604744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Sreenivasan U, Gong D-W, O’Connell KA, Dabkowski ER, Hecker PA, Ionica N, Konig M, Mahurkar A, Sun Y, et al. (2013). Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction1[S]. J. Lipid Res. 54, 953–965. 10.1194/jlr.M032466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Zhao Y, Gao X, Li L, Yuan Y, Liu F, Zhang L, Wu J, Hu P, Zhang X, et al. (2014). Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Preprint at Hepatology, In Press. 10.1002/hep.27409. [DOI] [PubMed] [Google Scholar]
- 19.Kimmel AR, and Sztalryd C (2014). Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr. Opin. Lipidol. 25, 110–117. 10.1097/MOL.0000000000000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan Y, Jin Y, Wang Q, Huang J, Wu X, and Ren Z (2019). Perilipin 5 protects against cellular oxidative stress by enhancing mitochondrial function in HepG2 cells. Cells 8, 1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mason RR, Mokhtar R, Matzaris M, Selathurai A, Kowalski GM, Mokbel N, Meikle PJ, Bruce CR, and Watt MJ (2014). Plin5 deletion remodels intracellular lipid composition and causes insulin resistance in muscle. Mol. Metab. 3, 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montgomery MK, Mokhtar R, Bayliss J, Parkington HC, Suturin VM, Bruce CR, and Watt MJ (2018). Perilipin 5 deletion unmasks an endoplasmic reticulum stress–fibroblast growth factor 21 Axis in skeletal muscle. Diabetes 67, 594–606. 10.2337/db17-0923. [DOI] [PubMed] [Google Scholar]
- 23.Keenan SN, Meex RC, Lo JCY, Ryan A, Nie S, Montgomery MK, and Watt MJ (2019). Perilipin 5 deletion in hepatocytes remodels lipid metabolism and causes hepatic insulin resistance in mice. Diabetes 68, 543–555. 10.2337/db18-0670. [DOI] [PubMed] [Google Scholar]
- 24.Benador IY, Veliova M, Liesa M, and Shirihai OS (2019). Mitochondria bound to lipid droplets: where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab. 29, 827–835. 10.1016/j.cmet.2019.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Najt CP, Khan SA, Heden TD, Witthuhn BA, Perez M, Heier JL, Mead LE, Franklin MP, Karanja KK, Graham MJ, et al. (2020). Lipid droplet-derived monounsaturated fatty acids traffic via PLIN5 to allosterically activate SIRT1. Mol. Cell 77, 810–824.e8. 10.1016/j.molcel.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang E, Cui W, Lopresti M, Mashek MT, Najt CP, Hu H, and Mashek DG (2020). Hepatic PLIN5 signals via SIRT1 to promote autophagy and prevent inflammation during fasting. J. Lipid Res. 61, 338–350. 10.1194/jlr.RA119000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallardo-Montejano VI, Saxena G, Kusminski CM, Yang C, McAfee JL, Hahner L, Hoch K, Dubinsky W, Narkar VA, and Bickel PE (2016). Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC-1α/SIRT1-dependent transcriptional regulation of mitochondrial function. Nat. Commun. 7, 12723. 10.1038/ncomms12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Najt CP, Devarajan M, and Mashek DG (2022). Perilipins at a glance. J. Cell Sci. 135, jcs259501. 10.1242/jcs.259501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freyre CAC, Rauher PC, Ejsing CS, and Klemm RW (2019). MIGA2 links mitochondria, the ER, and lipid droplets and promotes de novo lipogenesis in adipocytes. Mol. Cell 76, 811–825.e14. 10.1016/j.molcel.2019.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Vance JE (2014). MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta 1841, 595–609. 10.1016/j.bbalip.2013.11.014. [DOI] [PubMed] [Google Scholar]
- 31.Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera-Alberni M, Combe R, Zorzano A, and Cantó C (2017). Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 36, 1543–1558. 10.15252/embj.201694914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW, et al. (2021). MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547. 10.1093/nar/gkaa1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nesteruk M, Hennig EE, Mikula M, Karczmarski J, Dzwonek A, Goryca K, Rubel T, Paziewska A, Woszczynski M, Ledwon J, et al. (2014). Mitochondrial-related proteomic changes during obesity and fasting in mice are greater in the liver than skeletal muscles. Funct. Integr. Genomics 14, 245–259. 10.1007/s10142-013-0342-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang B, Wang W, Bates M, and Zhuang X (2008). Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319, 810–813. 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adhikari S, Moscatelli J, Smith EM, Banerjee C, and Puchner EM (2019). Single-molecule localization microscopy and tracking with red-shifted states of conventional BODIPY conjugates in living cells. Nat. Commun. 10, 3400. 10.1038/s41467-019-11384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adhikari S, Banerjee C, Moscatelli J, and Puchner EM (2020). Conventional BODIPY conjugates for live-cell super-resolution microscopy and single-molecule tracking. J. Vis. Exp. 10.3791/60950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adhikari S, Moscatelli J, and Puchner EM (2021). Quantitative live-cell PALM reveals nanoscopic Faa4 redistributions and dynamics on lipid droplets during metabolic transitions of yeast. Mol. Biol. Cell 32, 1565–1578. 10.1091/mbc.E20-11-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mancebo A, Mehra D, Banerjee C, Kim D-H, and Puchner EM (2021). Efficient cross-correlation filtering of one- and two-color single molecule localization microscopy data. Front. Bioinform. 1, 739769. 10.3389/fbinf.2021.739769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang S, Wang Y, Cui L, Deng Y, Xu S, Yu J, Cichello S, Serrero G, Ying Y, and Liu P (2016). Morphologically and functionally distinct lipid droplet subpopulations. Sci. Rep. 6, 29539. 10.1038/srep29539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anastasia I, Ilacqua N, Raimondi A, Lemieux P, Ghandehari-Alavijeh R, Faure G, Mekhedov SL, Williams KJ, Caicci F, Valle G, et al. (2021). Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Rep. 34, 108873. 10.1016/j.celrep.2021.108873. [DOI] [PubMed] [Google Scholar]
- 41.Cieri D, Vicario M, Giacomello M, Vallese F, Filadi R, Wagner T, Pozzan T, Pizzo P, Scorrano L, Brini M, and Cal`ı, T. (2018). SPLICS: a split green fluorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 25, 1131–1145. 10.1038/s41418-017-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Theurey P, and Rieusset J (2017). Mitochondria-associated membranes response to nutrient availability and role in metabolic diseases. Trends Endocrinol. Metab. 28, 32–45. 10.1016/j.tem.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Gilles JF, Dos Santos M, Boudier T, Bolte S, and Heck N (2017). DiAna, an ImageJ tool for object-based 3D co-localization and distance analysis. Methods 115, 55–64. 10.1016/j.ymeth.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 44.Geisler CE, Hepler C, Higgins MR, and Renquist BJ (2016). Hepatic adaptations to maintain metabolic homeostasis in response to fasting and refeeding in mice. Nutr. Metab. 13, 62. 10.1186/s12986-016-0122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen MD, Ekberg K, and Landau BR (2001). Lipid metabolism during fasting. Am. J. Physiol. Endocrinol. Metab. 281, E789–E793. 10.1152/ajpendo.2001.281.4.E789. [DOI] [PubMed] [Google Scholar]
- 46.Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, and Pinton P (2009). Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590. 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi T, Rizzuto R, Hajnoczky G, and Su TP (2009). MAM: more than just a housekeeper. Trends Cell Biol. 19, 81–88. 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poston CN, Krishnan SC, and Bazemore-Walker CR (2013). In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteomics 79, 219–230. 10.1016/j.jprot.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 49.Hung V, Lam SS, Udeshi ND, Svinkina T, Guzman G, Mootha VK, Carr SA, and Ting AY (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6, e24463. 10.7554/eLife.24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peppiatt CM, Collins TJ, Mackenzie L, Conway SJ, Holmes AB, Bootman MD, Berridge MJ, Seo JT, and Roderick HL (2003). 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium 34, 97–108. 10.1016/s0143-4160(03)00026-5. [DOI] [PubMed] [Google Scholar]
- 51.Mironov SL, Ivannikov MV, and Johansson M (2005). [Ca2+]i signaling between mitochondria and endoplasmic reticulum in neurons is regulated by microtubules: FROM MITOCHONDRIAL PERMEABILITY TRANSITION PORE TO Ca2+-INDUCED Ca2+ RELEASE. J. Biol. Chem. 280, 715–721. 10.1074/jbc.M409819200. [DOI] [PubMed] [Google Scholar]
- 52.Rieusset J, Fauconnier J, Paillard M, Belaidi E, Tubbs E, Chauvin M-A, Durand A, Bravard A, Teixeira G, Bartosch B, et al. (2016). Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 59, 614–623. 10.1007/s00125-015-3829-8. [DOI] [PubMed] [Google Scholar]
- 53.Brewer JW, Hendershot LM, Sherr CJ, and Diehl JA (1999). Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc. Natl. Acad. Sci. USA 96, 8505–8510. 10.1073/pnas.96.15.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, Nomura DK, and Olzmann JA (2017). DGAT1-Dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev. Cell 42, 9–21.e5. 10.1016/j.devcel.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stone SJ, Levin MC, Zhou P, Han J, Walther TC, and Farese RV (2009). The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem. 284, 5352–5361. 10.1074/jbc.M805768200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rusiñol AE, Cui Z, Chen MH, and Vance JE (1994). A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 269, 27494–27502. [PubMed] [Google Scholar]
- 57.Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, and Hotamisligil GS (2014). Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 20, 1427–1435. 10.1038/nm.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jin C, Felli E, Lange NF, Berzigotti A, Gracia-Sancho J, and Dufour JF (2022). Endoplasmic reticulum and mitochondria contacts correlate with the presence and severity of NASH in humans. Int. J. Mol. Sci. 23, 8348. 10.3390/ijms23158348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, Chauvin MA, Ji-Cao J, Zoulim F, Bartosch B, Ovize M, et al. (2014). Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63, 3279–3294. 10.2337/db13-1751. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, Li G, Goode J, Paz JC, Ouyang K, Screaton R, Fischer WH, Chen J, Tabas I, and Montminy M (2012). Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 485, 128–132. 10.1038/nature10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rambold AS, Cohen S, and Lippincott-Schwartz J (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692. 10.1016/j.devcel.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eynaudi A, Dáz-Castro F, Bórquez JC, Bravo-Sagua R, Parra V, and Troncoso R (2021). Differential effects of oleic and palmitic acids on lipid droplet-mitochondria interaction in the hepatic cell line HepG2. Front. Nutr. 8, 775382. 10.3389/fnut.2021.775382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahmadian M, Duncan RE, Varady KA, Frasson D, Hellerstein MK, Birkenfeld AL, Samuel VT, Shulman GI, Wang Y, Kang C, and Sul HS (2009). Adipose overexpression of desnutrin promotes fatty acid use and attenuates diet-induced obesity. Diabetes 58, 855–866. 10.2337/db08-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haemmerle G, Moustafa T, Woelkart G, Büttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, et al. (2011). ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 17, 1076–1085. 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khan SA, Sathyanarayan A, Mashek MT, Ong KT, Wollaston-Hayden EE, and Mashek DG (2015). ATGL-catalyzed lipolysis regulates SIRT1 to control PGC-1α/PPAR-α signaling. Diabetes 64, 418–426. 10.2337/db14-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ong KT, Mashek MT, Bu SY, Greenberg AS, and Mashek DG (2011). Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 53, 116–126. 10.1002/hep.24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bosma M, Sparks LM, Hooiveld GJ, Jorgensen JA, Houten SM, Schrauwen P, Kersten S, and Hesselink MKC (2013). Overexpression of Plin5 in skeletal muscle promotes oxidative gene expression and intrayocellular lipid content without compromising insulin sensitivity. Biochim. Biophys. Acta 1831, 844–852. [DOI] [PubMed] [Google Scholar]
- 68.Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Croce MA, Gropler MC, Varma V, Yao-Borengasser A, Rasouli N, Kern PA, et al. (2006). OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 55, 3418–3428. [DOI] [PubMed] [Google Scholar]
- 69.Seibert JT, Najt CP, Heden TD, Mashek DG, and Chow LS (2020). Muscle lipid droplets: cellular signaling to exercise physiology and beyond. Trends Endocrinol. Metab. 31, 928–938. 10.1016/j.tem.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kien B, Kolleritsch S, Kunowska N, Heier C, Chalhoub G, Tilp A, Wolinski H, Stelzl U, and Haemmerle G (2022). Lipid droplet-mitochondria coupling via perilipin 5 augments respiratory capacity but is dispensable for FA oxidation. J. Lipid Res. 63, 100172. 10.1016/j.jlr.2022.100172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu Y, Zhang X, Zhang L, Zhang M, Li L, Luo D, and Zhong Y (2019). Perilipin5 protects against lipotoxicity and alleviates endoplasmic reticulum stress in pancreatic β-cells. Nutr. Metab. 16, 50. 10.1186/s12986-019-0375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Atshaves BP, Petrescu AD, Starodub O, Roths JB, Kier AB, and Schroeder F (1999). Expression and intracellular processing of the 58 KDa SCP-x/3-oxoacyl-CoA thiolase in transfected mouse L cells. J. Lipid Res. 40, 610–622. [PubMed] [Google Scholar]
- 73.Muratore KA, Najt CP, Livezey NM, Marti J, Mashek DG, and Arriaga EA (2018). Sizing lipid droplets from adult and geriatric mouse liver tissue via nanoparticle tracking analysis. Anal. Bioanal. Chem. 410, 3629–3638. 10.1007/s00216-018-1016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chaanine AH, Higgins L, Markowski T, Harman J, Kachman M, Burant C, Navar LG, Busija D, and Delafontaine P (2021). Multi-omics approach profiling metabolic remodeling in early systolic dysfunction and in overt systolic heart failure. Int. J. Mol. Sci. 23, 235. 10.3390/ijms23010235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rappsilber J, Ishihama Y, and Mann M (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670. 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 76.Käll L, Storey JD, and Noble WS (2008). Non-parametric estimation of posterior error probabilities associated with peptides identified by tandem mass spectrometry. Bioinformatics 24, i42–i48. 10.1093/bioinformatics/btn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658. 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 78.Shadforth IP, Dunkley TPJ, Lilley KS, and Bessant C (2005). i-Tracker: for quantitative proteomics using iTRAQ. BMC Genom. 6, 145. 10.1186/1471-2164-6-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oberg AL, Mahoney DW, Eckel-Passow JE, Malone CJ, Wolfinger RD, Hill EG, Cooper LT, Onuma OK, Spiro C, Therneau TM, and Bergen HR 3rd. (2008). Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 7, 225–233. 10.1021/pr700734f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 81.Najt CP, Senthivinayagam S, Aljazi MB, Fader KA, Olenic SD, Brock JRL, Lydic TA, Jones AD, and Atshaves BP (2016). Liver-specific loss of Perilipin 2 alleviates diet-induced hepatic steatosis, inflammation, and fibrosis. Am J Physiol Gastro Liver Physiol 310, G726–G738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shang L, Aughey E, Kim H, Heden TD, Wang L, Najt CP, Esch N, Brunko S, Abrahante JE, Macchietto M, et al. (2022). Systemic lipolysis promotes physiological fitness in Drosophila melanogaster</i>. Aging 14, 6481–6506. 10.18632/aging.204251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Hoon MJL, Imoto S, Nolan J, and Miyano S (2004). Open source clustering software. Bioinformatics 20, 1453–1454. 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 84.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gene Ontology Consortium (2021). The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 49, D325–D334. 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ge SX, Jung D, and Yao R (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huynh FK, Green MF, Koves TR, and Hirschey MD (2014). Chapter twenty - measurement of fatty acid oxidation rates in animal tissues and cell lines. In Methods in Enzymology, Galluzzi Land Kroemer G, eds. (Academic Press; ), pp. 391–405. 10.1016/B978-0-12-416618-9.00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heden TD, Chen C, Leland G, Mashek MM, Najt CP, Shang L, Chow LS, and Mashek DG (2022). Isolated and combined impact of dietary olive oil and exercise on markers of health and energy metabolism in female mice. J. Nutr. Biochem. 107, 109040. 10.1016/j.jnutbio.2022.109040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rauckhorst AJ, Borcherding N, Pape DJ, Kraus AS, Scerbo DA, and Taylor EB (2022). Mouse tissue harvest-induced hypoxia rapidly alters the in vivo metabolome, between-genotype metabolite level differences, and (13)C-tracing enrichments. Mol. Metab. 66, 101596. 10.1016/j.molmet.2022.101596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aldridge GM, Podrebarac DM, Greenough WT, and Weiler IJ (2008). The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J. Neurosci. Methods 172, 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Willenborg M, Schmidt CK, Braun P, Landgrebe J, von Figura K, Saftig P, and Eskelinen E-L (2005). Mannose 6-phosphate receptors, Niemann-Pick C2 protein, and lysosomal cholesterol accumulation. J. Lipid Res. 46, 2559–2569. [DOI] [PubMed] [Google Scholar]
- 93.Hassan J, Feighery C, and Whelan A (1987). Staining of molecular weight markers on nitrocellulose using Ponceau S. J. Clin. Lab. Immunol. 24, 104. [PubMed] [Google Scholar]
- 94.Romero-Calvo I, Ocón B, Martínez-Moya P, Suárez MD, Zarzuelo A, Martínez-Augustin O, and de Medina FS (2010). Reversible Ponceau staining as a loading control alternative to actin in Western blots. Anal. Biochem. 401, 318–320. [DOI] [PubMed] [Google Scholar]
- 95.Gilda JE, and Gomes AV (2013). Stain-Free total protein staining is a superior loading control to β-actin for Western blots. Anal. Biochem. 440, 186–188. 10.1016/j.ab.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sander H, Wallace S, Plouse R, Tiwari S, and Gomes AV (2019). Ponceau S waste: Ponceau S staining for total protein normalization. Anal. Biochem. 575, 44–53. 10.1016/j.ab.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mancebo A, DeMars L, Ertsgaard CT, and Puchner EM (2020). Precisely calibrated and spatially informed illumination for conventional fluorescence and improved PALM imaging applications. Methods Appl. Fluoresc. 8, 025004. 10.1088/2050-6120/ab716a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fujimoto Y, Itabe H, Kinoshita T, Homma KJ, Onoduka J, Mori M, Yamaguchi S, Makita M, Higashi Y, Yamashita A, and Takano T (2007). Involvement of ACSL in local synthesis of neutral lipids in cytoplasmic lipid droplets in human hepatocyte HuH7. J. Lipid Res. 48, 1280–1292. 10.1194/jlr.M700050-JLR200. [DOI] [PubMed] [Google Scholar]
- 99.Thomas J, Deerinck EAB, Ellisman MH, and Thor A (2022). Preparation of Biological Tissues for Serial Block Face Scanning Electron Microscopy (SBEM), Protocols.io. 10.17504/protocols.io.36wgq7je5vk5/v2. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The two-color single molecule cross-correlation-colocalization filtering code has been up loaded to GitHub (PuchnerLab GitHub).
The proteomics raw data and datasets have been deposited on Massive: https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.