

Abstract
Novel enzymatic methods are poised to become the dominant processes for de novo synthesis of DNA, promising functional, economic, and environmental advantages over the longstanding approach of phosphoramidite synthesis. Before this can occur, however, enzymatic synthesis methods must be parallelized to enable production of multiple DNA sequences simultaneously. As a means to this parallelization, we report a polymerase-nucleotide conjugate that is cleaved using electrochemical oxidation on a microelectrode array. The developed conjugate maintains polymerase activity toward surface-bound substrates with single-base control and detaches from the surface at mild oxidative voltages, leaving an extendable oligonucleotide behind. Our approach readies the way for enzymatic DNA synthesis on the scale necessary for DNA-intensive applications such as DNA data storage or gene synthesis.
Keywords: DNA nanotechnology, molecular data storage, microarrays, electrochemistry, terminal deoxynucleotidyl transferase
1. Introduction
Most synthetic DNA produced today is generated via phosphoramidite synthesis, a chemical method originally developed during the 1980s.1 Adoption of high-throughput synthesis methods such as deposition, maskless photoarrays, and microelectrode arrays has made phosphoramidite synthesis an enabling technology for work across the biological disciplines.2−5 It is, however, a mature technology with well-characterized drawbacks, including poor yields for longer oligonucleotides and reliance on expensive, environmentally hazardous organic solvents.6,7 These drawbacks provide an ultimate limit on the scaling of phosphoramidite synthesis, leaving it poorly suited to meet the demands of emerging DNA-hungry applications such as DNA data storage and gene synthesis.1,8
Enzymatic DNA synthesis stands as the apparent successor to phosphoramidite synthesis, and several processes have reached early stage development.9−13 Although alternative approaches have been reported, the majority of these methods employ cycles of untemplated extension and deblocking in a fashion analogous to phosphoramidite synthesis. Terminal deoxynucleotidyl transferase (TdT), a template-independent polymerase, is typically used for extension of the growing single-stranded DNA with a deoxynucleotide triphosphate (dNTP).14−16 Reversible modification of the dNTP substrate with a blocking group minimizes the addition of multiple dNTP and allows for control of the synthesized sequence with single-base control.
Mirroring the development of large-scale phosphoramidite synthesis, a natural next step in the development of enzymatic synthesis methods is parallelization to enable production of multiple DNA sequences simultaneously. Parallelization allows for increased synthesis throughput and amortizes reagent costs during production.17 These benefits scale with the density of synthesis sites employed by the synthesis platform, and the highest density demonstrated to date was achieved using a microelectrode array synthesis platform.18 There is therefore substantial motive for development of enzymatic DNA synthesis methods compatible with electrochemical deblocking. Methods reported to date, however, have generally relied on blocking groups cleaved via chemical or photochemical means.
Recently, Jung and co-workers took a first step toward remedying this shortfall, demonstrating spatially selective deblocking of a 3′-aminoxy-protected oligonucleotide with nitrous acid produced along an electrochemically generated pH gradient.19 Originally developed for use in sequencing-by-synthesis applications, the 3′-aminoxy group was shown by Lu and co-workers to be incorporable in solution phase into de novo synthesized oligonucleotides by engineered TdT.13,20 Combined, these achievements lay out a feasible pathway to realization of electrochemically parallelized enzymatic DNA synthesis, although challenges remain. At the enzymatic extension step, single-step dNTP incorporation yields with the reported TdT mutants are below those observed for phosphoramidite synthesis. At the electrochemical deblocking step, nitrous acid-induced deamination of guanosine, adenosine, and cytidine bases is a known side reaction, and care must be taken to tightly regulate local nitrosamine concentrations and exposure times to avoid base-substitutions in the synthesized oligonucleotide. Additional engineering of the mutant TdT polymerase and development of the deprotection system is required to synthesize oligonucleotides of useful lengths.
Here, we develop a novel electrochemically cleavable polymerase-nucleotide conjugate and demonstrate spatially selective cleavage of the conjugate on a microelectrode array. This method utilizes a cleavable linker to covalently attach a template-independent polymerase, such as TdT, to its deoxynucleotide triphosphate substrate.10 This covalent attachment alters the TdT-catalyzed extension reaction in two key ways. First, it accelerates the polymerase-catalyzed condensation of the base-modified nucleotide by decreasing the entropic penalty associated with an intermolecular reaction.21 Second, it covalently attaches the polymerase to the growing oligonucleotide, preventing diffusion into the bulk solution. In the absence of this covalent tethering, TdT displays distributive kinetics, releasing its oligonucleotide substrate after each base addition.16 The presence of the bound polymerase minimizes binding of additional equivalents of polymerase-nucleotide conjugate, limiting the occurrence of multiple base additions. A general scheme for extension and cleavage of an initiating oligonucleotide is presented in Figure 1.
Figure 1.

General working scheme. (a) An initiating oligonucleotide is extended at the 3′-terminus by a polymerase-nucleotide conjugate. The polymerase remains covalently attached to the extended oligonucleotide, preventing further extension by a second conjugate. (b) A positive voltage is applied to the electrode, resulting in electrochemical cleavage of the linker and allowing the polymerase to freely diffuse into solution. (c) The array is washed to clear residual unbound polymerase, completing a synthesis cycle. (d) Additional synthesis cycles may affect further elongation.
Unlike methods employing 3′-modified nucleotides, the developed polymerase-nucleotide conjugate utilizes a minimally modified TdT, capitalizing on the inherently high activity of native TdT and avoiding the need for targeted evolution of the polymerase specific to each blocking strategy.13,22 We deblock with direct electrochemical oxidation in simple phosphate buffer, avoiding the use of environmentally hazardous species such as quinones and nitrite salts during the DNA synthesis process and easing the logistics of colocation with existing infrastructure.23,24 The developed conjugate maintains polymerase activity toward surface-bound substrates with single-base control and detaches from the surface at mild oxidative voltages, leaving an extendable oligonucleotide behind, therein achieving a complete synthesis cycle of extension and deblocking. Altogether, electrochemically cleavable polymerase-nucleotide conjugates provide a differentiated technology with which parallelized enzymatic DNA synthesis may be realized to provide a high-throughput, low-cost, and environmentally benign alternative to phosphoramidite synthesis.
2. Results and Discussion
2.1. Linker Design
Compatibility of polymerase-nucleotide conjugates with microelectrode arrays hinges foremost on the linker moiety, which contains differentiable chemical functionality allowing cleavage in the presence of the growing oligonucleotide. We therefore set out to identify electrochemically cleavable functionality which could be incorporated with minimal disruption to the other roles of the linker. Efforts began with an evaluation of the enzyme-nucleotide conjugates previously reported by Palluk et al.10 The disulfide and nitrobenzyl chemical functionalities present in these conjugates have been demonstrated to be cleavable under reductive conditions.25−28 However, evaluation of model systems containing these functionalities by cyclic voltammetry indicated that electrochemical reduction was unlikely to proceed within the typical electrochemical window of aqueous buffers for either of these functionalities (Figure S1). With the existing enzyme-nucleotide conjugate incompatible with parallelization on microelectrode arrays, we elected to generate a novel conjugate by altering the linker.
A literature review of linker structures with known biological compatibility produced several potentially electrochemically cleavable options (Table S1). Among the identified linkers, we initially considered those cleavable under acidic conditions, as the acid-catalyzed removal of dimethoxytrityl protecting groups for spatially controlled phosphoramidite synthesis has been previously implemented on microelectrode arrays.5,18,29 We were concerned by the potential for acid-catalyzed depurination of the growing oligonucleotide under the strongly acidic conditions likely to be necessary for rapid linker cleavage, however, and thus did not pursue this path.30,31 Also potentially attractive were linkers cleavable under basic conditions. Unfortunately, we found that the voltage necessary to affect basicity by reduction of water were incompatible with our microelectrode arrays (Figure S2a).
Having examined and ruled out an acid/base-catalyzed cleavage mechanism, we turned our attention to redox cleavable linkers and took inspiration from 1, a linker developed by van der Veken et al. for peptide conjugation in phosphoproteomics studies (Figure 2a).32 The para-alkoxy benzyl functionality provides 1 modest acid lability; however, its similarity to commonly employed para-methoxy benzyl (PMB) protecting groups,33 typically cleaved using chemical oxidants but also electrochemically cleavable, suggests potential oxidative lability.34,35 Additionally exciting was the similarity of the para-alkoxy benzyl functionality to the ortho-nitrobenzyl photocleavage motif present in a previously reported conjugate,10 which we speculated increased the odds of compatibility with the polymerase in the conjugate system.
Figure 2.

Cleavable linker design process. (a) Acid-cleavable linker 1 inspired the design of oxidatively cleavable linker 4. (b) Cyclic voltammograms of alcohols 2 and 3. Pt disc working electrode (2 mm diameter), platinum wire counter electrode, and Ag/AgCl reference electrode. Induction period of 3 s at initial voltage prior to sweep at 10 mV/s. Alcohol (20 mM) in 60 vol % 0.5 M potassium phosphate buffer, pH 7.4, 40 vol % acetonitrile. Oxidation of 2 was observed to be irreversible with E1/2 = +1.18 V. A substantial increase in current is observed, suggesting catalytic oxidation of water. Oxidation of 3 was observed to be reversible with E1/2 = +0.81 V. (c) Structure of conjugate 5.
We hypothesized that additional electron-donating substituents would need to be placed on the para-alkoxy benzyl moiety to bring the redox potential into the electrochemical window of a suitable aqueous buffer system and to mitigate competitive oxidation of guanosine residues,36,37 as the redox potential of PMB ester derivatives are known to be higher than the corresponding PMB ethers.38 Inspired by the dimethoxybenzyl protecting group, meta-methoxy groups were selected for addition,39 and alcohols 2 and 3 were synthesized to evaluate the effects of meta-methoxy introduction. Cyclic voltammetry placed the half-wave potential of 2, bearing a single meta-methoxy group, at +1.18 V versus Ag/AgCl, with the irreversible oxidation wave merging with water oxidation at the edge of the electrochemical window. An additional meta-methoxy group dropped the half-wave potential of 3 to +0.81 V versus Ag/AgCl, a reversible wave that minimizes production of radical oxygen species by staying well within the electrochemical window and falls below the oxidation potential of +0.93 V versus Ag/AgCl reported for guanosine residues by Oliveira-Brett and co-workers (Figure 2b).40
We were pleased to have brought the redox potential into a useful range but wary of the reversibility of the oxidation. The switch from an irreversible to a reversible wave is indicative of additional stability afforded the oxidized species by the second meta-methoxy group; stability that implies an increased likelihood of undesired fragmentation of the linker. With this trade-off in mind, the addition of substituents to further lower the redox potential was not attempted, and the aromatic core of alcohol 3 was integrated to yield a final linker 4.
2.2. Polymerase-Nucleotide Conjugation
To attach linker 4 to the polymerase, we followed the successful strategy of Palluk et al., employing a TdT mutant with all but a single cysteine residue removed.10 Devising a strategy for attachment of a nucleotide to linker 4 proved more interesting, as the choice of attachment location and chemistry determines what chemical residue, or scar, remains attached to the nucleotide after linker cleavage. We quickly settled on the nucleobase as the attachment point so as to maximize synthetic tractability and narrowed our focus to three potential scars, each theoretically capable of attachment to linker 4: propargylamine, hydroxymethyl, and a propargylamine-derived β-hydroxyamide (Figure S3). To aid in selecting between the three, we employed each alongside native TdT in the uncontrolled extension of a single-stranded oligonucleotide. From this assay, it was determined that while the presence of each of the three scars slows nucleotide addition relative to unmodified dTTP, the β-hydroxyamide scar does so to a much greater degree. Seeking to balance inhibition of addition against predicted stability and synthetic accessibility, we elected to move forward with the propargylamine scar. Having determined the linker structure alongside attachment strategies for both the polymerase and the nucleotide, the three components were combined to form polymerase-nucleotide conjugate 5 as previously described by Palluk et al. (Figure 2c).10
2.3. Characterization of Conjugate Activity
We next wished to establish that polymerase-nucleotide conjugate 5 retained both template-free polymerase activity and selectivity for single nucleotide addition in solution-phase extension reactions. Treatment of a short deoxyoligonucleotide with conjugate 5 across a range of stoichiometric excesses and reaction times proved the conjugate both active and selective (Figure 3). With a small excess of 3.6 equivalent of conjugate 5, complete extension of the deoxyoligonucleotide was observed after just 30 s. With exposure increased to 240 equivalent of conjugate 5 for 480 s, an identical product peak was observed without the appearance of multiple nucleotide addition products. Combined, these results confirmed compatibility of linker 4 with the general polymerase-nucleotide conjugate strategy.
Figure 3.
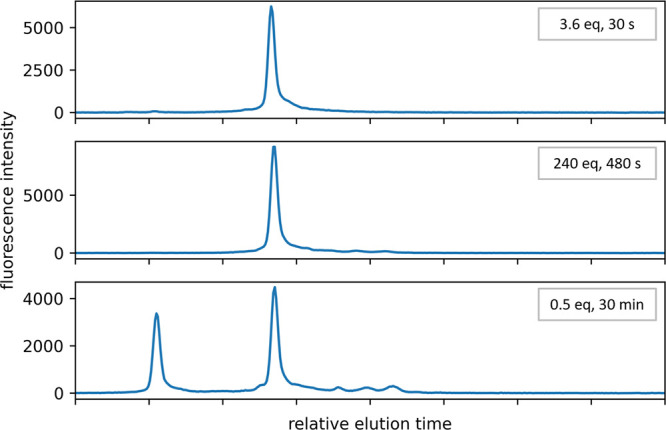
Solution-phase activity of conjugate 5. Conjugate 5 was added to a synthetic deoxyoligonucleotide 40 nucleotides in length in solution at the indicated stoichiometry and exposure time. The disulfide bond connecting the enzyme to the linker was cleaved, leaving the intact linker attached to the extended deoxyoligonucleotide, and the product mixture analyzed by capillary electrophoresis. Addition of conjugate 5 to a 2-fold excess of deoxyoligonucleotide provides a partial conversion control.
Additionally, we sought to probe the stability of the conjugate-oligonucleotide addition product, both to the expected usage conditions and to the acidic conditions used for cleavage of inspiring linker 1. To this end, we treated a short oligonucleotide with conjugate 5, heat-denatured the TdT, then acidified the reaction mixture and allowed it to incubate. After incubation, the reaction mixtures were loaded onto a denaturing polyacrylamide gel which effectively separated TdT-linked DNA from free DNA (Figure S4). No cleavage of the polymerase from the oligonucleotide was observed under either neutral or acidic conditions at room temperature, and at elevated temperature fragmentation of the polymerase was prominent, with only minimal release of free DNA. These experiments suggest that linker 4 is a robust point of attachment, capable of withstanding multiple rounds of addition and deprotection as required in enzymatic DNA synthesis. Satisfied with the solution-phase behavior of conjugate 5, we turned our attention to extension of surface-bound oligonucleotides.
2.4. Extension of Surface-Bound Initiators with TdT
We began by coating the surface of a microelectrode array with surface-bound oligonucleotide initiators suitable for extension by conjugate 5. For this task, we utilized spatially controlled phosphoramidite synthesis to pattern an array with a (dT)5 homopolymer as previously described.18 Briefly, the microelectrode array was treated with DMT-dT phosphoramidite, resulting in uniform coating of the silicon dioxide surface with a 4,4′-dimethoxytrityl (DMT) protected residue by reaction with the native silanol layer. After oxidation and capping, electrochemical acid generation was used to selectively remove the DMT protecting group around a chosen subset of electrodes. Repetition of this addition, oxidation, capping, and cleavage cycle generated the patterned (dT)5 homopolymer. To create an initiator extendable by TdT, so-called “reverse” phosphoramidite reagents were used, giving an initiator tethered to the surface by the 5′-end and leaving the 3′-end free for extension.41
Next, the array was exposed to a solution of TdT and Texas Red X-modified dUTP, incubated in sodium dodecyl sulfate solution to remove nonspecifically bound species, and imaged by fluorescent microscopy to reveal patterned fluorescence over the activated group of electrodes, indicative of successful extension of the (dT)5 homopolymer initiators by TdT (Figure 4a). Interestingly, while the patterned regions were significantly brighter, fluorescence from the background regions of the array was not reduced to the limit of detection. This nonzero level of background fluorescence is consistent with loss of the DMT protecting group and extension of the single-base layer by TdT. Fluorescent patterning can still be observed despite this loss of specificity, as the (dT)5 homopolymer is extended more quickly than the single dTTP residue.42
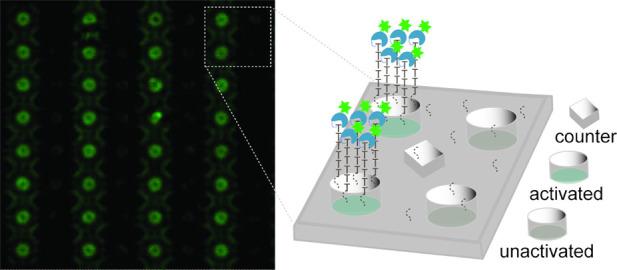
Figure 4.

TdT extension of surface-bound initiators. Electrodes are depicted in one of three states, representative of their role during electrochemical reactions. These states are: activated, held at positive voltage; unactivated, excluded from the circuit; or counter, held at ground voltage. Initiators are present on the silicon dioxide walls of the cylindrical electrode wells but not on the electrode surface. Diffraction-limited imaging may therefore yield either a disc or a torus. (a) An array was coated in a single dT residue with (dT)5 initiator synthesized at one working electrode. The initiators were extended with TdT/Texas Red X-dUTP. Greater fluorescent intensity over the activated electrode is indicative of incorporation of Texas Red X-dUTP. (b) An array was coated in a single C12 linker with (C12)4(dT)5 initiator synthesized at two working electrodes. The initiators were extended with conjugate 5 then stained with NHS-FAM. Greater fluorescent intensity over the activated electrodes is indicative of extension by conjugate 5. A control array not treated with conjugate 5 emitted no detectable fluorescence.
While this kinetic differential was sufficient to allow differentiation with an uncontrolled TdT extension, extension of the single-nucleotide background presented a challenge for visualizing addition of conjugate 5 to the initiators, where a single nucleotide addition was expected regardless of initiator length. To work around this, a hydrocarbon spacer was inserted as the first synthesis cycle between the surface and the (dT)5 homopolymer initiator, generating a surface uniformly coated in long-chain alcohols then patterned with oligonucleotide initiators. Schaudy et al. have demonstrated that arbitrary alcohols may be effective initiators for TdT extension; however, their rate of extension is substantially lower.42 Employing this more kinetically differentiated surface allowed for observation of spatially controlled addition of a polymerase-nucleotide conjugate. Treatment of the surface with conjugate 5 and subsequent staining of the conjugated TdT using fluorescein N-hydroxysuccinimide ester gave a patterned fluorescent surface, indicative of selective addition of the conjugate to the (dT)5 initiators (Figure 4b).
2.5. Cleavage of Linker Conjugate on Microelectrode Array
Satisfied that conjugate 5 maintained polymerase activity, generated durable linkages to the initiator strand, and could be applied to the extension of surface-bound initiator strands, we turned to the question of linker cleavage. Using phosphoramidite chemistry, the silicon dioxide surface of a microelectrode array was coated in “reverse” dT residues, presenting a uniform layer for extension. The initiator-coated microelectrode array was treated with conjugate 5, then the array was incubated in phosphate buffer and a constant cell potential of +1.8 V was applied between a single working electrode the counter electrodes for 90 s. While staining the conjugate with fluorescein N-hydroxysuccinimide ester as above was effective, staining with an Alexa Fluor 488-labeled antibody specific for a 6x-His tag present at the N-terminus of the employed TdT mutant was found to yield improved signal-to-noise. To visualize loss of conjugate 5, the microelectrode array was thus blocked with bovine serum albumin then antibody stained. Fluorescent microscopy showed the silicon dioxide surface to be generally fluorescent, save the area immediately adjacent to a single working electrode (Figure 5a). This patterning is consistent with uniform addition of conjugate 5 to the surface-bound initiators with spatially selective cleavage at the activated working electrode. To verify that the lack of fluorescence around the activated working electrode was due to the positive applied cell potential, rather than shear force applied by migratory flux to the electrode surface, the experiment was repeated, changing the applied cell potential to −1.8 V. With the electrode polarity inverted, fluorescence was retained around the activated electrode (Figure 5b).
Figure 5.

Cleavage of conjugate 5 addition product. dT initiators were extended with conjugate 5 and a cell potential applied to one of the four groups of working electrodes. The array surfaces were then stained with 6x-His antibody. Uniform fluorescent intensity is indicative of the presence of conjugate 5 across the surface, and a lack of fluorescence is indicative of the removal of conjugate 5 from that region. (a) +1.8 V was applied to single working electrode for 90 s. (b) −1.8 V was applied to single working electrode for 90 s. (c) The array was incubated in cleavage buffer for 90 s with no potential applied. (d) +1.0 V was applied to a single working electrode for the indicated time.
The maximum tolerated potential applicable between two electrodes on a microelectrode array is directly related to the transistor size in the controlling circuitry.43 While an applied cell potential of +1.8 V is compatible with the microelectrode arrays used in this study, more feature-dense arrays developed in the future are likely to have stricter requirements. We thus sought to demonstrate cleavage of conjugate 5 at lower applied cell potential and selected +1.0 V as a target. Measurement of the observed current versus applied cell potential during conjugate cleavage showed this decrease in cell potential to affect a two order-of-magnitude decrease in current density (Figure S2b). The previously described conjugate cleavage experiment was repeated save for reduction of the applied cell potential to +1.0 V. After passing current for 90 s, some loss of fluorescence over the activated working electrode was observed, and fluorescence loss continued with additional passage of current, replicating the results observed at +1.8 V by 360 s (Figure 5d). These observations suggest that conjugate 5 can be successfully cleaved with applied cell voltages compatible with substantially smaller microelectrode arrays.
Next, we sought to verify that cleavage of conjugate 5 resulted in an oligonucleotide further extendable by TdT. For this purpose, the silicon dioxide surface of a microelectrode array was once again coated in “reverse” dT residues, with (dT)10 homopolymers selectively placed around three of the four working electrodes. The initiator-coated microelectrode array was treated with conjugate 5, then a constant cell potential was applied over successive 90 s intervals between two of the working electrodes and the counter electrodes, +1.8 V to the first and +1.0 V to the second. The array was incubated in a solution of TdT and Texas Red X-modified dUTP overnight to extend free surface-bound initiators then imaged by fluorescent microscopy. Fluorescence was observed around two of the four working electrodes in the expected pattern, suggesting successful cleavage of conjugate 5 to an extendable oligonucleotide at both applied cell potentials as well as inhibition of TdT addition by uncleaved conjugate 5 across the remainder of the array (Figure 6).
Figure 6.
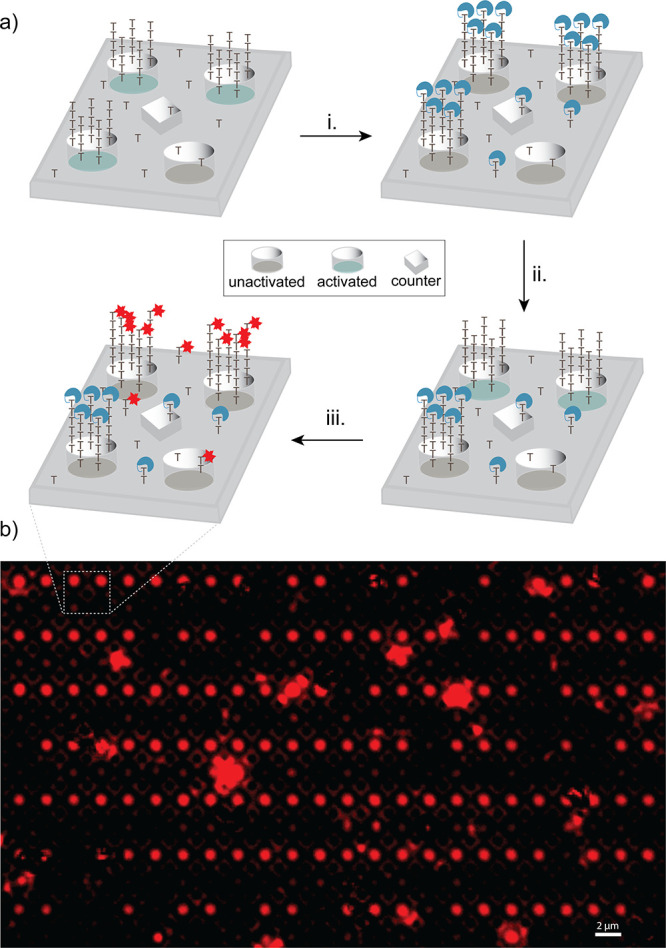
Cleavage of conjugate 5 addition product and subsequent extension with TdT. Electrodes are depicted in one of three states, representative of their role during electrochemical reactions. These states are: activated, held at positive voltage; unactivated, excluded from the circuit; or counter, held at ground voltage. An array was coated in a single dT residue with (dT)10 initiators synthesized at three working electrodes. The initiators were extended with conjugate 5 (i) followed by application of +1.8 V to one working electrode for 90 s then application of +1.0 V to a second working electrode for 90 s (ii). The cleaved conjugate 5 addition products were then extended with TdT/Texas Red X-dUTP (iii). Greater fluorescent intensity over the activated electrodes is indicative of incorporation of Texas Red X-dUTP. The lack of fluorescence over a subset of the activated electrodes is believed to be the result of failures in the circuitry preventing the intended application of voltage. The anomalous fluorescent splotches observed are believed to be precipitated aggregates of Texas Red X-dUTP.
As a final verification of complete conjugate addition and cleavage process, we altered the initiator structure to allow for collection from the microelectrode array surface for characterization by capillary electrophoresis. Compelled by the desire for an initiator producible at bulk scale, we designed a strategy employing a uniform coating of presynthesized oligonucleotides. First, a plasma-cleaned microelectrode array was silanized under vacuum with 3-(glycidoxyproypl)trimethoxysilane to give a coating of reactive epoxide groups. Then, a single-stranded oligonucleotide containing a 60 bp hybridization region and a 3′-terminal amine functionality was added to the surface by acid-catalyzed nucleophilic addition to give a uniform coating of anchor strands. Finally, the array was incubated in an 80 bp single-stranded oligonucleotide containing a 60 bp hybridization region and 5′-terminal fluorescein functionality, leaving a 20 bp overhang from the anchor strands to serve as the initiator for TdT extension. Following this strategy, a microelectrode array was coated in double-stranded DNA initiators and treated with conjugate 5. A cell potential of +1.0 V was applied between the four working electrodes and the counter electrodes for 90 s, after which the initiator strands were recovered from the surface by incubation in Milli-Q water at 90 °C. Capillary electrophoresis of the recovered material showed single-base extension of the initiator strand (Figure 7).
Figure 7.

Capillary electropherogram of conjugate 5 addition product collected from microelectrode array surface. A microelectrode array was functionalized with 80 bp oligonucleotide initiators by hybridization to a covalent-anchored complementary oligonucleotide. Conjugate 5 was added to the initiators and cleaved by applying +1.0 V for 90 s. The initiators were heat-denatured from the surface, collected, and analyzed by capillary electrophoresis. Control experiments showed that intact oligonucleotide-polymerase complexes cannot be observed in the electropherogram.
2.6. Conclusions
Enzymatic methods for DNA synthesis promise to overcome the limitations of the widely used phosphoramidite synthesis, but fully capturing the functional, economic, and environmental benefits of the enzymatic process requires parallelization to increase synthesis throughput and minimize per-sequence costs.7,12 To bridge this gap, we have developed a novel cleavable polymerase-nucleotide conjugate 5 and demonstrated spatially selective electrochemical deblocking at the submicron scale using a microelectrode array. Conjugate 5 maintains polymerase activity, inhibits extension by additional polymerase equivalents, and deblocks to release an oligonucleotide compatible with further enzymatic extension. In anticipation of future advances to microelectrode array technology, we have demonstrated cleavage of conjugate 5 at applied cell voltages well below the maximum tolerated by today’s arrays, ensuring compatibility with the decreased feature sizes and stricter tolerances of state-of-the-art microelectrode arrays for the foreseeable future.
There are questions that remain to be answered. First, the propargylamine scars left after cleavage of the polymerase and linker from the growing oligonucleotide complicate direct use of the synthesized strands. While these scars can be removed by inclusion of an amplification step prior to long-term storage or gene assembly, an iteration on the linker that avoids the scar would be a more elegant solution.1 Second, the relatively narrow gap in oxidation potential between alcohol 3 and guanosine residues in the growing oligonucleotide presents the possibility of oxidative damage if the electrochemical cleavage conditions are poorly controlled. The use of platinum working electrodes helps to inhibit direct guanosine oxidation, but a greater separation of oxidation potentials may be necessary to prevent intramolecular electron transfer.44,45 Further studies addressing these questions over multiple cycles of nucleotide addition and deblocking are ultimately required.
In many ways, however, electrochemically cleavable polymerase-nucleotide conjugates compare favorably to previously reported methods. Compared to parallelized enzymatic DNA synthesis methods that rely on kinetic competitions to mediate TdT activity, conjugate 5 allows for controlled, single-base additions to the synthesized oligonucleotides, a requirement for synthesis of arbitrary sequences.46,47 Compared to the prospective system of 3-aminoxy-protected nucleotides, nitrous acid-mediated deblocking, and a compatible TdT mutant extrapolable from the work of Jung and Lu,13,19 conjugate 5 avoids the use of environmentally hazardous compounds such as quinones and nitrite salts at the point of synthesis,23,24 simplifies future development by capitalizing on the native activity of TdT, and has been demonstrated to function over a complete synthesis cycle of nucleotide addition and deblocking on the surface of a microelectrode array. Given these advantages, electrochemically cleavable polymerase-nucleotide conjugates present an alternative route to the realization of enzymatic DNA synthesis on the scale necessary for applications such as DNA data storage or gene synthesis.
3. Methods
3.1. Preparation of Polymerase-Nucleotide Conjugate 5
A TdT variant engineered to contain only one cysteine residue, thereby enabling site-specific labeling, was expressed and purified following the protocol outlined in Palluk et al.10 The sequence of the variant may be found in the Supporting Information.
To label the TdT variant, 100 nmol of linker-nucleotide complex was combined with 33 nmol of TdT in 1× KP buffer and incubated overnight at 4 °C under gentle rotation of the tube. The labeled TdT was then purified from unreacted linker-nucleotide complex using size exclusion chromatography as follows. The entire labeling reaction was applied to a Superdex 200 10/30 GL column equilibrated with 1× TP6.5 buffer. The fractions of purified conjugate 5 were collected, combined, and then concentrated using a 30 kDa MWCO Pierce Protein Concentrator PES. Aliquots of conjugate 5 were frozen in liquid nitrogen and stored at 0.5 mg/mL in 1× TP6.5 buffer with 0.1 vol % Cytiva Surfactant P20.
An active site titration was performed to measure the amount of active conjugate. Extension reactions were performed in 1× TP6.5 buffer supplemented with 10 mM Mg acetate and 0.25 mM CoCl2. Four different solutions containing a mixture of a synthetic deoxyoligonucleotide (5′-/56-FAM/TGT GCC GTG AGA CCT GGC TCC TGA CGA TAT GGA TGG ATT T-3′) labeled with FAM at the 5′-terminus (100 nM) and increasing amounts of unlabeled deoxyoligonucleotides of the same sequence but without FAM (0, 100, 200, and 300 nM) were prepared. A second solution containing conjugate 5 (200 nM) was also prepared for each of the four mixes, then the deoxyoligonucleotide and conjugate 5 solutions were combined in equal volumes at room temperature to start the reaction. Extension was allowed to proceed for 20 min after which a 2 μL aliquot of each reaction was combined with 8 μL of quenching solution (40 mM ethylenediaminetetraacetic acid in 75 vol % HiDi formamide and 25 vol % Milli Q water). A second 2 μL aliquot was quenched after 30 min total reaction time to verify complete turnover of the enzyme by comparison of the 20 and 30 min time points. The final concentration of conjugate was 100 nM for each of the reactions, the ssDNA concentrations were 50, 100, 150, and 200 nM, respectively. A 2 μL aliquot of the quenched reaction was diluted with HiDi formamide (13.3 μL), Milli Q water (4 μL), 1 M dithiothreitol (0.2 μL), and GeneScan 600 LIZ dye Size Standard v2.0 (0.5 μL) then analyzed by capillary electrophoresis. Dithiothreitol was added to the analytical solution to hydrolyze the disulfide bond between the linker and the cysteine residue of the protein. The extent of TdT labeling was found to be approximately 90% (Figure S6).
3.2. Solution-Phase DNA Extension with dUTP-Scar Nucleotides
For each dUTP-scar nucleotide, an aqueous solution containing a synthetic deoxyoligonucleotide (5′-GTT TCA CAC CGT TCG TCC TA-3′) (100 nM) and dUTP-scar nucleotide (100 μM) was prepared including 10× TdT buffer (5 μL), 2.5 mM CoCl2 (5 μL), and terminal transferase (M0315) (10 units) for a total volume of 50 μL. The solution was vortex mixed, heated to 37 °C for 30 min, then chilled to room temperature. Ten μL aliquots of each reaction were diluted with Alfa Aesar TBE-urea sample buffer (2×), loaded onto a 5% TBE-urea PAGE gel, and run at 175 V in 1× TBE buffer. The completed gel was stained with SYBR Gold and imaged using a blue light imager.
3.3. Solution-Phase DNA Extension with Conjugate 5
A synthetic deoxyoligonucleotide (5′-/56-FAM/TGT GCC GTG AGA CCT GGC TCC TGA CGA TAT GGA TGG ATT T-3′) labeled with FAM at the 5′-terminus (100 nM) was used to measure DNA extension. Two solutions were prepared: one containing 100 nM deoxyoligonucleotide, 20 mM MgOAc, and 0.5 mM CoCl2 in 1× TP6.5 buffer and another containing conjugate 5 in 1× TP6.5 buffer with 0.01 vol % Cytiva Surfactant P20. The deoxyoligonucleotide and conjugate 5 solutions were combined at room temperature in a 1:1 ratio to start the reaction. A 2 μL aliquot of the reaction was quenched at 0.5, 1, 2, 4, 8, and 16 min in 8 μL of quenching solution (40 mM ethylenediaminetetraacetic acid in 75 vol % HiDi formamide and 25 vol % Milli Q water). The final concentration of DNA for each of the four reactions was 50 nM, and the final concentration of conjugate 5 was 0.1, 0.0625, 0.03125, and 0.0156 mg/mL, respectively. A 2 μL aliquot of the quenched reaction was diluted with HiDi formamide (13.3 μL), Milli Q water (4 μL), 1 M dithiothreitol (0.2 μL), and GeneScan 600 LIZ dye Size Standard v2.0 (0.5 μL) then analyzed by capillary electrophoresis. Dithiothreitol was added to the analytical solution to hydrolyze the disulfide bond between the linker and the cysteine residue of the protein.
3.4. Acid Stability of Conjugate 5 Adducts
An aqueous solution was prepared from Milli Q water containing a synthetic deoxynucleotide (5′-T60-3′) labeled with FAM at the 5′-terminus (100 nM), 1× TP8 buffer, and 2× metal mix. This solution (20 μL) was mixed by pipetor for 15 s with an 0.2 μg/μL solution of conjugate 5 in 1× TP8 buffer (20 μL). The mixture was allowed to stand at room temperature for an additional 45 s, heated to 70 °C for 10 min to denature active TdT, then cooled to room temperature.
Four 5 μL aliquots were pulled from the mixture, treated with 1 μL of 1 M hydrochloric acid, 1 M acetic acid, 1 M sulfuric acid, or DI water, and allowed to stand at room temperature for 30 min. The aliquots were diluted to 20 μL in 1× TGS buffer, loaded onto a 4–20% TGX precast PAGE gel, and run at 200 V in 1× TGS buffer. The completed gel was imaged using a fluorescent scanner.
The experiment was repeated, and a second set of five 5 μL aliquots were pulled from the resulting mixture then treated with 1 μL of 1 M hydrochloric acid. Three aliquots were allowed to stand at room temperature for 30 min, 2 h, or 4 h. The remaining two aliquots were heated to 40 or 80 °C for 30 min. The aliquots were diluted to 20 μL in 1× TGS buffer, loaded onto a 4–20% TGX precast PAGE gel, and run at 200 V in 1× TGS buffer. The completed gel was imaged using a fluorescent scanner.
3.5. Extension of Surface-Bound Initiators with Native TdT
A microelectrode array was treated with a single dT phosphoramidite then patterned over a single working electrode with 5′-(dT)5-3′ initiators using spatially controlled phosphoramidite synthesis as previously described.18 “Reverse” phosphoramidites were used to leave an unmodified 3′ terminus suitable for extension by TdT, and the array was treated with 30% ammonium hydroxide for 1 h then washed with DI water and blown dry under a stream of compressed air to deprotect the synthesized initiators prior to use.
A TdT extension solution was prepared from 10× TdT buffer (5 μL), 2.5 mM CoCl2 (5 μL), 1 mM ChromaTide Texas Red-12-dUTP (1 μL), terminal transferase (M0315) (20 units), and 38 μL Milli Q water. The modified array was treated with TdT extension solution for 30 min then washed with DI water and incubated in 0.1 wt % aqueous sodium dodecyl sulfate overnight. The array was washed with DI water, blown dry under a stream of compressed air, and imaged by fluorescence microscopy.
3.6. Extension of Surface-Bound Initiators with Conjugate 5
A microelectrode array was treated with a single C12 phosphoramidite then patterned over a single working electrode with 5′-(C12)4(dT)10-3′ initiators using spatially controlled phosphoramidite synthesis as previously described.18 “Reverse” phosphoramidites were used to leave an unmodified 3′ terminus suitable for extension by TdT, and the array was treated with 30% ammonium hydroxide for 1 h then washed with DI water and blown dry under a stream of compressed air to deprotect the synthesized initiators prior to use.
A conjugate 5 extension solution was prepared from 10× TP8 buffer (5 μL), 10× metal mix (5 μL), 5 μL 2 mg/mL conjugate 5, and 35 μL Milli Q water. The microelectrode array was treated with conjugate 5 extension solution for 30 min then washed with DI water, 0.1 wt % aqueous sodium dodecyl sulfate solution, and DI water and blown dry under a stream of compressed air.
A staining solution was prepared from 10× TP8 buffer (10 μL), 1 mg/mL fluorescein N-hydroxysuccinimide ester in dimethyl sulfoxide (1 μL), and 89 μL DI water. The microelectrode array was treated with staining solution for 30 min then washed with DI water, 0.1 wt % aqueous sodium dodecyl sulfate solution, and DI water and blown dry under a stream of compressed air. The array was imaged by fluorescence microscopy.
3.7. Cleavage of Conjugate 5 on Microelectrode Array
A microelectrode array was treated with a single dT phosphoramidite using unpatterned phosphoramidite chemistry. “Reverse” phosphoramidites were used to leave an unmodified 3′ terminus suitable for extension by TdT, and the array was treated with 30% ammonium hydroxide for 1 h then washed with DI water and blown dry under a stream of compressed air to deprotect the synthesized initiators prior to use.
A conjugate 5 extension solution was prepared from 10× TP8 buffer (5 μL), 10× metal mix (5 μL), 5 μL 2 mg/mL conjugate 5, and 35 μL Milli Q water. The microelectrode array was treated with conjugate 5 extension solution for 30 min then washed with DI water, 0.1 wt % aqueous sodium dodecyl sulfate solution, and DI water and blown dry under a stream of compressed air.
The microelectrode array was covered with 50 mM potassium phosphate buffer (pH 7.4), and a voltage of +1.8 V was applied between one working electrode and the counter electrodes for 90 s. The array was washed with DI water, 0.1 wt % aqueous sodium dodecyl sulfate solution, and DI water and blown dry under a stream of compressed air.
The microelectrode array was incubated in a 30 mg/mL solution of bovine serum albumin in 1× PBST buffer for 1 h and then washed with DI water and blown dry under a stream of compressed air. The array was then incubated in a solution of 1 mg/mL Alex Fluor 488-labeled 6x-His tag monoclonal antibody (1 μL) in 1× TE buffer (500 μL) for 1 h, washed with DI water, and blown dry under a stream of compressed air. The array was imaged by fluorescence microscopy.
3.8. Extension of Conjugate 5 Cleavage-Product with TdT
A microelectrode array was treated with a single dT phosphoramidite then patterned over three working electrodes with 5′-(dT)10-3′ initiators using spatially controlled phosphoramidite synthesis as previously described.18 “Reverse” phosphoramidites were used to leave an unmodified 3′ terminus suitable for extension by TdT, and the array was treated with 30% ammonium hydroxide for 1 h then washed with DI water and blown dry under a stream of compressed air to deprotect the synthesized initiators prior to use.
Surface-bound initiators on the patterned array were extended with conjugate 5 as described above. The microelectrode array was covered with 50 mM potassium phosphate buffer (pH 7.4), and a voltage of +1.8 V was applied between one patterned working electrode and the counter electrodes for 90 s. A voltage of +1.0 V was applied between the second patterned working electrode and the counter electrodes for 90 s. The array was washed with DI water, 0.1 wt % aqueous sodium dodecyl sulfate solution, and DI water and blown dry under a stream of compressed air.
A TdT extension solution was prepared from 10× TdT buffer (10 μL), 2.5 mM CoCl2 (10 μL), 1 mM ChromaTide Texas Red-12-dUTP (1 μL), 10 mM dNTP mix (0.5 μL), terminal transferase (M0315) (80 units), and 72 μL Milli Q water. The modified array was treated with TdT extension solution overnight then washed with DI water, blown dry under a stream of compressed air, and imaged by fluorescence microscopy.
3.9. Recovery of Conjugate-Extended Surface-Bound Initiators
A microelectrode array was placed in a glass jar alongside a crumpled Kimtech Kimwipe. A solution of (3-glycidoxy)trimethoxysilane (2 μL) in toluene (98 μL) was added to the Kimwipe, then the jar was sealed with a screw top cap and placed in an oven at 150 °C for 2 h. The jar was removed from the oven and allowed to reach room temperature before removal of the microelectrode array.
An oligonucleotide anchoring solution was prepared from 100 μM Anchor_T10_Amine (3 μL), 100 mM sulfuric acid (17 μL), 2× PBS buffer (50 μL), and DI water (30 μL). The silanized microelectrode array was incubated in the oligonucleotide anchoring solution overnight, washed with DI water, 0.1 wt % aqueous sodium dodecyl sulfate solution, and DI water and blown dry under a stream of compressed air. The array was then incubated in a solution of 1 μM FAM_Anchor*_Init in 1× PBS buffer for 2 h and again washed with DI water, 0.1 wt % aqueous sodium dodecyl sulfate solution, and DI water and blown dry under a stream of compressed air.
Surface-bound initiators on the resulting array were extended with conjugate 5 as described above. The microelectrode array was covered with 50 mM potassium phosphate buffer (pH 7.4), and a voltage of +1.0 V was applied between four working electrodes and the counter electrodes for 90 s. The array was washed with DI water, 0.1 wt % aqueous sodium dodecyl sulfate solution, and DI water and blown dry under a stream of compressed air.
To collect the initiators from the surface, the microelectrode array was placed into a jig, covered in 300 μL Milli Q water, and incubated in an oven at 90 °C for 15 min. The covering water was collected then the array and jig washed with an additional 300 μL Milli Q water. The combined covering water and wash were concentrated on a SpeedVac vacuum concentrator, and the resulting residue was resuspended in 20 μL Milli Q water. A 2 μL aliquot of the recovered material was diluted with HiDi formamide (13.3 μL), Milli Q water (4.2 μL), and GeneScan 600 LIZ dye Size Standard v2.0 (0.5 μL) and then analyzed by capillary electrophoresis.
Acknowledgments
The authors thank D. Carmean, L. Ceze, Y.-J. Chen, J. Mulligan, H.-Y. Parker, and C. Zhang for discussion throughout the project. Additionally, we thank E. Jensen and D. Covarrubias for assistance with fragment analysis. Finally, we thank the Molecular Information Systems Lab at the University of Washington for their support of experiments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.3c00044.
Additional data and methods, including protein and DNA sequences, descriptions of general methods, buffer preparation, and synthetic protocols, unprocessed gel and microscopy images, and characterization data (PDF)
Author Contributions
J.A.S. performed experiments. J.A.S. and B.H.N. wrote the manuscript. J.A.S., B.H.N., R.C., and K.S. performed data analysis. J.A.S., B.H.N., S.P., and D.H. designed the linker structure. J.G.B. performed protein expression, enzyme conjugation, and characterized solution-phase activity. S.P. and D.H. designed solution-phase experiments. B.H.N., R.C., S.P., D.H., and K.S. reviewed and edited the manuscript. B.H.N., R.C., and K.S. conceived the project.
This work was funded by Microsoft Corporation.
The authors declare the following competing financial interest(s): J.A.S., B.H.N., R.C., and K.S. were employed by or consulted for Microsoft for the duration of this work. J.G.B., S.P., and D.H.A. were employed by Ansa Biotechnologies for the duration of this work. Microsoft Technology Licensing LLC has filed patent application US20210238577 covering aspects of this work. S.P. and D.H.A. have financial interest in IP around polymerase-nucleotide conjugates owned by UC Berkeley. The authors declare no other competing interests.
Supplementary Material
References
- Hughes R. A.; Ellington A. D. Synthetic DNA Synthesis and Assembly: Putting the Synthetic in Synthetic Biology. Cold Spring Harbor Perspectives in Biology 2017, 9, a023812. 10.1101/cshperspect.a023812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Liu B.; Pei B.; Chen J.; Zhou D.; Peng J.; Zhang X.; Jia W.; Xu T. Inkjet Bioprinting of Biomaterials. Chem. Rev. 2020, 120, 10793–10833. 10.1021/acs.chemrev.0c00008. [DOI] [PubMed] [Google Scholar]
- Pawloski A. R.; et al. Photolithographic synthesis of high-density DNA probe arrays: Challenges and opportunities. Journal of Vacuum Science & Technology B: Microelectronics and Nanometer Structures 2007, 25, 2537. 10.1116/1.2794325. [DOI] [Google Scholar]
- Negrete O. D.; Cerrina F. Step-and-scan maskless lithography for ultra large scale DNA chips. Microelectron. Eng. 2008, 85, 834–837. 10.1016/j.mee.2008.01.014. [DOI] [Google Scholar]
- Egeland R. D. Electrochemically directed synthesis of oligonucleotides for DNA microarray fabrication. Nucleic Acids Res. 2005, 33, e125–e125. 10.1093/nar/gni117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeProust E. M.; Peck B. J.; Spirin K.; McCuen H. B.; Moore B.; Namsaraev E.; Caruthers M. H. Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Res. 2010, 38, 2522–2540. 10.1093/nar/gkq163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B. H.; Sinistore J. C.; Smith J. A.; Praneet Singh A.; Johnson L. M.; Kidman T.; diCaprio T. J.; Carmean D. M.; Strauss K.. Architecting Datacenters for Sustainability: Greener Data Storage using Synthetic DNA; Electronics Goes Green, 2020. [Google Scholar]
- Ceze L.; Nivala J.; Strauss K. Molecular digital data storage using DNA. Nat. Rev. Genet. 2019, 20, 456–466. 10.1038/s41576-019-0125-3. [DOI] [PubMed] [Google Scholar]
- Horspool D. R.; Coope R. J.; Holt R. A. Efficient assembly of very short oligonucleotides using T4 DNA Ligase. BMC Res. Notes 2010, 3, 291. 10.1186/1756-0500-3-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palluk S.; Arlow D. H.; de Rond T.; Barthel S.; Kang J. S.; Bector R.; Baghdassarian H. M.; Truong A. N.; Kim P. W.; Singh A. K.; Hillson N. J.; Keasling J. D. De novo DNA synthesis using polymerase-nucleotide conjugates. Nat. Biotechnol. 2018, 36, 645–650. 10.1038/nbt.4173. [DOI] [PubMed] [Google Scholar]
- Hoff K.; Halpain M.; Garbagnati G.; Edwards J.; Zhou W. Enzymatic Synthesis of Designer DNA Using Cyclic Reversible Termination and a Universal Template. ACS synthetic biology 2020, 9, 283–293. 10.1021/acssynbio.9b00315. [DOI] [PubMed] [Google Scholar]
- Eisenstein M. Enzymatic DNA synthesis enters new phase. Nat. Biotechnol. 2020, 38, 1113–1115. 10.1038/s41587-020-0695-9. [DOI] [PubMed] [Google Scholar]
- Lu X.; et al. Enzymatic DNA Synthesis by Engineering Terminal Deoxynucleotidyl Transferase. ACS Catal. 2022, 12, 2988–2997. 10.1021/acscatal.1c04879. [DOI] [Google Scholar]
- Motea E. A.; Berdis A. J. Terminal deoxynucleotidyl transferase: The story of a misguided DNA polymerase. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2010, 1804, 1151–1166. 10.1016/j.bbapap.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarac I.; Hollenstein M. Terminal Deoxynucleotidyl Transferase in the Synthesis and Modification of Nucleic Acids. ChemBioChem. 2019, 20, 860–871. 10.1002/cbic.201800658. [DOI] [PubMed] [Google Scholar]
- Jensen M. A.; Davis R. W. Template-Independent Enzymatic Oligonucleotide Synthesis (TiEOS): Its History, Prospects, and Challenges. Biochemistry 2018, 57, 1821–1832. 10.1021/acs.biochem.7b00937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosuri S.; Church G. M. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 2014, 11, 499–507. 10.1038/nmeth.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B. H.; Takahashi C. N.; Gupta G.; Smith J. A.; Rouse R.; Berndt P.; Yekhanin S.; Ward D. P.; Ang S. D.; Garvan P.; Parker H.-Y.; Carlson R.; Carmean D.; Ceze L.; Strauss K. Scaling DNA data storage with nanoscale electrode wells. Sci. Adv. 2021, 7, eabi6714 10.1126/sciadv.abi6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. S.; Jung W.-B.; Wang J.; Abbott J.; Horgan A.; Fournier M.; Hinton H.; Hwang Y.-H.; Godron X.; Nicol R.; Park H.; Ham D. CMOS electrochemical pH localizer-imager. Sci. Adv. 2022, 8, eabm6815 10.1126/sciadv.abm6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter D.; Kim M.-J.; Karalkar N.; Leal N. A.; Chen F.; Guggenheim E.; Visalakshi V.; Olejnik J.; Gordon S.; Benner S. A. Labeled Nucleoside Triphosphates with Reversibly Terminating Aminoalkoxyl Groups. Nucleosides, Nucleotides & Nucleic Acids 2010, 29, 879–895. 10.1080/15257770.2010.536191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page M. I.; Jencks W. P. Entropic Contributions to Rate Accelerations in Enzymic and Intramolecular Reactions and the Chelate Effect. Proc. Natl. Acad. Sci. U. S. A. 1971, 68, 1678–1683. 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamme M.; Hanlon S.; Marzuoli I.; Püntener K.; Sladojevich F.; Hollenstein M. Evaluation of 3′-phosphate as a transient protecting group for controlled enzymatic synthesis of DNA and XNA oligonucleotides. Commun. Chem. 2022, 5, 68. 10.1038/s42004-022-00685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enguita F. J.; Leitão A. L. Hydroquinone: Environmental Pollution, Toxicity, and Microbial Answers. BioMed. Research International 2013, 2013, 1–14. 10.1155/2013/542168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toxicological Profile for Nitrate and Nitrite; Agency for Toxic Substances and Disease Registry (ATSDR), 2017. [PubMed]
- Nicolardi S.; Giera M.; Kooijman P.; Kraj A.; Chervet J.-P.; Deelder A. M.; van der Burgt Y. E. M. On-Line Electrochemical Reduction of Disulfide Bonds: Improved FTICR-CID and -ETD Coverage of Oxytocin and Hepcidin. J. Am. Soc. Mass Spectrom. 2013, 24, 1980–1987. 10.1007/s13361-013-0725-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraj A.; Brouwer H.-J.; Reinhoud N.; Chervet J.-P. A novel electrochemical method for efficient reduction of disulfide bonds in peptides and proteins prior to MS detection. Anal. Bioanal. Chem. 2013, 405, 9311–9320. 10.1007/s00216-013-7374-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y.; Banno A.; Kitagawa H.; Higashi S.; Kitade Y.; Shibata A.; Ikeda M. Reduction-Responsive DNA Duplex Containing O6-Nitrobenzyl-Guanine. ACS Omega 2018, 3, 9267–9275. 10.1021/acsomega.8b01177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakami N.; Higashi S. L.; Kawaki Y.; Kitamura Y.; Shibata A.; Ikeda M. Construction of a reduction-responsive oligonucleotide via a post-modification approach utilizing 4-nitrophenyl diazomethane. Polym. J. 2021, 53, 741–746. 10.1038/s41428-021-00464-4. [DOI] [Google Scholar]
- Maurer K.; Cooper J.; Caraballo M.; Crye J.; Suciu D.; Ghindilis A.; Leonetti J. A.; Wang W.; Rossi F. M.; Stöver A. G.; Larson C.; Gao H.; Dill K.; McShea A. Electrochemically Generated Acid and Its Containment to 100 Micron Reaction Areas for the Production of DNA Microarrays. PLoS One 2006, 1, e34 10.1371/journal.pone.0000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An R.; Jia Y.; Wan B.; Zhang Y.; Dong P.; Li J.; Liang X. Non-Enzymatic Depurination of Nucleic Acids: Factors and Mechanisms. PLoS One 2014, 9, e115950 10.1371/journal.pone.0115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Septak M. Kinetic studies on depurination and detritylation of CPG-bound intermediates during oligonucleotide synthesis. Nucleic Acids Res. 1996, 24, 3053–3058. 10.1093/nar/24.15.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veken P.; Dirksen E. H. C.; Ruijter E.; Elgersma R. C.; Heck A. J. R.; Rijkers D. T. S.; Slijper M.; Liskamp R. M. J. Development of a Novel Chemical Probe for the Selective Enrichment of Phosphorylated Serine- and Threonine-Containing Peptides. ChemBioChem. 2005, 6, 2271–2280. 10.1002/cbic.200500209. [DOI] [PubMed] [Google Scholar]
- Horita K.; Yoshioka T.; Tanaka T.; Oikawa Y.; Yonemitsu O. On the selectivity of deprotection of benzyl, mpm (4-methoxybenzyl) and dmpm (3,4-dimethoxybenzyl) protecting groups for hydroxy functions. Tetrahedron 1986, 42, 3021–3028. 10.1016/S0040-4020(01)90593-9. [DOI] [Google Scholar]
- Weinreb S. M.; Epling G. A.; Comi R.; Reitano M. Efficacious cleavage of the benzyl ether protecting group by electrochemical oxidation. Journal of Organic Chemistry 1975, 40, 1356–1358. 10.1021/jo00897a043. [DOI] [Google Scholar]
- Green R. A.; Jolley K. E.; Al-Hadedi A. A. M.; Pletcher D.; Harrowven D. C.; De Frutos O.; Mateos C.; Klauber D. J.; Rincón J. A.; Brown R. C. D. Electrochemical Deprotection of para -Methoxybenzyl Ethers in a Flow Electrolysis Cell. Org. Lett. 2017, 19, 2050–2053. 10.1021/acs.orglett.7b00641. [DOI] [PubMed] [Google Scholar]
- Reipa V.; Atha D. H.; Coskun S. H.; Sims C. M.; Nelson B. C. Controlled potential electro-oxidation of genomic DNA. PLoS One 2018, 13, e0190907 10.1371/journal.pone.0190907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprun E. V.; Kutdusova G. R.; Khmeleva S. A.; Radko S. P. Towards deeper understanding of DNA electrochemical oxidation on carbon electrodes. Electrochem. Commun. 2021, 124, 106947. 10.1016/j.elecom.2021.106947. [DOI] [Google Scholar]
- Howard K. T.; Chisholm J. D. Preparation and Applications of 4-Methoxybenzyl Esters in Organic Synthesis. Organic preparations and procedures international 2016, 48, 1–36. 10.1080/00304948.2016.1127096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa Y.; Tanaka T.; Horita K.; Yoshioka T.; Yonemitsu O. DMPM (3,4-dimethoxybenzyl) protecting group for hydroxy function more readily removable than MPM (P-methoxybenzyl) protecting group by DDQ oxidation. Tetrahedron Lett. 1984, 25, 5393–5396. 10.1016/S0040-4039(01)91293-6. [DOI] [Google Scholar]
- Oliveira-Brett A.; Piedade J.; Silva L.; Diculescu V. Voltammetric determination of all DNA nucleotides. Anal. Biochem. 2004, 332, 321–329. 10.1016/j.ab.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Glen Report 6.14: 5′ to 3′ Synthesis. https://www.glenresearch.com/reports/gr6-14.
- Schaudy E.; Lietard J.; Somoza M. M. Sequence Preference and Initiator Promiscuity for De Novo DNA Synthesis by Terminal Deoxynucleotidyl Transferase. ACS Synth. Biol. 2021, 10, 1750–1760. 10.1021/acssynbio.1c00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadami O.; Lu H.; Chan M.; Tan M.; Chung S.; Lee S. H.; Holden M. T.; Ridder R. d.; Merriman B.; Hall D. A.. Helix: An Electrochemical CMOS DNA Synthesizer. In 2022 IEEE Symposium on VLSI Technology and Circuits (VLSI Technology and Circuits); IEEE: Honolulu, HI, 2022; pp 66–67.
- Chatterjee S.; Chen A. Facile electrochemical approach for the effective detection of guanine. Electrochem. Commun. 2012, 20, 29–32. 10.1016/j.elecom.2012.03.044. [DOI] [Google Scholar]
- Pivoňková H.; Horáková P.; Fojtová M.; Fojta M. Direct Voltammetric Analysis of DNA Modified with Enzymatically Incorporated 7-Deazapurines. Anal. Chem. 2010, 82, 6807–6813. 10.1021/ac100757v. [DOI] [PubMed] [Google Scholar]
- Lee H. H.; Kalhor R.; Goela N.; Bolot J.; Church G. M. Terminator-free template-independent enzymatic DNA synthesis for digital information storage. Nat. Commun. 2019, 10, 2383. 10.1038/s41467-019-10258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Wiegand D. J.; Griswold K.; Punthambaker S.; Chun H.; Kohman R. E.; Church G. M. Photon-directed multiplexed enzymatic DNA synthesis for molecular digital data storage. Nat. Commun. 2020, 11, 5246. 10.1038/s41467-020-18681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.