

Abstract
Therapies with genetically modified T cells that express chimeric antigen receptors (CARs) specific for CD19 or B cell maturation antigen (BCMA) are approved to treat certain B cell malignancies. However, translating these successes into treatments for patients with solid tumours presents various challenges, including the risk of clinically serious on-target, off-tumour toxicity (OTOT) owing to CAR T cell-mediated cytotoxicity against non-malignant tissues expressing the target antigen. Indeed, severe OTOT has been observed in various CAR T cell clinical trials involving patients with solid tumours, highlighting the importance of establishing strategies to predict, mitigate and control the onset of this effect. In this Review, we summarize current clinical evidence of OTOT with CAR T cells in the treatment of solid tumours and discuss the utility of preclinical mouse models in predicting clinical OTOT. We then describe novel strategies being developed to improve the specificity of CAR T cells in solid tumours, particularly the role of affinity tuning of target binders, logic circuits and synthetic biology. Furthermore, we highlight control strategies that can be used to mitigate clinical OTOT following cell infusion such as regulating or eliminating CAR T cell activity, exogenous control of CAR expression, and local administration of CAR T cells.
Introduction
Chimeric antigen receptor (CAR) T cells have emerged as an effective cancer treatment modality. This approach relies on genetically engineering patient-derived or donor-derived T cells to express a synthetic CAR that can recognize a tumour cell-surface molecule in a major histocompatibility complex (MHC)-independent manner1. CAR designs are continuously evolving to improve the sensitivity, specificity, efficacy and persistence of engineered T cells2. Antigen recognition by CAR T cells is mediated via a specific monoclonal antibody-derived single-chain variable fragment (scFv) fused to transmembrane and intracellular signalling domains. CD3ζ is the required domain for T cell activation, and co-stimulatory endodomains, such as CD28 and/or 4–1BB, are pertinent for CAR T cell proliferation, differentiation and persistence2. To date, all six FDA-approved CAR T cell products target either CD19 or B cell maturation antigen (BCMA) and utilize a second-generation CAR design3,4,5,6,7,8,9 (Fig.1).
Figure 1. CAR architecture.
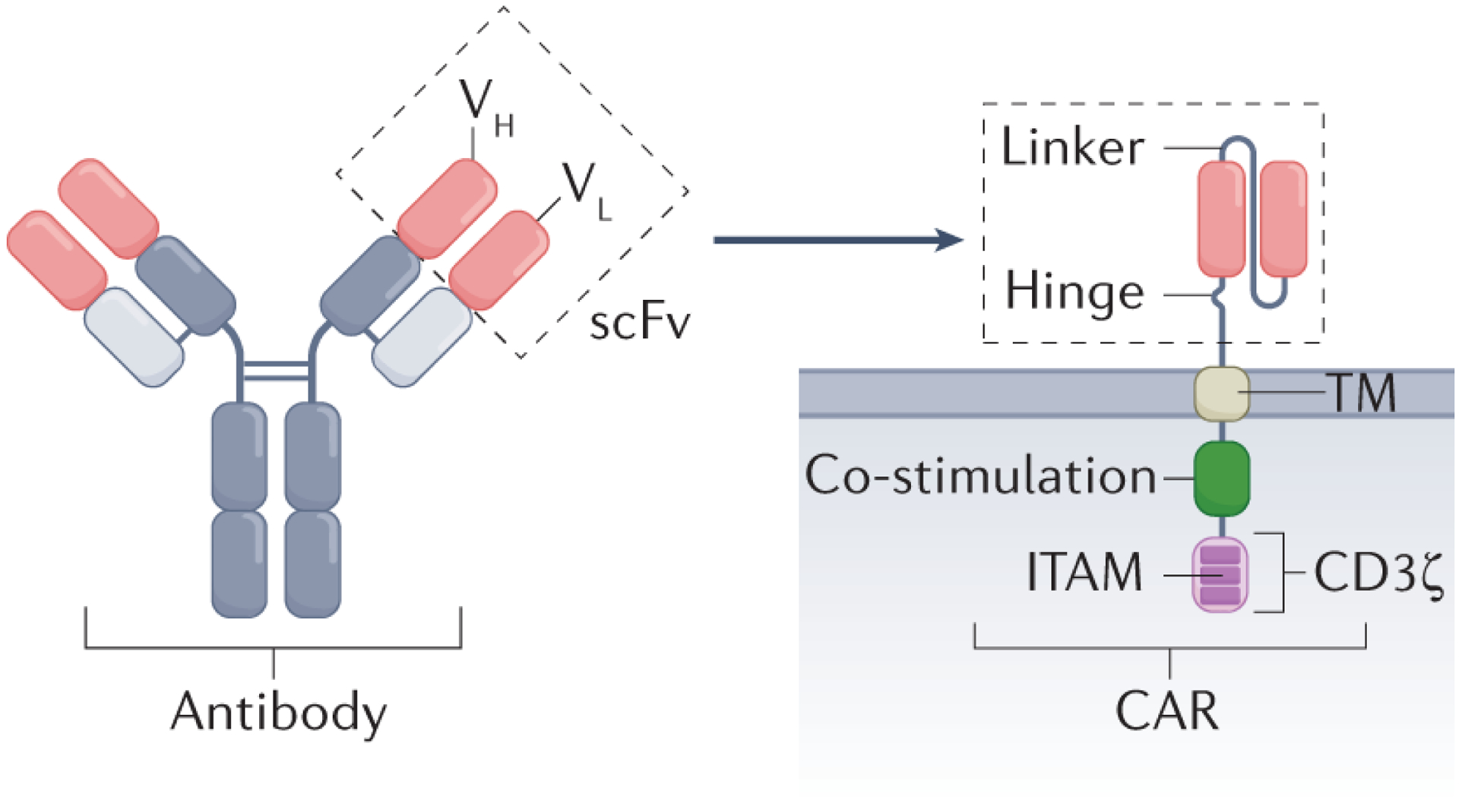
Standard second-generation chimeric antigen receptor (CAR) with a single-chain variable fragment (scFv) derived from a monoclonal antibody linked via a transmembrane domain (TM) to a co-stimulatory signalling domain (for example, from CD28 or 4–1BB) and an intracellular CD3ζ signalling domain. CD28 allows rapid expansion but less durability165, and 4–1BB promotes sustained effector function and persistence166. ITAM, immunoreceptor tyrosine-based activation motif; VH, heavy chain variable region; VL, light chain variable region.
B cell malignancies and multiple myelomas are particularly responsive to CAR T cells, mainly owing to the abundant expression of lineage-derived antigens such as CD19 and BCMA. Anti-CD19 and anti-BCMA CAR T cells can deplete endogenous non-malignant B cells and plasma cells that also express the target molecules, leading to hypogammaglobulinaemia. This toxicity is generally tolerable and can be corrected with intravenous immunoglobulin infusions10,11. Other toxicities, such as cytokine-release syndrome and immune effector cell-associated neurotoxicity syndrome are primarily linked to acute cytokine production following CAR T cell infusion and are manageable in a majority of patients12.
The success of CAR T cells in the treatment of haematological malignancies2,3,4,5,6,7,8has provided the impetus for investigating this approach in solid tumours. A considerable barrier to the development of CAR T cells for patients with solid tumours is that most candidate target antigens are often co-expressed on non-malignant tissues, creating a substantial risk of morbidities owing to on-target, off-tumour toxicity (OTOT). OTOT of varying severity has been reported in both preclinical13,14,15,16 and clinical17,18,19,20,21,22,23,24 studies using CAR T cells targeting antigens shared by malignant and non-malignant tissues19,23,25. In this Review, we examine the implications of OTOT on the development of CAR T cell therapies targeting solid tumours, summarize OTOT evidence in preclinical and clinical studies, and discuss advances in CAR T cell engineering that might help to overcome OTOT in the clinic.
Risks and mechanisms of OTOT
OTOT stems from CAR T cell-mediated recognition and lysis of non-malignant tissues expressing the target antigen, potentially causing severe adverse events23,26. Upon recognition of a target antigen, CAR T cell activation leads to the formation of an immune synapse between the CAR and the target cell27, triggering effector functions (Fig. 2). The release of perforin and granzymes28 is assumed to be a principal mechanism of CAR T cell-mediated cytotoxicity. However, other mechanisms, such as upregulation of T cell-surface molecules to induce target apoptosis (such as FAS ligand)29 or secretion of cytokines, including IFNγ and/or TNF, may also contribute to tissue destruction29,30,31 (Fig. 2).
Figure 2. CAR T cell cytolytic mechanisms and paracrine effects.
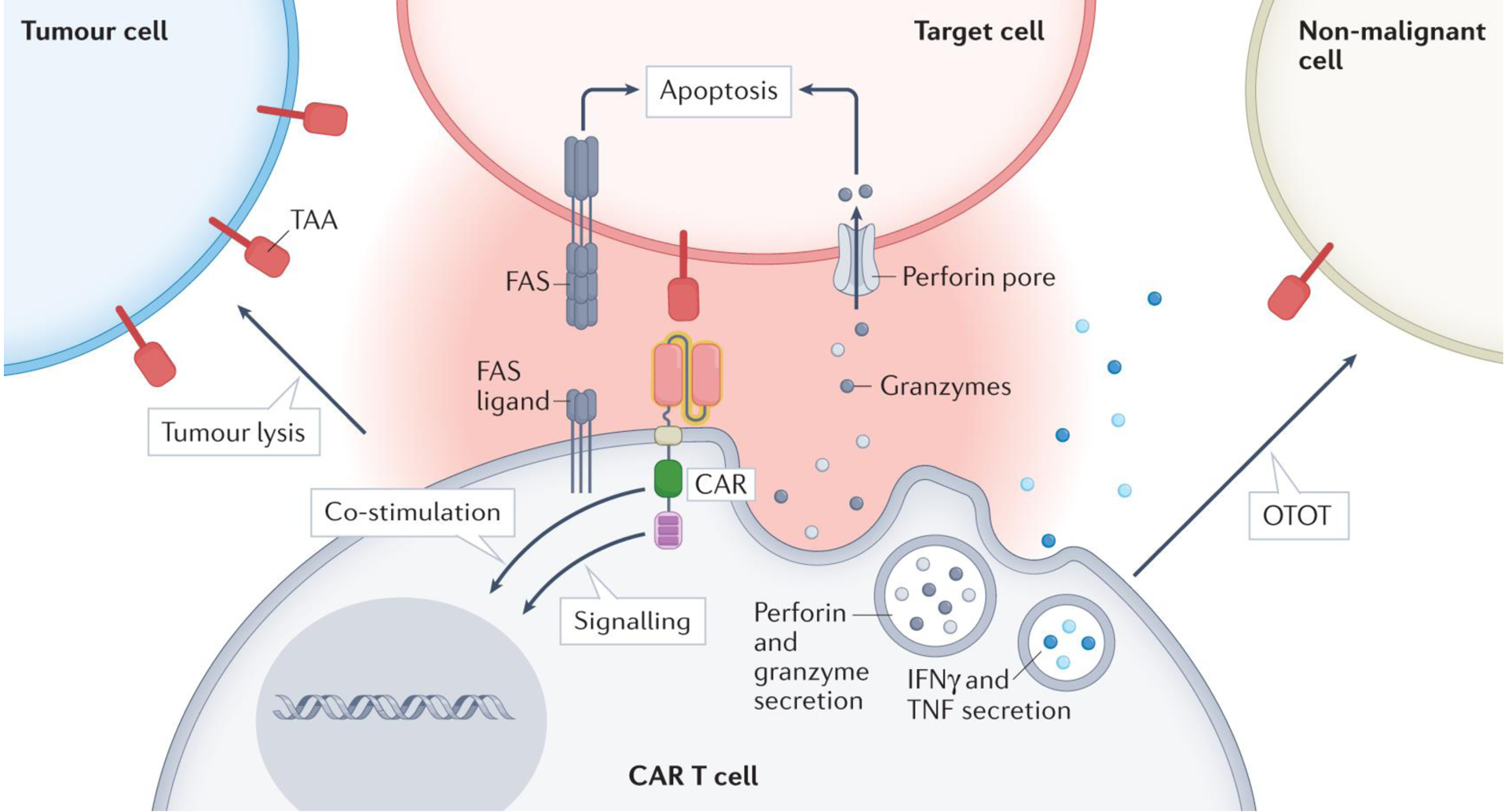
Upon recognition of a tumour-associated antigen (TAA) by a chimeric antigen receptor (CAR) T cell167, an immune synapse is formed, followed by CAR T cell activation. Subsequently, cytotoxic granules containing perforin and granzymes are released, with the latter entering the target cell via perforin channels, triggering intrinsic apoptosis by damaging mitochondria and activating caspases28,30. Additionally, CAR T cells upregulate FAS ligand, which engages the FAS receptor on target cells, triggering the extrinsic pathway of apoptosis and caspase-mediated targeted cell death29. Further, CAR T cells release IFNγ and TNF that activate immune cells such as macrophages168. CAR T cells might recognize TAAs on non-malignant cells, leading to undesired lysis of healthy tissues. Next-generation CAR T cells can also be equipped with additional effector functions (such as IL-12 secretion) that extend the immune response to include endogenous T cells169,170. OTOT, on-target, off-tumour toxicity.
To generate CAR T cells that are both safe and effective in patients with solid tumours, target antigen selection is crucial. Optimal antigen candidates, referred to as neoantigens, should be exclusively expressed on malignant cells and not on non-malignant cells. Such antigens could arise from tumour-specific non-synonymous mutations, insertions or deletions that alter the amino acid sequence of cell-surface proteins, aberrant expression of oncofetal antigens, or tumour-specific post-translational modifications32,33,34,35,36,37. However, cell-surface neoantigens are rare, particularly in tumours with a low mutational burden38. EGFRvIII, found in 24–67% of glioblastomas, is one of the few identified examples39,40. Consequently, the majority of CAR T cell therapy targets for solid tumours are tumour-associated antigens (TAAs) that are also expressed on non-malignant tissues (Fig. 3). Examples of TAAs include EGFR19,20,21,25, HER2 (refs.41,42,43,44), CAIX17,45, B7-H3 (refs.13,46,47), mesothelin48,49,50,51 and GD2 (refs.52,53,54,55,56).
Figure 3. Publicly available protein expression densities of selected solid tumour TAAs on non-malignant tissues.
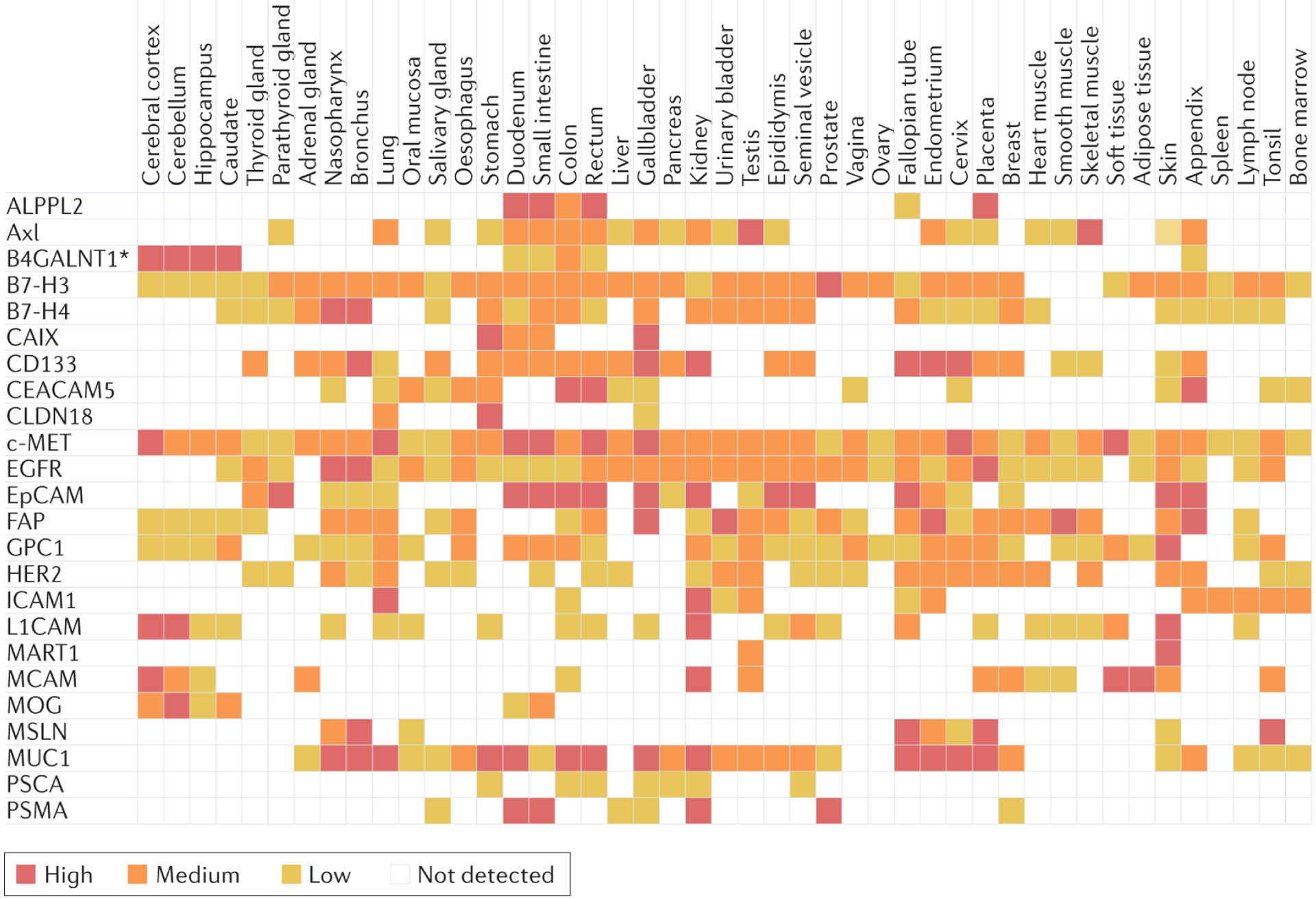
Expression scores were established using immunohistochemistry and are based on staining intensity, the fraction of stained cells (<25%, 25–75% and >75%) and subcellular localization58,171. TAA, tumour-associated antigen. *B4GALNT1 is analysed as a proxy for GD2 expression (B4GALNT1 is involved in the biosynthesis of GD2)172. Data are correct as of October 2022.
Evidence for CAR T cell-associated OTOT
Clinical manifestations
OTOT was observed in a clinical trial in which patients with metastatic renal cell carcinoma received anti-CAIX first-generation CAR T cells (Supplementary Table 2). Grade 2–4 liver toxicities were observed in all three patients and liver biopsy revealed discrete cholangitis with CAIX expression on the bile duct epithelium17,18,57. Similarly, a phase I trial testing anti-CEACAM5 CAR T cells in patients with advanced-stage CEACAM5-positive solid tumours led to clinically serious adverse events (tachypnoea, pulmonary infiltrates or respiratory distress), with one patient requiring intensive care23. The overall unfavourable efficacy and safety profile resulted in closure of the trial. The possibility of gastrointestinal toxicity was anticipated given that CEACAM5 is expressed in the intestines but the pulmonary toxicity was unexpected. The investigators identified intermediate-to-strong CEACAM5 expression in non-malignant lung resection samples from five of eight patients (63%)23. This finding is supported by proteomic data, published elsewhere58, showing low levels of CEACAM5 expression on type I and type II alveolar cells (Fig. 3), suggesting that OTOT might have driven the observed adverse clinical manifestations.
CAR T cells targeting HER2 have been tested for several indications in patients with advanced-stage solid tumours. In a case report, a single infusion of high-dose (1010) anti-HER2 CAR T cells led to acute respiratory distress followed by death 5 days after infusion, in a 39-year-old individual with metastatic colon cancer44 (Supplementary Table 2). Similarly, mild skin pruritus and one case of severe upper gastrointestinal haemorrhage were observed in a phase I study of anti-HER2 CAR T cells in patients with advanced-stage biliary tract or pancreatic cancer22. Altogether, clinical data have suggested a moderate toxicity profile for lower doses of anti-HER2 CAR T cells, with evidence of clinical responses41,43,59,60. Differences in CAR T cell dose, scFv properties, CAR design, tumour location and other variables in the different clinical trials preclude definitive conclusions regarding OTOT in patients receiving anti-HER2 CAR T cells, suggesting the need for additional data to validate HER2 as a CAR T cell target22,41,43,59,60.
Despite widespread target expression of EGFR58 (Fig. 3), OTOT described with EGFR-directed CAR T cells has been manageable19. Mild or moderate dermal OTOT was reported following infusion of an anti-EGFR CAR T cell product, which included lichen striatus-like skin rashes, loss of partial epidermis and vacuolar degeneration of basal cells19. In a phase I study testing anti-EGFR CAR T cells in patients with advanced-stage biliary tract cancers, potential OTOT was observed in the form of oral mucositis, oral ulcers, gastrointestinal haemorrhage, desquamation and pruritus (all of grade 1–2)20. More severe mucosal and cutaneous adverse events (some of grade 3–4) were observed in 6–13% of patients with metastatic pancreatic cancer receiving anti-EGFR CAR T cells in a phase I trial21. The investigators were able to control these adverse events in one patient using methylprednisolone21.
In another phase I trial, one patient with advanced-stage gastric cancer receiving anti-CLDN18.2 CAR T cells developed grade 3 mucosal toxicity and a further five developed less severe (grade 1 and 2) forms of this toxicity. The authors characterized this as OTOT owing to high levels of CLDN18.2 expression on differentiated gastric mucosal cells24. Nevertheless, the safety profile in this cohort of 37 patients was deemed acceptable, with no dose-limiting toxicities reported24.
Collectively, these phase I clinical data demonstrate the likelihood of severe OTOT in patients with solid tumours receiving CAR T cells. This highlights the importance of understanding the expression of TAAs on non-malignant tissues in order to accurately attribute CAR T cell-related adverse events to OTOT. These data indicate that OTOT is potentially influenced by a myriad of factors, including the number of transfused cells, antigen expression densities on both the tumour and non-malignant tissues, CAR design, and the mode of administration17,23,44,61.
Two reports suggest that CAR T cells might also cause unexpected OTOT in patients with haematological malignancies. The findings of a single-cell RNA sequencing analysis suggested that certain neurotoxicities, including rare cases of fatal cerebral oedema observed in patients with B cell malignancies receiving anti-CD19 CAR T cells, might be related to OTOT against CD19-positive pericytes in the brain62. Furthermore, OTOT against BCMA-expressing neurons and astrocytes in the basal ganglia has been suggested as a cause of progressive movement disorder and parkinsonism described 3 months after infusion of anti-BCMA CAR T cells in patients with multiple myeloma63.
Preclinical predictability of OTOT
The risk of OTOT with CAR T cells has been investigated in preclinical models. Histological analysis of mouse organs can be performed to examine CAR T cell infiltration, the extent of both tumour and normal tissue necrosis, and levels of TAA expression at inflammatory sites both surrounding and distant from the tumour14,26,46,64,65. Immunohistochemistry can be used to verify possible off-tumour TAA expression and CAR T cell infiltration, thus providing clear evidence of OTOT14,26,46,64. For example, the detection of late-onset OTOT following infusion of anti-B7-H4 CAR T cells (45–48 days after infusion) was feasible in a mouse model15. Immunohistochemical staining of non-malignant mouse tissues revealed widespread B7-H4 expression that was comparable to human B7-H4 protein distribution in non-malignant tissues15. Moreover, luciferase-tagging of CAR T cells in mice can also be used to monitor CAR T cell trafficking and activation, thus improving the understanding of CAR T cell migration and off-tumour interactions16,66.
Despite the use of mouse models to study OTOT, differences in the expression of target antigens between non-malignant mouse and human tissues warrant caution in predicting clinical OTOT (Supplementary Tables 1 and 2). For example, lethal central nervous system (CNS) toxicities reported using a high-affinity variant of the 14G2a scFv-based anti-GD2 CAR in five of eight neuroblastoma xenograft mice were interpreted as OTOT against GD2-expressing regions of the mouse brain26. The role of OTOT in these toxicities was debated by others who did not observe such effects even with a high-affinity CAR67. These preclinical findings are also in conflict with the clinical toxicity profiles of anti-GD2 CAR T cells featuring the original 14G2a scFv CAR construct52,54,68. Moreover, the gastrointestinal OTOT seen in patients receiving anti-CLDN18.2 CAR T cells was not observed in mouse models most probably owing to several factors, including limited expression of CLDN18.2 in non-malignant mouse tissues and rapid tissue regeneration by CLDN18.2-negative gastric stem cells24. Thus, OTOT in mice26 might not translate into OTOT in clinical settings53,54,55,68, mainly owing to differences in the antigen structure of human and mouse CLDN18.2 homologues, target expression patterns, or distinct tissue microenvironments and/or anatomical niches69.
The predictiveness of mouse models can be enhanced using different approaches. Examples include genetic engineering to replace mouse genes with their human homologues14 or engrafting a cell line into mice to emulate off-tumour TAA expression in non-malignant tissues70,71,72. Nonetheless, these approaches are rarely used and still might not accurately reflect the complex expression patterns of TAAs on non-malignant human tissues. Modern organ-on-a-chip technologies designed to simulate in vivo microarchitectures and physiological processes within tissues in a fully humanized model are currently being considered. These models cannot entirely mimic the complexity of human tissue antigen expression73,74, although they might help to address the deficiencies of more traditional models.
Engineering strategies to mitigate OTOT
Toxicities observed in clinical CAR T cell trials have encouraged the development of technologies designed to improve safety while maintaining antitumour efficacy. Drawing on synthetic biology, new approaches are emerging to better restrict the potent cytotoxic activity of CAR T cells to tumours, thereby avoiding OTOT.
Fine-tuning CAR domains
Affinity tuning via modification of the scFv of the CAR has emerged as an interesting approach to achieve an affinity sufficient for tumour cell recognition, whilst sparing non-malignant cells with limited TAA expression65,75. This can be achieved by mutagenesis of an existing scFv or by screening scFv libraries to identify an alternative binder with a different affinity. High-affinity CAR T cells might permit better reactivity against tumour cells with a low density of antigen expression but could also lead to recognition of target antigens present on off-tumour tissues26 (Fig. 3). ScFvs with a very high affinity for target antigens might limit the potential for CAR T cell proliferation as a result of activation-induced cell death76. Conversely, low-affinity CAR T cells might lack antitumour activity owing to their inability to sufficiently recognize and/or lyse tumour cells with lower levels of TAA expression71. Furthermore, a lower-affinity scFv increases the risk of low antigen-density tumour cells escaping CAR T cell recognition77,78, which has been observed in patients with haematological malignancies79.
The impact of scFv affinity on OTOT was examined in a mouse model in which human HER2 was ectopically expressed. The investigators found that high-affinity anti-HER2 CAR T cells cause more liver damage than low-affinity CAR T cells targeting the same antigen. This was attributed to the faster clearance of low-affinity CAR T cells from the liver14. Elsewhere, investigators testing anti-EGFR CAR T cells in mouse glioblastoma xenograft models showed that OTOT is strongly dependent on CAR affinity. Mice infused with high-affinity anti-EGFR CAR T cells had increased toxicities and reached a 53-day survival rate of 57% compared to 100% for those infused with low-affinity CAR T cells71. Indeed, anti-HER2 and anti-EGFR CAR constructs with variable scFv affinities conferred robust antitumour responses without OTOT both in vitro and in mouse xenograft models75.
Beyond affinity tuning of the scFv, modifications to the hinge and transmembrane domain (H/T)80 as well as the number of immunoreceptor tyrosine-based activation motifs (ITAMs)81 can alter the antigen-density threshold of a CAR (Fig. 1). For example, 4–1BB-CD3ζ CAR T cells with a CD8-H/T have a higher antigen-density threshold compared to those with a CD28-H/T and form a less stable immunological synapse81. Moreover, reducing the number of ITAMs in the CD3ζ domain reduces the cytotoxicity of CAR T cells against cells with a limited antigen density, while maintaining cytotoxicity against high antigen-density targets81. Loss-of-function mutations in one or more of the three ITAMs in CD3ζ can also be used to calibrate activation and differentiation programmes82, and the position of the mutated ITAMs can determine CAR T cell functionality, differentiation and antitumour activity82.
Logic-gated CAR T cells
Boolean logic-gating approaches, which refer to the mathematical operators ‘IF/THEN’, ‘AND’, ‘OR’ and ‘NOT’, can be applied to control the activation of CAR T cells. Some of these approaches can be used to increase the specificity of cell killing and reduce the potential for OTOT (Fig. 4).
Figure 4. Principles of Boolean logic-gating to circumvent OTOT.
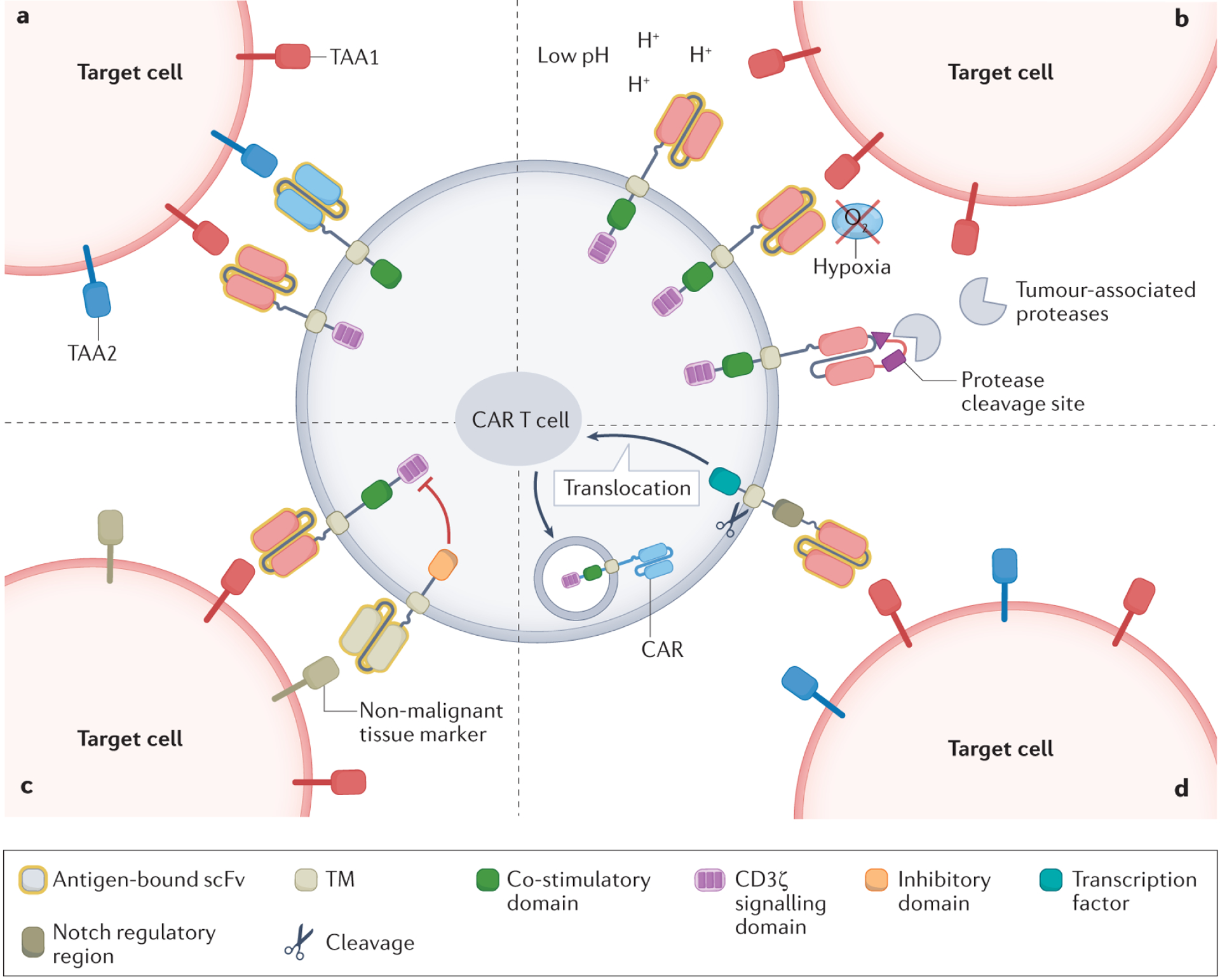
a, AND-logic chimeric antigen receptor (CAR) T cells are activated by binding to two different tumour-associated antigens (TAAs) on target cells. b, IF/THEN tumour microenvironment-based logic-gated CAR T cells can only be activated when certain tumour microenvironment-specific characteristics are present. Examples include pH-restricted binding of TAAs, hypoxia-dependent expression of CARs or protease-dependent liberation of the antigen-binding site. c, NOT-logic CAR T cells co-express a stimulatory CAR and an inhibitory CAR targeting distinct antigens. The inhibitory CAR suppresses T cell activation when the target cell expresses selected ligands associated with non-malignant tissues. d, In IF/THEN TAA-based logic circuits, CAR T cells become activated only when encountering two different TAAs on target cells. The recognition of one TAA by a constitutively expressed synthetic Notch receptor subsequently induces the expression of a CAR targeting the other TAA. scFv, single-chain variable fragment; TM, transmembrane domain.
Uncoupling CAR T cell activation signals using AND-logic circuits
The different CAR T cell activation signals (Fig. 5a) can be uncoupled via so-called AND-logic circuits to avoid OTOT in certain scenarios. Dual AND-logic CAR T cells express two distinct synthetic receptors (one containing CD3ζ and the other CD28 or 4–1BB signalling domains), each targeting a different TAA in a split approach83,84 (Figs. 4a and 5b). This dual control theoretically reduces the risk of OTOT because both TAAs are required to be expressed on non-malignant tissues to enable complete CAR T cell activation, while ligation of only one receptor would result in attenuated signalling. AND-logic-gated CAR T cells targeting CEA and mesothelin have shown specific cytotoxicity against dual-TAA-positive cells without recognition of single-TAA-positive cells in mice85. A separate study demonstrated more potent killing of dual-TAA-positive cells using this approach84. They also found that engagement of the CAR containing only CD3ζ could lead to leakiness84. In this scenario, the logic circuit was activated by only one signal, inducing single-TAA-positive cell lysis and an increased risk of OTOT.
Figure 5. Examples of logic-gating strategies tested in CAR T cells.
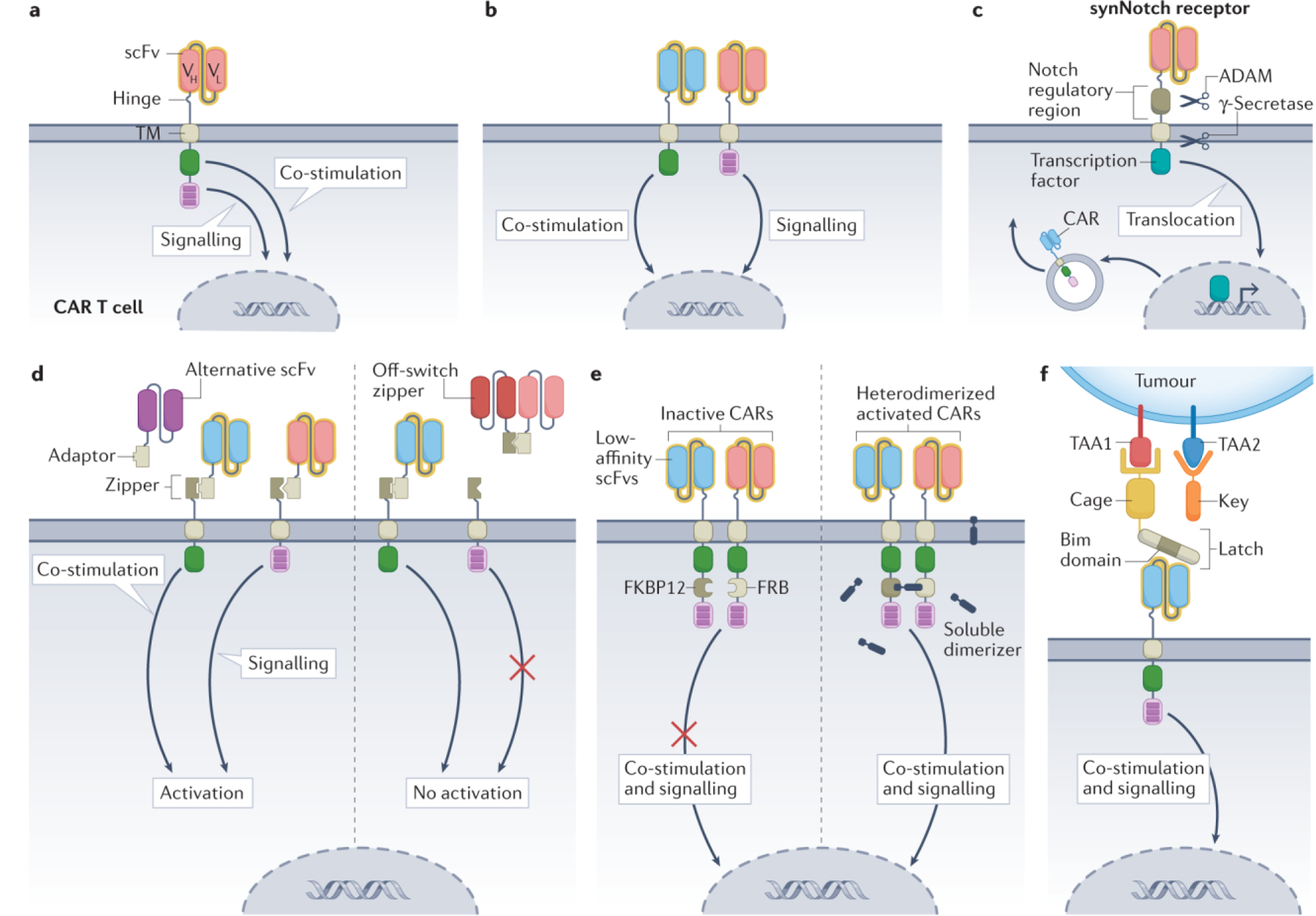
a, Conventional second-generation chimeric antigen receptor (CAR) T cells combine the two signals required for optimal CAR T cell function within a single construct. b, Dual AND-logic CAR T cells accomplish signalling and co-stimulation via two independent receptors that bind distinct tumour-associated antigens (TAAs)84. c, Synthetic Notch (synNotch) CAR T cells constitutively express a synNotch receptor. Antigen binding by this receptor leads to exposure of protein cleavage sites in the Notch regulatory region and transmembrane domains, allowing sequential cleavage via the enzymes ADAM and γ-secretase. This releases a tethered, intracellular transcription factor, which promotes CAR expression. The expressed CAR can recognize its own cognate antigen and trigger a cytotoxic response97. d, Split, universal and programmable (SUPRA) CAR T cells express a leucine zipper as an extracellular binding domain. The leucine zipper can bind a leucine adaptor molecule carrying a single-chain variable fragment (scFv). This soluble adaptor–scFv endows the CAR T cells with antigen recognition capabilities toward a specific target. As the adaptor–scFv conjugate is administered independently to the CAR T cells, infusing a different scFv allows in vivo switching of the CAR target. A soluble zipper–scFv can competitively bind the adaptor–scFv, preventing CAR T cell signalling. This system can also be designed using the AND-logic split architecture as in b108. e, AND-logic avidity-controlled CAR T cells contain low-affinity scFvs that rely on avidity for TAA encounter. CAR T cell activation is dependent on antigen recognition and dimerization of two low-affinity CARs in the presence of a soluble dimerizer109. f, The Co-LOCKR system uses cage and key intermediary proteins to recognize and bind to target cells. The cage and key molecules carry a TAA binding domain, whereas only the cage carries a latch that contains a hidden Bim domain (binding site). Once the cage and key bind their cognate antigens, the latch is released, exposing the Bim domain and allowing recognition by CAR T cells119. Co-stimulation is either via CD28 or 4–1BB. TF, transcription factor; TM, transmembrane domain; VH, heavy chain variable region; VL, light chain variable region.
Rather than a second cell-surface antigen, other tumour-associated characteristics can be targeted via AND-logic. For example, the combined presence of the tumour antigen PSCA and the immunosuppressive cytokines TGFβ and IL-4, which are exclusive to the pancreatic carcinoma tumour microenvironment (TME), has been leveraged into an AND-logic system86. Researchers generated anti-PSCA CAR T cells co-expressing two transgenic cytokine receptors capable of inverting the immunosuppressive effects of TGFβ and IL-4 into immunostimulatory signals. This approach enhanced the potency and safety of CAR T cells and showed selective antitumour effects in mouse models86. Such a strategy reduces the probability of OTOT, but leakiness by the CD3ζ-containing CAR remains a possibility. Overall, the dependence of AND-logic circuits on more than one TAA increases the risk of antigen escape86.
Sensing of the TME facilitates IF/THEN-logic gating
IF/THEN-logic gating offers another strategy to restrict CAR T cell killing to the TME with a reduced risk of leakiness84,87 (Fig. 4b). Engineering pH-sensitive CAR T cells that optimally recognize TAAs in an acidic environment provides one example of IF/THEN-logic-based targeting. This approach capitalizes on the Warburg effect, which is prevalent in many tumours and refers to accelerated glycolysis and increased lactate production that results in an acidic intratumoural environment88. A HER2-targeting CAR with a pH-restricted binding domain preferentially detected HER2 in an acidic TME, leading to robust CAR T cell expansion and regression of high antigen-density HER2-positive tumour cells in mouse models87.
Hypoxia sensing can also restrict CAR expression and activity to the TME (Fig. 4b). In a proof-of-concept study, the hypoxia-sensing subdomain of HIF1α was fused to the intracellular C-terminal end of a multichain anti-CD19 CAR89. Under normoxia, the HIF1α subdomain and the CAR are degraded. However, under hypoxic conditions in the TME, HIF1α degradation does not occur, enabling local CAR expression89. A similar approach has been applied to a pan-anti-ErbB CAR that prevented lethal toxicity in mice (Supplementary Table 1). The T cells selectively expressed CARs only under stringent hypoxia (0.1% oxygen), resulting in tumour control in an ovarian adenocarcinoma mouse xenograft model, without systemic toxicity66.
Another approach used a protease-sensitive linker to attach a masking peptide to the antigen-binding site of an anti-EGFR CAR (Fig. 4b). The antigen-binding site of the CAR is only liberated by protease cleavage of the masking peptide. This strategy restricts CAR function to the TME, which has high local concentrations of membrane-type serine protease 1 (MT-SP1), urokinase-type plasminogen activator (uPA) and the lysosomal enzyme legumain90,91,92. This protease-dependent CAR provides a level of antitumour activity equivalent to that observed with unmasked CARs, but in a more selective fashion90.
An important caveat of these approaches is that certain non-malignant tissues might share the targeted features of the TME, resulting in the activation of TME-sensing CAR T cells in anatomical locations distant from the tumour. For example, the renal medulla and the gut mucosa are hypoxic under physiological conditions93,94,95. Therefore, target antigen selection for T cell therapies featuring hypoxia-induced or pH-induced CAR expression and/or function should focus on TAAs that are not expressed in non-malignant hypoxic and/or acidic tissues.
SynNotch circuits facilitate multiple antigen sensing using IF/THEN-logic gating
IF/THEN-logic-gated CAR T cells using synthetic Notch (synNotch) circuits rely on multi-antigen sensing, overcoming the trade-off between affinity and selectivity (Fig. 4d). By restricting CAR T cell effector function to the presence of multiple tumour targets, this approach enables the use of more aggressive, higher-affinity CARs that otherwise have the potential to cause OTOT. SynNotch CAR T cells are activated in two steps with CAR transgene expression induced only after synNotch receptor recognition of the so-called priming antigen (Fig. 5c). In the initial reports describing synNotch CAR T cells, expression of the CAR and T cell activation were dependent on the recognition of two different TAAs96,97,98. SynNotch CAR T cells were able to clear dual-TAA-positive tumours whilst cells expressing only one of the two TAAs survived97. Other groups have also investigated synNotch circuits and demonstrated potent in vitro antitumour activity with no signs of OTOT13,46.
The synNotch CAR T cell circuit has shown enhanced specificity in a preclinical model of ALPPL2-expressing solid tumours99. It is worth noting that superior tumour control was achieved in this model by preventing CAR-mediated tonic signalling99. In a synNotch CAR T cell that is endowed with a tandem CAR, the same group showed precise tumour targeting in a xenograft model of EGFRvIII-expressing glioblastoma (Supplementary Table 1) without evidence of OTOT100.
CAR T cell designs that combine synNotch circuits with affinity tuning, including a low-affinity anti-HER2 synNotch receptor and a high-affinity anti-HER2 CAR, have also been described72. This system allows synNotch CAR T cells to identify targets based on a sigmoidal antigen-density threshold, where CAR T cells are inactive at low levels of HER2 expression but confer a substantial increase in cytotoxicity once the HER2 density surpasses a specific threshold72. These cooperative effects enable robust antitumour activity that is subject to a high antigen density, thereby reducing the risk of OTOT.
The versatility of synNotch circuits, including the option to use multiple synNotch receptors101, offers the possibility of designing more specific and less toxic CAR T cells. Nevertheless, the likelihood of host immune responses to synNotch proteins102 and the complexity and size of the transgenes pose challenges for clinical applications of this technology103. To overcome the latter, researchers re-engineered the underlying synNotch construct and established the ‘SyNthetic Intramembrane Proteolysis Receptors’ (SNIPRs) system, which is more compact and compatible with human transcription factors103. This approach was successfully tested in a proof-of-principle preclinical study involving an ALPPL2-HER2-expressing ovarian cancer xenograft model103. Contrary to AND-logic, IF/THEN gates are not associated with the risk of leakiness owing to the standalone cytotoxic effects of the CD3ζ signal84,97. However, migration of primed synNotch CAR T cells out of the tumour and into co-localized non-malignant tissues expressing the CAR antigen remains a potential risk13,97.
Versatile CAR designs with AND-logic gating allow exogenous control
Synthetic biology enables the engineering of more complex logic systems that include tuneable control of CAR T cell activity after infusion104,105,106,107. For example, a split, universal and programmable (termed SUPRA) CAR circuit can be modified to combine different logic-gating strategies108. The fully functional SUPRA CAR contains two elements: (1) the transmembrane and endodomain of a CAR connected to an extracellular leucine zipper, and (2) a soluble scFv attached to a leucine adaptor (adaptor–scFv). SUPRA CAR T cells are activated only when the adaptor–scFv binds the TAA and the adaptor attaches to the CAR via the zipper. To avoid OTOT, a SUPRA circuit can be further programmed to include an AND-logic gate (Fig. 5d). In this case, two SUPRA CARs are designed so that CD3ζ signalling and co-stimulation are delivered by two independent receptors to achieve antitumour activity108. Further, administering an adaptor–scFv that binds a different TAA allows in vivo switching of the CAR target. Moreover, to prevent CAR T cell signalling, a soluble zipper–scFv can be administered that competitively binds the adaptor–scFv and prevents its antigen recognition and CAR T cell activation.
Another design uses the concept of CAR T cell avidity, in which an immune response relies on multiple low-affinity CAR–antigen interactions109. In this so-called avidity-controlled CAR (AvidCAR) system, activation is achieved only when two low-affinity CARs independently recognize the target antigen and become dimerized in the presence of a specific small-molecule dimerizer. This design can be modified to incorporate CARs with scFvs targeting different TAAs, thus turning this system into an avidity-dependent AND-logic circuit (Fig. 5e). Various extensions to this system have been reported, including an AND-logic gate exploiting VEGF as the dimerizer. Although also found in non-malignant tissues, VEGF is a key mediator of tumour angiogenesis109,110 and can therefore enable CAR T cell activation within the TME.
The ability to engineer AND-logic gated CAR T cells using SUPRA and AvidCAR offers a method of reducing the risk of OTOT whilst also providing an element of exogenous control. The switchable nature of SUPRA CAR T cells can also reduce the risk of antigen escape by administration of an adaptor–scFv targeting alternative TAAs without having to re-engineer the CAR T cells ex vivo. This might be particularly useful for constructs using AND-logic circuits given that dependence on multiple TAAs increases the risk of antigen escape86. Moreover, establishing a system that permits exogenous control of CAR T cell activity is an important safety measure that enables a response to any toxicities as they arise.
NOT-logic gating can prevent CAR T cell activation
AND-logic and IF/THEN-logic circuits regulate the activity of stimulatory CARs that, upon antigen recognition, trigger a cytotoxic response. By contrast, NOT-logic gating contains a stimulatory CAR that induces a cytotoxic response and an inhibitory CAR (iCAR) that triggers a potent inhibitory signal that interprets a marker expressed on non-malignant cells as a signal to prevent killing70 (Fig. 4c). The first example of a NOT-logic circuit was a PSMA-targeting iCAR tested in a mouse xenograft model in which PSMA was used as a proxy antigen for non-malignant tissue and CD19 as the target antigen70. The iCAR T cells successfully inhibited the elimination of PSMA-positive NALM6 cells that co-expressed CD19, but did not affect the response against PSMA-negative counterparts70. A similar proof-of-concept approach has been investigated elsewhere, in which OTOT was successfully averted in vitro using an anti-CD93 CAR/anti-CD19 iCAR construct111.
iCARs have also been used to target clonal loss of heterozygosity (LOH) to reduce OTOT112. LOH occurs when irreversible genetic alterations affect only one chromosome, resulting in non-malignant cells being predisposed to the development of cancer113. LOH in HLA molecules observed in various cancers acts as a mechanism of tumour immune evasion113,114,115 and has been leveraged by several research groups as a tumour-specific marker112,116,117,118 (Supplementary Table 1). Targeting HLA molecules with an inhibitory receptor would hamper CAR T cell signalling when encountering non-mutated cells whilst allowing cytotoxicity against LOH-affected cells117.
Another NOT-logic approach based on downstream inhibition of T cell function provides an alternative to iCARs. In this system, a synNotch receptor is designed to recognize an antigen found predominantly on non-malignant tissues, which then activates expression of the pro-apoptotic factor truncated BH3-interacting domain death agonist (tBID), inducing CAR T cell apoptosis101. As noted by the authors, finding the balance between off-tumour apoptosis and on-tumour expansion will be vital for the clinical success of such an approach101.
Adaptable logic gating with intermediary proteins permits exogenous control
A multi-antigen sensing system that can perform AND-logic and NOT-logic gating, termed Co-LOCKR, has also been developed (Fig. 5f). In this approach, CAR T cells do not directly bind to the target antigen but rather to two soluble intermediary proteins (named ‘cage’ and ‘key’) that facilitate CAR T cell and target-cell binding119 (Fig. 5f). The TAA-bound cage contains a latch with a Bim domain that is only revealed when the co-localized key protein binds its respective antigen. Subsequently, an anti-Bim CAR T cell can recognize the Bim domain of the cage and trigger CAR T cell cytotoxicity119. Furthermore, administering a decoy protein that can bind a specific antigen expressed on non-malignant tissues prevents the CAR T cell from binding the Bim domain and averts CAR T cell activation. This NOT-logic circuit is expected to increase specificity and reduce the risk of OTOT119.
Other strategies to limit OTOT in vivo
Additional modifications to CAR T cells are being developed to avert or reduce the severity of OTOT following infusion. Some strategies have been tested clinically, although most have been limited to preclinical models.
Controlling CAR T cells after infusion
Control with clinically approved drugs
Systemic immunosuppression using corticosteroids is currently the go-to strategy in managing most immunotherapy-related toxicities. However, administering corticosteroids might negatively affect the antitumour effects and the persistence of CAR T cells120,121. Reversible (or temporary) control of CAR T cells might be achieved with the tyrosine kinase inhibitor dasatinib, which acts as an immediate off switch for CAR T cell signalling, without affecting cell viability122,123,124 (Fig. 6a). Although not yet tested in the clinic, dasatinib might provide a useful method of controlling acute CAR T cell toxicities, particularly considering its ability to cross the blood–brain barrier and thus manage neurotoxicities125. However, given that CAR T cell viability and activity are maintained after dasatinib is discontinued123, this agent might not be ideal for the long-term management of OTOT.
Figure 6. Control switches in CAR T cells.
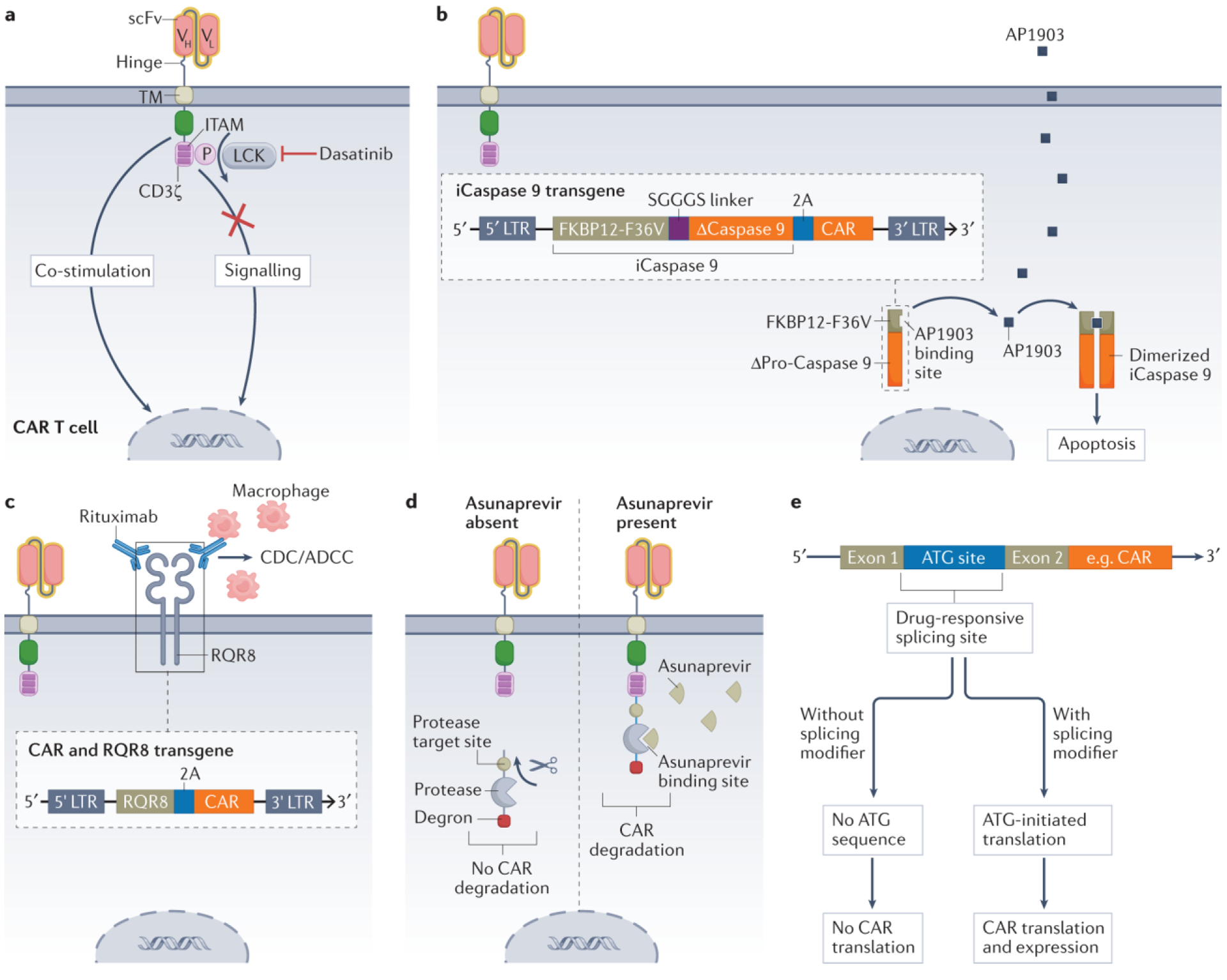
a, Reversible inhibition of the tyrosine kinase LCK with dasatinib prevents phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) within CD3ζ, interrupting a necessary signal for chimeric antigen receptor (CAR) T cell effector functions122,123. b, Administration and binding of AP1903 to FKBP12-F36V leads to dimerization and activation of an inducible caspase 9-based suicide construct (iCaspase 9), which triggers apoptosis of the CAR T cell131. c, The CAR transgene can be engineered to also encode the selection marker RQR8, which can be targeted with rituximab to trigger complement-dependent cytotoxicity (CDC) and/or antibody-dependent cell-mediated cytotoxicity (ADCC) and thus depletion of the CAR T cells129. d, A protease target site, a protease and a degron are fused to the intracellular C terminus of the CAR. The protease can cleave itself and the degron from the CAR, preventing CAR degradation and maintaining its expression on the cell surface. However, inhibition of the protease with asunaprevir prevents cleavage, leading to retention of the degron and thus degradation of the entire CAR construct138. e, The inclusion or exclusion of exons during mRNA splicing can be altered using so-called splicing modifier drugs. By inserting the start codon (ATG) of the CAR into a site that is responsive to a splicing modifier, CAR translation and expression can be controlled. In the presence of this drug, the start codon of the CAR remains part of the mRNA. This allows translation of the mRNA and permits assembly and expression of the CAR. In the absence of the drug, the start codon is removed and CAR translation is not initiated150. LTR, long terminal repeat; scFv, single-chain variable fragment; TM, transmembrane domain; VH, heavy chain variable region; VL, light chain variable region.
Control via suicide switches
Suicide switches126,127,128 and other strategies129,130 designed to eliminate and/or inactivate CAR T cells in vivo are important safety measures to treat potentially life-threatening adverse events. Various strategies have been tested126,131,132, including the use of an inducible caspase 9-based suicide construct (iCaspase 9). This construct can be activated through administration of the small molecule AP1903, inducing apoptosis in CAR T cells within minutes of exposure131 (Fig. 6b). Data from a clinical study published in 2021 demonstrate that iCaspase 9 can be exploited to safely ameliorate clinically serious neurotoxicities in patients receiving anti-CD19 CAR T cells133.
CAR T cells can also be engineered to express an additional cell-surface molecule that can then be targeted using a monoclonal antibody. This allows CAR T cell clearance via complement-dependent and/or antibody-dependent cell-mediated cytotoxicity129. A polypeptide sequence derived from CD34 and CD20 (termed RQR8) and recognized by rituximab, has been developed for this purpose (Fig. 6c). CAR-transduced splenocytes coexpressing RQR8 were not detected in peripheral blood samples 7 days after the administration of rituximab in a mouse splenocyte engraftment model129. Similarly, anti-CD19 CAR T cells that co-express a truncated version of EGFR (EGFRt) can be targeted using cetuximab, enabling elimination of CAR T cells and reversal of B cell aplasia130,134.
Despite the potential of this approach, several factors might affect the efficiency of antibody-mediated CAR T cell elimination. Limited trafficking of monoclonal antibodies to the CNS might hinder CAR T cell elimination in this location135. Furthermore, recognition of the antibody target on non-malignant cells might cause additional toxicities136. Moreover, the selection pressures induced by monoclonal antibodies might lead to the expansion of a CAR T cell population that lacks sufficient antibody-targetable cell-surface marker and thereby evades control137. Finally, the kinetics of this form of T cell elimination might not be sufficiently rapid to abrogate acute damage to vital organs.
Control by regulating CAR expression and cytotoxicity
Irreversible elimination of CAR T cells using suicide switches prior to complete tumour eradication might limit clinical efficacy. An alternative could be designing reversible off/on-switches that permit temporal control of CAR T cell activation and preserve antitumour function138,139,140. Several designs have been proposed to date, many of which rely on the administration of small molecules109,141,142 or antibodies143, among other methods144,145,146.
In an off-switch system, control of CAR surface expression was demonstrated using the protease inhibitor asunaprevir138. To achieve this effect, a protease target site, a protease and a degron are fused to the C terminus of the CAR. In the absence of asunaprevir, the protease cleaves the degron from the CAR construct, resulting in the maintenance of CAR surface expression. Upon asunaprevir administration, the protease is inhibited and the degron cannot be cleaved, leading to CAR degradation138 (Fig. 6d). Similarly, a system in which the CAR is bound to a ligand-induced degradation domain has been developed. This approach enables proteasomal degradation of the CAR upon administration of the ligand, and thus confers tuneable control of CAR expression in vivo147.
In an on-switch approach, investigators introduced a protease cleavage site between the transmembrane and signalling domains of the CAR construct. Further, they integrated a membrane-bound NS3 protease expressed in trans to the CAR. Administration of the protease inhibitor grazoprevir inhibited the constitutive cleavage of CAR signalling domains, allowing CAR functionality in mouse models. Cessation of drug delivery reversed the CAR T cell-related OTOT seen with high doses of grazoprevir, demonstrating that drug dosage can tune CAR T cell activity to achieve tumour control while maintaining an acceptable safety profile16. Another approach that allows remote control of CAR T cells is the previously discussed AvidCAR (Fig. 5e). Suggested dimerizers in AvidCARs and other on-switch systems include AP20187 (ref.109), lenalidomide141, A1120 (ref.148) and rapamycin149. Data on a system that uses drug-induced splicing to regulate the expression of therapeutic proteins has also been reported and could potentially be adapted as an on-switch for CAR T cells150 (Fig. 6e). Whether CAR T cells incorporating drug-induced safety switches will be successful in patients with solid tumours might depend on drug tolerability and drug trafficking to the site of CAR T cell activity150.
Control of CAR expression via sound or light
Ultrasonography can be used to control the transcription and subsequent expression of CARs. An ultrasound-sensitive Piezo1 ion channel was shown to trigger a signalling cascade, leading to the transcriptional upregulation of anti-CD19 CAR molecules in vitro151. In August 2021, researchers published a method in which expression of an anti-PSMA or anti-CD19 CAR is placed under the regulatory control of a heat shock protein (Fig. 7a). This method enabled in vivo control of CAR expression by applying focused ultrasound for approximately 5 min, which raised the temperature within tumours to around 43 °C. This induced a tumour-confined cytotoxic response while limiting off-tumour cytotoxicity152.
Figure 7. Regional restriction of CAR T cell activity.
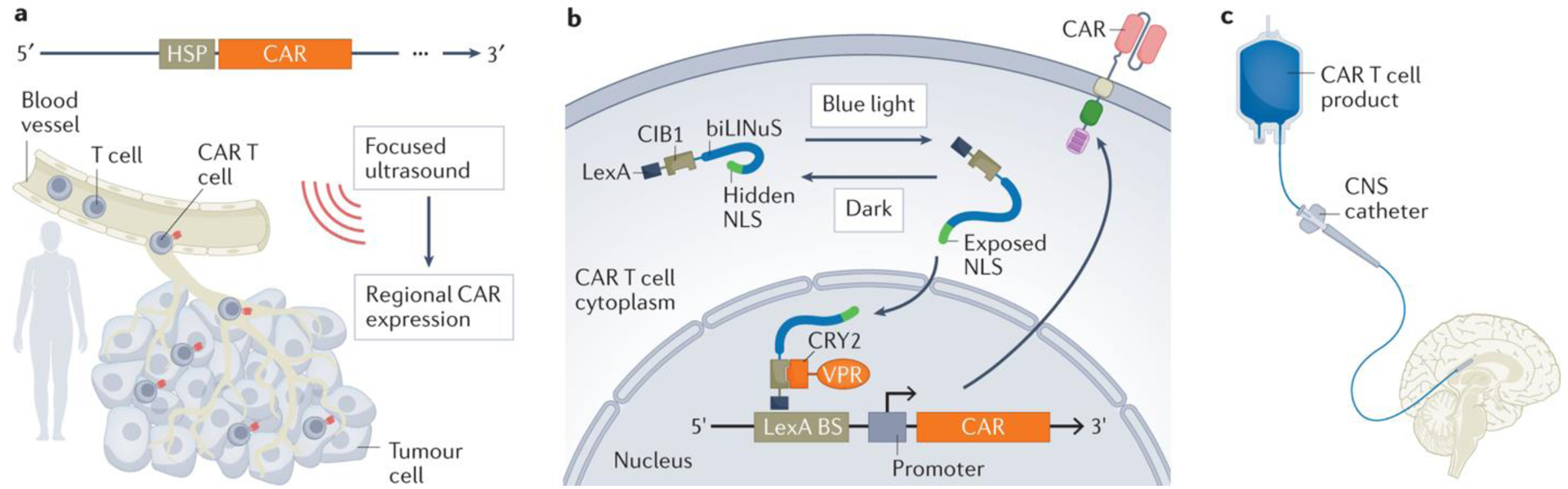
a, The chimeric antigen receptor (CAR) transgene can be placed under regulatory control of a heat shock protein (HSP), such that focused ultrasound can be used to generate a localized, transient increase in temperature. This activates the HSP and thus reversibly upregulates CAR transcription and expression152. b, Blue light-induced CAR transcription can be achieved using the 3-component light-inducible nuclear translocation and dimerization (LINTAD) system. In addition to the CAR transgene, the T cells express a LexA-CIB1-biLINuS (LCB) construct. The biLINuS-responsive element is activated upon localized application of blue light, resulting in exposure of a nuclear localization sequence (NLS) and translocation of the LCB construct into the nucleus. LexA (as part of LCB) can bind to a LexA binding sequence (BS) engineered in the CAR transgene. The CIB1 component of LCB can then recruit a co-expressed CRY2–VPR fusion construct that can target the minimal promoter and trigger the expression of the CAR153. c, Locoregional intraventricular administration of CAR T cells as an alternative to systemic intravenous infusion41. CNS, central nervous system.
Light-responsive CAR T cells that resemble an on-switch system can be used to induce localized CAR expression. The introduction of a light-inducible nuclear translocation and dimerization (LINTAD) system was shown to control CAR T cell-mediated tumour cell killing in vitro and in mice153 (Fig. 7b). Elsewhere, a light-responsive universal CAR T cell has been developed that relies on the UV-sensitive small molecule CMNB (5-carboxymethoxy-2-nitrobenzyl) for TAA recognition. In vitro tumour cell binding and killing were dependent on the presence of UV light154. Spatial and temporal induction of CAR expression using sound-controlled or light-controlled CAR T cells might prevent systemic CAR T cell activation and thus reduce the potential for OTOT.
Local administration of CAR T cells
CAR T cells are traditionally administered systemically, presenting challenges both for adequate CAR T cell trafficking to tumour sites and minimal trafficking to non-malignant tissues. Where possible, regional155 or local156,157 administration could help to overcome these challenges. For instance, intraventricular and intrathecal administration of CAR T cells targeting CNS tumours has shown promising preclinical and early clinical results61,158,159,160,161. Intracranial delivery of anti-IL13Rα2 CAR T cells resulted in transient antitumour responses in two of three patients with glioblastoma. Adverse events were manageable, although grade 3 neurotoxicities were observed in a patient with high IL13Rα2-expressing tumour cells61. Similarly, anti-HER2 CARs were infused directly into the tumour cavity or ventricular system of paediatric patients with recurrent and/or refractory CNS tumours in a phase I trial41 (Fig. 7c). Preliminary data from this trial suggest that repeated local administration is well tolerated and leads to localized immune responses41. In March 2022, investigators described the successful intracerebroventricular administration of anti-GD2 CAR T cells in patients with diffuse intrinsic pontine glioma, a highly lethal paediatric CNS tumour. Three out of four patients had clinical and radiographic improvements with no OTOT observed despite GD2 expression on normal brain tissues68.
Local delivery of anti-FAP CAR T cells to the pleural cavity has also been performed in three patients with malignant pleural mesothelioma (MPM)157. This trial demonstrated the feasibility of local delivery in an organ-confined tumour, such as MPM157. Similarly, anti-mesothelin CAR T cells were administered via intrapleural infusion in 27 patients with malignant pleural disease. Although CAR T cells were detected in peripheral blood samples from 39% of patients for >100 days, no signs of OTOT were reported162.
Anti-B7-H3 CAR T cells have been studied in several tumour types with a specific focus on delivery routes. When delivered locoregionally, anti-B7-H3 CAR T cells resulted in more-effective antitumour responses in mouse xenograft models of atypical teratoid/rhabdoid tumours161. However, data from a study using an immunocompetent mouse model suggest that B7-H3 CAR T cells are safe and effective regardless of the route of administration163. Ongoing early phase clinical trials of anti-B7-H3 CAR T cells involving paediatric patients with brain tumours (NCT04185038) and other solid tumours (NCT04483778, NCT04897321) might provide additional insights into the implications of delivery routes on antitumour activity and OTOT. Although local administration of CAR T cells can limit cytotoxicity to the specific tissue compartment164, a possibility remains that these cells could enter the systemic circulation162.
Conclusion
CAR T cell therapies with additional modifications that augment their potency are being developed for solid tumours, and are steadily moving towards clinical testing. The identification of numerous TAAs has laid the groundwork for testing CAR T cells in patients with solid tumours, although their expression on non-malignant tissues might portend an increased risk of OTOT. Strategies for assessing such risks are imperfect as observed by data from several CAR T cell clinical trials, where OTOT is inconsistent with preclinical data. An important area of research is to refine animal models and/or advance in vitro systems to more accurately predict these toxicities.
Intensive research has focused on developing more specific CAR systems, including those that permit exogenous control of T cell function or survival. Tuning the affinity of scFvs and altering the CAR architecture can potentially mitigate the risk of OTOT. Various protein-based logic-circuit strategies aiming to further restrict CAR T cell activation and killing to the tumour site have been explored in preclinical models. Other attempts to control CAR T cell-mediated OTOT include engineering approaches designed to exogenously control CAR T cell activity or that lead to timely CAR T cell elimination. Locoregional administration of CAR T cells to concentrate antitumour activity within the tumour milieu might avoid OTOT and has already been tested in early phase clinical studies. Addressing the risk of OTOT during the development of novel CAR T cells for solid tumours is imperative to guide the successful development of safe and effective therapeutic strategies in the clinic.
Supplementary Material
Key points.
Chimeric antigen receptor (CAR) T cell therapies have led to on-target, off-tumour toxicity (OTOT) in clinical trials involving patients with solid tumours.
Preclinical mouse models might provide inaccurate predictions of OTOT in patients, reflecting the need for better models to perform preclinical safety assessments.
Logic-gating circuits and synthetic biology approaches to CAR T cell engineering have demonstrated specificity in mice, although many of these approaches remain untested in clinical studies.
Methods of controlling CAR T cell activity and responding to unexpected OTOT have the potential to improve safety after infusion.
We advocate for robust preclinical analyses of the risks of OTOT and the implementation of control strategies capable of regulating CAR T cell activity in patients.
Acknowledgements
The authors thank K. T. Roybal (University of California, San Francisco, USA) for his helpful comments on an earlier draft of the manuscript. They thank A. Cadinanos-Garai (University of Southern California, USA) for her critical review of the manuscript and figures. The work of G.K. is supported by R01NS121249 from NINDS and the Assisi Foundation of Memphis. The work of M.A. is supported, in part, by award P30CA014089 from the US NIH National Cancer Institute (NCI).
Competing interests
R.G.M. has acted as an adviser and/or consultant of Aptorum Group, Arovella Therapeutics, Immunai, Innervate Radiopharmaceuticals, Link Cell Therapies, Lyell Immunopharma, NKarta, Syncopation Life Sciences, and Zai lab and is a co-founder of and holds equity in Syncopation Life Sciences and Link Cell Therapies. G.K. has patent applications in the field of immunotherapy. G.D. has acted as a scientific adviser and/or consultant of Bellicum Pharmaceutical and Catamaran and Tessa Therapeutics and holds patents in the field of CAR T cells. S.R.R. has acted as a scientific adviser of Adaptive Biotechnologies and Juno Therapeutics, is a co-founder of and has intellectual property licensed to Lyell Immunopharma and Juno Therapeutics, and holds shares in and has received research funding from Lyell Immunopharma. C.L.F., D.L.W. and M.A. declare no competing interests.
Footnotes
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41571-022-00704-3.
References
- 1.June CH & Sadelain M Chimeric antigen receptor therapy. N. Engl. J. Med 379, 64–73(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larson RC & Maus MV Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 21, 145–161 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.FDA. ABECMA (idecabtagene vicleucel). https://www.fda.gov/vaccines-blood-biologics/abecma-idecabtagene-vicleucel (2021).
- 4.FDA. BREYANZI (lisocabtagene maraleucel). https://www.fda.gov/vaccines-bloodbiologics/cellular-gene-therapy-products/breyanzi-lisocabtagene-maraleucel (2021).
- 5.FDA. KYMRIAH (tisagenlecleucel). https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel (2021).
- 6.FDA. TECARTUS (brexucabtagene autoleucel). https://www.fda.gov/vaccines-bloodbiologics/cellular-gene-therapy-products/tecartus-brexucabtagene-autoleucel (2021).
- 7.FDA. YESCARTA (axicabtagene ciloleucel). https://www.fda.gov/vaccines-bloodbiologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel (2021).
- 8.FDA. CARVYKTI (ciltacabtagene autoleucel). https://www.fda.gov/vaccines-bloodbiologics/carvykti (2022). [PubMed]
- 9.Amini L et al. Preparing for CAR T cell therapy: patient selection, bridging therapies and lymphodepletion. Nat. Rev. Clin. Oncol 19, 342–355 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Logue JM et al. Immune reconstitution and associated infections following axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma. Haematologica 106, 978–986 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uy NF et al. Hypogammaglobulinemia and infection risk in chronic lymphocytic leukemia (CLL) patients treated with CD19-directed chimeric antigen receptor T (CAR-T) cells. Blood 136, 30–32 (2020). [Google Scholar]
- 12.Schubert ML et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann. Oncol 32, 34–48 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Srivastava S et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 35, 489–503.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellarin M et al. A rational mouse model to detect on-target, off-tumor CAR T cell toxicity. JCI Insight 5, e136012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith JB et al. Tumor regression and delayed onset toxicity following B7-H4 CAR T cell therapy. Mol. Ther 24, 1987–1999 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labanieh L et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 185, 1745–1763 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamers CH et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol 24, e20–e22 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Lamers CHJ et al. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells–a completed study overview. Biochem. Soc. Trans 44, 951–959 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Feng K-C et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol 10, 4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Y et al. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin. Cancer Res 24, 1277–1286 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Liu Y et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: a phase I clinical trial. Cytotherapy 22, 573–580 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Feng K et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 9, 838–847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thistlethwaite FC et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother 66, 1425–1436 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qi C et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat. Med 28, 1189–1198 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol 147, 3725–3734 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richman SA et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol. Res 6, 36–46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davenport AJ et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl Acad. Sci. USA 115, E2068–E2076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meiraz A, Garber OG, Harari S, Hassin D & Berke G Switch from perforin-expressing to perforin-deficient CD8+ T cells accounts for two distinct types of effector cytotoxic T lymphocytes in vivo. Immunology 128, 69–82 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong LK et al. CD30-redirected chimeric antigen receptor T cells target CD30+ and CD30- embryonal carcinoma via antigen-dependent and Fas/FasL interactions. Cancer Immunol. Res 6, 1274–1287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benmebarek M-R et al. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int. J. Mol. Sci 20, 1283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dufva O et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 135, 597–609 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schietinger A et al. A mutant chaperone converts a wild-type protein into a tumor-specific antigen. Science 314, 304–308 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Wong AJ et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl Acad. Sci. USA 89, 2965–2969 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Posey AD Jr et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 44, 1444–1454 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heitzeneder S et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell 40, 53–69.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bosse KR et al. Identification of GPC2 as an oncoprotein and candidate immunotherapeutic target in high-risk neuroblastoma. Cancer Cell 32, 295–309.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith CC et al. Alternative tumour-specific antigens. Nat. Rev. Cancer 19, 465–478 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Li G & Wong AJ EGF receptor variant III as a target antigen for tumor immunotherapy. Expert Rev. Vaccines 7, 977–985 (2008). [DOI] [PubMed] [Google Scholar]
- 40.O’Rourke DM et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med 9, eaaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vitanza NA et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat. Med 27, 1544–1552 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Ahmed N et al. Autologous HER2 CMV bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a Phase I trial. J. ImmunoTher. Cancer 3, O11 (2015). [Google Scholar]
- 43.Hegde M et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun 11, 3549 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan RA et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther 18, 843–851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ivanov S et al. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am. J. Pathol 158, 905–919 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moghimi B et al. Preclinical assessment of the efficacy and specificity of GD2-B7H3 SynNotch CAR-T in metastatic neuroblastoma. Nat. Commun 12, 511 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Du H et al. Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell 35, 221–237.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beatty GL et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 155, 29–32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haas AR et al. Phase I study of lentiviral-transduced chimeric antigen receptor modified T cells recognizing mesothelin in advanced solid cancers. Mol. Ther 27, 1919–1929 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maus MV et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res 1, 26–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beatty GL et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res 2, 112–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heczey A et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol. Ther 25, 2214–2224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Straathof K et al. Antitumor activity without on-target off-tumor toxicity of GD2–chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med 12, eabd6169 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Pule MA et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med 14, 1264–1270 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Louis CU et al. Antitumor activity and long-term fate of chimeric antigen receptor positive T cells in patients with neuroblastoma. Blood 118, 6050–6056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gargett T et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol. Ther 24, 1135–1149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lamers CH et al. Treatment of metastatic renal cell carcinoma with CAIX CARengineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther 21, 904–912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uhlén M et al. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Ahmed N et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol 33, 1688–1696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahmed N et al. HER2-specific chimeric antigen receptor–modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 3, 1094–1101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brown CE et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res 21, 4062–4072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parker KR et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell 183, 126–142.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Oekelen O et al. Neurocognitive and hypokinetic movement disorder with features of parkinsonism after BCMA-targeting CAR-T cell therapy. Nat. Med 27, 2099–2103 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mount CW et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med 24, 572–579 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park S et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep 7, 14366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kosti P et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med 2, 100227 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Majzner RG, Weber EW, Lynn RC, Xu P & Mackall CL Neurotoxicity associated with a high-affinity GD2 CAR-letter. Cancer Immunol. Res 6, 494–495 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Majzner RG et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 603, 934–941 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kato D et al. GPC1 specific CAR-T cells eradicate established solid tumor without adverse effects and synergize with anti-PD-1 Ab. eLlife 9, e49392 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fedorov VD, Themeli M & Sadelain M PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med 5, 215ra172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caruso HG et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 75, 3505–3518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hernandez-Lopez RA et al. T cell circuits that sense antigen density with an ultrasensitive threshold. Science 371, 1166–1171 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cui X et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. eLlife 9, e52253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ingber DE Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet 23, 467–491 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu X et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 75, 3596–3607 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watanabe K et al. Excessively high-affinity single-chain fragment variable region in a chimeric antigen receptor can counteract T-cell proliferation. Blood 124, 4799 (2014). [Google Scholar]
- 77.Majzner RG & Mackall CL Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 8, 1219–1226 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Hegde M et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest 126, 3036–3052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fry TJ et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med 24, 20–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alabanza L et al. Function of Novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol. Ther 25, 2452–2465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Majzner RG et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 10, 702–723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feucht J et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med 25, 82–88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wilkie S et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol 32, 1059–1070 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Kloss CC, Condomines M, Cartellieri M, Bachmann M & Sadelain M Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol 31, 71–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang E et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J. Hematol. Oncol 11, 102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sukumaran S et al. Enhancing the potency and specificity of engineered T cells for cancer treatment. Cancer Discov. 8, 972–987 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang W et al. Abstract PO074: Logic-gating HER2 CAR-T to the tumor microenvironment mitigates on-target, off-tumor toxicity without compromising cytotoxicity against HER2-over-expressing tumors. Cancer Immunol. Res 9, PO074 (2021). [Google Scholar]
- 88.Vaupel P & Multhoff G Revisiting the Warburg effect: historical dogma versus current understanding. J. Physiol 599, 1745–1757 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Juillerat A et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep 7, 39833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Han X et al. Masked chimeric antigen receptor for tumor-specific activation. Mol. Ther 25, 274–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu C, Sun C, Huang H, Janda K & Edgington T Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res. 63, 2957–2964 (2003). [PubMed] [Google Scholar]
- 92.Liu G, Shuman MA & Cohen RL Co-expression of urokinase, urokinase receptor and PAI-1 is necessary for optimum invasiveness of cultured lung cancer cells. Int. J. Cancer 60, 501–506 (1995). [DOI] [PubMed] [Google Scholar]
- 93.Singhal R & Shah YM Oxygen battle in the gut: Hypoxia and hypoxia-inducible factors in metabolic and inflammatory responses in the intestine. J. Biol. Chem 295, 10493–10505 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang JL et al. Measurement of renal tissue oxygenation with blood oxygen level-dependent MRI and oxygen transit modeling. Am. J. Physiol. Ren. Physiol 306, F579–F587 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dumas SJ et al. Phenotypic diversity and metabolic specialization of renal endothelial cells. Nat. Rev. Nephrol 17, 441–464 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roybal KT et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 167, 419–432.e416 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roybal KT et al. Precision tumor recognition by T cells with combinatorial antigensensing circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morsut L et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 164, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hyrenius-Wittsten A et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci. Transl. Med 13, eabd8836 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Choe JH et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med 13, eabe7378 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Williams JZ et al. Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science 370, 1099–1104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wagner DL et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol 18, 379–393 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhu I et al. Modular design of synthetic receptors for programmed gene regulation in cell therapies. Cell 185, 1431–1443 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feldmann A et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget 8, 31368–31385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Albert S et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology 6, e1287246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mitwasi N et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget 8, 108584–108603 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jureczek J et al. Highly efficient targeting of EGFR-expressing tumor cells with UniCAR T cells via target modules based on cetuximab(®). OncoTargets Ther. 13, 5515–5527 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cho JH, Collins JJ & Wong WW Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 173, 1426–1438.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Salzer B et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat. Commun 11, 4166 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carmeliet P VEGF as a key mediator of angiogenesis in cancer. Oncology 69, 4–10 (2005). [DOI] [PubMed] [Google Scholar]
- 111.Richards RM et al. NOT-Gated CD93 CAR T cells effectively target AML with minimized endothelial cross-reactivity. Blood Cancer Discov. 2, 648–665 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sandberg ML et al. A carcinoembryonic antigen-specific cell therapy selectively targets tumor cells with HLA loss of heterozygosity in vitro and in vivo. Sci. Transl. Med 14, eabm0306 (2022). [DOI] [PubMed] [Google Scholar]
- 113.McGranahan N et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171, 1259–1271.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.De Mattos-Arruda L et al. The genomic and immune landscapes of lethal metastatic breast cancer. Cell Rep. 27, 2690–2708.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dong L-Q et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J. Hepatol 72, 896–908 (2020). [DOI] [PubMed] [Google Scholar]
- 116.Hamburger AE et al. Engineered T cells directed at tumors with defined allelic loss. Mol. Immunol 128, 298–310 (2020). [DOI] [PubMed] [Google Scholar]
- 117.Tokatlian T et al. Mesothelin-specific CAR-T cell therapy that incorporates an HLA-gated safety mechanism selectively kills tumor cells. J. Immunother. Cancer 10, e003826 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hwang MS et al. Targeting loss of heterozygosity for cancer-specific immunotherapy. Proc. Natl Acad. Sci. USA 118, e2022410118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lajoie MJ et al. Designed protein logic to target cells with precise combinations of surface antigens. Science 369, 1637–1643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brudno JN & Kochenderfer JN Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321–3330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thompson JA New NCCN guidelines: recognition and management of immunotherapy-related toxicity. J. Natl Compr. Canc. Netw 16, 594–596 (2018). [DOI] [PubMed] [Google Scholar]
- 122.Weber EW et al. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 3, 711–717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mestermann K et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med 11, eaau5907 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Weber EW et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372, eaba1786 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Porkka K et al. Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood 112, 1005–1012 (2008). [DOI] [PubMed] [Google Scholar]
- 126.Straathof KC et al. An inducible caspase 9 safety switch for T-cell therapy. Blood 105, 4247–4254 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stavrou M et al. A rapamycin-activated Caspase 9-based suicide gene. Mol. Ther 26, 1266–1276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Duong MT et al. Two-dimensional regulation of CAR-T cell therapy with orthogonal switches. Mol. Ther. Oncolytics 12, 124–137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Philip B et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 124, 1277–1287 (2014). [DOI] [PubMed] [Google Scholar]
- 130.Wang X et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 118, 1255–1263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Di Stasi A et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med 365, 1673–1683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.de Witte MA et al. An inducible caspase 9 safety switch can halt cell therapy-induced autoimmune disease. J. Immunol 180, 6365–6373 (2008). [DOI] [PubMed] [Google Scholar]
- 133.Foster MC et al. Utility of a safety switch to abrogate CD19.CAR T-cell-associated neurotoxicity. Blood 137, 3306–3309 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Paszkiewicz PJ et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest 126, 4262–4272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Villaseñor R et al. Trafficking of endogenous immunoglobulins by endothelial cells at the blood-brain barrier. Sci. Rep 6, 25658 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wehler TC et al. Cetuximab-induced skin exanthema: prophylactic and reactive skin therapy are equally effective. J. Cancer Res. Clin. Oncol 139, 1667–1672 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Koristka S et al. Anti-CAR-engineered T cells for epitope-based elimination of autologous CAR T cells. Cancer Immunol. Immunother 68, 1401–1415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Juillerat A et al. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 19, 44–44 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sahillioglu AC, Toebes M, Apriamashvili G, Gomez R & Schumacher TN CRASH-IT switch enables reversible and dose-dependent control of TCR and CAR T-cell function. Cancer Immunol. Res 9, 999–1007 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li HS et al. High-performance multiplex drug-gated CAR circuits. Cancer Cell 10.1016/j.ccell.2022.08.008 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jan M et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci. Transl. Med 13, eabb6295 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu CY, Roybal KT, Puchner EM, Onuffer J & Lim WA Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lohmueller J et al. Post-translational covalent assembly of CAR and synNotch receptors for programmable antigen targeting. Preprint at 10.1101/2020.01.17.909895 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mata M et al. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. 7, 1306–1319 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kotter B et al. Titratable pharmacological regulation of CAR T cells using zinc finger-based transcription factors. Cancers 13, 4741 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hotblack A et al. Tunable control of CAR T cell activity through tetracycline mediated disruption of protein-protein interaction. Sci. Rep 11, 21902 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Richman SA et al. Ligand-induced degradation of a CAR permits reversible remote control of CAR T cell activity in vitro and in vivo. Mol. Ther 28, 1600–1613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zajc CU et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc. Natl Acad. Sci. USA 117, 14926 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Leung W-H et al. Sensitive and adaptable pharmacological control of CAR T cells through extracellular receptor dimerization. JCI Insight 5, e124430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Monteys AM et al. Regulated control of gene therapies by drug-induced splicing. Nature 596, 291–295 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pan Y et al. Mechanogenetics for the remote and noninvasive control of cancer immunotherapy. Proc. Natl Acad. Sci. USA 115, 992–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wu Y et al. Control of the activity of CAR-T cells within tumours via focused ultrasound. Nat. Biomed. Eng 5, 1336–1347 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Huang Z et al. Engineering light-controllable CAR T cells for cancer immunotherapy. Sci. Adv 6, eaay9209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kobayashi A et al. Light-controllable binary switch activation of CAR T cells. ChemMedChem 17, e202100722 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Priceman SJ et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2+ breast cancer metastasis to the brain. Clin. Cancer Res 24, 95–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tchou J et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol. Res 5, 1152–1161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hiltbrunner S et al. Local delivery of CAR T cells targeting fibroblast activation protein is safe in patients with pleural mesothelioma: first report of FAPME, a phase I clinical trial. Ann. Oncol 32, 120–121 (2021). [DOI] [PubMed] [Google Scholar]
- 158.Donovan LK et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med 26, 720–731 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nellan A et al. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 6, 30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Brown CE et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med 375, 2561–2569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Theruvath J et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med 26, 712–719 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Adusumilli PS et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discov. 11, 2748–2763 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Haydar D et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. NeuroOncology 23, 999–1011 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Srivastava S & Riddell SR Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. J. Immunol 200, 459–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Savoldo B et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest 121, 1822–1826 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Imai C et al. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 4, 676–84 (2004). [DOI] [PubMed] [Google Scholar]
- 167.Xiao Q et al. Size-dependent activation of CAR-T cells. Sci. Immunol 7, eabl3995 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Alizadeh D et al. IFNγ is critical for CAR T cell-mediated myeloid activation and induction of endogenous immunity. Cancer Discov. 11, 2248–2265 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chmielewski M, Kopecky C, Hombach AA & Abken H IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 71, 5697–5706 (2011). [DOI] [PubMed] [Google Scholar]
- 170.Zhang L et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol. Ther 19, 751–759 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Uhlen M et al. Towards a knowledge-based human protein atlas. Nat. Biotechnol 28, 1248–1250 (2010). [DOI] [PubMed] [Google Scholar]
- 172.Nagata Y et al. Expression cloning of beta 1,4 N-acetylgalactosaminyltransferase cDNAs that determine the expression of GM2 and GD2 gangliosides. J. Biol. Chem 267, 12082–12089 (1992). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.