

Polygenic scores quantify inherited risk by integrating information from many common DNA variants. In studies of middle-aged individuals, genome-wide polygenic scores for coronary heart disease (GPSCHD) associate with CHD with effect size comparable – or exceeding – that of traditional risk factors such as blood pressure or cholesterol.(1, 2) Here, we study the relationship between a previously described GPSCHD, coronary artery calcification, and incident CHD events in the Coronary Artery Risk Development in Young Adults (CARDIA) study.
The CARDIA study enrolled White and Black young adults free of CHD beginning in 1985 with follow-up assessment of coronary artery calcium (CAC)—at Year 15, 20, and 25, study visits—and incident CHD events (myocardial infarction, coronary revascularization, and death from coronary causes).(3) Regression models were adjusted for age, sex, 5 principal components of ancestry, and study recruitment center, with subsequent adjustment for 7 clinical risk factors—systolic blood pressure, total cholesterol, HDL cholesterol, body mass index, diabetes, current smoking, and parental history of myocardial infarction. Informed consent was obtained by CARDIA investigators and analysis approved by the Institutional Review Board of Mass General Brigham (Boston, MA).
The previously described GPSCHD was calculated in 1,663 White and 955 Black participants with genetic information available.(4) Median age at time of enrollment was 25 years and 56% were female. 61% of the participants had none of the 7 clinical risk factors listed above at time of enrollment.
Among White participants, presence of CAC > 0 at the Year 15 visit ranged from 5.2% to 17.9% across GPSCHD quintiles, corresponding to an adjusted odds ratio for the highest versus lowest quintiles of 4.2 (95%CI 2.2—8.1; p<0.001). By the Year 25 study visit, prevalence had increased to 20.9% for those in the lowest quintile versus 45.1% for those in the highest quintile—odds ratio 3.9 (95%CI 2.5—6.0; p<0.001). These effect estimates were minimally attenuated after adjustment for 7 clinical risk factors—odds ratios of 3.8 (95% CI 1.9 to 7.4; p = 1.3 × 10−4) and 3.7 (95%CI 2.3 to 5.8; p = 2.4 × 10−8) respectively. Among Black participants, there was no statistically significant relationship between GPSCHD and CAC > 0, with presence ranging from 20.0% to 27.7% at the Year 25 study visit (p=0.09).
Over a median follow-up of 31.9 years, an incident CHD event was noted in 66 (4.0%) of White participants and 28 (2.9%) of Black participants. Among White participants, risk was significantly increased for those in the highest versus lowest quintile (Figure 1)—hazard ratio of 7.1 (95% CI 2.8—18.3; p<0.0001). This estimate was minimally affected by additional adjustment for 7 clinical risk factors—hazard ratio 7.7 (95% CI 2.7—22.0; p=0.0001). When GPSCHD expressed as a continuous variable was added to a model of age, sex, 5 principal components of ancestry, study center and 7 clinical risk factors, the C-statistic increased from 0.843 to 0.872 (+0.029; 95%CI 0.008—0.053; p=0.013). Similar results for improvement in C-statistic were obtained when using five-fold cross-validation (0.0316 95% CI 0.002, 0.061; p=0.03).
Figure 1. Cumulative incidence of CHD events by polygenic score and CAC.
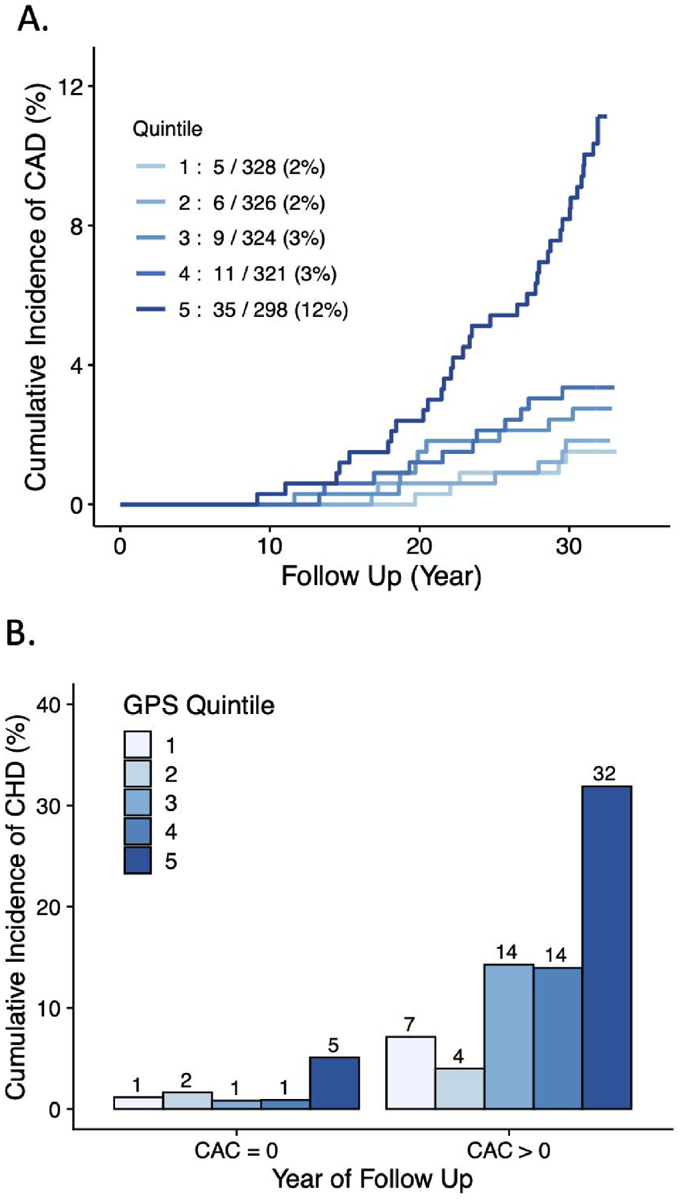
(A) Cumulative incidence of coronary heart disease events in White participants according to quintile of the GPSCHD. (B) Cumulative incidence of coronary heart disease events in White participants stratified according to presence of CAC > 0 at the Year 15 study visit.
Whether a GPSCHD is associated with incident CHD among individuals stratified by the presence of CAC has been largely unexplored. We analyzed the relationship between GPSCHD and incident disease in an analysis stratified by presence of CAC>0 at the Year 15 visit (Figure 1). Among 1,173 White participants with no CAC present, a 4.2-fold (95%CI 1.1—15.1; p=0.03) increased risk was noted for those in the highest versus lowest quintile. A similar risk gradient—hazard ratio 4.1 (95%CI 0.5—32.1; p=0.18) was noted in those with CAC>0, but this result was not statistically significant.
In this study, a substantial gradient in risk of future CAC and CHD events was noted according to GPSCHD assessed in young adulthood, with new evidence suggesting utility even among individuals with no detectable CAC in middle age. These results warrant replication in studies with a larger number of CHD events and highlight the importance of developing new polygenic scores with improved performance in non-European ancestral groups.(5)
Acknowledgements
The Coronary Artery Risk Development in Young Adults Study (CARDIA) is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with the University of Alabama at Birmingham (HHSN268201800005I & HHSN268201800007I), Northwestern University (HHSN268201800003I), University of Minnesota (HHSN268201800006I), and Kaiser Foundation Research Institute (HHSN268201800004I). CARDIA was also partially supported by the Intramural Research Program of the National Institute on Aging (NIA) and an intra‐agency agreement between NIA and NHLBI (AG0005). Genotyping was funded as part of the NHLBI Candidate-gene Association Resource (N01-HC-65226) and the NHGRI Gene Environment Association Studies (GENEVA) (U01-HG004729, U01-HG04424, and U01-HG004446). This manuscript has been reviewed and approved by CARDIA for scientific content.
Funding support was provided by grants 1K08HG010155 and 1U01HG011719 (to A.V.K.) from the National Human Genome Research Institute, a Hassenfeld Scholar Award from Massachusetts General Hospital (to A.V.K.), and a Merkin Institute Fellowship from the Broad Institute of MIT and Harvard (to A.V.K.).
Competing Financial Interests
CAE is an employee of Foresite Labs and holds equity in Foresite Labs; has served as a consultant or received honoraria from Acceleron Pharma, Deerfield Management, Navitor Pharma, Nference, Novartis and Verve Therapeutics. A.V.K. is an employee and holds equity in Verve Therapeutics; has served as a scientific advisor to Amgen, Maze Therapeutics, Navitor Pharmaceuticals, Sarepta Therapeutics, Novartis, Silence Therapeutics, Korro Bio, Veritas International, Color Health, Third Rock Ventures, Illumina, Foresite Labs, and Columbia University (NIH); received speaking fees from Illumina, MedGenome, Amgen, and the Novartis Institute for Biomedical Research; received a sponsored research agreement from IBM Research, and is listed as a co-inventor on a patent application for use of imaging data in assessing body fat distribution and associated cardiometabolic risk. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
REFERENCES
- 1.Inouye M, Abraham G, Nelson CP, et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. Journal of the American College of Cardiology 2018;72:1883–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hindy G, Aragam KG, Ng K, et al. Genome-Wide Polygenic Score, Clinical Risk Factors, and Long-Term Trajectories of Coronary Artery Disease. Arterioscler Thromb Vasc Biol 2020:ATVBAHA120314856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman GD, Cutter GR, Donahue RP, et al. CARDIA: study design, recruitment, and some characteristics of the examined subjects. J Clin Epidemiol 1988;41:1105–16. [DOI] [PubMed] [Google Scholar]
- 4.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nature genetics 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fahed AC, Aragam KG, Hindy G, et al. Transethnic Transferability of a Genome-Wide Polygenic Score for Coronary Artery Disease. Circ: Genomic and Precision Medicine 2021;14:e003092. [DOI] [PMC free article] [PubMed] [Google Scholar]