

Abstract
Scp160p is a 160 kDa protein in the yeast Saccharomyces cerevisiae that contains 14 repeats of the hnRNP K-homology (KH) domain, and demonstrates significant sequence homology to a family of proteins collectively known as vigilins. As a first step towards defining the function of Scp160p, we have characterized the subcellular distribution and in vivo interactions of this protein. Using sucrose gradient fractionation studies we have demonstrated that Scp160p in cytoplasmic lysates is predominantly associated with polyribosomes. Furthermore, we have found that Scp160p is released from polyribosomes by EDTA in the form of a large complex of ≥1300 kDa that is sensitive both to RNase and NaCl. Using affinity-chromatography to isolate these complexes, we have identified two protein components other than Scp160p: poly(A) binding protein, Pab1p, and Bfr1p. The presence of Pab1p confirms these complexes to be mRNPs. The presence of Bfr1p is intriguing because the null phenotype for this gene is essentially the same as that reported for scp160-null cells: increased cell size and aberrant DNA content. These results demonstrate that Scp160p associates with polyribosome-bound mRNP complexes in vivo, implicating a role for this protein in one or more levels of mRNA metabolism in yeast.
INTRODUCTION
Scp160p is a 1222 amino acid Saccharomyces cerevisiae protein that contains 14 copies of the hnRNP K homology (KH) domain, a highly conserved motif found in many proteins involved in RNA metabolism (1). KH domain-containing proteins appear to have diverse functions and have been identified in all kingdoms of life, including the ribosomal protein S3 from Escherichia coli (1), Mer1p from S.cerevisiae, a meiosis-specific splicing factor (1), MEX-3 from Caenorhabditis elegans, presumably involved in mRNA localization during development (2), and FMRP, the fragile-X mental retardation protein in humans (3). A partial clone of Scp160p, known as HX, was one of the first multiple-KH proteins identified (1).
Whole cell immunofluorescence studies have demonstrated that Scp160p localizes to the cytoplasm, with enrichment around the nuclear envelope, and what appears to be the endoplasmic reticulum (3). Deletion of the SCP160 locus in yeast is not lethal, but results in a complex phenotype, including increased DNA content per cell, missegregation of genetic markers during sporulation, and abnormal cell morphology, including increased size and irregular shape (4). Observation of this phenotype led to the hypothesis that Scp160p may function in regulating ploidy during cell division. More recently, Weber and colleagues demonstrated in vitro RNA binding activity of Scp160p using northwestern blot analyses; the protein was found to bind efficiently to ribohomopolymers and rRNA, but not to tRNA (5). Cell fractionation studies revealed that a large percentage of Scp160p associates with membrane-pellets, and is released by treatment with either 10 mM EDTA or 500 mM NaCl (5). While these authors interpreted these results to suggest that the nuclear envelope/ER localization of Scp160p was due primarily to interactions of the protein with membrane-bound polyribosomes, clear evidence of this association has not been reported (5). Currently, the relationship between the phenotype of scp160 null mutants and the RNA-binding activity of Scp160p remains unclear.
Scp160p demonstrates significant sequence homology (~23% identity and ~40% similarity at the amino acid level) to a class of vertebrate KH-domain proteins collectively known as vigilins. First identified in chicken, vigilin homologues have now been found in human (6), Xenopus laevis (7), Drosophila melanogaster (8), C.elegans (5) and Schizosaccharomyces pombe. While all of the vigilin proteins studied to date are reported to bind nucleic acid, both the type of nucleic acid bound and the functional significance of these interactions remain unclear. For example, Kruse and colleagues reported from their work with human cells in culture that vigilin may be involved in the binding and transport of tRNA (9–11). In contrast, Dodson and Shapiro concluded from their work with Xenopus vigilin that, in response to estrogen, the protein bound specifically to the 3′ UTR of vitellogenin mRNA (7,12), potentially stabilizing the message (13). Lastly, DDP1, the Drosophila homolog of vigilin, was reported recently to interact with the dodeca-satellite repeat regions of centromeric heterochromatin in embryonic and larval cell nuclei, suggesting a possible role for this protein in heterochromatin structure (8).
The goal of the present study was to begin elucidating the function of Scp160p in yeast by characterizing the subcellular distribution and macromolecular interactions of this protein. We have demonstrated that Scp160p in cytoplasmic lysates is associated predominantly with polyribosomes, and that following treatment with EDTA, Scp160p remains in an RNase/NaCl-sensitive complex of apparent molecular weight approximately ≥1300 kDa. Affinity purification of this complex revealed the presence of poly(A)-binding protein (Pab1p), a well-characterized component of eukaryotic mRNPs (14,15). Finally, a third abundant protein component of this complex was identified as Bfr1p, a protein not previously reported to associate with mRNPs. While the function of Bfr1p remains unknown, gene deletion reportedly leads to a phenotype remarkably similar to that of scp160 deletion (16). These results indicate a role for Scp160p in mRNA metabolism in yeast, and by extension, support results seen with Xenopus vigilin in its interactions with mRNA. To our knowledge, the data reported here demonstrate Scp160p to be the first example of a KH-domain protein that functions as a component of polyribosome-associated mRNP complexes in the yeast, S.cerevisiae.
MATERIALS AND METHODS
Plasmids, yeast strains and culture conditions
All recombinant DNA manipulations were performed according to standard techniques and utilized E.coli strain XL1-Blue (Stratagene). The yeast strain JFy1511, expressing FLAG–Scp160p, was derived by two-step gene replacement from strain yJJ52 (MATα gal7Δ102 ura3-52 trp1-289 ade1 lys1 leu2-3, 112; generously donated by Drs Mark Parthun and Judith Jaehning, University of Colorado Health Sciences Center), and confirmed by PCR. The wild-type SCP160 coding sequence was obtained by PCR amplification from a genomic DNA preparation from yJFK1 (MATα gal7Δ102 ura3-52 trp1-289 ade1 lys1 leu2-3, 112 ΔGAL80::URA3), using a 16:1 mixture of Taq (Fisher Biotech) and Pfu (Stratagene) DNA polymerases and the following primers: Scp160F1 (5′-GCCGGTCGA-CTAACTGCAATGTCTGAAGAACAAACCGCTATTG-3′) and Scp160R1 (5′-GCGCGTCGACGAGCTTGTCTATCTT-CTTAAGG-3′). A wild-type SCP160 genomic clone, containing 1 kb upstream and 300 bp downstream sequence was PCR amplified from yJFK1 genomic DNA using the primers Scp160F0 (5′-GCCGAGCTCACACCAGCTTTGTCCTGG-3′) and Scp160R2 (5′-GCGCAAGCTTGTGCGGTA-TCCCAGTCTATG-3′). The resultant clone was confirmed by dideoxy sequencing. The N-terminal FLAG and HA tags were added using PCR with the primers ScpFLAGF1(5′-CCAT-TATAACTGCAATGGACTACAAGGACGACGACGACGAC-AAGATGTCTGAAGAACAAACCGCTATTG-3′) and ScpHAF1 (5′-CCCCCTCCTGTCGACATTATAACTGCAATGCACCA-TCACCATCACCATTCTGAAGAACAAACCGCTATTG-3′) respectively. For integration, constructs containing the 1 kb upstream and 300 bp downstream sequence were subcloned into the plasmid YIplac211 (17) using SacI and HindIII restriction sites. The HA-tagged construct was subcloned into the low copy number plasmid, YCplac22 using SacI and HindIII restriction sites. The SCP160 deletion construct was made by removal of a 3.4 kb ApaI–KpnI fragment from the coding region of this construct, followed by treatment with Klenow fragment and re-ligation following attachment of BamHI linker oligonucleotides. Genomic integration of the plasmid sequences and subsequent removal of the endogenous SCP160 allele were achieved by standard two-step gene replacement techniques (18) and confirmed by PCR. The N-terminally HA-tagged allele of BFR1 was generated by PCR-amplifiction of the BFR1 locus from wild-type (W303) yeast genomic DNA using the primers BFR1HAF1 (5′-CCGCGGATCCATGTACC-CATACGACGTCCCAGACTACGCTATGTCCTCCCAAC-AACACAA-3′) and BFR1HINDR1 (5′-CCGCAAGCTTGTCG-ACTATTTCATATGCCACAGGAAACAG-3′), and subcloned into YIPlac211. The BFR1 promoter region was PCR-amplified in a similar manner using the primers BFR1SACF1 (5′-CCGCGAG-CTCAGCATTAAGCATTCACGAGC-3′) and BFR1BAMR1 (5′-CCGCGGATCCGGCAATGGCTGTGTTGTTAGA-3′) and subcloned into the appropriate position upstream of the HA–Bfr1p open reading frame in the plasmid backbone. The entire open reading frame was confirmed by dideoxy sequencing. Finally, the HA–BFR1 allele was substituted into the yeast genome in place of the native allele using linearization with SphI and standard two-step gene replacement techniques. All yeast transformations and culture manipulations were performed according to standard protocols as described elsewhere (19).
Confirmation of genomic integrations
All genomic integrations were confirmed by PCR amplifications from purified yeast genomic DNA. For scp160 deletion mutants, the primers Scp160PF (5′-GATTTCCTAACTTTCC-GTCTA-3′) and Scp160R5 (5′-GCGCAAGCTTCACCGCCTTATAACGAAGAC-3′) that flanked the deleted region were used.
Similarly, epitope tags on Scp160p were confirmed by PCR using primers that flanked the tag sequence. Positive clones were further confirmed by western blot analysis of crude cell lysates using the appropriate anti-tag antibodies. The presence of HA–Bfr1p in cells was confirmed by western blot analysis of soluble cell lysates using 12CA5 mAb (Boehringer Mannheim).
Polyribosome isolation
Polyribosomes were isolated using a combination of protocols described by Stansfield and colleagues (20) and by Dr Maurice Swanson (personal communication). In brief, a 100 ml culture of yeast was grown in either YEPD or Hartwell synthetic medium, to early-log phase (OD600 = 1.0), at which time cyclohexamide (Sigma) was added directly to the culture to a final concentration of 100 µg/ml. The culture was incubated on ice for 15 min, and cells were harvested by centrifugation (4000 r.p.m./10 min). Following two washes in 10 ml of water containing 100 µg/ml cyclohexamide, cells were lysed by vortex agitation with an equal volume of glass beads in 1 ml lysis buffer (25 mM Tris pH 7.2, 50 mM KCl, 30 mM MgCl2, 5 mM β-mercaptoethanol, 200 µg/ml cyclohexamide, 2 µg/ml aprotonin, 1 mM PMSF, 0.5 µg/ml leupeptin, 2.9 µg/ml E64, 1 µg/ml antipain, 0.2 µg/ml chymostatin). Lysate was transferred to a clean microfuge tube, and centrifuged 10 min at 3000 g at 4°C. The supernatant was again transferred to a clean microfuge tube and centrifuged at 12 000 g for 15 min at 4°C, and then assayed for absorbance at 260 nm.
Approximately 12 OD260 units were loaded onto a 11 ml 15–45% sucrose gradient made in 10 mM Tris, pH 7.4, 70 mM NH4Cl, 4 mM MgOAc, using a Gradient Master automatic system, and the gradient was centrifuged in a SW41ti rotor (Beckman) at 39 000 r.p.m. for 2.5 h. 0.5 ml fractions were collected using an Isco gradient fractionator, and gradient profiles were determined by monitoring absorbance at 254 nm. For EDTA controls, lysis buffer containing 5 mM MgCl2 was used, and 30 mM EDTA was added to the sample before loading onto the gradient. Where indicated, addition of 50 U/ml of RNase One (Promega) was performed prior to loading the sample onto the gradient.
Gel filtration chromatography
Gel filtration chromatography was performed using a 120 ml Hi-Prep S-300 Sephacryl column (Pharmacia) with a cut-off of 1300 kDa, attached to an FPLC system (Pharmacia). The column had been calibrated previously using the following molecular weight standards (Sigma): blue dextran (2000 kDa), thyroglobulin (669 kDa), apoferritin (443 kDa), alcohol dehydrogenase (150 kDa), and bovine serum albumin (66 kDa). Yeast were lysed as described for polyribosome analysis, with an additional clarification by passage through a 0.2 µm syringe-tip filter (Acrodisc) and run over the column at a rate of 0.5 ml/min in polyribosome lysis buffer following treatment with the indicated reagents. Fractions (2.0 ml) were collected, from which 12 µl were combined with sample buffer (2% SDS, 10% glycerol, 100 mM dithiothreitol, 60 mM Tris pH 6.8, 0.001% bromophenol blue) and analyzed by western blot using the indicated antibodies.
α-FLAG affinity chromatography
For most experiments, one liter yeast cultures were grown to early log-phase and harvested by centrifugation. Cells were washed twice in T75 buffer (25 mM Tris pH 7.5, 75 mM NaCl) and then were lysed by vortex agitation with an equal volume of glass beads in 4 ml T75 buffer containing 30 mM EDTA. Lysate was transferred to a clean microfuge tube, and centrifuged for 10 min at 3000 g at 4°C. The supernatant was again transferred to a clean microfuge tube and centrifuged at 12 000 g for 15 min at 4°C, and finally passed through a 0.2 µm syringe filter. Lysate was then pre-purified by running over the S-300 gel-filtration column in T75 buffer, with pooling of the void volume fractions (~10 ml total). For several experiments (Fig. 5A, B, E and F), a low concentration (<10 µg/ml) of FLAG peptide (N-DYKDDDDK-C) was added to this sample. This void material was then loaded onto a 1 ml M2 α-FLAG column (Sigma); column flow-through was passed over the column a second time. The column was then washed extensively with 75 ml T75 buffer; material bound to the column was eluted with 5 ml T75 containing 184 mg/ml FLAG peptide. The peptide solution was allowed to incubate on the column for 20 min prior to collection of the first 1 ml fraction; subsequent fractions were collected every 10 min. Samples of crude material, S-300 void, α-FLAG flow-through, first and last wash, and eluate fractions were analyzed by western blot using the indicated antibodies. For Figure 5A and E, samples of first and last wash and eluate fractions were concentrated, run on SDS–PAGE and stained with colloidal G250 Coomassie.
Figure 5.

α-FLAG affinity purification of Scp160p-containing complexes. Cell lysates expressing the indicated alleles of Scp160p were first passed over an S-300 gel filtration column, followed by an α-FLAG immunoaffinity column as described in the Materials and Methods section. Lanes: crude, starting material loaded onto S-300 column; void, pooled fractions containing void volume collected after S-300 run; FT, flow-through material after two passes over α-FLAG column; E1-4, 1 ml elution fractions collected after adding FLAG peptide. (A) Top, α-FLAG western blot, showing that FLAG–Scp160p appears predominantly in the first several fractions following treatment with elution buffer. Bottom, colloidal G250 Coomassie stained gel of concentrated samples. (B) α-Pab1p western, demonstrating co-isolation of Pab1p with Scp160p. (C) RNase treatment prior to α-FLAG purification disrupts the Pab1p interaction. Upper panel, α-FLAG western; lower panel, α-Pab1p western. (D) α-Pub1p western, demonstrates that this protein does not co-purify with Scp160p. (E) Negative control showing colloidal G250 Coomassie stained gel of fractions from a wild-type (non-FLAG–Scp160p) purification. (F) α-Pab1p western of negative control fractions, showing no detectable Pab1p signal. (G) Background levels of Pab1p non-specifically isolated during α-FLAG purification from yeast expressing an unrelated protein, FLAG–GALT. Since FLAG–GALT exists primarily as an 88 kDa dimer in vivo, lysates were not pre-purified over the S-300 column.
Concentration of samples
Where indicated, samples were concentrated using the method of Traub et al. (21). 400 µl of sample were transferred to a microfuge tube, and combined with 400 µl of methanol, and 100 µl of chloroform. Samples were then vortexed vigorously, and centrifuged for 5 min at 12 000 g. The supernatant was discarded, leaving the interface intact, and an additional 400 µl of methanol was added to each tube. Samples were then inverted several times, and centrifuged again for 5 min. The supernatant was discarded, and the protein pellet was air-dried and resuspended in 1× sample buffer.
Western blot analysis
Western blot analysis was performed essentially as described previously (19). Briefly, samples to be analyzed were mixed with sample buffer, boiled, electrophoresed through a 10% SDS–polyacrylamide gel, and electro-blotted onto nitrocellulose (Bio-Rad). FLAG–Scp160p fusion protein was detected by incubation of the filter with mouse M2 anti-FLAG monoclonal antibody (10 µg/ml final concentration), followed by HRP-conjugated sheep anti-mouse secondary antibody (Amersham), diluted 1:5000 as per the manufacturer’s instructions, and ECL reagent (Amersham), followed by exposure to X-ray film. HA-tagged Scp160p and HA-tagged Bfr1p were detected using the 12CA5 mAb (Boehringer Mannheim) at a final concentration of 0.8 µg/ml; Pab1p and Pub1p were detected using 1G1 mAb at 1:5000, and 4C3 mAb at 1:1000, respectively, both generous gifts from Dr Maurice Swanson (22,23). Where appropriate, films were analyzed by scanning densitometry (Molecular Dynamics), and quantitated using ImageQuant software (Molecular Dynamics).
Colloidal G250 coomassie staining
Procedure used was that of Neuhoff (24). Briefly, a one liter stock of staining solution was prepared containing 1 g Coomassie brilliant blue G-250 (Sigma), 100 g ammonium sulfate (Sigma), and 11.76 ml 85% phosphoric acid (Fisher). Following SDS–PAGE, gels were fixed in 40% methanol, 10% acetic acid for 10 min. Gels were then rinsed several times in water, then stained using 40 ml of the stock staining solution mixed with 10 ml methanol, for 2 h at room temperature. Stain was then poured off, and residual stain was removed by rinsing in water. By performing a standard analysis using bovine serum albumin, the stain was able to detect ~15 ng total protein per lane.
Pichia expression system
An N-terminally HIS6/FLAG-tagged allele of the SCP160 coding sequence was blunt-end sub-cloned into the BamHI/SnaBI sites of the Pichia expression vector pPIC3.5K (Invitrogen). The construct was then linearized using SalI and integrated into the genome of the Pichia strain GS115 (Invitrogen). High expression transformants were selected initially on histidine-deficient medium followed by selection on increasing concentrations of G418 (US Biological). Crude lysates of the resultant transformants were then confirmed by western blot analysis with the anti-FLAG antibody M2, and the best expressing strain was cultured in a fermenter, harvested, and lysed by agitation with glass beads. To purify Scp160p, 2 ml of crude lysate was first diluted to 10 ml with 25 mM Tris, pH 7.5, 1 M NaCl, then twice passed over a 1 ml α-FLAG affinity column. The column was then washed with 75 ml of 25 mM Tris, pH 7.5, 1 M NaCl, and then eluted with 184 µg/ml FLAG peptide prepared in the same buffer.
RESULTS
Expression of tagged Scp160p in yeast
An N-terminal FLAG-tagged form of Scp160p was created to facilitate detection of the protein in cells and extracts (Fig. 1A). To probe functionality of this fusion protein, we used two-step gene replacement (18) to substitute the modified allele into the SCP160 locus of haploid yeast, and then tested the morphological phenotype of the resultant cells. All strains were confirmed by PCR analysis of the SCP160 locus with appropriate primers (Materials and Methods), and expression of the tagged protein was confirmed by western blot analysis with the appropriate antibody (M2αFLAG) (Fig. 1B). In all cases, yeast expressing tagged Scp160p in place of the native protein appeared indistinguishable from the corresponding wild-type strains (data not shown). As a negative control, we also deleted almost the entire SCP160 coding region from the genomes of these yeast strains (Fig. 1A), and confirmed the expected mutant phenotype (data not shown).
Figure 1.

Epitope-tagged and deletion alleles of Scp160p expressed in yeast. (A) Diagram of FLAG-tagged and deletion alleles of SCP160 substituted into the yJJ52 haploid genome in place of the endogenous SCP160 allele. Replacement of the endogenous allele in both cases was confirmed by PCR of isolated genomic DNA as described in Materials and Methods. (B) Western blot analysis using anti-FLAG monoclonal antibody M2. Lane 1, cellular extract from normal yJJ52. Lane 2, an extract from yJJ52 cells modified to encode FLAG–Scp160p in place of their endogenous Scp160p (JFy1511). The solid arrow indicates the position of the ~160 kDa Scp160p band; the open arrow indicates an abundant, anti-FLAG cross-reacting yeast protein of unknown identity that is used as an internal control in several experiments.
Scp160p associates with polyribosomes
To test the hypothesis that Scp160p associates with polyribosomes we used sucrose gradient ultracentrifugation to size-fractionate subcellular components of lysates prepared from yeast expressing the N-terminal FLAG-tagged Scp160p protein. Western blot analyses of gradient fractions with an α-FLAG antibody (M2, Boehringer Mannheim), revealed a 160 kDa band that was most abundant in the denser fractions, consistent with the location of polyribosomes (>80S) (Fig. 2A, solid arrow). A convenient internal control for these gradients was provided by an unknown endogenous yeast cross-reacting protein at ~100 kDa, that exemplified the migration pattern of a free protein, appearing only in the upper fractions of the gradient (Fig. 2A, open arrow).
Figure 2.

Scp160p associates with polyribosomes. (A) Top panel shows OD254 profile of the 15–45% sucrose gradient. Arrows indicate peaks representing small (40S) and large (60S) ribosomal subunits, single ribosomes (80S) and polyribosomes (>80S). Bottom panel shows an α-FLAG western blot of gradient fractions concentrated and run on a 10% SDS–PAGE gel. Position of the 160 kDa band is indicated (solid arrow), as well as the non-specific. 100 kDa band which serves as an internal control (open arrow). (B) Gradient profile and western blot analysis of a sample pre-treated with 30 mM EDTA. (C) Gradient profile and western blot analysis of a sample pretreated with 50 U/ml RNase One.
To determine whether the migration pattern of Scp160p in these sucrose gradients truly reflected association with yeast polyribosomes, lysates were pre-treated with either 30 mM EDTA or 50 U/ml RNase One (Promega) immediately prior to sucrose gradient fractionation. EDTA chelates Mg2+ cations, resulting in the dissociation of the small and large ribosomal subunits, reflected in gradient profiles (OD254) by the disappearance of 80S monosomes and polyribosomes along with a marked increase in the abundance of free ribosomal subunits (Fig. 2B). Under these conditions, the greatest intensity of FLAG–Scp160p signal was seen only in the upper-most fractions of the gradient (Fig. 2B). Alternatively, pre-treatment of lysates with RNase, which results in inter-ribosomal severing of translating messages, gave rise to a large pool of single 80S ribosomes (Fig. 2C). Again, FLAG–Scp160p was shifted to the uppermost fractions of these gradients. Although some of the Scp160p signal was detected in fractions larger than 40S, the migration pattern of the 100 kDa cross-reacting protein in these experiments indicated that RNase treatment caused an apparent diffusion of material in the upper portion of the gradient.
Characterization of Scp160p-containing complexes following EDTA and RNase treatment
Gel-filtration chromatography was used to determine the apparent molecular weight of Scp160p following its release from polyribosomes by both EDTA and RNase treatment. Lysates were prepared as described above for sucrose gradient analysis, but were instead size fractionated over a Sephacryl S-300 Hi-Prep column, with an inclusion cut-off of 1300 kDa. As expected, Scp160p from untreated lysates eluted in the void volume, confirming its association with large complexes, ostensibly polyribosomes (Fig. 3A, solid arrow). In EDTA treated lysates, all Scp160p signal was still detected in the void volume, indicating it remained in a complex of >1300 kDa (Fig. 3B, solid arrow). In contrast, limited RNase treatment (10 min) resulted in the appearance of an Scp160p-containing species of ~450 kDa, in addition to a fraction still visible in the void (Fig. 3C, solid arrow). More extensive RNase treatment (30 min) led to complete conversion to the 450 kDa species (Fig. 3D, solid arrow). Sequential treatment, first with RNase, then with EDTA, also resulted in a 450 kDa species (data not shown), suggesting that all components of this apparent 450 kDa complex were also present following EDTA treatment alone. As before, the 100 kDa, endogenous α-FLAG cross-reacting protein (Fig. 3, open arrows) served as a convenient internal control, eluting from the column at a volume consistent with its expected monomeric size.
Figure 3.
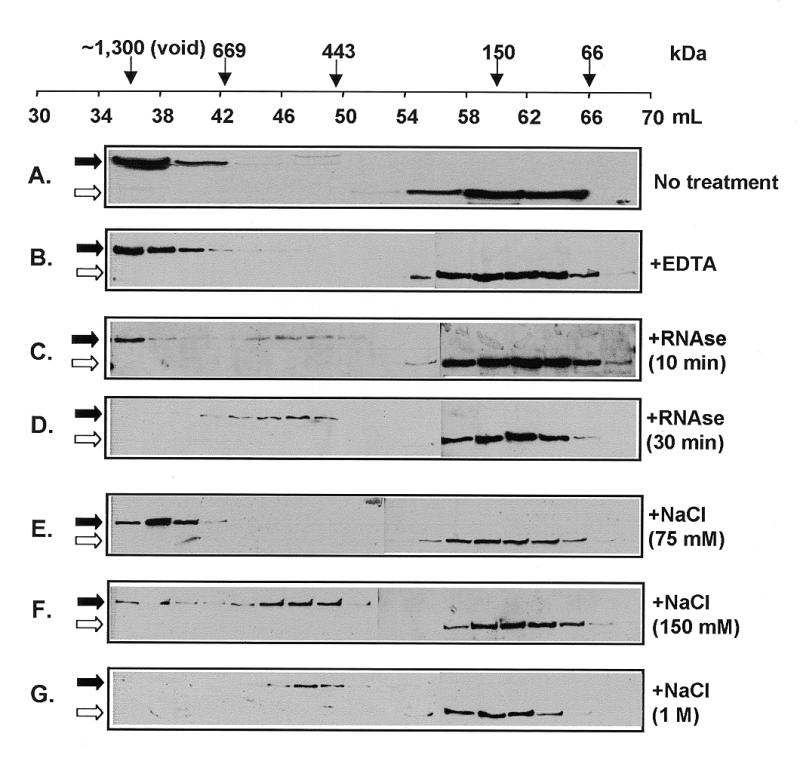
Gel-filtration analysis of Scp160p-containing complexes. Cell lysates from yeast expressing FLAG–Scp160p were passed over a 120 ml S-300 Sephacryl column, and 2 ml fractions [except (A) where fractions were 4 ml] collected between 35 and 70 ml were assayed by α-FLAG western. Solid arrows indicate the position of the 160 kDa Scp160p band, the open arrows indicate the 100 kDa endogenous cross-reacting band. (A) No treatment, showing presence of FLAG–Scp160p in the void fractions. (B) Treatment with 30 mM EDTA, showing retention of Scp160p in the void fractions. (C) Treatment with 50 U/ml RNase for 10 min at room temperature, partially releasing a ~450 kDa complex. (D) Thirty minute RNase treatment, showing only the 450 kDa complex. (E) Treatment with 75 mM NaCl (F). Treatment with 150 mM NaCl. (G) Treatment with 1 M NaCl.
To characterize further the stability of the large (apparent molecular weight >1300 kDa), EDTA-resistant complex, lysates were treated with increasing concentrations of NaCl prior to size fractionation (Fig. 3, panels E, F and G). As shown in Figure 3E, at 75 mM NaCl, the large complex remained intact. However, at a NaCl concentration of 150 mM, the Scp160p complex was partially reduced to 450 kDa, with some signal still remaining in the void (Fig. 3F, solid arrow). Following treatment with 1 M NaCl, Scp160p was only visible as the 450 kDa species (Fig. 3G, solid arrow). The fact that RNase treatment and high salt both generated Scp160p species of similar apparent molecular weight suggests that these high salt concentrations resulted in the disassociation of RNA, and perhaps other components, from Scp160p.
Pab1p and Bfr1p are present in EDTA-resistant Scp160p-containing complexes
To determine if the >1300 kDa Scp160p-containing complexes remaining after EDTA treatment were mRNPs, we assayed for the presence of the yeast poly(A) binding protein, Pab1p, following α-FLAG affinity purification as illustrated in Figure 4. Pab1p is an abundant and well-characterized component of mRNP complexes in yeast, as well as higher eukaryotes (14,15,25). As seen in Figure 5A and B, Pab1p did co-purify with FLAG–Scp160p; moreover, treatment with RNase immediately prior to FLAG-purification completely abolished this interaction (Fig. 5C), indicating an RNA-dependant association between these two proteins.
Figure 4.

Schematic representation of the FLAG–Scp160p purification protocol.
We also probed the isolated complexes for the presence of another abundant mRNP-component protein, Pub1p (23,26). Pub1p, as opposed to Pab1p, is not reported to associate with polyribosomes, and is hypothesized to bind a pool of non-translatable mRNAs (23). As predicted, Pub1p did not co-purify with Scp160p (Fig. 5D). The absence of Pub1p in Scp160p-containing complexes served as a negative control, demonstrating that abundant RNA-binding proteins did not co-purify non-specifically by this protocol. In addition, we performed mock purifications using lysates from yeast expressing the wild-type allele of SCP160 without the FLAG epitope (Fig. 5F), or with an unrelated FLAG-tagged protein, human galactose 1-phosphate uridylyltransferase (GALT) (Fig. 5G, lower panel). In neither case did Pab1p bind and elute from the FLAG affinity column. Finally, no bands were visible by colloidal G250 Coomassie staining of samples from the mock (wild-type) preparations (Fig. 5E), further demonstrating the specificity of this procedure.
By colloidal G250 Coomassie staining (Materials and Methods) of the FLAG-purified complex, we observed in addition to Scp160p, two major bands, at ~70 kDa and ~55 kDa, which ostensibly represent co-purifying proteins (Fig. 5A, bottom panel). Western blot analysis suggested that the ~70 kDa species is Pab1p. To identify the 55 kDa protein, that band was excised from the gel, and sent to the Keck microchemical facility at Yale University for analysis by in gel tryptic digestion, followed by MALDI-mass spectrometry. The results of these analyses clearly identified the unknown protein as Bfr1p. To confirm this result, an N-terminal HA tag was engineered onto the genomic BFR1 locus in strains expressing FLAG-tagged or wild-type Scp160p. Scp160p complexes were then isolated from both of these strains using the protocol described above. As seen in Figure 6, an HA-tagged protein of ~55 kDa (center panel) appears in α-FLAG column elutions only when FLAG–Scp160p is also present (top panel). No signal is visible in mock purifications of extracts where Scp160p is not FLAG-tagged (bottom panel).
Figure 6.
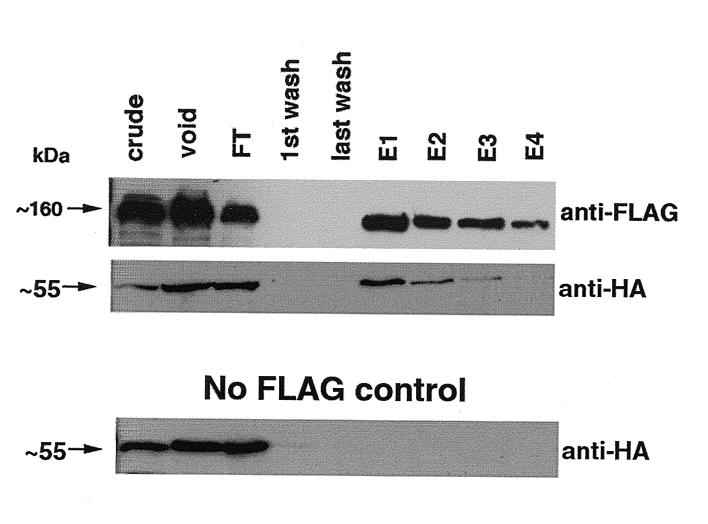
Bfr1 associates with Scp160p complexes. Yeast expressing either FLAG–Scp160p and HA–Bfr1p together, or HA–Bfr1p in the presence of native, untagged Scp160p, were lysed and subjected to the purification protocol described in Figure 4. Top, α-FLAG western blot. Center, α-HA western blot showing co-elution of HA–Bfr1. Bottom, α-HA western blot showing the absence of HA–Bfr1p in elutions from lysates lacking FLAG–Scp160p.
Monomeric Scp160p migrates as a ~450 kDa protein under native conditions
As a first step to characterize the post RNase/NaCl 450 kDa Scp160p species we compared it with Scp160p over-expressed and purified from an exogenous host, the yeast Pichia pastoris. The purified N-terminal FLAG/hexahistidine-tagged Scp160p was run over an S300 gel filtration column; 2 ml fractions were collected and analyzed by α-FLAG western blot. As seen in Figure 7A, purified Scp160p eluted at a volume consistent with a >443 kDa protein. To confirm that no other proteins were associated with the purified Scp160p, 12 µl of the protein was run on SDS–PAGE and subjected to colloidal G250 Coomassie staining before loading on the S-300 column (Fig. 7B). As shown, no bands other than Scp160p were visible.
Figure 7.
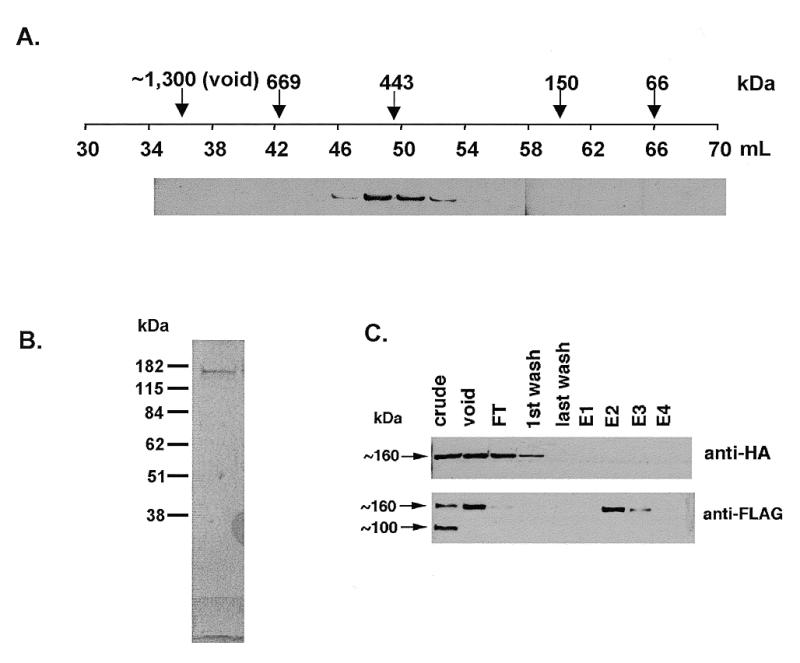
Characterization of the 450 kDa Scp160p species. (A) α-FLAG western of FLAG–His6–Scp160p expressed and purified from Pichia, and then run on the S-300 gel filtration column. (B) Twelve microliters of purified FLAG–His6–Scp160p was run on SDS–PAGE and stained with colloidal G250 Coomassie. As shown, no other proteins were detected in stoichiometric quantities with Scp160p. (C) Top, α-HA western blot of purified extract from yeast co-expressing both FLAG–Scp160p and HA–Scp160p. No HA–Scp160p appears to co-isolate during the α-FLAG purification. Bottom, α-FLAG western blot of the same samples showing that FLAG–Scp160p was successfully isolated.
Several KH-domain proteins, including Sam68 and FMRP, have been reported to form homodimers in vivo (27). Therefore, we explored the possibility that the ~450 kDa Scp160p species might contain two or more copies of Scp160p. To test this hypothesis, yeast expressing FLAG–Scp160p were transfected with a centromeric plasmid, YCplac22–HA–Scp160p, encoding a distinct, HA-tagged allele of the protein. The functionality of this tagged protein had previously been demonstrated by polyribosome association as well as complementation of the null morphological phenotype in cells expressing only HA–Scp160p (data not shown). As seen in Figure 7C, when yeast co-expressing the two alleles were lysed and the FLAG-tagged protein was isolated as described above, no HA–Scp160p co-purified with the FLAG–Scp160p. These data strongly suggest that Scp160p does not form homo-dimers or higher-order multimers in vivo, and that the ~450 kDa species of Scp160p observed is simply the monomeric protein.
DISCUSSION
The data presented here demonstrate two main points regarding the biochemical associations and function of Scp160p in yeast. First, the sucrose gradient fractionation data clearly demonstrate that Scp160p exists primarily associated with large complexes, likely to be polyribosomes. To our knowledge, this is the first time a vigilin family member has been shown to associate with polyribosomes in this manner. Interestingly, Weber and colleagues did not observe sucrose gradient fractionation data consistent with polyribosome association of Scp160p (5). These authors hypothesized that Scp160p associates only with membrane-bound polyribosomes, and that Scp160p is not found complexed with cytosolic polyribosomes (5). Our data do not support this hypothesis, since unlike Weber and colleagues, we were able to detect significant amounts of Scp160p associated with polyribosomes without using detergents during preparation. There are several possible explanations for this discrepancy. First, the lysate buffer utilized by that group contained 100 mM NaCl, whereas our buffer contained 50 mM KCl. Our data shown in Figure 3 suggest that 100 mM NaCl may have caused an instability in the Scp160p–RNA interaction, leading to release from polyribosomes during their procedure. Secondly, Weber’s lysate buffer reportedly contained heparin to inhibit ribonucleases; a number of reports (28,29) suggest that heparin can disrupt RNA–protein interactions. Although we have not in the past included heparin in any of our experiments, we have observed disruption of the polyribosome association in buffer containing residual diethylpyrocarbonate, another inhibitor of ribonucleases (data not shown).
Second, our data provide compelling evidence that Scp160p is released from polyribosomes as a component of an mRNP complex. Since Scp160p does not remain associated with either single ribosomes or ribosomal subunits following treatment with EDTA or RNase, it seems unlikely that Scp160p is a constitutive component of the translational machinery. Partial purification of the EDTA-resistant Scp160p complexes allowed us to demonstrate the presence of Pab1p but not Pub1p in these preparations, which is consistent with the idea that Scp160p associates with polyribosomes as a component of mRNP complexes. Previously, Scp160p had been shown to bind ribohomopolymers and rRNA in vitro, although the in vivo nucleic acid targets of Scp160p were unknown (5). The presence of Pab1p in RNase-sensitive Scp160p complexes indicates that Scp160p is primarily bound to polyadenylated RNAs. Whether or not Scp160p binds only specific sets of mRNAs will be the subject of future studies.
In addition to Scp160p and Pab1p, we have identified a third component of the complex, the protein Bfr1p. The gene, BFR1, was originally identified in a screen for high-copy suppressors of Brefeldin-A induced lethality, suggesting a role in the secretory pathway (16). Interestingly, however, bfr1 null mutants do not demonstrate any defects in the secretory pathway, but rather display similar phenotypes to scp160 null mutants, most notably increased ploidy and increased cell size (16). It is therefore unclear whether Bfr1p is capable of functioning in both RNA metabolism and secretion directly, or if the Scp160p–Bfr1p complex regulates the expression of one or more secretory genes at the post-transcriptional level. Additionally, two-hybrid analysis indicated an interaction of Bfr1p with the protein Bbp1p, a component of the mitotic spindle apparatus (30). While Bbp1p is an essential gene, overexpression leads to a phenotype similar to both bfr1 and scp160 null strains (30). Future work will address the functional relationship between Scp160p and Bfr1p including the possibility that these proteins represent a regulatory mechanism connecting the translational machinery with cell division and/or the secretory pathway.
Following treatment with either RNase or >150 mM NaCl, Scp160p remained as an apparent ~450 kDa species. Considering that purified Scp160p alone also migrates at this size, we conclude that this apparent complex may consist only of Scp160p, although the presence of other small components cannot be ruled out at this time. Furthermore, by utilizing yeast co-expressing two distinct epitope-tagged versions of Scp160p, we have ruled out the possibility of self-association of Scp160p. Although both tagged proteins appear functional, it remains a formal possibility that the tags somehow prevented formation of FLAG–HA hetero-complexes. Nonetheless, it seems most likely that native Scp160p monomers may simply migrate aberrantly under native conditions due to an unusual shape or some other physical property.
In conclusion we have shown convincing evidence that Scp160p exists in yeast cytoplasmic extracts primarily associated with polyribosomes. We have purified Scp160p following disruption of polyribosomes with EDTA, and identified two associated proteins: Pab1p and Bfr1p. The presence of Pab1p suggests that Scp160p associates with polyribosomes as a component of an mRNP, demonstrating Scp160p to be the first S.cerevisiae multiple-KH domain protein characterized to function in this way.
Acknowledgments
ACKNOWLEDGEMENTS
We gratefully acknowledge the assistance of the following people: Dr Maurice Swanson for Pab1p and Pub1p monoclonal antibodies; Gunther Boekhoudt for assistance in constructing the SCP160 deletion allele; Dr Yue Feng and Devin Absher for assistance in polyribosome analysis; Dr Keith Wilkinson for materials and assistance in α-FLAG purification; Heather Brewster, Melanie Marson and Kristen Riehman for excellent technical assistance; Keely Solomon and Deanna Greene for Pichia expressed protein; Drs Anita Corbett, Yue Feng and Gerald Shadel for critical reading of the manuscript; Dr Steve Warren, Dr Maurice Swanson, Travis Wohlers, Dr Lance Wells, Dr Boots Quimby, Dr Anita Corbett and Dr Yue Feng for many helpful discussions. This research was supported by NIH grant 1P01HD35576-010006 to Dr Steve Warren (Program Director) and J.F.K. (PI). B.D.L. was supported in part by PHS predoctoral training grant GM08367.
REFERENCES
- 1.Siomi H., Matunis,M.J., Michael,W.M. and Dreyfuss,G. (1993) Nucleic Acids Res., 21, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Draper B.W., Mello,C.C., Bowerman,B., Hardin,J. and Priess,J.R. (1996) Cell, 87, 205–216. [DOI] [PubMed] [Google Scholar]
- 3.Ashley C.T. Jr, Wilkinson,K.D., Reines,D. and Warren,S.T. (1993) Science, 262, 563–566. [DOI] [PubMed] [Google Scholar]
- 4.Wintersberger U., Kuhne,C. and Karwan,A. (1995) Yeast, 11, 929–944. [DOI] [PubMed] [Google Scholar]
- 5.Weber V., Wernitznig,A., Hager,G., Harata,M., Frank,P. and Wintersberger,U. (1997) Eur. J. Biochem., 249, 309–317. [DOI] [PubMed] [Google Scholar]
- 6.Plenz G., Kugler,S., Schnittger,S., Rieder,H., Fonatsch,C. and Muller,P.K. (1994) Hum. Genet., 93, 575–582. [DOI] [PubMed] [Google Scholar]
- 7.Dodson R.E. and Shapiro,D.J. (1997) J. Biol. Chem., 272, 12249–12252. [DOI] [PubMed] [Google Scholar]
- 8.Cortes A., Huertas,D., Fanti,L., Pimpinelli,S., Marsellach,F.X., Pina,B. and Azorin,F. (1999) EMBO J., 18, 3820–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klinger M.H.F. and Kruse,C. (1996) Ann. Anat., 178, 331–335. [DOI] [PubMed] [Google Scholar]
- 10.Kruse C., Grunweller,A., Notbohm,H., Kugler,S., Purschke,W.G. and Muller,P.K. (1996) Biochem. J., 320, 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kruse C., Grunweller,A., Willkomm,D.K., Pfeiffer,T., Hartmann,R.K. and Muller,P.K. (1998) Biochem. J., 329, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodson R.E. and Shapiro,D.J. (1994) Mol. Cell. Biol., 14, 3130–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brock M.L. and Shapiro,D.J. (1983) Cell, 34, 207–214. [DOI] [PubMed] [Google Scholar]
- 14.Adam S.A., Nakagawa,T., Swanson,M.S., Woodruff,T.K. and Dreyfuss,G. (1986) Mol. Cell. Biol., 6, 2932–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sachs A.B., Bond,M.W. and Kornberg,R.D. (1986) Cell, 45, 827–835. [DOI] [PubMed] [Google Scholar]
- 16.Jackson C.L. and Kepes,F. (1994) Genetics, 137, 423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gietz R.D. and Sugino,A. (1988) Gene, 74, 527–534. [DOI] [PubMed] [Google Scholar]
- 18.Guthrie C. and Fink,G. (eds) (1991) Guide to Yeast Genetics and Molecular Biology. Vol. 194. Methods in Enzymology. Academic Press, Inc., San Diego, CA. [PubMed]
- 19.Fridovich-Keil J.L., Quimby,B.B., Wells,L., Mazur,L.A. and Elsevier,J.P. (1995) Biochem. Mol. Med., 56, 121–130. [DOI] [PubMed] [Google Scholar]
- 20.Stansfield I., Grant,C.M., Akhmaloka and Tuite,M.F. (1992) Mol. Microbiol., 6, 3469–3478. [DOI] [PubMed] [Google Scholar]
- 21.Traub L.M., Kornfeld,S. and Ungewickell,E. (1995) J. Biol. Chem., 270, 4933–4942. [DOI] [PubMed] [Google Scholar]
- 22.Anderson J.T., Wilson,S.M., Datar,K.V. and Swanson,M.S. (1993) Mol. Cell. Biol., 13, 2730–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson J.T., Paddy,M.R. and Swanson,M.S. (1993) Mol. Cell. Biol., 13, 6102–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neuhoff V., Arold,N., Taube,D. and Ehrhardt,W. (1988) Electrophoresis, 9, 255–262. [DOI] [PubMed] [Google Scholar]
- 25.Sachs A. (1990) Curr. Opin. Cell Biol., 2, 1092–1098. [DOI] [PubMed] [Google Scholar]
- 26.Matunis M.J., Matunis,E.L. and Dreyfuss,G. (1993) Mol. Cell. Biol., 13, 6114–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen T., Damaj,B.B., Herrera,C., Lasko,P. and Richard,S. (1997) Mol. Cell. Biol., 17, 5707–5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwon Y.K., Murray,M.T. and Hecht,N.B. (1993) Dev. Biol., 158, 99–100. [DOI] [PubMed] [Google Scholar]
- 29.Ladomery M. and Sommerville,J. (1994) Nucleic Acids Res., 22, 5582–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue Z., Shan,X., Sinelnikov,A. and Melese,T. (1996) Genetics, 144, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]