

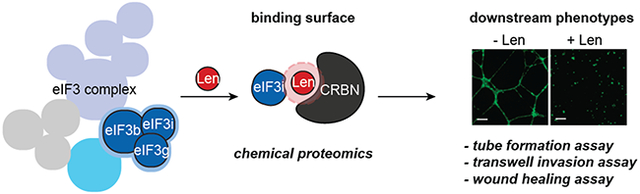
Abstract
Lenalidomide is a ligand of the E3 ligase substrate adapter cereblon (CRBN) that achieves its clinical effects in part by promotion of substrate recruitment and degradation. In contrast to prior targets, eIF3i is recruited but not degraded upon complex formation with lenalidomide and CRBN, although the structural details and mechanistic outcomes of this interaction are unresolved. Here, we characterize the structural basis and mechanistic outcomes of lenalidomide-induced sequestration of eIF3i from the eIF3 complex. Identification of the binding interface on eIF3i by a covalent lenalidomide probe and mass spectrometry rationalizes the sequestration event. We further connect eIF3i and CRBN to lenalidomide-driven effects on angiogenic markers, Akt1 phosphorylation, and associated antiangiogenesis phenotypes. Finally, we find that eIF3i sequestration is observed in MM.1S and MOLM13 cells after the degradation of other substrates, such as IKZF1. The defined binding interface elucidated by chemical proteomics, and the observation of eIF3i sequestration as a lenalidomide function opens future directions in designing new chemical adapters for protein sequestration as a strategy to selectively control protein functions.
Keywords: cereblon, lenalidomide, targeted protein degradation, chemical proteomics
Graphical Abstract
INTRODUCTION
Lenalidomide (Len) is an analog of thalidomide that is used to treat multiple myeloma and del(5q) myelodysplastic syndrome. Lenalidomide and its derivatives achieve their clinical activity in part by engagement of the E3 ligase substrate adapter CRBN, which promotes the recruitment and degradation of target proteins, including IKZF1, IKZF3 and CK1α.1-3 The growing definition of mechanistic targets of lenalidomide has driven parallel growth in the application of CRBN ligands in targeted protein degradation strategies and the engagement of CRBN in the clinic for novel therapies.4, 5 Lenalidomide and its analogs additionally possess immunosuppressive, immune co-stimulatory and antiangiogenetic effects, whose mechanistic targets are only partially described.6, 7, 8
In our pursuit to discover additional targets of lenalidomide, we recently reported the eukaryotic translation initiation factor 3 subunit I (eIF3i) as a novel target for lenalidomide in epithelial cells, including HEK293T, HeLa and HepG2 cells using a photoaffinity labeling (PAL)-assisted chemical proteomics strategy.9 eIF3i is a part of the eIF3 complex, which is composed of 13 subunits and involved in the protein translation initiation process, including regulation of ribosomal complexes such as 40S and 60S prior to initiation, and 43S pre-initiation complex in mRNA scanning and recognition.10-12 We previously found that eIF3i is stabilized in a complex with CRBN and lenalidomide in a dose-dependent manner, and unlike other known targets of lenalidomide, is not degraded upon recruitment to CRBN. However, the structural underpinning of the interaction, the functional outcome on the eIF3 complex, and the contribution of eIF3i recruitment to the overall effects of lenalidomide remained to be defined (Figure 1a). Structurally, eIF3i is composed of a helical WD40 wheel, which is unique from other recruited substrates that bear a beta-loop as the recognition motif.13 Loss of eIF3i may affect the functions of the eIF3 complex in translation initiation and elongation,14 and thus impact the regulation on angiogenic marker vascular endothelial growth factor A (VEGFA).15 Other reported roles of eIF3i may also be impaired, such as promoting the constitutive phosphorylation on Akt1.16, 17 These phenotypes associated with eIF3i loss are reminiscent of phenotypic effects that are measured for lenalidomide.18, 19
Figure 1 ∣. Lenalidomide sequesters eIF3i from the eIF3 complex.

(a) Overview of the structural and mechanistic foci in this study. Coimmunoprecipitation of (b) endogenous CRBN, (c) endogenous eIF3i, and (d) endogenous eIF3b from HUVECs. (e) Representative sucrose gradient analysis of purified eIF3i-Flag complexes with or without lenalidomide treatment. Samples were separated to 11 fractions, and the eIF3i-eIF3b and CRBN–lenalidomide–eIF3i complex were examined by immunoblotting.
Here, we report the structural features and mechanistic outcomes of lenalidomide-mediated eIF3i recruitment. We find that eIF3i is sequestered from the eIF3 complex by lenalidomide, which is rationalized by characterization of the binding interface using an electrophilic covalent probe of lenalidomide developed herein. By using biochemical assays, we found that sequestration of eIF3i by lenalidomide is connected to changes in translational profiles, VEGFA sensitivity and Akt1 phosphorylation in epithelial cells, as well as antiangiogenetic effects in human umbilical vein endothelial cells (HUVECs). Cellular time course studies further demonstrate that the interaction of eIF3i with CRBN and lenalidomide is prevalent in different cell types including multiple myeloma MM.1S cells. Therefore, this interaction may contribute to the physiological functions of lenalidomide, especially during extended exposure time.
RESULTS
Lenalidomide sequesters eIF3i from the eIF3 complex
Previously, we observed a dose-dependent interaction of CRBN and eIF3i in the presence of lenalidomide or photolenalidomide in HEK293T cells stably expressing Flag-CRBN (HEK293T-CRBN) over 10–50 μM concentrations,9 which are concentrations relevant to the anti-inflammatory and antiangiogenetic properties of lenalidomide in cellular assays.18-20 To study the effect of lenalidomide in a model of angiogenesis, we therefore examined human umbilical vein endothelial cells (HUVECs) and found a similar association between CRBN and eIF3i that is dependent on lenalidomide (50 μM, Figure 1b). Reciprocal immunoprecipitation of eIF3i confirmed that the CRBN–eIF3i interaction is enhanced in the presence of lenalidomide (Figure 1c). We next evaluated the effect of lenalidomide on the eIF3 complex, where eIF3i directly interacts with eIF3b and eIF3g in the (b:g:i) subcomplex.14, 21, 22 As expected, both eIF3b and eIF3g coimmunoprecipitated with eIF3i; however, these associations are decreased after addition of lenalidomide (Figure 1c). Likewise, immunoprecipitation of endogenous eIF3b from HUVECs showed a strong interaction with eIF3i that decreased in the presence of lenalidomide, whereas association of eIF3b with eIF3g appears unaffected (Figure 1d). The interaction between eIF3i and eIF3b was similarly disrupted in HEK293T and HEK293T-CRBN cells by lenalidomide treatment (Figure S1a, S1b). Collectively, these data indicate that eIF3i is sequestered from the eIF3 complex upon treatment with lenalidomide.
We then assessed the fraction of eIF3i associated with eIF3b or with CRBN on lenalidomide treatment. We performed a sucrose gradient analysis of the eIF3i-Flag immunoprecipitated from HEK293T cells to separate the complexes (Figure 1e). We found that eIF3i is associated with eIF3b in the untreated cells but co-elutes in two complexes in the presence of lenalidomide. After treatment with lenalidomide, the majority of eIF3i was observed in complexation with CRBN, without association to eIF3b, while a residual amount of eIF3i co-eluted in later fractions. This partial sequestration may be related to the relatively higher expression level and copy number of eIF3i compared to those of CRBN.23 When evaluating the protein levels upon treatment at different time points, we found that the protein levels of eIF3i, eIF3b, eIF3g and CRBN appeared steady over the course of a 5 d incubation with lenalidomide in HEK293T cells (Figure S2). Evaluation of a panel of CRBN ligands showed that sequestration of eIF3i is primarily induced by lenalidomide and potentiated by pomalidomide and thalidomide to a lesser degree (Figure S3a, S3b). By contrast, larger CRBN ligands, such as CC-885, CC-220, or dBET6 do not recruit eIF3i to a complex with CRBN (Figure S3a, S3b). Taken together, these data demonstrate that lenalidomide selectively sequesters eIF3i from the eIF3 complex to reassociate with CRBN in epithelial cells.
Mapping the binding interface of eIF3i and lenalidomide by chemical proteomics
Intrigued by the sequestration of eIF3i with lenalidomide and CRBN, we next sought to evaluate the binding surface of the ternary complex. Previously, we reported photolenalidomide (pLen) as a probe to measure protein interactions of lenalidomide in cells and map a binding site on CRBN; however, eIF3i was impervious to our attempts to map a binding site with photolenalidomide.9 Given that activity-based covalent labeling chemistries provide and alternative method for labeling proteins,24, 25 we designed and tested a covalent probe, termed SL1, that contains an acrylamide moiety as an electrophilic warhead for nucleophilic cysteines (Figure 2a).
Figure 2 ∣. Definition of the binding interface of eIF3i and lenalidomide via chemical proteomics.

(a) The structures of photolenalidomide and the covalent probe SL1. (b) Schematic demonstration of direct covalent labelling using SL1 and the covalent competitive displacement photoaffinity labelling workflow using photolenalidomide and SL1. (c) Mutated Cys residues highlighted on the structure of eIF3i (AlphaFold: AF-Q13347-F1-model_v2). eIF3i is in light blue with the region of interest highlighted in dark blue. C120 is colored in red. Other Cys residues mutated as controls are colored in orange. (d) Gel-based covalent competitive displacement of photolenalidomide with SL1. (e) Coimmunoprecipitation of wild-type (WT) eIF3i or eIF3i mutants in the presence of lenalidomide from HEK293T cells. (f) Proposed binding surface of lenalidomide-CRBN complex on eIF3i. eIF3i is colored in blue with residues mutated in this study (C120, Y118, G117) highlighted in yellow.
SL1 was first evaluated as a suitable covalent probe for lenalidomide (Figure S4). Like lenalidomide and photolenalidomide, SL1 degrades IKZF3 in MM.1S cells after 8 h in a NEDDlyation-dependent manner and promotes engagement of IKZF1 with endogenous CRBN in MM.1S cells (Figure S4a, S4b). When coimmunoprecipitating endogenous CRBN from HEK293T cells, SL1 enriched more eIF3i with CRBN as compared to lenalidomide (Figure S4c), suggesting greater stabilization of the complex with the covalent probe. Quantification by AlphaScreen assay showed that SL1 also generated a stronger ternary complex in vitro between GST-eIF3i and His-CRBN (Figure S4d), which additionally confirms that the direct interaction between eIF3i and CRBN occurs without contribution of additional cellular components.
We next focused on mapping the binding site of SL1 to eIF3i. We first performed the direct covalent labeling where recombinant His-CRBN and GST-eIF3i were incubated with 50 μM SL1 for 30 min at 37 °C before the proteins were precipitated with acetone and digested with trypsin (Figure 2b). By searching against His-CRBN, GST-eIF3i and common contaminant proteins, tandem MS analysis identified a cysteine residue C120 on eIF3i being labeled by SL1 (Figure S5, Table S1). The eIF3i residue C120 is located on the loop of the bottom face of the blade 3 on the WD40 wheel (AlphaFold: AF-Q13347-F1-model_v2, Figure 2c). Interestingly, this region on eIF3i is the same surface that engages eIF3b in the eIF3 complex,26, 27 which may explain why the interaction of eIF3i with eIF3b is impaired upon eIF3i sequestration by CRBN.
To visualize and quantify the binding event, we used photolenalidomide as a probe to visualize engagement of CRBN and eIF3i, and performed a competitive displacement of photolenalidomide using SL1 (Figure 2b). In brief, Flag-CRBN and eIF3i-Flag were separately purified from HEK293T cells and incubated with DMSO or different concentrations of SL1 for 30 min at 37 °C before 25 μM photolenalidomide was added and incubated for additional 30 min at 37 °C. After UV irradiation (30 s), the proteins were precipitated with acetone, resuspended, and conjugated to AlexaFluor 488 azide (AF488) for visualization by in-gel fluorescence. As expected, both eIF3i and CRBN are labeled by photolenalidomide and the labeling is competitively displaced by SL1 in a dose-dependent manner, with almost complete competition at 50 μM (Figure 2d). Labeling of eIF3i is dependent on the presence of CRBN, indicating that the interaction and labeling event is mediated by CRBN.
We then mutated eIF3i C120 and used the purified eIF3i-Flag (C120A) to evaluate whether C120 is the primary contributing residue to SL1 labeling. Three other Cys residues were also included as controls including C57, C76 and C160 (residues highlighted in orange, Figure 2c).27 The competitive displacement of photolenalidomide by SL1 (50 μM) revealed that only the eIF3i C120A mutation rescued the labelling displacement with SL1 competition as expected. (Figure 2d). These data indicate that the C120 residue is essential for SL1 engagement with CRBN and that this region of eIF3i may be critical for the binding interaction between eIF3i, lenalidomide, and CRBN (Figure 2c).
In parallel, we performed tryptophan-scanning mutagenesis on the bottom-face loop of blade 3 on eIF3i (residues 120-115) to gain structural insight to the eIF3i binding surface with CRBN and lenalidomide (Figure 2c, 2e). We found that two eIF3i mutants, Y118W and G117W, selectively escape engagement of CRBN and lenalidomide by coimmunoprecipitation (Figure 2e). Mutations on M116 and Q115 also mitigated binding ability, but not mutations on C120 or Q119, possibly due to their relatively distal position from the binding pocket with lenalidomide. Mapping of the eIF3i Y118W and G117W mutations on eIF3i indicates that these mutations may sterically block engagement with the CRBN–lenalidomide complex and strengthens the identification of blade 3 on eIF3i as the primary binding interface mediating this interaction (Figure 2f, Figure S6a-c).
eIF3i sequestration affects the functions of both eIF3i and the eIF3 complex
We next sought to explore the downstream effect of eIF3i sequestration by lenalidomide. Since eIF3i is part of the eIF3 translational complex, we first used polysome profiling to evaluate if sequestration of eIF3i will disturb the function of the eIF3 complex. Translational initiation mediated by the eIF3 complex starts by promoting the joining to 40S with 60S to form the 80S ribosome,10 which aligns with the role of the eIF3 complex in the elongation process and polysome generation.28 Using a sucrose density gradient, we fractionated whole cell lysates from HEK293T cells treated with or without lenalidomide and found an increase in the 80S monosome peak and a slight decrease in the 40S and 60S complexes when overlaying the lenalidomide-treated profile with the control profile (Figure 3a). Polysome peaks, which are indicative of well-translated mRNAs, were slightly decreased, suggesting a change in the translation initiation:elongation ratio driven by lenalidomide treatment. Protein levels of eIF3i, eIF3b and eIF3g in HEK293T cells and mRNA levels of eIF3i and eIF3b in both HEK293T cells and in HUVECs were not significantly different over the course of these experiments (Figure S2, S7).
Figure 3 ∣. Effects of lenalidomide treatment polysome profiles and Akt1 phosphorylation.

(a) Polysome profiling of HEK293T cells with or without lenalidomide analyzed by sucrose gradient separation and analysis. The elution profiles of the indicated fractions were determined by absorbance at 280 nm and normalized. The migration of the 40S, 60S and 80S ribosomes are highlighted. (b) Akt1 phosphorylation levels (Ser473, Thr308) with or without lenalidomide treatment by immunoblotting. (c) Akt1 phosphorylation level (Ser473) with or without lenalidomide treatment by ELISA assay.
In addition to participating in translation as a part of the eIF3 complex, eIF3i possesses other reported functions, such as promoting constitutive Akt1 activation.16 We evaluated the phosphorylation of Akt1 on both Ser473 and Thr308 by immunoblotting. Lenalidomide inhibited Akt1 phosphorylation as expected.18, 19 We found that lenalidomide inhibited phosphorylation of Akt1 in HEK293T cells in a manner that is dependent on CRBN and eIF3i (Figure 3b). We further quantified Akt1 phosphorylation at Ser473 by ELISA and found that the inhibition of pAkt1 is dose-dependent in HEK293T cells over 1–10 μM concentrations of lenalidomide, which was similarly dependent on CRBN and eIF3i levels (Figure 3c, Figure S8a). As expected, CRBN overexpression (OE) enhanced the response and eIF3i OE rescued Akt1 phosphorylation. eIF3i mutant identified in the structural studies (G117W) does not engage lenalidomide in the ternary complex, thus not responding to lenalidomide treatment. These results indicate that the decrease in Akt1 phosphorylation on lenalidomide treatment is mediated by both CRBN and eIF3i. CRBN or eIF3i knock-out (KO) and OE was validated by qRT-PCR (Figure S8b).
eIF3i sequestration is associated with lenalidomide antiangiogenesis in cells
We next evaluated the phenotypic effects of the CRBN–lenalidomide–eIF3i complex. Since ternary complex formation was observed in epithelial cells and HUVECs, which is a cellular model for angiogenesis, we next evaluated if eIF3i and Len-mediated eIF3i sequestration is relevant to lenalidomide antiangiogenesis.
To evaluate whether the antiangiogenic properties of lenalidomide may be due to eIF3i sequestration, we measured a proangiogenic factor, the vascular endothelial growth factor A (VEGFA) in HEK293T cells (Figure 4a). Previous studies have established that upon lenalidomide treatment, VEGFA levels decrease.18, 29 We found that lenalidomide significantly decreased VEGFA mRNA levels in HEK293T cells, in a manner that was CRBN- and eIF3i-dependent (Figure 4a). A similar dependence on CRBN and eIF3i was observed with expression of another angiogenesis marker, the basic fibroblast growth factor (bFGF), which is also inhibited in response to lenalidomide treatment (Figure S9).
Figure 4 ∣. Lenalidomide and eIF3i are associated with antiangiogenesis effects.

(a) VEGFA mRNA levels in HEK293T cells with or without lenalidomide by qRT-PCR. (b) Fluorescent images of tube formation assay with HUVECs with or without lenalidomide treatment for 24 h. (c) Fluorescent images of wound healing migration assay with HUVECs with or without lenalidomide for 24 h.
We then evaluated whether the antiangiogenetic phenotypes of lenalidomide are dependent on both CRBN and eIF3i and performed both two-dimensional (2D) cell migration and three-dimensional (3D) cell invasion assays with HUVECs to measure angiogenesis. As a vital process for cancer cell proliferation, angiogenesis involves multiple steps including endothelial cell migration, invasion, and differentiation into capillaries.30 HUVEC differentiation was first confirmed with capillary tube formation of HUVECs in a 3D Matrigel matrix culture environment (Figure 4b, Figure S10b). We found that the tube formation ability of HUVECs was significantly decreased in the presence of lenalidomide (Figure 4b). HUVECs were not sensitive to lenalidomide treatment when CRBN was knocked-out (KO), while eIF3i KO resembled lenalidomide-treated WT-HUVECs. To visualize HUVEC invasiveness, we performed a 3D transwell assay and the number of invasive cells that passed through the transwell membrane was quantified and compared (Figure S10). Inhibition of HUVECs invasiveness by lenalidomide was similarly dependent on eIF3i and CRBN. Furthermore, we performed a wound healing assay where a monolayer of HUVECs was scratched and allowed to migrate over a period of 24 h (Figure 4c). CRBN KO decreased the responsiveness to lenalidomide inhibition significantly, indicating that CRBN is necessary for the antiangiogenesis properties of lenalidomide. The migration of HUVECs was also inhibited on eIF3i KO. Taken together, these data suggest that lenalidomide exerts antiangiogenetic functions in HUVECs in a CRBN- and eIF3i-dependent manner.
eIF3i is a direct target of lenalidomide in MM.1S cells after degradation of other targets
Finally, we returned to evaluate the sequestration of eIF3i in hematopoietic cell lines. Previously, we observed the sequestration of eIF3i in epithelial cell lines, but not in multiple myeloma MM.1S cells, potentially due to the stronger recruitment of IKZF1 to CRBN by lenalidomide.9 However, as IKZF1 is depleted within the first 8 h by the CRBN–lenalidomide complex and cell viability studies extend to 5 d incubation,2, 9, 31 we hypothesized that the CRBN–lenalidomide complex may recruit eIF3i following the degradation of IKZF1.
We first monitored the protein levels of IKZF1, IKZF3 and eIF3i in MM.1S cells over time (Figure S11). Levels of IKZF1 and IKZF3 significantly decreased starting at 4 h and are almost completely depleted after 8–12 h, while eIF3i levels were steady up to 48 h. We therefore compared the interactions of CRBN by coimmunoprecipitation in the presence of lenalidomide at 0 h and 24 h of lenalidomide incubation in MM.1S cells, which represent time points before and after IKZF1 degradation (Figure 5a, Table S1). As expected, IKZF1 was significantly coimmunoprecipitated in the presence of lenalidomide [enrichment ratio (Len/DMSO) = 2.746] in MM.1S cells prior to the degradation of IKZF1. By contrast, enrichment of IKZF1 was decreased after 24 h [enrichment ratio (Len/DMSO) = 1.258], and instead eIF3i was one of the most enriched proteins [enrichment ratio (Len/DMSO) = 7.411]. We also observed histone H2A enrichment in both datasets. The preferential immunoprecipitation of IKZF1 at 0 h and eIF3i after 24 h incubation was further validated by Western blot (Figure S12). These data indicate that CRBN favors a ternary complex with IKZF1 in the presence of lenalidomide, but as protein levels shift in response to degradation, complexation with eIF3i can subsequently accumulate.
Figure 5 ∣. eIF3i complexes with CRBN on lenalidomide treatment in MM.1S cells after the degradation of other substrates.

(a) CRBN interactome before IKZF1 degradation (time = 0 h) or after IKZF1 degradation (time = 48 h, 10 μM lenalidomide) by coimmunoprecipitation of endogenous CRBN with 50 μM lenalidomide. The significantly enriched region of proteins from MM.1S at time = 0 h is shaded in red. The significantly enriched region of proteins from MM.1S at time = 24 h is shaded in blue. (b) Endogenous CRBN coimmunoprecipitation with or without lenalidomide in MM.1S before and after lenalidomide-mediated target degradation. (c) Schematic model of CRBN–lenalidomide–eIF3i interaction in MM.1S after IKZF1 is degraded.
In order to validate this finding, we further performed a time-course coimmunoprecipitation experiment where the MM.1S cells were treated with lenalidomide and endogenous CRBN was immunoprecipitated from each sample (Figure 5b). As expected, IKZF1 levels decreased over the first 8 h while eIF3i and CRBN levels were largely unchanged over the 48 h experiment. Similarly, IKZF1 coimmunoprecipitated with CRBN in the presence of lenalidomide over the first 8 h in proportion to the overall decreasing levels of IKZF1. By contrast, eIF3i coimmunoprecipitated with CRBN to a greater degree over time. This shift in complexation is observable after 12 h, and appears to plateau between 24–48 h. This shift in complexation as a function of time was likewise observed in MOLM13 cells, which also express IKZF1 (Figure S13). Coimmunoprecipitation of CRBN recruited IKZF1 in a lenalidomide-dependent manner, while eIF3i is instead recruited to the complex after 24 h. Taken together, these data indicate that eIF3i is a direct target of CRBN and lenalidomide in MM.1S and MOLM13 cells, and that this interaction becomes observable after other targets are degraded (e.g., IKZF1, Figure 5c).
DISCUSSION
Here we report the functional, structural, and phenotypic outcomes of eIF3i sequestration by lenalidomide to a complex with CRBN. We find that the recruitment of eIF3i by CRBN and lenalidomide dissociates eIF3i from the eIF3 complex, and by developing a new covalent probe SL1, we revealed the binding surface on eIF3i that interacts with lenalidomide and CRBN in a ternary complex. We further showed that sequestration of eIF3i is associated with the antiangiogenic effects of lenalidomide treatment in epithelial cells, such as HEK293T cells and HUVECs, and occurs after longer dosing regimens for MM.1S or MOLM13 cells, where recruitment of eIF3i to CRBN by lenalidomide increases over longer treatment times after degradation of other targets like IKZF1. These findings highlight the prevalence of eIF3i engagement with CRBN in the presence of lenalidomide in different cell types. Future efforts to characterize the relevance of this interaction at physiologically relevant concentrations would strengthen these observations made in tissue culture. Additionally, examination of the recruitment and stabilization of substrates by other lenalidomide analogs or bifunctional degraders after the degradation of desired targets may reveal additional targets and mechanisms.
The successful identification of the binding interface between eIF3i, lenalidomide, and CRBN with the covalent probe SL1 indicates that distinct protein folds are recruited in addition to the beta-loop commonly recognized in other chemically-induced substrates.13 In the future, a crystal structure of the complex would further enhance the structural information reported by chemical proteomics. The chemical proteomics approach adapted in this study may also be useful to map the binding interface of CRBN binding targets with undefined structures, including ARID2 and p63,32, 33 or lead to the discovery of additional targets in the global proteome that are recruited in a complex with CRBN by SL1. Application of chemical proteomics using photolenalidomide or SL1 may additionally provide a complementary approach to the identification of interactions detected by proximity labeling enzymes.34 These future efforts may identify other targets of lenalidomide or derivatives that share homology with the WD40 wheel structure found in eIF3i. Additional studies to reveal why eIF3i is not degraded upon engagement or to engineer an eIF3i that is degradable may also open new directions in precise control of eukaryotic translation. Development of novel eIF3i-selective chemical moieties can also serve as an alternative strategy to alter transcriptional regulation. Given our observation that lenalidomide can induce interaction with eIF3i in MM.1S cells, eIF3i-selective compounds would enable further study on whether the interaction with eIF3i may be contributing to physiologically relevant phenotypes and illuminate the translational outcomes associated with eIF3i sequestration.
Supplementary Material
ACKNOWLEDGMENT
We thank Y. Amako, Y. Ge, W. Xu for insightful discussions; B. Budnik, R. A. Robinson, and J. X. Wang Harvard University Mass Spectrometry and Proteomics Resource Laboratory; J. Yu from Mazeed lab; C. M. Whilden from Whipple lab; T. J. Chang and C. B. Hartmann from Bauer Core Facility; E. Cronin-Furman with Olympus for technical support.
Funding Sources
Support from the Ono Pharma Foundation (C.M.W.), Sloan Research Foundation (C.M.W.), Camille–Dreyfus Foundation (C.M.W.), Blavatnik Accelerator at Harvard University (C.M.W.), and the National Institutes of Health (R01GM141406) is gratefully acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supplementary figures, uncropped data, synthetic schemes and materials and methods (PDF)
Complete list of proteins and peptide spectral matches from proteomics experiments (XLSX)
The authors declare no competing financial interest.
REFERENCES
- 1.Kronke J; Hurst SN; Ebert BL, Lenalidomide induces degradation of IKZF1 and IKZF3. Oncoimmunology 2014, 3 (7), e941742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu G; Middleton RE; Sun H; Naniong M; Ott CJ; Mitsiades CS; Wong KK; Bradner JE; Kaelin WG Jr., The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343 (6168), 305–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petzold G; Fischer ES; Thoma NH, Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature 2016, 532 (7597), 127–30. [DOI] [PubMed] [Google Scholar]
- 4.Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE, Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348 (6241), 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler James D.; Crew Andrew P.; Coleman K; Crews Craig M.; Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 2015, 22 (6), 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rehman W; Arfons LM; Lazarus HM, The rise, fall and subsequent triumph of thalidomide: lessons learned in drug development. Ther Adv Hematol 2011, 2 (5), 291–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beedie SL; Huang PA; Harris EM; Strope JD; Mahony C; Chau CH; Vargesson N; Figg WD, Role of cereblon in angiogenesis and in mediating the antiangiogenic activity of immunomodulatory drugs. FASEB J 2020, 34 (9), 11395–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo HM; Sun L; Yang L; Liu XJ; Nie ZY; Luo JM, Microvesicles shed from bortezomib-treated or lenalidomide-treated human myeloma cells inhibit angiogenesis in vitro. Oncol Rep 2018, 39 (6), 2873–2880. [DOI] [PubMed] [Google Scholar]
- 9.Lin Z; Amako Y; Kabir F; Flaxman HA; Budnik B; Woo CM, Development of Photolenalidomide for Cellular Target Identification. J Am Chem Soc 2022, 144 (1), 606–614. [DOI] [PubMed] [Google Scholar]
- 10.Hinnebusch AG, eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem Sci 2006, 31 (10), 553–62. [DOI] [PubMed] [Google Scholar]
- 11.Masutani M; Sonenberg N; Yokoyama S; Imataka H, Reconstitution reveals the functional core of mammalian eIF3. EMBO J 2007, 26 (14), 3373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun C; Todorovic A; Querol-Audi J; Bai Y; Villa N; Snyder M; Ashchyan J; Lewis CS; Hartland A; Gradia S; Fraser CS; Doudna JA; Nogales E; Cate JH, Functional reconstitution of human eukaryotic translation initiation factor 3 (eIF3). Proc Natl Acad Sci U S A 2011, 108 (51), 20473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sievers QL P. G; Bunker RD; Renneville A; Slabicki M; Liddicoat BJ; Abdulrahman W; Mikkelsen T; Ebert BL; Thoma NH, Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362 (6414), eaat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simonetti A; Brito Querido J; Myasnikov AG; Mancera-Martinez E; Renaud A; Kuhn L; Hashem Y, eIF3 Peripheral Subunits Rearrangement after mRNA Binding and Start-Codon Recognition. Mol Cell 2016, 63 (2), 206–217. [DOI] [PubMed] [Google Scholar]
- 15.Yuan Y; Zhang Y; Yao S; Shi H; Huang X; Li Y; Wei Y; Lin S, The translation initiation factor eIF3i up-regulates vascular endothelial growth factor A, accelerates cell proliferation, and promotes angiogenesis in embryonic development and tumorigenesis. J Biol Chem 2014, 289 (41), 28310–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang YW; Lin KT; Chen SC; Gu DL; Chen CF; Tu PH; Jou YS, Overexpressed-eIF3I interacted and activated oncogenic Akt1 is a theranostic target in human hepatocellular carcinoma. Hepatology 2013, 58 (1), 239–50. [DOI] [PubMed] [Google Scholar]
- 17.Ma S; Dong Z; Cui Q; Liu JY; Zhang JT, eIF3i regulation of protein synthesis, cell proliferation, cell cycle progression, and tumorigenesis. Cancer Lett 2021, 500, 11–20. [DOI] [PubMed] [Google Scholar]
- 18.Dredge K; Horsfall R; Robinson SP; Zhang LH; Lu L; Tang Y; Shirley MA; Muller G; Schafer P; Stirling D; Dalgleish AG; Bartlett JB, Orally administered lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitro. Microvasc Res 2005, 69 (1-2), 56–63. [DOI] [PubMed] [Google Scholar]
- 19.Lu L; Payvandi F; Wu L; Zhang LH; Hariri RJ; Man HW; Chen RS; Muller GW; Hughes CC; Stirling DI; Schafer PH; Bartlett JB, The anti-cancer drug lenalidomide inhibits angiogenesis and metastasis via multiple inhibitory effects on endothelial cell function in normoxic and hypoxic conditions. Microvasc Res 2009, 77 (2), 78–86. [DOI] [PubMed] [Google Scholar]
- 20.Mahony C; Erskine L; Niven J; Greig NH; Figg WD; Vargesson N, Pomalidomide is nonteratogenic in chicken and zebrafish embryos and nonneurotoxic in vitro. Proc Natl Acad Sci U S A 2013, 110 (31), 12703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.des Georges A; Dhote V; Kuhn L; Hellen CU; Pestova TV; Frank J; Hashem Y, Structure of mammalian eIF3 in the context of the 43S preinitiation complex. Nature 2015, 525 (7570), 491–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eliseev B; Yeramala L; Leitner A; Karuppasamy M; Raimondeau E; Huard K; Alkalaeva E; Aebersold R; Schaffitzel C, Structure of a human cap-dependent 48S translation pre-initiation complex. Nucleic Acids Res 2018, 46 (5), 2678–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wisniewski JR; Hein MY; Cox J; Mann M, A "proteomic ruler" for protein copy number and concentration estimation without spike-in standards. Mol Cell Proteomics 2014, 13 (12), 3497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cravatt BF; Wright AT; Kozarich JW, Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem 2008, 77, 383–414. [DOI] [PubMed] [Google Scholar]
- 25.Nomura DK; Dix MM; Cravatt BF, Activity-based protein profiling for biochemical pathway discovery in cancer. Nat Rev Cancer 2010, 10 (9), 630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erzberger JP; Stengel F; Pellarin R; Zhang S; Schaefer T; Aylett CHS; Cimermancic P; Boehringer D; Sali A; Aebersold R; Ban N, Molecular architecture of the 40S·eIF1·eIF3 translation initiation complex. Cell 2014, 159 (5), 1227–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brito Querido J; Sokabe M; Kraatz S; Gordiyenko Y; Skehel JM; Fraser CS; Ramakrishnan V, Structure of a human 48S translational initiation complex. Science 2020, 369 (6508), 1220–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin Y; Li F; Huang L; Polte C; Duan H; Fang J; Sun L; Xing X; Tian G; Cheng Y; Ignatova Z; Yang X; Wolf DA, eIF3 Associates with 80S Ribosomes to Promote Translation Elongation, Mitochondrial Homeostasis, and Muscle Health. Mol Cell 2020, 79 (4), 575–587 e7. [DOI] [PubMed] [Google Scholar]
- 29.Qu Z; Jiang C; Wu J; Ding Y, Lenalidomide induces apoptosis and inhibits angiogenesis via caspase3 and VEGF in hepatocellular carcinoma cells. Mol Med Rep 2016, 14 (5), 4781–4786. [DOI] [PubMed] [Google Scholar]
- 30.Huinen ZR; Huijbers EJM; van Beijnum JR; Nowak-Sliwinska P; Griffioen AW, Anti-angiogenic agents - overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol 2021, 18 (8), 527–540. [DOI] [PubMed] [Google Scholar]
- 31.Zhu YX; Braggio E; Shi CX; Kortuem KM; Bruins LA; Schmidt JE; Chang XB; Langlais P; Luo M; Jedlowski P; LaPlant B; Laumann K; Fonseca R; Bergsagel PL; Mikhael J; Lacy M; Champion MD; Stewart AK, Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124 (4), 536–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asatsuma-Okumura T; Ando H; De Simone M; Yamamoto J; Sato T; Shimizu N; Asakawa K; Yamaguchi Y; Ito T; Guerrini L; Handa H, p63 is a cereblon substrate involved in thalidomide teratogenicity. Nat Chem Biol 2019, 15 (11), 1077–1084. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto J; Suwa T; Murase Y; Tateno S; Mizutome H; Asatsuma-Okumura T; Shimizu N; Kishi T; Momose S; Kizaki M; Ito T; Yamaguchi Y; Handa H, ARID2 is a pomalidomide-dependent CRL4(CRBN) substrate in multiple myeloma cells. Nat Chem Biol 2020, 16 (11), 1208–1217. [DOI] [PubMed] [Google Scholar]
- 34.Yamanaka S; Horiuchi Y; Matsuoka S; Kido K; Nishino K; Maeno M; Shibata N; Kosako H; Sawasaki T, A proximity biotinylation-based approach to identify protein-E3 ligase interactions induced by PROTACs and molecular glues. Nat Commun 2022, 13 (1), 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.