

Abstract
Antisense oligonucleotide (ASO) therapeutics are being investigated for a broad range of neurological diseases. While ASOs have been effective in the clinic, improving productive ASO internalization into target cells remains a key area of focus in the field. Here, we investigated how the delivery of ASO-loaded lipid nanoparticles (LNPs) affects ASO activity, subcellular trafficking, and distribution in the brain. We show that ASO-LNPs increase ASO activity up to 100-fold in cultured primary brain cells as compared to non-encapsulated ASO. However, in contrast to the widespread ASO uptake and activity observed following free ASO delivery in vivo, LNP-delivered ASOs did not downregulate mRNA levels throughout the brain after intracerebroventricular injection. This lack of activity was likely due to ASO accumulation in cells lining the ventricles and blood vessels. Furthermore, we reveal a formulation-dependent activation of the immune system post dosing, suggesting that LNP encapsulation cannot mask cellular ASO backbone-mediated toxicities. Together, these data provide insights into how LNP encapsulation affects ASO distribution as well as activity in the brain, and a foundation that enables future optimization of brain-targeting ASO-LNPs.
Keywords: MT: Delivery Strategies, antisense oligonucleotide, delivery, gymnosis, lipid nanoparticles, central nervous system, distribution, trafficking, immunoelectron microscopy
Graphical abstract
Here, Byrnes and colleagues examine how lipid nanoparticle (LNP) encapsulation affects antisense oligonucleotide (ASO) delivery to the brain. Compared with free ASO uptake, LNP delivery differentially regulated ASO distribution, activity, and immune cell function. Thus, this study reveals remaining challenges and opportunities for treatment of neurodegenerative disorders using ASO-LNPs.
Introduction
The use of oligonucleotides as therapeutic agents has enabled unprecedented growth in the development of new treatments for patients with devastating central nervous system (CNS) disorders. Of the platforms being investigated for use in the brain, antisense oligonucleotides (ASOs) are particularly promising due to their capacity to differentially regulate genes, ability to enter cells without transfection reagents, and broad brain distribution following injection.1 However, many basic mechanistic questions regarding ASO internalization, subcellular trafficking, distribution, and modes of enhancing delivery remain largely unaddressed.
ASOs are synthetic, chemically modified single-stranded nucleic acids ranging from 12 to 30 nucleotides in length. Their structure and sequence facilitate RNA binding, which promotes ASO design-dependent changes in gene expression. ASOs with fully modified sugar rings typically regulate non-degradative mechanisms such as translational inhibition or alternative splicing.2 For example, SPINRAZA (nusinersen) induces alternative splicing of SMN2 to produce functional protein and was approved for the treatment of spinal muscular atrophy.3 Alternatively, ASO gapmers typically promote RNase H1 recruitment, which targets RNA for degradation and reduces protein expression.2 Recent years have seen a marked increase in ASO development for the treatment of various neurodegenerative diseases.4 This trend is partially driven by advances in backbone and sugar ring modifications that enhance ASO stability, circulation time, and protein binding.5 Another major advantage of ASOs over other oligonucleotide platforms is their ability to enter cells and tissues without the need for delivery vehicles.6 In cell culture models, naked ASOs are taken up by gymnosis and traffic through the endolysosomal pathway, ultimately destined for accumulation in lysosomes.7 While ASOs are actively endocytosed, only a small fraction of internalized ASO escapes the endolysosomal system to become effective for RNA degradation in the cytoplasm or nucleus.8,9 Thus, understanding how ASOs traffic within cells and increasing ASO endosomal escape are key for further therapeutic development.
Lipid nanoparticle (LNP) encapsulation can be used to enhance oligonucleotide delivery and potency.10 LNPs are composed of four major components—ionizable lipids, helper phospholipids, cholesterol, and polyethylene glycol-lipids (PEG-lipids)—that can be screened in various proportions to create oligonucleotide-particle formulations with ideal drug delivery properties.11 When optimally formulated, LNPs not only prevent cargo degradation and increase circulation time, but can also promote oligonucleotide uptake, enhance intracellular transport, and facilitate endosomal escape.12 LNPs have historically been used to deliver cell-impenetrable oligonucleotides such as short-interfering RNA (siRNA) and mRNA to a range of cell types as well as tissues, including the brain. While there currently are no brain-targeting oligonucleotide-LNP formulations in the clinic, the potential of siRNA- and mRNA-LNP formulations were realized upon approval of ONPATTRO and the SARS-CoV-2 vaccines, respectively.13,14 Brain delivery studies to date have focused on the development of siRNA-LNPs,15,16 with particular emphasis on creating blood-brain barrier (BBB) penetrant formulations.17,18,19 Accordingly, little is known about how encapsulating ASOs in LNPs affects ASO activity, delivery, and distribution in the brain. Furthermore, whether LNP encapsulation prevents ASO-mediated immune stimulation and inflammation has not been actively investigated.20,21 As ASOs are currently delivered at high doses intrathecally (i.t.) in patients, a comprehensive study examining the impact of LNP delivery on ASO cellular uptake, trafficking, and potency in biologically relevant CNS cell types would be invaluable for advancing ASO delivery strategies.
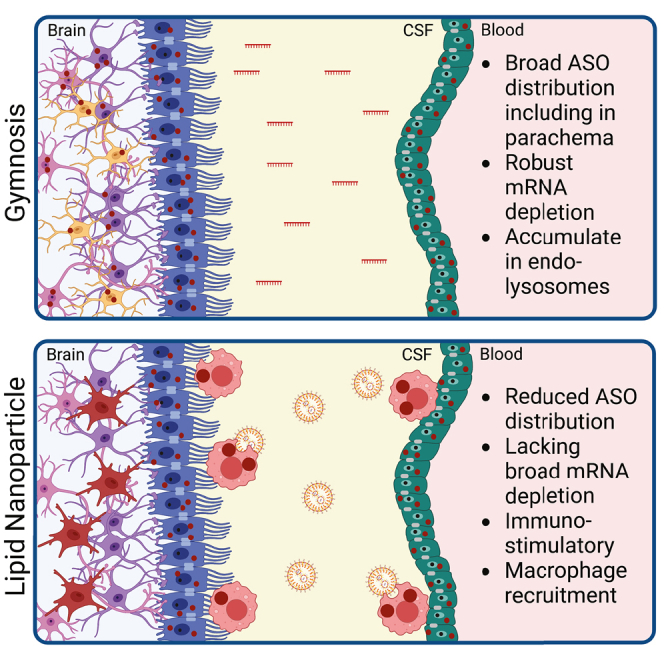
Here, we investigated how LNP delivery affects ASO trafficking, activity, and distribution in the brain. We found that ASO potency was dramatically increased in four CNS cell types in vitro after LNP encapsulation using a formulation optimized for neuronal siRNA delivery. This enhancement was mediated by a cell-type-specific decrease in lysosomal trafficking or enhanced uptake. We then screened ASO-LNP formulations containing unique combinations of ionizable and PEG-lipids in cells, and selected top-performing candidates for injection in mice. ASO delivery via both gymnosis and LNPs triggered an ASO-mediated microgliosis response that was evident by increased Iba1 signal and transcriptional activation of inflammatory pathways in the brain. These data indicate that ASO backbone toxicities are not easily masked by LNP encapsulation. While ASO-LNPs were internalized in a formulation-dependent manner, mRNA transcript levels were not downregulated in bulk brain tissue. Immunohistochemistry (IHC) and immunoelectron microscopy (immuno-EM) analysis of fixed brain samples revealed that this inactivity was likely due to changes in cellular ASO distribution within the tissue. While gymnotically internalized ASO was distributed broadly throughout the brain, LNP-delivered ASOs accumulated in cells lining the blood vessels and ventricles. Together, these data provide a framework for understanding how LNP encapsulation affects ASO brain delivery and reveal challenges in using ASO-LNPs for treating neurodegenerative disorders.
Results
Gymnotic TA1 delivery promotes mRNA knockdown in major brain cell types
The development of ASO therapies for the treatment of CNS disorders has focused on optimizing ASO delivery in the absence of carriers or conjugation, termed gymnosis. While these studies have been vital to translating ASO therapeutics into the clinic, a comprehensive study examining how delivery vehicles impact ASO function is lacking. Thus, we focused this study on determining how ASO cellular internalization, trafficking, and distribution affect mRNA knockdown activity, and whether LNP encapsulation differentially regulates these processes. We utilized two TMEM106b-targeting ASOs, TA1 and TA2, as tool compounds. While these ASOs have similar potencies in vitro and in vivo (Figures S1A and S1B), TA2 is moderately toxic in vivo with mice showing a mild reduction in movement corresponding to an acute toxicity score of two and modest weight loss (Figures S1C and S1D). Thus, we utilized TA1 to examine how ASO gymnosis correlates with cell-type-specific activity in mice, and TA2 to test how LNP delivery affects ASO activity and backbone-mediated toxicities in vivo.
We first examined gymnotic ASO distribution and activity in the brain following a single intracerebroventricular (i.c.v.) bolus injection of saline or 100 μg TA1 in mice (Figures 1A–1J). Treated mice tolerated TA1 well post-administration with normal grooming behaviors noted, mild lethargy observed, and normal weight gain measured relative to saline-injected animals (Figures 1B and 1C). We then characterized the distribution of TA1 in the brain by immunofluorescence (IF) using an anti-ASO antibody recognizing the phosphorothioate backbone (Figures 1D and 1E). As expected, TA1 distributed throughout the brain quickly with broad ASO distribution observed 2 days post injection (Figure 1D). In addition, TA1 colocalized with both neurons and glial cells 14 days post injection, indicating that ASO was internalized and maintained in multiple CNS cell types throughout the duration of this study (Figures 1E and S1E). These data are consistent with previous studies showing that locked nucleic acid (LNA)-ASOs are stable, which correlates with a long half-life in mouse tissues.22 To assess whether cellular ASO internalization correlates with ASO activity, we coupled fluorescence-activated cell sorting (FACS) with qRT-PCR gene expression analysis. Fourteen days post i.c.v. injection of saline or TA1 (100 μg), we FACS sorted pure populations of neurons, microglia, and astrocytes using NeuN, CD11b, and GFAP as cell-type-specific markers, respectively, and compared TMEM106b mRNA expression levels with unsorted brain samples (Figure 1F, final gates shown). qRT-PCR analysis on unsorted cells revealed that TMEM106b mRNA was reduced by 73% following bolus injection (Figure 1G). While TMEM106b mRNA expression was similarly reduced in sorted neuron and astrocyte samples (53% vs. 49%, respectively), microglia showed a trend of increased ASO activity with a 65% reduction in mRNA measured (Figures 1H–1J). Taken together, these results show that gymnotic ASO delivery leads to long-lasting, widespread ASO distribution and the concomitant reduction of mRNA transcript levels in all major brain cell types in vivo.
Figure 1.

ASOs taken up via gymnosis are active in neurons, microglia, and astrocytes following i.c.v. injection
(A) C57BL/6N mice were i.c.v. injected with saline or 100 μg TA1, and analyzed over 14 days. Illustration created using BioRender.com. (B) Acute toxicity 1 h post injection. (C) Percent body weight change normalized to day 0, showing no acute toxicity over the course of the experiment. Error bars cannot be visualized as plotted. (D and E) Representative IF images of brain sections stained for TA1. (D) Coronal brain section stained 2 days post injection of 100 μg TA1. Scale bars, 2 mm and 500 μm (inset). (E) TA1 colocalizes with NeuN (left) as well as Iba1 (right) 14 days post injection of 100 μg TA1. Scale bars, 100 μm. (F–J) Neurons, microglia, and astrocytes were FACS sorted and analyzed for TMEM106b mRNA expression levels 14 days post saline (n = 4) or TA1 (100 μg, n = 4) i.c.v. injection. (F) Gating strategy for cell-specific FACS sorting. (G–J) Relative TMEM106b mRNA expression levels in unsorted brain (G), and sorted neurons (H), microglia (I), or astrocytes (J) as assessed by qRT-PCR. Data were normalized to actin- and saline-treated samples. For all panels, mean ± SEM are shown. Unpaired two-tailed Student’s t test, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Gymnotic TA2 delivery leads to efficient mRNA knockdown in primary cortical neurons, microglia, astrocytes, and OPCs
To further understand the mechanisms underlying free ASO uptake and trafficking in the brain, we cultured and assayed TA2-treated primary murine cortical neurons, microglia, astrocytes, and oligodendrocyte precursor cells (OPCs). qRT-PCR analysis of TMEM106b transcript levels revealed TA2 (10 μM) specifically targeted and reduced TMEM106b mRNA expression in all CNS cell types examined after 3 days of treatment as compared to non-targeting ASO NC1 (Figure 2A). Furthermore, TA2 activity was dose- and time-dependent, with microglia being the most sensitive to TA2 (Figures 2B and 2C). These data are consistent with our in vivo findings, and support the use of in vitro primary cell culture model systems to study ASO cellular uptake and activity. To determine where ASO localizes after free uptake, we treated cells with a high dose (EC90-95) of TA2 for 3 days and analyzed ASO localization by IF using an anti-ASO antibody (Figures 2D and S1F). TA2 localized in discrete intracellular puncta resembling endolysosomal vesicles in all cell types examined. While TA2 puncta were predominantly clustered perinuclear in microglia and astrocytes, OPCs had more uniformly distributed ASO signal. Interestingly, TA2-positive vesicles were found in neuronal cell bodies and dendrites with only little accumulation observed in axons. Given that ASO therapeutics are used for a broad range of neurodegenerative disease models, we next asked whether modulating cellular activity affects ASO potency (Figures 2E–2J). Neurons were pre-treated with bicuculline, tetrodotoxin (TTX), potassium chloride (KCl), or glutamate before TA2 addition at increasing doses. We then assessed TMEM106b expression after 3 days of TA2 treatment to evaluate potential changes in dose-response. However, no dramatic change in TA2 potency was observed when compared with non-treated neurons (Figures 2E–2H). Interestingly, there was also little change in TA2 dose-response in microglia after M1 or M2 polarization with either LPS or IL-4+IL-13 treatment, respectively (Figures 2I and 2J). In addition, microglia were not activated after NC1 or TA2 treatment as measured using Luminex cytokine release panels (Figure S2). These data reveal that ASO potency may be mostly regulated by cell type rather than cellular activity or polarization state.
Figure 2.

LNP delivery increases ASO potency in murine primary CNS cell cultures
(A–C) TMEM106b mRNA expression in cortical neurons (teal), microglia (burgundy), astrocytes (blue), and OPCs (salmon) as assessed by qRT-PCR. (A) Cells were treated with 10 μM NC1 or TA2, and TMEM106b mRNA expression was analyzed after 3 days. (B) Dose-response curves analyzing TMEM106b mRNA expression after 3 days of TA2 treatment. (C) TMEM106b mRNA expression after 0, 4, 8, 16, 24, 48, 72, 144, and 192 h of treatment with 500 nM TA2. (D) Representative z-projection IF images of cells treated with 5 μM TA2 for 3 days (EC90-95). Scale bars, 25 μm. (E–H) TA2 dose-response curves generated in neurons treated for 3 days with 20 μM bicuculline (E), 1 μM TTX (F), 5 mM KCl (G), or 5 μM glutamate (H) and compared with non-treated (NT) cultures (teal). (I and J) TA2 dose-response curves generated in microglia treated for 3 days with 1× LPS (I) or 20 ng/mL IL-4+IL-13 (J) and compared with NT cultures (burgundy). (K) Cells were treated with 2, 4, 20, 100, and 500 nM TA2-LNPs (dashed) and compared with gymnosis controls (solid), showing increased potency with LNP delivery. TMEM106b mRNA expression was analyzed by qRT-PCR after 3 days. (L) Representative z-projection IF images of cells treated with 100 nM TA2-LNPs for 3 days (EC90-95). Scale bars, 25 μm. Gymnosis dose-response curves were generated using 2, 4, 20, 100, 500, 2,500, 5,000, and 10,000 nM TA2. All mRNA expression data were normalized to GAPDH- and PBS-treated control cells. Three internal replicates were averaged and experiments were completed in triplicate, mean ± SEM are shown.
LNP delivery increases TA2 potency in primary cultured CNS cells
We next examined how LNP encapsulation affects TA2 activity and trafficking in vitro in primary cortical neurons, microglia, astrocytes, and OPCs. As these cell types are notoriously difficult to transfect, we modified a formulation system optimized for neuronal siRNA delivery—the Neuro9 siRNA Spark Kit from Precision Nanosystems—to increase the probability of producing active ASO-LNP particles. We replaced siRNA throughout the protocol with equimolar ASO concentrations and formulated ASO-LNPs using the Spark NanoAssemblr, which utilizes a microfluidic laminar mixing procedure to produce homogeneous LNPs. Despite this formulation mixture being optimized specifically for neuronal delivery, we observed dramatically increased TA2 potency with ASO-LNPs as compared to gymnosis in all CNS cell types studied after 3 days of treatment (Figure 2K). TMEM106b mRNA levels remained stable after treatment with empty LNPs (Figure S3A), suggesting that the observed response was mediated by TA2 encapsulation and not the presence of external lipids. While LNPs increased TA2-mediated gene silencing, TA2 localization 3 days after LNP delivery showed a very similar intracellular punctate pattern to TA2 delivered via gymnosis (Figures 2L and S1F). These results demonstrate that LNPs can be used as an efficient method to increase ASO potency and mRNA knockdown in major CNS cell types in vitro.
LNPs increase TA2 potency in vitro by decreasing lysosomal trafficking or increasing ASO uptake
To further understand the mechanisms underlying LNP-mediated increases in ASO potency, we investigated ASO uptake and localization in primary CNS cell cultures. We first compared TA2 localization along the endolysosomal pathway after gymnosis or LNP delivery using IF. We treated CNS cell types with TA2 or TA2-LNPs at EC90-95 for 3 days to ensure that internalized ASO could be detected by anti-ASO antibodies, and analyzed the degree of ASO colocalization with endolysosomal compartments—early endosomes, late endosomes/lysosomes, as well as recycling endosomes as labeled by EEA1, LAMP1, and GRASP1, respectively—using Mander’s coefficients that range from zero (no colocalization) to one (full colocalization) (Figures 3A–3E). After 3 days of gymnotic delivery, TA2 associated strongly with LAMP1-positive vesicles in all cell types (M = 0.68 for neurons, 0.72 for microglia, 0.82 for astrocytes, and 0.72 for OPCs) (Figures 3B–3E). These data agree with previous studies showing that ASOs traffic to lysosomes within a few hours after gymnosis and accumulate in these compartments over time.7 Interestingly, TA2-LAMP1 colocalization was dramatically decreased following LNP delivery in neurons, astrocytes, and OPCs after 3 days (Figures 3B, 3D, and 3E), suggesting there is an inverse correlation between ASO potency (Figure 2K) and late endosome/lysosomal accumulation in these specific cell types. In contrast, microglia showed no delivery-dependent difference in TA2-LAMP1 accumulation (Figure 3C), suggesting that LNP delivery has a unique effect on ASO activity in these cells. While out of the scope of this study, examining ASO trafficking at earlier timepoints could provide additional insights into why ASO potency increases in cells treated with ASO-LNPs. We next asked whether ASO localization within LAMP1-positive vesicles could explain LNP-mediated enhancements in ASO activity. However, there was no detectable change in ASO puncta localization within or on LAMP1-positive membranes after LNP delivery in microglia or cortical neurons (Figure 3F). To determine whether changes in ASO uptake may regulate ASO potency, we measured internalized TA2 concentrations in microglia and neurons 24 h following gymnosis or LNP delivery using modified oligonucleotide ligation PCR (MOL-PCR) (Figure 3G). MOL-PCR quantifies the concentration of ASO in a sample using a synthetic ASO-binding DNA ligation probe that is amplified and compared with a reference standard curve. LNP delivery increased internalized ASO concentrations 3-fold over gymnosis in microglia, while neurons internalized the same amount of TA2 during both delivery methods (Figure 3G). Taken together, these findings suggest that the mechanism ASO-LNPs use to increase potency is CNS cell-type-specific and facilitated by increased ASO uptake in microglia or decreased lysosomal trafficking in neurons. These data are consistent with previous work showing that mRNA-LNP efficacy is dependent on both uptake efficiency and changes in endolysosomal trafficking, which is formulation-dependent.23
Figure 3.

LNP uptake differentially affects ASO delivery and trafficking in primary CNS cells
(A) Representative z-projection IF images of cortical neurons, microglia, astrocytes, and OPCs treated for 3 days with 5 μM TA2 or 100 nM TA2-LNPs (EC90-95) showing ASO localization (green) with endolysosomal markers EEA1, LAMP1, and GRASP1 (red). Scale bars, 10 μm and 5 μm (inset). (B–E) Mean Mander’s coefficients quantifying the fraction of overlap/colocalization between TA2 and EEA1, LAMP1, or GRASP1 after 3 days of gymnosis (solid) or LNP delivery (checked) in cortical neurons (B), microglia (C), astrocytes (D), or OPCs (E). ASO signal within and on the membrane was analyzed from single-plane manually thresholded images. Unpaired two-tailed Mann-Whitney test, ∗∗∗∗p < 0.0001. (F) Representative 2D deconvolved IF images showing ASO colocalization with LAMP1 in cortical neurons (left) and microglia (right) after 3 days of gymnosis or LNP delivery. Scale bars, 2 μm. (G) Internalized TA2 concentrations in cortical neurons and microglia after gymnosis or ASO-LNP delivery as determined by MOL-PCR. Cells were treated with 100 nM final ASO concentration for 24 h, mean ± SEM are shown (n = 3). Unpaired two-tailed Student’s t test, ∗∗∗p < 0.001.
Unique ASO-LNP formulations differentially modulate ASO activity in vitro
As ASO-LNPs proved to be a valuable tool for increasing ASO potency in culture, we next sought to determine the translatability of these results in vivo. Scaling up the previously characterized formulation was not feasible as it was designed and optimized for cell culture use, so we prepared six TA2-LNPs at physiologically relevant ASO concentrations for in vitro characterization before administration in mice. In particular, we assessed LNPs containing unique combinations of ionizable and PEG-lipids as these two components are vital for cargo release at endosomal pH and prolonging in vivo LNP circulation time, respectively (Figures 4A and S3B). The six TA2-LNPs were then screened for TMEM106b mRNA suppression in our in vitro primary CNS cell culture systems to determine which ASO-LNPs were active (Figures 4B and 4C). While formulations 2–5 did not alter TMEM106b mRNA levels in neurons or microglia 24 h after treatment as compared to gymnosis, formulations 1 (GenVoy-ILM) and 6 (MC3 with 2% DMG-PEG lipids) significantly increased ASO potency. These data were reproduced at a higher dose, validating the enhanced activity of these ASO-LNPs (Figures S3C and S3D). We next characterized mRNA degradation dynamics in microglia and neurons treated with formulations 1 or 6, hereafter named GenVoy and MC3, respectively. As expected, TMEM106b expression decreased exponentially over time following treatment with 200 nM TA2-GenVoy or TA2-MC3, and both LNP formulations were more potent than TA2 alone (Figures S3E and S3F). TMEM106b mRNA levels remained stable after treatment with empty LNPs (dashed lines), suggesting that the observed response was mediated by TA2 encapsulation. As TA2-GenVoy and TA2-MC3 were both active in vitro, we next characterized the physicochemical properties of these formulations before in vivo administration. We scaled up the formulations and analyzed LNP characteristics using cryoelectron microscopy (cryo-EM) (Figure 4D). Cryo-EM analyses revealed that the unloaded and TA2-loaded LNP samples were monodisperse, containing similar size distributions. Furthermore, all formulations had internal multilamellar stacks as expected. Supporting these cryo-EM findings, dynamic light scattering (DLS) measurements confirmed that the LNPs had similar diameters and polydispersity indices (Figures 4E and 4F). Given that these LNP formulations enhanced ASO activity in primary CNS cells in vitro and had desirable physicochemical characteristics, these particles were moved forward for further characterization in mice.
Figure 4.

ASO-LNPs induce gliosis in mice following i.c.v. injection
(A) TA2-LNP formulations screened for TMEM106b mRNA regulation in vitro. (B and C) TMEM106b mRNA expression in cortical neurons (B) and microglia (C) as determined by qRT-PCR. Cells were treated with 20 nM TA2 (leftmost bar) or TA2-LNP formulations (1–6) for 24 h. Data were normalized to GAPDH- and PBS-treated control cells. Three internal replicates were averaged and the experiments were completed in triplicate. One-way ANOVA with multiple comparisons Dunnett’s test (TA2 control), ∗p < 0.03, ∗∗p < 0.002, ∗∗∗p < 0.0002. (D) Representative cryo-EM images of unloaded, and TA2-loaded GenVoy (formulation 1) and MC3 (formulation 6) LNPs showing the internal structures of the formulations selected for in vivo delivery. Scale bars, 200 nm. (E and F) Particle sizes (E) and polydispersity measurements (F) of unloaded and TA2-loaded LNPs as determined by dynamic light scattering. (G) C57BL/6N mice were i.c.v. injected with saline (n = 6), TA2 (100 or 25 μg, n = 6 per group), TA2-LNPs (25 μg, n = 6 per group), or empty LNPs (n = 3 per group) and analyzed over 14 days. Illustration created using BioRender.com. (H) Acute toxicity 1 h post injection. (I) Percent body weight change normalized to day 0. GenVoy-treated groups showed signs of toxicity and were monitored closely post injection. One mouse in the TA2-GenVoy group did not survive the duration of the experiment. (J) Representative Iba1 IHC images from brain sections obtained 14 days post i.c.v. injection. Scale bars, 500 μm. (K and L) IHC quantification for total (K) and clustered (L) Iba1 signals. For all in vivo statistical analyses, one-way ANOVA with multiple comparisons Dunnett’s test (saline control) was used, ∗p < 0.03, ∗∗p < 0.002, ∗∗∗p < 0.0002, ∗∗∗∗p < 0.0001. Individual group comparisons (empty vs. TA2-loaded LNPs) were analyzed using unpaired two-tailed Student’s t test, ∗p < 0.05. For all panels, mean ± SEM are shown and the 100 μg TA2 dose is represented by bold font.
TA2 and TA2-LNPs induce microglia cell activation in vivo
We next administered a single i.c.v. bolus injection of free TA2 or TA2-LNPs to evaluate delivery-dependent ASO activity and distribution in the CNS (Figure 4G). Our pilot studies showed that free TA2 reduces TMEM106b mRNA expression in the brain by ∼60% when delivered at high doses (100 μg) while concomitantly inducing acute toxicity post injection as well as mild weight loss in some animals (Figures S1B–S1D). Thus, to test whether LNP encapsulation increases ASO potency and mitigates ASO-mediated toxicity, we administered free TA2 and TA2-LNPs at a low dose (25 μg) in addition to dosing a group of mice with 100 μg free TA2 as a control. The mice tolerated the administered doses well in the hours post injection with little acute toxicity observed (Figure 4H). However, the mice dosed with TA2-loaded GenVoy LNPs had a lack of appetite, were hunched, and intermittently tremored in the days following injection. These mice lost 15% of their total body weight within a few days, which was maintained for the duration of the 14 day study (Figure 4I). We stained brain sections for Iba1 to examine gliosis as a potential explanation for these observations 14 days post dosing (Figure 4J, hippocampus shown). Iba1 IHC analyses on hemibrain coronal brain sections showed that both free TA2 (100 μg) and TA2-GenVoy (25 μg) activated microglia dramatically, despite being dosed at 4-fold different TA2 concentrations (Figures 4K and 4L). TA2-GenVoy also increased both total and clustered lysosomal CD68 levels (Figures S3G and S3H), confirming the microgliosis phenotype in these mice. This induction of microgliosis could not be fully explained by potential lipid toxicities as unloaded LNPs had no significant effect on total or clustered Iba1 and CD68 signals. Interestingly, while TA2-GenVoy administration activated microglia, TA2-MC3 did not alter microgliosis significantly above saline or unloaded MC3 control levels suggesting that this phenotype was formulation-dependent. To further examine this phenomenon, we assayed cytokine release from primary microglia cultures treated for 1 or 3 days with unloaded or TA2-loaded LNPs using Luminex (Figures S4A and S4B), and compared the results with release profiles obtained from free ASO-treated cultures (Figures S2A and S2B). While there was no induction of cytokine release from microglia treated with free ASOs, TA2-LNPs differentially induced microglia activation. Specifically, TA2-GenVoy-treated microglia released multiple proinflammatory cytokines during the 3 days of exposure; however, TA2-MC3-treated microglia did not (Figure S4B). These data are consistent with our observations in mice, suggesting that screening cytokine release from ASO-LNP-treated microglia could eliminate immunostimulatory formulations before in vivo administration.
In vivo transcriptomics reveals inflammatory profiles in response to TA2 and TA2-LNPs
We next investigated the transcriptomic changes associated with free TA2, unloaded LNP, and TA2-LNP administration in the brain. We first assessed delivery-induced transcriptome effects as compared to saline-injected animals (Figures 5A, 5B, and S5A–S5D). Both TA2-GenVoy and TA2-MC3 induced large, genome-wide (FDR < 0.05) transcriptional responses in the brain 2 weeks post injection (Figures S5C and S5D). Free TA2 administration also resulted in a similar scale of transcriptional response when dosed at 100 μg (Figures 5A and S5E). Gene ontology (GO) analyses revealed a significant (FWER < 0.1) enrichment for immune response terms, which was less pronounced in animals administered free TA2 (25 μg) or unloaded LNPs (Figure S5F). As treatment with unloaded LNPs resulted in a transcriptomic response in the brain (Figures S5A and S5B), we next used the matched delivery vehicle as the control for each comparison—saline for free TA2 and the respective unloaded particles for both TA2-LNPs—to directly assess the impact of TA2 (Figure 5). For all comparisons, the genome-wide profile was primarily defined by increased gene expression. Free TA2, TA2-GenVoy, and TA2-MC3 dosed at 25 μg ASO resulted in a smaller genome-wide differential gene expression profile than the higher (100 μg) free TA2 dose (Figures 5A–5F). Despite the similarities in scale from the genome-wide perspective, the specific gene sets significantly (p < 0.05) altered by TA2-LNPs were more similar to the higher free TA2 dose, and this was more pronounced in TA2-GenVoy than TA2-MC3 (Figures 5G–5K). Free TA2 (100 μg) and TA2-GenVoy both resulted in a significant increase in the expression of microglial genes. These data are consistent with our IHC results showing that TA2 promotes brain microgliosis in free TA2 (100 μg)- and TA2-GenVoy-dosed animals (Figures 4K and 4L). Interferon-related genes were also significantly increased in all samples, including the 2′-5′-oligoadenylate synthetase (OAS) enzymes. The OAS enzymes are interferon-inducible sensors of cytosolic double-stranded RNA, which synthesize secondary messengers that activate RNase L in order to degrade viral RNA.24 Also relevant to the detection of viral RNA were genes related to trafficking and processing of endosomal Toll-like receptors (TLRs), which were upregulated in all formulations except for TA2-MC3. Endosomal TLRs function as sensors of non-self nucleic acids25 and TLR7 is involved in the recognition of single-stranded viral RNA.26 Nucleic acid-sensing TLRs are also present in the lysosome and lysosomal genes were significantly increased in both free TA2 (100 μg) and TA2-Genvoy. While there was a clear TA2-dependent upregulation of similar gene sets across all TA2-injected animals, suggesting that ASO was successfully delivered to the tissue, in particular free TA2 (100 μg) and TA2-GenVoy, TMEM106b mRNA only showed a genome-wide significant (FDR < 0.05) decrease in the 100 μg TA2 dose (Figure 5L). Together, these results suggest that TA2 ASO, not TMEM106b knockdown, results in an immune response related to the detection of non-host RNA both after gymnosis and LNP delivery.
Figure 5.

Transcriptomic analysis characterizing ASO-LNP-mediated gliosis
(A–D) Volcano plots of differential gene expression profiles from pairwise comparisons of TA2 (100 μg) vs. saline (A), TA2 (25 μg) vs. saline (B), TA2-GenVoy(1) vs. GenVoy(1) (C), and TA2-MC3(6) vs. MC3(6) (D). Red dots represent FDR < 0.05 and absolute log fold change (logFC) > 1, blue dots represent FDR < 0.05 and absolute logFC < 1, green dots represent FDR > 0.05 and absolute logFC > 1, and gray dots represent FDR > 0.05 and absolute logFC < 1. (E) UpSet plot of direction stratified overlaps between significant (FDR < 0.05 and absolute logFC > 1) differentially expressed genes (DEGs) from all pairwise comparisons. (F) Heatmap of hierarchical clustering analysis of significant (FDR < 0.05 and absolute logFC > 1) DEGs from all pairwise comparisons. (G–K) Gene set scores and select genes for microglia (G), interferon (H), OAS antiviral response (I), trafficking and processing of endosomal TLR (J), and lysosome (K). (L) TMEM106b mRNA expression levels. Gene set score comparisons were analyzed using unpaired two-tailed Student’s t test, ∗p < 0.05. Individual gene comparisons were calculated using voom-limma, ∗FDR < 0.05.
TA2-LNP delivery reduces mRNA knockdown in vivo
Despite observing an ASO-mediated microgliosis response following both TA2 gymnosis and LNP delivery, our transcriptomic analysis of TA2- and TA2-LNP-treated brains revealed that TMEM106b transcript levels only decreased in animals dosed with 100 μg free TA2 (Figure 5L). As these results were unexpected, we next sought to confirm these findings by specifically analyzing TMEM106b mRNA levels in bulk brain tissue using qRT-PCR (Figure 6A). While both free TA2 doses triggered a concentration-dependent decrease in TMEM106b transcript levels 14 days after administration, no significant change was observed in TA2-LNP-treated animals. However, MOL-PCR analyses detected ASO in the brain for all TA2-containing formulations studied (Figure 6B), which supported our Iba1 IHC and transcriptomic results. In particular, animals dosed with 25 μg free TA2 or TA2-GenVoy had similar ASO accumulation in bulk brain tissue despite having different TMEM106b mRNA expression levels (Figures 6A and 6B). Our previous results using TA1 revealed that widespread ASO brain distribution correlates with TMEM106b mRNA depletion in CNS cell types (Figure 1). Thus, these data suggest that changes in ASO tissue distribution may explain the lack of TA2-LNP activity in bulk brain tissue readouts.
Figure 6.

ASOs have formulation-dependent brain distribution following i.c.v. injection
(A) TMEM106b mRNA expression levels 14 days post i.c.v. injection as assessed by qRT-PCR. Data were normalized to GAPDH- and saline-treated controls. One-way ANOVA with multiple comparisons Dunnett’s test (saline control), ∗∗p < 0.002, ∗∗∗∗p < 0.0001. (B) Internalized TA2 concentration 14 days post i.c.v. injection as determined by MOL-PCR. (C and D) Representative anti-ASO IHC brain sections of free TA2 (100 μg)- or TA2-GenVoy(1) (25 μg)-treated animals 4 days following i.c.v. injection. (C) Hemibrain sections stained for TA2 (red staining). Scale bars, 2,000 μm. (D) TA2 IHC staining showing ASO accumulation in piriform cortical neurons, endothelial cells, and the choroid plexus (red staining). Scale bars, 20 μm. (E) Representative cryo-SEM images showing subcellular TA2 localization, red arrows, in piriform cortical neurons, endothelial cells, and ciliated ependymal cells 4 days after administration. Scale bars, 1 μm. (F) Representative cryo-SEM images showing TA2 localization in macrophages lining the ventricle 4 days following TA2-GenVoy(1) administration, red circles. Scale bars, 2 μm. For all panels, mean ± SEM are shown and the 100 μg TA2 dose is represented by bold font.
TA2-LNP concentrates in cells lining the ventricles and blood vessels in vivo
To better understand how delivery affects tissue- and cell-type-specific ASO distributions in the CNS, we next examined TA2 localization in the brain. We compared free TA2 (100 μg)- and TA2-GenVoy (25 μg)-dosed animals as these treatments had differential TMEM106b mRNA expression levels 14 days post injection, yet both showed a TA2-dependent induction of microgliosis despite being administered at 4-fold different ASO concentrations. Tissues were analyzed 4 days following injection to ensure maximum TA2 retention in the brain. First, we performed anti-ASO IHC to determine the specific CNS cell types that internalize TA2 via gymnosis and LNP delivery (Figures 6C, 6D, and S6A). When dosed at 100 μg free TA2, gymnotic uptake was observed in multiple cell types as indicated by broad ASO staining throughout the brain (Figure 6C). A robust TA2 signal was seen in neuronal cell populations across the tissue including in the hippocampus and cortex. However, astrocytes and microglia are also present in these brain regions and thus may also contribute to some of the ASO-containing cellular signal. Endothelial and ependymal cells in the choroid plexus also had ASO accumulation (Figure 6D). This strong ASO staining pattern is consistent with our earlier results revealing that free TA1 (100 μg) administration promotes widespread ASO distribution and TMEM106b mRNA knockdown in major CNS cell types (Figure 1). In contrast, ASO signal following TA2-GenVoy dosing did not penetrate widely throughout the brain tissue, but was instead enriched in the cells lining the ventricles and blood vessels (Figure 6C). While both delivery methods had appreciable ASO signal within hippocampal and cortical neurons, TA2-GenVoy sections showed preferential TA2 accumulation in ependymal cells, endothelial cells, and pericytes (Figure 6D). These data suggest that in vivo LNP delivery prevents ASO accumulation into cells outside of the ventricles and blood vessels, which likely accounts for the lack of widespread ASO activity in TA2-LNP-treated animals.
TA2 accumulates in phagosomes and endolysosomes in vivo
To better understand how delivery-mediated ASO distribution differences correlate with subcellular ASO accumulations in CNS cells in vivo, we next examined TA2 subcellular localization using anti-ASO immuno-EM 4 days after injection. Based on our IHC results, we focused our analysis on three key brain regions that had delivery- and concentration-independent ASO accumulation—a representative layer of piriform cortical neurons, endothelial cells lining blood vessels, and ependymal cells in the ventricle as well as the choroid plexus (Figures 6E, 6F, and S6B). Single gold-labeled TA2 particles were readily detected in all brain regions examined, as expected. For both delivery methods, subcellular TA2 molecules localized mainly to membrane-bound endolysosomal vesicles with little accumulation observed in the cytoplasm or nucleus (Figure 6E). These data support our in vitro colocalization data and previous studies showing that oligonucleotides quickly traffic through the endolysosomal pathway after gymnotic or LNP-mediated internalization, and suggest that few molecules escape these compartments.7,8,9,27 Overall, ASO-positive vesicles were more frequently observed after gymnosis, likely due to the 4-fold increase in dose; however, TA2 could also be detected in similar structures following LNP delivery (Figure 6E). These data align with our previous findings showing that high free ASO doses promote greater uptake into the brain (Figures 6B and 6C). While this phenomenon was consistent across most CNS cell types examined, we found non-ciliated macrophages lining the ventricles in TA2-GenVoy-dosed animals (Figure S6B). Not only were these cells more prevalent in TA2-GenVoy sections, they also contained large subcellular TA2-filled vacuoles resembling phagosomes (Figure 6F, red circles). These data suggest that macrophages may be recruited and activated following exposure to specific ASO-LNP formulations, which may promote ASO internalization, increase subcellular ASO accumulation, and could be responsible for triggering the inflammatory response observed in TA2-GenVoy-treated brains.
Discussion
The development of ASO therapies to treat neurodegenerative diseases has increased.4 While ASOs have been successful in the clinic,28 they require i.t. injections to cross the BBB and enter the tissue. However, ASOs also accumulate in other organs following cerebrospinal fluid (CSF) administration. This off-target delivery reduces ASO activity (or concentration) in the brain and contributes to toxicity.29 Accordingly, ASO delivery advancements that reduce ASO dosing, increase efficiency, and promote tissue-specific targeting would increase patient well-being. Here, we perform a comprehensive study analyzing ASO trafficking, distribution, and potency in the brain following two commonly used delivery methods—gymnosis and LNP encapsulation.
ASO gymnosis promotes efficient mRNA knockdown in CNS cells in vitro and in vivo
To date, ASO development has focused on optimizing gymnotic delivery, which promotes broad ASO distribution and activity in the brain.1 In this study, we utilized TMEM106b-targeting LNA ASOs, TA1 and TA2, to probe the mechanisms underlying ASO activity. TMEM106b is a lysosomal protein implicated as a risk modifier for FTLD-TDP30,31 and the C-terminal fragment forms amyloid fibrils in neurodegenerative diseases.32,33 Thus, understanding TMEM106b′s role in disease and regulating its expression may be efficacious. We reveal that free TMEM106b ASOs are active in vitro and in vivo. Specifically, ASO gymnosis correlates with robust mRNA degradation in neurons, microglia, astrocytes, and OPCs. These studies corroborate data showing that an MOE Malat1 ASO is active in major CNS cell types following i.c.v. administration in mice.34 These results reveal that ASO distribution and activity are target- and chemistry-independent. Furthermore, we show neuronal and microglia activity modulators do not affect ASO potency. Neurodegenerative diseases lead to cellular dysfunction and activity changes.35 Thus, our results suggest that gymnotically internalized ASOs are well-suited to treat neurological diseases that manifest in unique CNS cell types and brain regions. These data also support clinical work showing that ASOs modulate disease pathology in many neurodegenerative model systems.3,36,37
LNP encapsulation robustly increases ASO activity in CNS cell types in vitro
We also assessed how LNP delivery affects ASO activity. Historically, LNP research has focused on delivering cell-impermeable oligonucleotides. However, a renewed effort to utilize LNPs has emerged with the clinical success of ONPATTRO13 and the SARS-CoV-2 vaccines.14 Recent work shows that ASO-, siRNA-, and mRNA-LNPs have unique formulation and delivery challenges.38 Despite these obstacles, ASO-LNPs are efficacious in vitro when formulated with cell-penetrating peptides39 or bioreducible lipids.40 LNP encapsulation has also successfully delivered ASOs in zebrafish.41 However, few studies have examined ASO-LNP brain delivery in higher vertebrates. Of note are two studies: the first hijacked GLUT1 to deliver glucose-coated ASO-LNPs across the BBB42 and the second utilized neurotransmitter “lipidoids” to enhance brain delivery.43 Although these studies are promising, additional details are needed to understand how LNPs affect ASO localization, trafficking, and distribution in the brain.
We first needed to screen ASO-LNPs for activity. Assessing candidate formulations in vivo is time and cost intensive. Therefore, toxic and inactive combinations are commonly identified using screening cascades in immortalized cell lines. While these platforms are useful, we sought to determine if ASO-LNP potency could be easily assessed in primary CNS cell cultures. We treated cortical neurons, microglia, astrocytes, and OPCs with TA2-LNPs and monitored TMEM106b transcript levels using qRT-PCR. An LNP formulation known to increase siRNA delivery in neurons44 increased ASO activity in all cell types, confirming that these in vitro models are valuable delivery screening tools. We also developed and screened LNP formulations, showing that ASO-LNPs can be efficiently assessed for potency. Furthermore, since the completion of this study, we developed a high-throughput screening (HTS) cascade to evaluate ASO-LNP potency across a multitude of formulation parameters and cell culture systems.45
ASO-LNPs do not downregulate mRNA levels in the brain due to preferred internalization by cells lining the blood vessels and ventricles
As ASO-LNPs were active in vitro, we next assessed how LNP encapsulation affects ASO potency and distribution in the brain. We compared free TA2 with two LNP formulations, TA2-GenVoy and TA2-MC3, after i.c.v. injection in mice. While TA2 was detected in the brain for all ASO-containing formulations studied, neither ASO-LNP downregulated mRNA in bulk tissue. We determined that this was likely due to changes in ASO internalization. Specifically, gymnosis promoted broad ASO distribution in multiple brain regions and cell types, whereas LNP delivery stimulated ASO internalization in ependymal cells, endothelial cells, and pericytes. Single-cell transcriptomic analysis is out of the scope of this study, but we suspect that TMEM106b mRNA levels are reduced in all cell types that accumulate ASO. These data revealed that, while free ASO crosses the CSF-brain barrier, LNPs preferably deliver ASOs to cells exposed to the CSF, subarachnoid space (SAS), or are involved in CSF-blood turnover. Interestingly, this differential ASO distribution was stronger in TA2-GenVoy- vs. TA2-MC3-dosed animals, suggesting that ASO delivery is formulation-dependent. These findings are consistent with previous work showing that PEGylated, polystyrene LNPs distribute rapidly through the SAS, but do not penetrate into the brain after i.t. administration in healthy mice.46 Lack of distribution could also be due to many factors that regulate particle internalization including lipid composition,47 particle size,48 and shape.49 Thus, screening additional ASO-LNP parameters and administration routes will be necessary to circumvent this issue.
LNP encapsulation cannot mask ASO backbone-mediated immune activation in vivo
ASOs can induce immune activation in animals.20,21 Thus, we also investigated whether LNP encapsulation mitigates this toxicity. While no severe acute toxicity was observed in the hours following dosing, TA2-GenVoy-treated mice had rapid weight loss after injection. Furthermore, these mice showed a dramatic upregulation of genes involved in immune and defense response. These data revealed that LNPs cannot easily mask ASO-mediated immune responses against foreign RNA or DNA. Thus, screening candidate ASOs for activity as well as toxicity before formulation or conjugation will remain essential despite the use of advanced delivery technologies.
In addition to observing microglia activation in the parenchyma, immuno-EM analysis revealed an infiltration of non-ciliated macrophages in TA2-GenVoy-treated animals. These cells were focally localized near the ventricles and choroid plexus, and contained large phagosome-like structures filled with nanogold-labeled ASO. Coupled with our earlier results revealing that TA2-GenVoy induced microgliosis, these data suggest that this formulation stimulated a peripheral immune response that subsequently activated microglia despite TA2 not being internalized in these cells. The presence of ASO-filled macrophages also aligns with our in vitro results showing that primary microglia internalize more ASO when it is encapsulated in LNPs.
Screening strategies for ASO-LNP brain delivery
Our results show that ASO-LNP-exposed cells internalize and traffic ASOs within the endolysosomal system; however, LNP delivery to the parenchyma remains a major challenge. To overcome this obstacle, screening multiple formulations including those modified for cell-type-specific delivery is necessary. An HTS platform could provide insights into the formulation parameters required for efficient ASO-LNP brain delivery, yet also alleviate time- and cost-intensive strategies.
We suggest a two-pronged approach that utilizes in vitro and in vivo assays to determine non-toxic, active ASO-LNPs. First, screening formulations for microglia activation in vitro could eliminate toxic formulations before injection in animals. Second, using a barcoding strategy in a reporter mouse model that reads out ASO activity using fluorescence would allow for simultaneous ASO delivery and activity monitoring. Over the past decade, both mRNA- and siRNA-LNP in vivo barcoding approaches have successfully identified active formulations.50,51,52,53,54 Given that ASO-LNPs have unique formulation challenges,38 this method would advance the technology quickly as there are currently no published studies using similar strategies for ASO delivery.
Taken together, this study provides a framework for understanding the opportunity in delivering ASOs to the brain using LNPs, yet also illuminates remaining challenges. Thus, while free ASO delivery currently remains a superior method for delivering ASOs to the brain, future ASO-LNP mechanistic and HTS studies could improve ASO delivery to target cells thereby enhancing the overall benefit/risk ratio of ASO therapies.
Materials and methods
Oligonucleotides
ASOs were synthesized following standard phosphoramidite protocols as described previously.55 ASOs were formulated in RNase-free PBS. For in vivo studies, ASOs were diluted in PBS to the concentrations described before administration. ASOs used in this study were the following (ID: base sequence, sugar sequence): TA1: ETGTATTTCAAATETEA, LLLDDDDDDDDDDLLLL; NC1: ECAAATCTTATAATAAETAELD, LLLDDDDDDDDDDLLDLL; TA2: EEACACTGTATCTTEA, LLLDDDDDDDDDDDLL. E corresponds to 5-methyl C in the base sequences and in the sugar sequence L is LNA and D is DNA. All backbones are phosphorothioate.
Animals
Timed pregnant C57BL/6N mice and pups from Charles River Laboratories (Hollister, CA) were used to prepare primary cultures. C57BL/6J male mice from Jackson Laboratory (Ball Harbor, ME: stock no. 000664) between 3 and 6 months of age were used for in vivo i.c.v. dosing experiments. Mice were housed on a regular light/dark cycle (14:10 h) with ad libitum access to food (LabDiet 5010) and water. All injections and tissue collections were conducted during the light phase. All animal care/handling procedures were reviewed and approved by Genentech’s Institutional Animal Care and Use Committee, and were conducted in full compliance with regulatory statutes, Institutional Animal Care and Use Committee policies, and National Institutes of Health guidelines.
Intracerebroventricular injections
Injections (i.c.v.) were performed using a freehand method. In brief, animals were anesthetized with isoflurane, the skull was shaved, and the skin was cleaned with either antimicrobial betadine and 70% ethanol, or Chloraprep (260100, BD Biosciences). A skin incision was made between the occiput and forehead to identify bregma. Injection coordinates were 1–2 mm to the right of the midline and posterior −0.5 mm from bregma. The needle, fitted with cut polyethylene tubing so that no more than 3 mm of needle was available, was inserted through the skull to a depth of −2.8 mm and 1 min was given to allow for the brain to seal around the needle. A bolus 5 μL injection was given at a rate of 1 μL/s, using a 26-gauge 25 μL Hamilton syringe. One min after injection the needle was withdrawn. The incision was closed with tissue glue and buprenorphine was given for up to 3 days to reduce any pain caused by the procedure.
Mouse observations post i.c.v. dosing
Acute side effects scoring was performed at 30 min and 1 h post i.c.v. injection and an average score was calculated. Acute side effects observations include reduced movement, ataxia, and/or hunched body. These observations were scored as follows: 0, normal; 1, mild; 2, moderate; 3, severe. An even more severe score of 4 is given if seizures, death, or catatonic behavior is observed with no improvement after 2 h. Individual body weights were measured pre-i.c.v. injection as well as at 1 and 2 weeks post injection.
Tissue collection
Mice were anesthetized with 2.5% tribromoethanol (0.5 mL/25 g body weight) and transcardially perfused with PBS. For the in vivo ASO distribution and cell sorting study (Figure 1), animals were either also perfused with 4% PFA and brains were subsequently post-fixed in 4% PFA overnight before tissue sectioning/staining for IF analysis, or the cortices were processed for single-cell FACS without additional PFA perfusion. For the in vivo ASO-LNP delivery study (Figures 4, 5, and 6), the left hemisphere was drop-fixed in 4% PFA for 48 h and stored in PBS at 4°C until being sectioned/stained for IHC analysis. A 2 mm slice through the right hemibrain was collected just anterior to the injection site and the tissue was immediately placed into RNALater (Ambion) and stored at 4°C until qRT-PCR/transcriptomic analysis was performed. A second 2 mm slice through the right hemibrain was collected just posterior to the injection site, the tissue was weighed, immediately flash frozen in liquid nitrogen, and stored at −80°C until MOL-PCR analysis was completed. For the cellular and subcellular ASO distribution study (Figure 6), animals were also transcardially perfused with 4% PFA supplemented with 0.1% glutaraldehyde, and brains were stored in PBS at 4°C until being processed for anti-ASO IHC or immuno-EM analysis. See specific materials and methods sections, below, for additional tissue processing details.
Tissue sectioning, IF staining, and imaging
For hemibrain ASO distribution analysis (Figure 1D), PFA-fixed brain samples were coronally sectioned at 35 μm thickness. Brain sections were washed in PBS followed by PBST (0.3%), and blocked in PBST (0.3%) supplemented with 5% BSA and 5% normal donkey serum. Primary rat anti-ASO antibody (Roche) was diluted at 1:1,000 in PBST (0.3%) supplemented with 1% BSA and incubated overnight at 4°C. Sections were washed 3× with PBST (0.3%) and incubated for 2 h at room temperature (RT) with secondary antibody (donkey anti-Rat Alexa 488, Thermo Fisher Scientific) 1:1,000. Sections were washed 3× in PBST (0.3%) and rinsed 3× in PBS. Sections were then mounted onto slides with 0.1% gelatin in PBS and allowed to dry/adhere to the slide at RT. Coverslips were applied using ProLong Gold Antifade Mountant (P36930, Thermo Fisher Scientific). Imaging was performed at 200× magnification using a Nanozoomer S60 digital whole-slide scanner. The ideal exposure was determined based on samples with the brightest intensity and set for all slides to run as a batch.
For ASO colocalization studies (Figure 1E), brains were paraffined fixed and coronally sectioned at 10 μm thickness. Sections were washed in PBS followed by PBST (0.3%), and blocked in PBST (0.3%) supplemented with 5% BSA and 5% normal donkey serum. Primary antibodies were diluted in PBST (0.3%) supplemented with 1% BSA and incubated overnight at 4°C. Antibodies used in this study included rat anti-ASO (Roche) 1:1,000; rabbit anti-fox3 (ABN51, Millipore Sigma) 1:1,000; and rabbit anti-Iba1 (019–19741, FUJIFILM Wako Pure Chemical) 1:1,000. Sections were washed 3× in PBST (0.3%) and incubated for 2 h at RT with secondary antibodies including DAPI and donkey anti-rat Alexa 488 as well as donkey anti-rabbit Alexa 555 (Thermo Fisher Scientific) 1:1,000. Sections were washed 3× in PBST (0.3%) and rinsed 3× in PBS. Coverslips were applied using ProLong Gold Antifade Mountant (P36930, Thermo Fisher Scientific). Images were acquired on a Zeiss LSM 710 confocal microscope with Plan-Apochromat 20×/0.8 M27 objective. All images were processed in the software Fiji.56
Preparation and fixation of dissociated cell suspensions from adult mouse brain
Cell suspensions were prepared as described previously.57 In brief, mice were anesthetized as described above (see tissue collection) and cortices were dissected on ice, minced, and transferred to an Eppendorf tube containing Accutase (SCR005, Millipore Sigma). Cortices were rotated for 20 min at 4°C, centrifuged at 2,000 × g for 1 min at 4°C, and Hibernate A Low Fluorescence medium (BrainBits) was added. The tissue was triturated and incubated on ice for 10 s. The cloudy cell suspension was removed, 70 μm filtered, and Hibernate A was added. Titration and filtration steps were repeated 4×. The final cell suspension was separated using a discontinuous Percoll gradient (high density on bottom) centrifuged at 430 × g for 4 min. Cloudy liquid was discarded and the remaining liquid was centrifuged at 550 × g for 4 min. The cell pellet was resuspended in Hibernate A and ice-cold 100% ethanol was added drop-by-drop until a final concentration of 50% ethanol was reached. The ethanol-fixed cells were incubated on ice for 15 min and centrifuged at 550 × g for 5 min. The cell pellet was resuspended in Hibernate A and centrifugation was repeated. The purified cells were resuspended in Hibernate A and immunostained.
Cell immunostaining, FACS, and RNA isolation
Dissociated cells were immunostained, FACS sorted, and RNA isolated as described by Srinivasan et al.57 In brief, cells were rotated for 20 min at 4°C with DAPI and the following antibodies targeting neurons, astrocytes, and microglia, respectively: Alexa 488-conjugated anti-NeuN (MAB377X, Millipore Sigma) 1:1,000; PE-conjugated anti-GFAP (561483, BD Biosciences) 1:50; and APC-conjugated anti-CD11b (561690, BD Biosciences) 1:250. Stained cells were centrifuged at 2,000 × g for 2 min, washed 2×, resuspended in Hibernate A, and 40 μm filtered. Cell sorting was performed on a BD FACSAria Fusion equipped with 5 lasers (355, 405, 488, 561, and 638 nm). The instrument was set up with a nozzle size of 100 μm, a frequency of 32 kHz, and pressure of 20 psi. The "4-Way Purity" mode was used for coincident discrimination. DAPI+ signal was used as a parental gate to select for singlet, fixed cell bodies. Further gates were chosen based on isotype controls. CD11b+ cells were distinct from other cell populations and gated for microglia. CD11b– cells separated into NeuN+ and NeuN– clusters, of which neurons were gated from the NeuN+ population. Astrocytes were gated from GFAPhigh cells that were also NeuN–. All final gates were compared across each cell-specific marker described to assure that final cell populations were pure. Unsorted cells were also collected for downstream comparison of TMEM106b mRNA levels (see below). After sorting, cells were centrifuged at 2,600 × g for 7 min (neurons) or 6,600 × g for 7 min (astrocytes, microglia, and unsorted cells). RNA was extracted from the pellet using the RNeasy Plus Micro kit according to the manufacturer’s instructions (74034, QIAGEN).
Reverse transcription, preamplification, and Fluidigm qPCR analysis on sorted brain cells
RNA from sorted cells was reverse transcribed using the High-Capacity cDNA Reverse Transcription kit according to the manufacturer’s instructions (4368814, Applied Biosystems). For cDNA preamplification, assay probes were pooled and diluted as recommended by Applied Biosystems. Pooled probes were then combined with 2.5 μL cDNA, 10 μL 2× TaqMan PreAmp Master Mix (1410056, Applied Biosystems), and 2.5 μL TE buffer. Following enzyme activation for 10 min at 95°C, 14 cycles of PCR were conducted at 95°C for 15 s and 60°C for 4 min using a Veriti 96-well Fast Thermal Cycler (Applied Biosystems). Assay probes were obtained from Life Technologies (TaqMan Gene Expression Assays, Thermo Fisher Scientific): FAM-MGB mouse TMEM106b (Mm00510952_m1) and VIC-MGB mouse ActB (Mm00607939_s1).
Fluidigm was performed using 48 × 48 chips with two technical replicates included per experiment. Sample and assay mixtures were prepared independently, and were combined at equal volumes before running the assays. In 10 μL total volume, sample mixtures included 2.5 μL preamplification product from sorted cell samples or 1.5 μL undiluted reverse transcription product from whole tissue samples, 20× GE Sample Loading Reagent (85000735, Fluidigm), 2× PCR master mix (58003365-01, Applied Biosystems), and TE buffer. For assay mixtures, equal volumes of TaqMan assay and 2× Assay Loading Reagent (85000736, Fluidigm) were combined. Data from Fluidigm runs were manually assessed for reaction quality before analysis. Gene expression values were calculated using the comparative delta Ct (–ΔΔCt) method using a housekeeper gene, actin (ActB), Ct values, and saline-treated Ct values for normalization. The assay probes used for these studies were the same as for preamplification.
Dissociated primary murine cortical neuron cultures
Mouse embryonic cortical neurons were cultured as described previously.58 Cortices from day 15 C57BL/6N embryos (E15) were dissected, stripped of meninges, washed 3× with cold HBSS (Invitrogen), and incubated for 10 min at 37°C in HBSS supplemented with 0.25% trypsin (Invitrogen) and DNase I (Roche CustomBiotech). Tissue was washed 3× with HBSS and triturated in plating medium containing DNase I (Gibco Neurobasal Medium [Thermo Fisher Scientific], 20% heat-inactivated horse serum [Thermo Fisher Scientific], 25 mM sucrose, and 0.25% Gibco GlutaMAX [Thermo Fisher Scientific]). Dissociated cells were centrifuged at 125 × g for 5 min at 4°C, resuspended in a plating medium, and plated in poly-L-lysine (PLL)-coated (Millipore Sigma) plates. The plating medium was replaced with Neurobasal Medium supplemented with 1% B-27 (Thermo Fisher Scientific) and 0.25% Gibco GlutaMAX after 24 h. Cells were maintained at 37°C with 5% CO2 and the medium was renewed using 50% exchange every 3–4 days.
Dissociated primary murine mixed glial cultures (microglia, OPCs, and astrocytes)
Primary murine mixed glial cells were cultured from day 3 C57BL/6N pups (P3). Cortices were dissected, the meninges were removed, and the tissue was washed 3× with cold HBSS (Invitrogen). The tissue was incubated for 10 min at 37°C in Gibco 0.25% trypsin-EDTA (Thermo Fisher Scientific), washed 3× in glial culture medium (DMEM high glucose supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin [Thermo Fisher Scientific]), and triturated. Dissociated cells were filtered (70 μm) and centrifuged at 700 × g for 5 min at RT. The pellet was resuspended in glial culture medium and plated in PLL-coated 225 cm2 flasks at a density of 8 cortices/flask. Cultures were maintained at 37°C with 5% CO2. The flasks were washed 3× with PBS after 4 days and fresh glial culture medium was added. Primary mouse microglia, OPCs, and astrocytes were cultured using differential separation techniques, described below, after an additional 7 days.
Primary microglia were cultured as described previously with the following alterations.59,60 Mixed glial cultures containing suspended microglia were agitated on a rotary shaker at 200 rpm for 1 h at 37°C with 5% CO2. The resulting cell suspension was centrifuged at 700 × g for 5 min at RT. Cells were resuspended in glial culture medium supplemented with 10 ng/mL mCSF1 (416-ML-050, R&D Biosystems), counted, and plated in PLL-coated plates (Millipore Sigma).
OPCs and astrocytes were then separated from the flasks as described previously with the following alterations.61,62,63,64 Flasks were washed 3× with PBS, fresh glial culture medium was added, and agitation was initiated on a rotary shaker at 260 rpm for 24 h at 37°C with 5% CO2. The OPC-containing suspension was filtered through 30 μm nylon mesh onto untreated Petri dishes and incubated for 30–60 min at 37°C with 5% CO2 with swirling every 15 min. Non-adherent OPCs were removed from the dishes and the cell suspension was centrifuged at 700 × g for 5 min at RT. OPCs were resuspended in glial cell culture medium, counted, and plated in PLL-coated plates. After 3 h, the medium was replaced with Neurobasal Medium supplemented with 2% B-27 (Thermo Fisher Scientific) and 25 ng/mL PDGF AA (14-8989-80, Invitrogen, eBiosciences).
Astrocytes were harvested from the flasks by mild trypsinization following OPC removal and plating.64 In brief, flasks were washed 3× with PBS. Gibco 0.25% trypsin-EDTA (Thermo Fisher Scientific) was diluted 1:5 in DMEM/F12, added to the flasks, and incubated at 37°C with 5% CO2 until the confluent astrocyte layer was detached. The floating astrocyte layer was collected, excess trypsin was removed, and fresh 0.25% trypsin-EDTA (Thermo Fisher Scientific) was added. Astrocytes were incubated at 37°C with 5% CO2 with occasional manual agitation to promote cell detachment. Upon obtaining a single-cell suspension, cells were centrifuged at 700 × g for 5 min at RT. Astrocytes were resuspended in glial cell culture medium, counted, and plated in PLL-coated plates.
Preparation and characterization of ASO-LNPs
For the in vitro characterization of ASO-LNP uptake and potency (Figures 2 and 3), TA2-LNPs were prepared using the Neuro9 siRNA Spark Kit following the manufacturer’s instructions (Precision NanoSystems). siRNA was replaced with TA2 (ASO-LNPs) or nucleic acid storage buffer (empty LNPs) throughout the protocol and nanoparticles were formulated using the Spark NanoAssemblr. Due to limited volume loss during formulation, total ASO concentration was calculated using the final dilution factor after microfluidic mixing.
For ASO-LNP screening and the scale-up of active formulations for in vivo analysis (Figures 4, 5 and 6), the materials used were the following. The lipids 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000), and 1,2-dimyristoyl-rac-glycero-3-[methoxypolyethylene glycol-2000] (DMG-PEG2000) were purchased from Avanti Polar Lipids. The ionizable lipids dilinoleylmethyl-4-dimethylaminobutyrate (DLin-MC3-DMA, MC3), COATSOME SS-OP, and GenVoy-ILM were purchased from MedChemExpress, NOF, and Precision Nanosystems, respectively. Cholesterol was purchased from Millipore Sigma. All reagents were reagent grade and DNase/RNase free.
A microfluidic approach was used for ASO-LNP preparation as described previously.65 In brief, the total lipid concentration was set at 12.5 mM to maintain a nitrogen to phosphate ratio of 2 (tertiary amine in the ionizable lipid/phosphorothioate in TA2). The ionizable cationic lipids, DSPC, cholesterol, and PEG-DMG or PEG-DSPE were dissolved in ethanol at desired ratios and ASO TA2 was dissolved in citrate buffer (25 mM [pH 4.0]). The lipids were mixed with TA2 at a 1:3 volume ratio and a flow rate of 12 mL/min using a microfluidic laminar mixing device (NanoAssemblr Benchtop, Precision NanoSystems). The formulated TA2-LNPs were purified using centrifugal ultrafiltration (MWCO 10 kDa, Amicon, Millipore Sigma) to remove free ASO and lipids, and were buffer exchanged into RNase-free PBS. Purified TA2-LNP formulations were analyzed for mean particle diameters and size distributions (percent polydispersity, %PD) using a DynaPro plate reader III (Wyatt Technology). The ASO concentration was measured using hydrophilic interaction liquid chromatography (HILIC). HILIC analysis was performed to determine the total ASO concentration after dissolving the ASO-LNPs in 0.75% Triton solution. An ACQUITY UPLC BEH Amide column (3 × 50 mm, 1.7 μm, 130 Å) obtained from Waters was used with mobile phases containing 25 mM ammonium acetate (unadjusted pH) in acetonitrile/water solvent mixtures. The flow rate was set to 0.8 mL/min and the column heated at 40°C. The detection wavelength was set at 260 nm.
Primary cell culture treatments and qRT-PCR gene expression analysis
Cortical neurons, microglia, and OPCs were plated at 3 × 104 cells/well and astrocytes were plated at 1 × 104 cells/well in 96-well plates. For all studies, neurons, microglia, astrocytes, and OPCs were treated starting at DIV10 (DIV7 for time course), DIV1, DIV7, and DIV1, respectively. TA2 or TA2-LNPs were added at the described concentrations using 50% medium exchange and were incubated for 0–8 days. For dose-response ASO potency analysis, neurons were treated with bicuculline (40 μM, Tocris Bioscience), TTX (2 μM, Tocris Bioscience), KCl (10 mM, Millipore Sigma), or L-glutamic acid (glutamate, 10 μM, Tocris Bioscience) and microglia were treated with LPS (1×, Invitrogen, eBiosciences) or IL-4/IL-13 (40 ng/mL, R&D Biosystems) overnight followed by ASO addition during 50% medium exchange. For all studies, three internal replicates were averaged and the experiments were completed in triplicate. Additional treatment details are addressed in the figure legends.
Gene expression was analyzed using the TaqMan Fast Advanced Cells-to-CT Kit (A35378, Thermo Fisher Scientific) following the manufacturer’s instructions. Cells were washed with PBS and lysed. Lysis reactions were stopped and reverse transcription was performed at 37°C for 30 min followed by a 95°C 5-min inactivation using a Veriti 96-well Fast Thermal Cycler (Applied Biosystems). Following enzyme activation for 20 s at 95°C, 40 cycles of quantitative PCR (qPCR) were conducted at 95°C for 1 s and 60°C for 20 s using either a QuantStudio7 Flex or a ViiA 7 real-time PCR system (Applied Biosystems). Gene expression values were calculated using the –ΔΔCt method using a housekeeper gene (GAPDH) Ct values and control-treated Ct values for normalization. Assay probes were obtained from Life Technologies (TaqMman Gene Expression Assays, Thermo Fisher Scientific): FAM-MGB mouse TMEM106b (Mm00510952_m1) and VIC-MGB mouse GAPDH (Mm99999915_g1).
IF staining and imaging of primary cell cultures
Cells were plated in 24-well no. 1.5 coverslip plates (MatTek) coated with PLL (Millipore Sigma) at 1.5 × 105 cells/well or 5 × 104 cells/well for neurons, microglia, and OPCs, or astrocytes, respectively. Neurons (DIV10), microglia (DIV1), astrocytes (DIV7), and OPCs (DIV1) were treated with 5 μM TA2 or 100 nM TA2-LNPs. After 3 days, cells were washed 3× with PBS and fixed using 3% PFA (Electron Microscopy Sciences) for 20 min at 37°C. Fixed cells were washed 3× with PBS at RT, permeabilized/blocked for 1 h in PBS supplemented with 0.05% saponin (Millipore Sigma) and 2% BSA (Thermo Fisher Scientific), and incubated overnight at 4°C with primary antibodies. Antibodies used in this study included rabbit or rat anti-ASO (Roche) 1:10,000, mouse anti-MAPT (MAB361, Millipore Sigma) 1:200, chicken anti-MAP2 (ab5392, Abcam) 1:10,000, mouse anti-⍺-tubulin (DM1⍺, T6199, Millipore Sigma) 1:1,000, rabbit anti-Iba1 (019–19741, FUJIFILM Wako Pure Chemical Corporation) 1:500, rabbit anti-GFAP (ab7260, Abcam) 1:5000, rabbit anti-Olig2 (AB9610, Millipore Sigma) 1:500, rabbit anti-EEA1 (PA5-17228, Thermo Fisher Scientific) 1:100, rat anti-LAMP1 (ab25245, Abcam) 1:500, and rabbit anti-GRASP1 (PA5-34893, Thermo Fisher Scientific) 1:500. Cells were washed 3× with PBS containing 0.05% saponin (Millipore Sigma) and 2% BSA (Thermo Fisher Scientific), and secondary antibodies were added for 1 h at RT. Secondary antibodies used included donkey anti-rat Alexa 488/555, donkey anti-rabbit Alexa 488/555/647, donkey anti-chicken Alexa 555, and donkey anti-mouse Alexa 555/647 (Highly Cross-Adsorbed Secondary Antibodies, Molecular Probes) 1:500. Cells were washed 3× with PBS containing 0.05% saponin (Millipore Sigma) and 2% BSA (Thermo Fisher Scientific) before storage at 4°C in PBS.
Cells were imaged at RT with an HC PL APO 100× oil/1.49-NA objective (no. 11506318) on an Sp5 inverted laser scanning confocal microscope (Leica DMI6000CS) driven by LAS AF X software (Leica). Images were acquired simultaneously using a combination of photomultiplier tubes and hybrid detectors with photon boosting technology. To obtain higher-resolution images for colocalization analysis of ASOs with EEA1, LAMP1, and GRASP1, an additional 4× magnification was applied and z stacks were imaged across randomly chosen cell bodies.
Colocalization analysis
The mean overlap fraction between ASO and EEA1, LAMP1, or GRASP1 was calculated using Mander’s coefficients66 and the JACoP plugin in Fiji.56 A single z stack was used for quantification and manual thresholding was applied to remove background signal. Due to the presence of some larger donut-like vesicles in LAMP1 conditions, all ASO signal associated with the membrane or within vesicles was included in the analysis during manual thresholding. For each condition, 10 or more images were analyzed and the experiment was completed on two independent cell cultures. Mander’s coefficient varies from zero to one, with zero representing no overlap and one representing complete colocalization. All images were taken using identical microscope settings.
Confocal image deconvolution was completed using Huygens Professional v.21.10 (Scientific Volume Imaging, the Netherlands, http://svi.nl). Deconvolved images were imported into Imaris v.9.9 (Oxford Instruments). The Surfaces tool was used to mask lysosomes stained with LAMP1 and this mask was copied into a new channel. Images are single Z planes selected from the 3D image stack showing the new LAMP1 channel and the original ASO channel (green).
Cytokine and chemokine release assays
Microglia were plated at 3 × 104 cells/well in 96-well plates. At DIV1, cells were treated during 50% medium exchange, and incubated at 37°C with 5% CO2. After 1 or 3 days, the supernatant was removed and stored at −80°C until cytokine/chemokine release assays were performed. Microglia were treated with non-targeting ASO NC1 or TMEM106b-targeting ASO TA2 at 100 nM, 5 μM, or 10 μM final ASO concentration. For TA2-LNP studies, microglia were treated with 20 or 100 nM final ASO concentration, as determined and described previously. Control unloaded LNPs were added at the same final volume as their respective ASO-loaded counterparts. Three internal replicates were collected and the experiment was completed in triplicate.
Collected supernatants were analyzed for the concentration of IL-1ɑ, IL-3, IL-4, IL-6, IL-9, IL-10, G-CSF, IL-12 (p40), IL-12 (p70), MIP-1ɑ, MIP-1β, RANTES, Eotaxin, TNF-ɑ, KC, MCP-1, and INF-γ using Bio-Plex Pro reagents (Bio-Rad Laboratories) according to the manufacturer’s recommended protocol. Serial dilutions of reconstituted standards were made to increase the number of data points from 7 to 10 while maintaining original dynamic range. Fluorescence intensity was measured by xPONENT software v.4.2 on FlexMap 3D instruments (Luminex). Bio-Plex Manager v.6.2 (Bio-Rad Laboratories) was used to construct standard curves for each analyte on each plate using the median FI of at least 35 beads done in duplicate. A four- or five-point regression analysis was used to calculate best fit and cytokine levels. Measurements on internal replicates were averaged and three experimental replicates are plotted as mean ± SEM.
Cryo-EM imaging of ASO-LNPs
An undiluted 3 μL drop of sample was deposited onto a lacey carbon copper EM grid (L300CU100, Electron Microscopy Sciences). Vitrification was performed using a Vitrobot Mark IV (Thermo Fisher Scientific). With the inner chamber set to 100% humidity and 4°C, excess liquid was wicked away from the EM grid by blotting for 4 s on both sides before plunging into liquid ethane.
Grids were then transferred into a Glacios electron microscope (Thermo Fisher Scientific) for image acquisition. Images were taken using a K2 Summit camera (Gatan, Pleasanton, CA) with instrument settings at spot size 4, condenser aperture at 50 μm, and objective aperture at 100 μm. Images at various magnifications were collected as 15 s movies at 0.25 frames/s. Movies were then motion corrected and aligned within the Digital Micrograph software (Gatan).
Tissue sectioning, IHC, imaging, and analysis
Left hemispheres from the ASO-LNP delivery study were transferred from 4% PFA to PBS after 48 h of fixation, and sent to NeuroScience Associates (NSA) for sectioning/staining. Tissues were embedded into a gelatin matrix using MultiBrain Technology (NSA) and sectioned coronally at a thickness of 30 μm. Every 12th section, equally spaced at 360 μm intervals, was used for staining. IHC staining was performed using rabbit anti-Iba1 (ab178846, Abcam) 1:100,000 and rat anti-CD68 (mca1957b, AbD Serotec) 1:400. Binding was detected with biotin-conjugated secondary antibodies, followed by avidin-biotin-horseradish peroxidase complex as recommended by the manufacturer (VECTASTAIN Elite ABC, Vector, Burlingame, CA). Sections were then treated with chromogen:diaminobenzidine tetrahydrochloride (DAB) and hydrogen peroxide to create a visible reaction product. The Iba1 chromogen also included nickel (II) sulfate and the CD68-stained sections were counterstained with Nissl (0.05% thionine/0.08 M acetate buffer [pH 4.5]) to detect cell bodies.
For anti-ASO IHC, 4 μm paraffin sections were deparaffinized and pre-treated with Target Retrieval Solution (pH 6.0) (Dako, Agilent) before staining on the Dako Universal Autostainer (Agilent). Rabbit polyclonal antibody JW138-442 (Genentech) was diluted at 0.5 μg/mL in 3% BSA in PBS and incubated for 1 h at RT. For detection, PowerVision anti-rabbit-AP polymer (PV-6133, Leica) was followed by the Permanent Red Kit (PRD500, ScyTek Laboratories). Sections were dehydrated overnight, counterstained briefly with hematoxylin, washed with PBS, coverslips were added, and slides were stored until imaging was performed.
Whole-slide imaging of stained brain sections was performed on the TissueScope LE120 (Huron Digital Pathology, ON, Canada) slide scanner in 20× mode with a resolution of 0.40 μm/pixel. Image analysis was performed blinded on a high-performance computing cluster using MATLAB v.9.9 (The MathWorks) with the image processing, computer vision, and deep learning toolboxes. A trained DeepLabv3+ convolutional neural network was used to segment tissue from the background. Analysis of positive stain area was performed using color thresholds and morphological operations as described previously.67,68 Integrated optical density measurements were calculated by converting to grayscale intensity and applying Beer-Lambert law. The positive stain area and integrated OD was normalized to the tissue section area and averaged for nine sections/animal. All analyzed images and data were reviewed by a board-certified pathologist.
RNA extraction from brain and qPCR analysis
Tissues from the ASO-LNP delivery study were transferred from RNALater to RLT Buffer (QIAGEN) lysis reagent and lysed using a TissueLyser (QIAGEN). RNA was extracted using the RNeasy Mini QIAcube Kit (74116, QIAGEN) with a QIACUBE workstation. RNA concentrations were determined using a NanoDrop and each sample was diluted with water to yield 2 ng/μL solutions. RNA was heat denatured at 90°C for 40 s and qPCR was performed on an Applied Biosystems ViiA7 thermal cycler using qScript TM XLT One-Step qRT-PCR ToughMix Low ROX (95134-02K, QuantaBio). Gene expression values were calculated using the –ΔΔCt method using a housekeeper gene, GAPDH, Ct values, and saline-treated Ct values for normalization. Assay probes were obtained from Thermo Fisher Scientific: FAM-MGB TMEM106b (4351368) and VIC-MGB GAPDH (4352339E).
Transcriptomic analysis of bulk brain tissue
Purification of total RNA used for qPCR and RNA sequencing (RNA-seq) analyses were completed as described above (RNA extraction from brain and qPCR analysis). RNA concentration and purity was determined using a NanoDrop, and purified RNA was stored at −80°C. RNA (1,000 ng) per sample was used to prepare RNA-seq libraries.
Transcriptomic data were analyzed with the GSNAP aligner,69 HTSeqGenie,70 and voom-limma,71 as described previously.72 GO enrichment testing was performed using GOfuncR73,74 and redundant terms were reduced based on semantic similarity through rrvgo.75 REACTOME pathway76,77 testing was performed using CAMERA,78 which identified the OAS antiviral response and trafficking/processing of endosomal TLR pathways. The remaining gene sets were manually curated.72 Gene set scores were calculated by taking the average expression values of all genes in a set, which were first log-transformed and stabilized as log2(CPM).
MOL-PCR
For in vitro neuron and microglia ASO vs. ASO-LNP uptake analysis (Figure 4), cells were plated at 3 × 105 cells/well in 12-well plates. On DIV7 (neurons) or DIV1 (microglia), cells were treated with a final concentration of 100 nM TA2 or TA2-LNPs, and allowed to incubate at 37°C with 5% CO2. After 24 h, supernatant was removed, cells were washed 4× with PBS, lysed using RIPA buffer (150 μL, Thermo Fisher Scientific), and the cell lysates were stored at −80°C until analysis was performed. Right brain tissue was used to determine in vivo ASO uptake after gymnosis and LNP delivery (Figure 6) (see "tissue collection" section in materials and methods above for additional details on brain extraction and storage). A 3 mm stainless steel bead and RIPA (Thermo Fisher Scientific) were added to the tissue, which was then lysed using a TissueLyser II (QIAGEN) at 30 Hz for 3 min. After lysis, samples were incubated on ice for 20 min and subsequently centrifuged at 15,000 rpm for 20 min at 4°C. The supernatant was removed and stored at −80°C until analysis was performed.
MOL-PCR utilizes a synthetic DNA ligation probe to bind and quantify ASO concentration in samples using a standard curve. The ligation probe has a self-complementary region that creates a loop structure with a 5′ base overhang of its 3′ end. This overhang anneals to the 3′ end of the target ASO using a 5′ phosphate. The target ASO analyte is then ligated to the probe using T4 DNA ligase, which is amplified using qPCR. The ligation probe sequence used was 5’-/5Phos/GCCACCATTAATGCCTCATAGTACACAACTCGCTAGTCGGTGGCTGAAGA/3AmMO/-3′ and the primer sequences used were 5′-CGCCACACTGTATCTTC-3′ and 5′-CGAGTTGTGTACTATGAGGC-3′ (Integrated DNA Technologies). Standard curve samples were prepared in either untreated cell or brain lysate using 5-fold dilutions of a 50 nM ASO stock for a total of 11 points and a blank. The standard curve and experimental samples were then diluted 10× in RIPA buffer and heated to 75°C for 10 min. Then, 4 μL of sample was added to 4 μL ligation probe (100 nM stock, diluted in water) and incubated on ice, and 2 μL DNA ligase buffer, 6 μL PEG solution, 3.75 μL water, and 0.25 μL T4 DNA Ligase (30 U/μL, EL0013; Thermo Fisher Scientific) were added. Samples were mixed well and ligation was performed at 16°C for 40 min followed by ligase inactivation at 75°C for 5 min. The ligation product was diluted 100× in water before PCR amplification using the primers above (100 nM final concentration) in a standard SYBR-based qPCR reaction (Perfecta SYBR Green SuperMixes Low ROX, 95056-02K, VWR) with 40 cycles of 95°C for 15 s, 56°C for 30 s, and 58°C for 40 s, followed by generation of a melting curve. From the resulting standard curve, the background and assay window were determined and the concentration of target ASO analyte was analyzed using linear regression.
Tissue processing and image acquisition for immuno-EM
Mouse brains were fixed by perfusion fixation in 4% PFA supplemented with 0.1% glutaraldehyde buffered with 0.1 M cacodylate buffer. Brain slices (1 mm thick) were infiltrated with 50% dimethyl sulfoxide for freeze protection, then frozen in liquid nitrogen and freeze-substituted in dry acetone supplemented with 0.01% uranyl acetate and 0.5% glutaraldehyde for 72 h at −78°C (on dry ice). The freeze-substituted tissues were then warmed up to RT, washed in dimethylformamide, and embedded in LR White resin (Electron Microscopy Sciences).
Semithin sections (500 nm thickness) were cut with the UMC ultramicrotome (Leica) using a DIATOME diamond knife for histology (Electron Microscopy Sciences). Sections were transferred to carbon-coated histology glass slides and dried on a warm plate. Labeling was performed with a rabbit polyclonal antibody JW138-442 (Genentech), a secondary (donkey) anti-rabbit-biotinylated antibody (Jackson ImmunoResearch), followed by streptavidin-conjugated to 20 nm colloidal gold particles (Abcam). The primary antibody was omitted for a negative labeling control. Finally, sections were stained with 2% aqueous uranyl acetate for 15 min and 0.1% Reynold’s lead citrate for 1 min to enhance contrast. Sections were thoroughly rinsed with ultrapure water and dried on a heat plate before being transferred to the SEM.
Scanning electron microscopy (SEM) was performed using a Zeiss Gemini SEM 300 equipped with a field emission gun (Carl Zeiss AG). For operation of the GeminiSEM 300 microscope the application software SmartSEM (v.6.01) was used (Carl Zeiss AG). Imaging was carried out using a backscatter electron detector (BSD1) at 8.5 mm working distance, 60 μm aperture, 5 keV acceleration voltage, and with operation of the field emission gun in "high current" mode. For the majority of images, a scan speed of "4,″ noise reduction by 5× line averaging, and an image size of at least 4,096 × 3,072 (4k × 3k) pixels was chosen. For imaging of ultrastructural detail, pixel sizes between 2 and 5 nm were used. The greyscale of the images was inverted to achieve “TEM-like” representations.
Statistical analysis
Data plotting and statistical analyses were performed using Prism 9.2.0 (GraphPad Software). Results are presented as mean ± SEM and all experiments were completed in triplicate unless otherwise noted. For in vitro dose-response curves, data were analyzed using log (inhibitor concentration) vs. response, variable slope (four parameters) equations. Data involving multiple comparisons with saline or ASO treatment were analyzed using one-way ANOVA coupled with a multiple comparisons Dunnett’s test. All other data were analyzed using either unpaired two-tailed Student’s t tests or unpaired two-tailed Mann-Whitney tests, which are denoted in the figure legends. Unless otherwise noted, statistical significance was defined as p < 0.05.
Data availability
Sequencing data has been deposited in the GEO repository (GEO: GSE220777).
Acknowledgments
We thank D. Mellal and colleagues at Roche Innovation Center, Copenhagen, for synthesizing and formulating the ASOs required for this study, E. Koller (Roche) for gifting the rabbit and rat anti-ASO antibodies, C. Chalouni for confocal imaging support, J. Cupp, C. Poon, and members of the Flow Cytometry Core lab for technical help, W. Meilandt for training and troubleshooting insights, M. Gonzalez and M. Baca for assistance with tissue sectioning and staining, and B. Bingol, L. Kielpinski, H. Gylling, J. Lipka, and A. Vogl for discussions that were essential for propelling these studies forward.
Author contributions
A.E.B. and C.C.H. conceived the project, designed and/or executed experiments, analyzed the data, and wrote the manuscript. A.E.B., S.L.D., H.L., and A. Easton designed, executed, and analyzed data from the in vivo experiments. C.-W.Y., H.-C. L., A.G., J.B., J.L., and A. Estevez formulated and/or characterized the ASO-LNP formulations. B.I.L. and B.A.F. analyzed the transcriptomic data. O.F., M.R., K.H., and H.N. executed IHC and/or immunoEM studies, and helped interpret the data. M.S. assisted with IF analysis of fixed cells. C.S. executed and analyzed the MOL-PCR data. F.H. provided cortical neuron cell cultures for in vitro studies. All authors reviewed the manuscript and provided edits.
Declaration of interests
All authors are employees and shareholders of either F. Hoffmann-La Roche Ltd or Genentech, Inc., a member of the Roche group.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.05.005.
Supplemental information
References
- 1.Crooke S.T., Witztum J.L., Bennett C.F., Baker B.F. RNA-targeted therapeutics. Cell Metab. 2018;27:714–739. doi: 10.1016/j.cmet.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Schoch K.M., Miller T.M. Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron. 2017;94:1056–1070. doi: 10.1016/j.neuron.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finkel R.S., Mercuri E., Darras B.T., Connolly A.M., Kuntz N.L., Kirschner J., Chiriboga C.A., Saito K., Servais L., Tizzano E., et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017;377:1723–1732. doi: 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 4.Bennett C.F., Kordasiewicz H.B., Cleveland D.W. Antisense drugs make sense for neurological diseases. Annu. Rev. Pharmacol. Toxicol. 2021;61:831–852. doi: 10.1146/annurev-pharmtox-010919-023738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rinaldi C., Wood M.J.A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018;14:9–21. doi: 10.1038/nrneurol.2017.148. [DOI] [PubMed] [Google Scholar]
- 6.Geary R.S., Norris D., Yu R., Bennett C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015;87:46–51. doi: 10.1016/j.addr.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017;35:230–237. doi: 10.1038/nbt.3779. [DOI] [PubMed] [Google Scholar]
- 8.Juliano R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016;44:6518–6548. doi: 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varkouhi A.K., Scholte M., Storm G., Haisma H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Roberts T.C., Langer R., Wood M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020;19:673–694. doi: 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hou X., Zaks T., Langer R., Dong Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021;6:1078–1094. doi: 10.1038/s41578-021-00358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patravale V., Dandekar P., Jain R. Nanoparticulate systems as drug carriers: the need. Nanoparticulate Drug Delivery. 2012:1–28. [Google Scholar]
- 13.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.-C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L., et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 14.Teo S.P. Review of COVID-19 mRNA vaccines: BNT162b2 and mRNA-1273. J. Pharm. Pract. 2022;35:947–951. doi: 10.1177/08971900211009650. [DOI] [PubMed] [Google Scholar]
- 15.Rungta R.L., Choi H.B., Lin P.J., Ko R.W., Ashby D., Nair J., Manoharan M., Cullis P.R., Macvicar B.A. Lipid nanoparticle delivery of siRNA to silence neuronal gene expression in the brain. Mol. Ther. Nucleic Acids. 2013;2:e136. doi: 10.1038/mtna.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirunagi T., Sahashi K., Tachikawa K., Leu A.I., Nguyen M., Mukthavaram R., Karmali P.P., Chivukula P., Tohnai G., Iida M., et al. Selective suppression of polyglutamine-expanded protein by lipid nanoparticle-delivered siRNA targeting CAG expansions in the mouse CNS. Mol. Ther. Nucleic Acids. 2021;24:1–10. doi: 10.1016/j.omtn.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conceição M., Mendonça L., Nóbrega C., Gomes C., Costa P., Hirai H., Moreira J.N., Lima M.C., Manjunath N., Pereira de Almeida L. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials. 2016;82:124–137. doi: 10.1016/j.biomaterials.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 18.Kwon E.J., Skalak M., Lo Bu R., Bhatia S.N. Neuron-targeted nanoparticle for siRNA delivery to traumatic brain injuries. ACS Nano. 2016;10:7926–7933. doi: 10.1021/acsnano.6b03858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlsson J., Rui Y., Kozielski K.L., Placone A.L., Choi O., Tzeng S.Y., Kim J., Keyes J.J., Bogorad M.I., Gabrielson K., et al. Engineered nanoparticles for systemic siRNA delivery to malignant brain tumours. Nanoscale. 2019;11:20045–20057. doi: 10.1039/c9nr04795f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galbraith W.M., Hobson W.C., Giclas P.C., Schechter P.J., Agrawal S. Complement activation and hemodynamic changes following intravenous administration of phosphorothioate oligonucleotides in the monkey. Antisense Res. Dev. 1994;4:201–206. doi: 10.1089/ard.1994.4.201. [DOI] [PubMed] [Google Scholar]
- 21.Levin A.A. A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim. Biophys. Acta. 1999;1489:69–84. doi: 10.1016/s0167-4781(99)00140-2. [DOI] [PubMed] [Google Scholar]
- 22.Easton A., Jensen M.L., Wang C., Hagedorn P.H., Li Y., Weed M., Meredith J.E., Guss V., Jones K., Gill M., et al. Identification and characterization of a MAPT-targeting locked nucleic acid antisense oligonucleotide therapeutic for tauopathies. Mol. Ther. Nucleic Acids. 2022;29:625–642. doi: 10.1016/j.omtn.2022.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paramasivam P., Franke C., Stöter M., Höijer A., Bartesaghi S., Sabirsh A., Lindfors L., Arteta M.Y., Dahlén A., Bak A., et al. Endosomal escape of delivered mRNA from endosomal recycling tubules visualized at the nanoscale. J. Cell Biol. 2022;221:e202110137. doi: 10.1083/jcb.202110137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz S.L., Conn G.L. RNA regulation of the antiviral protein 2'-5'-oligoadenylate synthetase. Wiley Interdiscip. Rev. RNA. 2019;10:e1534. doi: 10.1002/wrna.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lind N.A., Rael V.E., Pestal K., Liu B., Barton G.M. Regulation of the nucleic acid-sensing Toll-like receptors. Nat. Rev. Immunol. 2022;22:224–235. doi: 10.1038/s41577-021-00577-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lund J.M., Alexopoulou L., Sato A., Karow M., Adams N.C., Gale N.W., Iwasaki A., Flavell R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilleron J., Querbes W., Zeigerer A., Borodovsky A., Marsico G., Schubert U., Manygoats K., Seifert S., Andree C., Stöter M., et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013;31:638–646. doi: 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- 28.Migliorati J.M., Liu S., Liu A., Gogate A., Nair S., Bahal R., Rasmussen T.P., Manautou J.E., Zhong X.B. Absorption, distribution, metabolism, and excretion of FDA-approved antisense oligonucleotide drugs. Drug Metab. Dispos. 2022;50:888–897. doi: 10.1124/dmd.121.000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dhuri K., Bechtold C., Quijano E., Pham H., Gupta A., Vikram A., Bahal R. Antisense oligonucleotides: an emerging area in drug discovery and development. J. Clin. Med. 2020;9:2004. doi: 10.3390/jcm9062004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Deerlin V.M., Sleiman P.M.A., Martinez-Lage M., Chen-Plotkin A., Wang L.S., Graff-Radford N.R., Dickson D.W., Rademakers R., Boeve B.F., Grossman M., et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat. Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finch N., Carrasquillo M.M., Baker M., Rutherford N.J., Coppola G., Dejesus-Hernandez M., Crook R., Hunter T., Ghidoni R., Benussi L., et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467–474. doi: 10.1212/WNL.0b013e31820a0e3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Y.X., Cao Q., Sawaya M.R., Abskharon R., Ge P., DeTure M., Dickson D.W., Fu J.Y., Ogorzalek Loo R.R., Loo J.A., Eisenberg D.S. Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature. 2022;605:304–309. doi: 10.1038/s41586-022-04670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schweighauser M., Arseni D., Bacioglu M., Huang M., Lövestam S., Shi Y., Yang Y., Zhang W., Kotecha A., Garringer H.J., et al. Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature. 2022;605:310–314. doi: 10.1038/s41586-022-04650-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jafar-Nejad P., Powers B., Soriano A., Zhao H., Norris D.A., Matson J., DeBrosse-Serra B., Watson J., Narayanan P., Chun S.J., et al. The atlas of RNase H antisense oligonucleotide distribution and activity in the CNS of rodents and non-human primates following central administration. Nucleic Acids Res. 2021;49:657–673. doi: 10.1093/nar/gkaa1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dugger B.N., Dickson D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017;9:a028035. doi: 10.1101/cshperspect.a028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tabrizi S.J., Leavitt B.R., Landwehrmeyer G.B., Wild E.J., Saft C., Barker R.A., Blair N.F., Craufurd D., Priller J., Rickards H., et al. Targeting huntingtin expression in patients with huntington's disease. N. Engl. J. Med. 2019;380:2307–2316. doi: 10.1056/NEJMoa1900907. [DOI] [PubMed] [Google Scholar]
- 37.Miller T., Cudkowicz M., Shaw P.J., Andersen P.M., Atassi N., Bucelli R.C., Genge A., Glass J., Ladha S., Ludolph A.L., et al. Phase 1-2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 2020;383:109–119. doi: 10.1056/NEJMoa2003715. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka H., Takata N., Sakurai Y., Yoshida T., Inoue T., Tamagawa S., Nakai Y., Tange K., Yoshioka H., Maeki M., et al. Delivery of oligonucleotides using a self-degradable lipid-like material. Pharmaceutics. 2021;13:544. doi: 10.3390/pharmaceutics13040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang S., Wang D., Sun Y., Zheng B. Delivery of antisense oligonucleotide using polyethylenimine-based lipid nanoparticle modified with cell penetrating peptide. Drug Deliv. 2019;26:965–974. doi: 10.1080/10717544.2019.1667453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang L., Ma F., Liu F., Chen J., Zhao X., Xu Q. Efficient delivery of antisense oligonucleotides using bioreducible lipid nanoparticles in vitro and in vivo. Mol. Ther. Nucleic Acids. 2020;19:1357–1367. doi: 10.1016/j.omtn.2020.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen L., Watson C., Morsch M., Cole N.J., Chung R.S., Saunders D.N., Yerbury J.J., Vine K.L. Improving the delivery of SOD1 antisense oligonucleotides to motor neurons using calcium phosphate-lipid nanoparticles. Front. Neurosci. 2017;11:476. doi: 10.3389/fnins.2017.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min H.S., Kim H.J., Naito M., Ogura S., Toh K., Hayashi K., Kim B.S., Fukushima S., Anraku Y., Miyata K., Kataoka K. Systemic brain delivery of antisense oligonucleotides across the blood–brain barrier with a glucose-coated polymeric nanocarrier. Angew. Chem. Int. Ed. Engl. 2020;59:8173–8180. doi: 10.1002/anie.201914751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma F., Yang L., Sun Z., Chen J., Rui X., Glass Z., Xu Q. Neurotransmitter-derived lipidoids (NT-lipidoids) for enhanced brain delivery through intravenous injection. Sci. Adv. 2020;6:eabb4429. doi: 10.1126/sciadv.abb4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fleck D., Phu L., Verschueren E., Hinkle T., Reichelt M., Bhangale T., Haley B., Wang Y., Graham R., Kirkpatrick D.S., et al. PTCD1 is required for mitochondrial oxidative-phosphorylation: possible genetic association with alzheimer's disease. J. Neurosci. 2019;39:4636–4656. doi: 10.1523/JNEUROSCI.0116-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarode A., Fan Y., Byrnes A.E., Hammel M., Hura G.L., Fu Y., Kou P., Hu C., Hinz F.I., Roberts J., et al. Predictive high-throughput screening of PEGylated lipids in oligonucleotide-loaded lipid nanoparticles for neuronal gene silencing. Nanoscale Adv. 2022;4:2107–2123. doi: 10.1039/d1na00712b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Householder K.T., Dharmaraj S., Sandberg D.I., Wechsler-Reya R.J., Sirianni R.W. Fate of nanoparticles in the central nervous system after intrathecal injection in healthy mice. Sci. Rep. 2019;9:12587. doi: 10.1038/s41598-019-49028-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Semple S.C., Akinc A., Chen J., Sandhu A.P., Mui B.L., Cho C.K., Sah D.W.Y., Stebbing D., Crosley E.J., Yaworski E., et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 48.Ohta S., Kikuchi E., Ishijima A., Azuma T., Sakuma I., Ito T. Investigating the optimum size of nanoparticles for their delivery into the brain assisted by focused ultrasound-induced blood–brain barrier opening. Sci. Rep. 2020;10:18220. doi: 10.1038/s41598-020-75253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shyam R., Ren Y., Lee J., Braunstein K.E., Mao H.Q., Wong P.C. Intraventricular delivery of siRNA nanoparticles to the central nervous system. Mol. Ther. Nucleic Acids. 2015;4:e242. doi: 10.1038/mtna.2015.15. [DOI] [PubMed] [Google Scholar]
- 50.Yaari Z., da Silva D., Zinger A., Goldman E., Kajal A., Tshuva R., Barak E., Dahan N., Hershkovitz D., Goldfeder M., et al. Theranostic barcoded nanoparticles for personalized cancer medicine. Nat. Commun. 2016;7:13325. doi: 10.1038/ncomms13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dahlman J.E., Kauffman K.J., Xing Y., Shaw T.E., Mir F.F., Dlott C.C., Langer R., Anderson D.G., Wang E.T. Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proc. Natl. Acad. Sci. USA. 2017;114:2060–2065. doi: 10.1073/pnas.1620874114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sago C.D., Lokugamage M.P., Paunovska K., Vanover D.A., Monaco C.M., Shah N.N., Gamboa Castro M., Anderson S.E., Rudoltz T.G., Lando G.N., et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. USA. 2018;115:E9944–E9952. doi: 10.1073/pnas.1811276115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guimaraes P.P.G., Zhang R., Spektor R., Tan M., Chung A., Billingsley M.M., El-Mayta R., Riley R.S., Wang L., Wilson J.M., Mitchell M.J. Ionizable lipid nanoparticles encapsulating barcoded mRNA for accelerated in vivo delivery screening. J. Control. Release. 2019;316:404–417. doi: 10.1016/j.jconrel.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sago C., Campbell E., Lutz B., Patwardhan N., Hamilton G., Wong C., Lee D., Keating S., Murray K., Singh M., Ciaramella G. Screening of chemically distinct lipid nanoparticles in vivo using DNA barcoding technology towards effectively delivering messenger RNA to hematopoietic stem and progenitor cells. Blood. 2021;138:2931. [Google Scholar]
- 55.Pendergraff H., Schmidt S., Vikeså J., Weile C., Øverup C., W Lindholm M., Koch T. Nuclear and cytoplasmatic quantification of unconjugated, label-free locked nucleic acid oligonucleotides. Nucleic Acid Ther. 2020;30:4–13. doi: 10.1089/nat.2019.0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srinivasan K., Friedman B.A., Larson J.L., Lauffer B.E., Goldstein L.D., Appling L.L., Borneo J., Poon C., Ho T., Cai F., et al. Untangling the brain's neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 2016;7:11295. doi: 10.1038/ncomms11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swarup V., Hinz F.I., Rexach J.E., Noguchi K.I., Toyoshiba H., Oda A., Hirai K., Sarkar A., Seyfried N.T., Cheng C., et al. Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat. Med. 2019;25:152–164. doi: 10.1038/s41591-018-0223-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Witting A., Möller T. Microglia cell culture: a primer for the novice. Methods Mol. Biol. 2011;758:49–66. doi: 10.1007/978-1-61779-170-3_4. [DOI] [PubMed] [Google Scholar]
- 60.Giulian D., Baker T.J. Characterization of ameboid microglia isolated from developing mammalian brain. J. Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCarthy K.D., de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J. Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Z., Watanabe M., Nishiyama A. Optimization of oligodendrocyte progenitor cell culture method for enhanced survival. J. Neurosci. Methods. 2005;149:50–56. doi: 10.1016/j.jneumeth.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 63.Schildge S., Bohrer C., Beck K., Schachtrup C. Isolation and culture of mouse cortical astrocytes. J. Vis. Exp. 2013;71:50079. doi: 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saura J., Tusell J.M., Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. doi: 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- 65.Fan Y., Yen C.W., Lin H.C., Hou W., Estevez A., Sarode A., Goyon A., Bian J., Lin J., Koenig S.G., et al. Automated high-throughput preparation and characterization of oligonucleotide-loaded lipid nanoparticles. Int. J. Pharm. 2021;599:120392. doi: 10.1016/j.ijpharm.2021.120392. [DOI] [PubMed] [Google Scholar]
- 66.Bolte S., Cordelières F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 67.Meilandt W.J., Ngu H., Gogineni A., Lalehzadeh G., Lee S.H., Srinivasan K., Imperio J., Wu T., Weber M., Kruse A.J., et al. Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the aβ42:aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP alzheimer's mouse model. J. Neurosci. 2020;40:1956–1974. doi: 10.1523/JNEUROSCI.1871-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Le Pichon C.E., Meilandt W.J., Dominguez S., Solanoy H., Lin H., Ngu H., Gogineni A., Sengupta Ghosh A., Jiang Z., Lee S.H., et al. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med. 2017;9:eaag0394. doi: 10.1126/scitranslmed.aag0394. [DOI] [PubMed] [Google Scholar]
- 69.Wu T.D., Reeder J., Lawrence M., Becker G., Brauer M.J. GMAP and GSNAP for genomic sequence alignment: enhancements to speed, accuracy, and functionality. Methods Mol. Biol. 2016;1418:283–334. doi: 10.1007/978-1-4939-3578-9_15. [DOI] [PubMed] [Google Scholar]
- 70.Pau G., Reeder J. 2021. HTSeqGenie: A NGS Analysis Pipeline. Bioconductor Version: Release (3.12) [Google Scholar]
- 71.Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Friedman B.A., Srinivasan K., Ayalon G., Meilandt W.J., Lin H., Huntley M.A., Cao Y., Lee S.H., Haddick P.C.G., Ngu H., et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of alzheimer's disease not evident in mouse models. Cell Rep. 2018;22:832–847. doi: 10.1016/j.celrep.2017.12.066. [DOI] [PubMed] [Google Scholar]
- 73.Prüfer K., Muetzel B., Do H.H., Weiss G., Khaitovich P., Rahm E., Pääbo S., Lachmann M., Enard W. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics. 2007;8:41. doi: 10.1186/1471-2105-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grote S. GOfuncR: gene ontology enrichment using FUNC. Bioconductor Release. 2021;3:13. [Google Scholar]
- 75.Sayols S. rrvgo: a Bioconductor package to reduce and visualize Gene Ontology terms. Bioconductor Release. 2020;3:13. doi: 10.17912/micropub.biology.000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R., et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498–D503. doi: 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu D., Smyth G.K. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012;40:e133. doi: 10.1093/nar/gks461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data has been deposited in the GEO repository (GEO: GSE220777).