


Abstract
Polyploidy, the presence of multiple sets of chromosomes that are similar but not identical, complicates both chromosome walking and assembly of sequence-ready contigs for many plant taxa including a large number of economically-significant crops. Traditional ‘dot-blot hybridization’ or PCR-based assays for identifying BAC clones corresponding to a mapped DNA landmark usually do not provide sufficient information to distinguish between allelic and non-allelic loci. A restriction fragment matching method using pools of BAC DNA in combination with dot-blots reveals the locus specificity of individual BACs that correspond to multi-locus DNA probes, in a manner that can efficiently be applied on a large scale. This approach also provides an alternative means of mapping DNA loci that exploits many advantages of ‘radiation hybrid’ mapping in taxa for which such hybrids are not available. The BAC-RF method is a practical and reliable approach for using high-density RFLP maps to anchor sequence-ready BAC contigs in highly-duplicated genomes, provides an alternative to high-density robotic gridding for screening BAC libraries when the necessary equipment is not available, and permits the expedient isolation of individual members of multigene or repetitive DNA families for a wide range of genetic and evolutionary investigations.
INTRODUCTION
Many angiosperm (flowering plant) lineages are polyploid, including most crops that feed, clothe and shelter humans. ‘Allopolyploids’ such as wheat, cotton and soybean contain two, three or more sets of ‘homoeologous’ chromosomes that are derived from common ancestral chromosomes and contain orthologous variants of the same genes, but do not normally pair or recombine. ‘Autopolyploids’ such as sugarcane can have 10 or more different homologs that can pair and recombine in many possible combinations. Parallel arrangements of genetic loci suggests duplication of some chromosomes or chromosome segments even in ‘diploids’ such as maize (1,2), sorghum (3,4), rice (5,6) and Arabidopsis (7).
Two decades of genomics research have yielded high-density maps of DNA markers for many major crops (cf. 8), providing a valuable foundation for basic research in genome organization and evolution, positional cloning of agriculturally important or developmentally interesting plant genes and, ultimately, complete sequencing of the genomes of the world’s leading crops. The facility of bacterial artificial chromosomes (BACs) as a large DNA cloning vector (9), together with the development of methods for high-throughput fingerprinting (10) and contig assembly (11,12), complement high-density genetic maps, helping to bridge gaps between DNA markers in physically large genomes. The emerging transition from genetic to physical mapping of crop genomes will efficiently provide the means for in silico chromosome walks rather than requiring costly case-by-case efforts in individual laboratories; afford the identification of minimal BAC ‘tiling paths’ that can be used to efficiently map large numbers of ESTs; and set the stage for exploratory or complete sequencing of chromosomal regions containing important genes.
Most flowering plant genomes are thought to have undergone one or more cycles of chromosomal duplication during their evolution (13). Identification of BACs corresponding to a mapped DNA landmark by using ‘dot blots’ or PCR amplification of short, well-conserved consensus sequences, usually fails to distinguish between BACs deriving from allelic or non-allelic loci. High levels of duplication of genes or chromosomal segments increases the propensity for ‘false joins’ among large DNA clones or contigs. Complex autopolyploids such as sugarcane will be especially problematic, as the four to eight different homologous chromosomes that might be found in an individual often include several allelic variants at a locus (14). By using traditional fingerprinting methods (10,11), allelic differences between homologs at restriction sites are confounded with differences in the genomic DNA content of the underlying BAC clones.
In polyploid genomes, in order to assemble a contig that truly represents differences in the genomic DNA content of the underlying BAC (or other) clones, it is important to have a priori knowledge that the clones derive from the same genetic locus. By utilizing DNA-level variation among homologous or homoeologous DNA sequences [‘alloalleles’—cf. (15)] that goes undetected by traditional ‘dot blot’ or PCR-based screening methods (16), individual BACs in polyploids can be assigned to their source loci. The BAC-RF method exploits the ease of purifying genomic DNA cloned into BACs to merge high-resolution mapping with sequence-ready contig assembly in highly duplicated genomes. DNA probes are directly hybridized both to restriction enzyme-digested pools of BAC DNA, and to ‘dot blots’, yielding both qualitative (dot) and quantitative (fragment size) data about positive BACs. BAC pools are similar to radiation hybrid cell lines (17,18), obviating the need for ‘DNA polymorphism’ to map loci to their chromosomal locations at fine-scale resolution. A two-step approach provides an economical alternative for screening of BAC libraries when robotic gridding is not available, first pre-selecting the subset of pools that contain positive BACs, then re-screening only the relevant subset of the BAC library. The BAC-RF method is made possible by the fact that a high degree of enrichment for genomic DNA cloned into BACs can be accomplished simply by plasmid isolation—it is made efficient by use of a pooling approach, acquiring data simultaneously from all members of a high-coverage BAC library. BAC-RF will facilitate contig assembly for many polyploid crops, resolving many of the complications introduced by gene and chromosome duplication. This approach also provides a facile means to identify and isolate individual members of multigene or repetitive DNA families for evolutionary studies.
MATERIALS AND METHODS
BAC libraries, pools and mapped DNA clones
A 6.6 genome-equivalent BAC library of Sorghum propinquum, including 38 016 clones with average insert size of 126 kb (19), was used. A 4.5-genome BAC library of Saccharum officinarum including 103 296 clones with average size of 130 kb (http://www.genome.clemson.edu/lib_frame.html ) was used. BAC DNA pools were prepared by inoculating liquid cultures (LB + 12.5 µg/ml CM) with 384 BACs from a single plate simultaneously. About 75 ml of sterile media was used to rinse cells from the tips of a 384-tooth replicator, then brought to 300 ml and incubated at 37°C with agitation. BACs were isolated en masse by alkaline lysis (20), with a typical yield of 10–20 µg DNA per pool. Genomic blot hybridization analysis typically employs 1–10 µg of plant DNA, or ~106 genome equivalents, to analyze low-copy DNA probes from genomes of 1–10 pg per nucleus. The equivalent titer of BAC pool DNA can be determined by the following equation:
Amount BAC DNA per lane = (mass of source DNA per blot/mass of DNA per source nucleus) × (mass of average cloned insert + 7 × 10–18 g mass of BAC vector) × (number of BACs per pool).
This study used 384 BACs per pool of average 126 kb (1.3 × 10–16 g), and 106 genome-equivalents of source DNA for comparison, equating to 52 ng of BAC DNA per pool. To avoid false-negatives due to variable growth rates of different BACs, we used a 3-fold excess or ~150 ng of BAC pool DNA per lane. The 300 ml cultures yielded ~10–20 µg BAC DNA, enough for about 100 blots. About 150 ng of BAC-pool DNA were digested with 10 U of HindIII (New England Biolabs), fractionated in 0.8% agarose gels immersed in neutral electrophoresis buffer at 18 V (0.36 V/cm) for 16–18 h and transferred to nylon membranes as described (3). Plasmids containing mapped sorghum DNA clones were isolated by alkaline lysis (20), digested with appropriate restriction enzyme(s), and the cloned insert recovered from low-melting-point agarose (BRL) using Gelase (Epicentre) according to the manufacturer’s instructions. Radioactive labelling and autoradiography were as described (3).
Fingerprinting of BAC clones
DNA of each positive BAC was extracted, digested with HindIII, and fragments separated on a 1.2% agarose gel using 100 ng BAC DNA per lane. A mixture of 1 kb ladder (Life Technologies) and Markers II and III (Boehringer-Mannheim) was loaded every fifth lane. After electrophoresis at 60 V (1.2 V/cm) and 16°C for 16 h, gels were stained with ethidium bromide, and imaged using a Kodak DC120 camera with a pixel size of 164 µm. Band mobilities (in pixels) were estimated with the Kodak 1D software, then reformatted with a custom-developed SAS/AF application (SAS, 1996) and input to FPC (12) to compute the probability that the number of matching bands of two clones could be explained by chance (Sulston’s score; see 12). The tolerance of FPC was set to 4, i.e. bands from different clones were considered to match if their relative mobilities were within four pixels (0.66 mm).
RESULTS
The BAC-RF method is illustrated in Figure 1, and its utility for efficiently assigning BACs to their source loci is illustrated in Figure 2a. Sorghum propinquum, the genotype used to make the BAC library, contains three restriction fragments that hybridize to the probe SH074 (Fig. 2a, lane 2), which had been previously mapped as an RFLP (3). In the 14 BAC pools shown (Fig. 2a, lanes 3–16: approximately one-genome coverage of sorghum), each of these restriction fragments are represented at least once (Fig. 2a, lane 15 for the 20 kb fragment; lane 4 for the 6.5 kb fragment, and lanes 8 and 14 for the 4 kb fragment). Hybridization of the labelled probe to dot-blots made by hand with a Nunc replicator confirmed that each positive pool contained a single BAC that hybridized to the probe (not shown). Traditional Southern blots of plant genomic DNA gave enough signal to map only the 4 kb S.propinquum fragment, allelic to the 9.4 kb Sorghum bicolor fragment (Fig. 2a, lane 1). However, since the 20, 6.5 and 4 kb S.propinquum fragments each correspond to different BACs, they must correspond to different genetic loci that have not been mapped. The relative signal intensity of genomic fragments (Fig. 2a, lane 2) is similar to that of BAC-RF fragments, suggesting that the DNA probe probably derives from the mapped locus, and that the two additional loci may be relatively ancient duplications. Coincidence of two or more restriction fragments of indistinguishable sizes in most or all of the same pools would indicate that the fragments are at a common genetic locus (or two closely-linked loci).
Figure 1.

BAC-RF method. Pooled BAC DNA is isolated from 384 BACs simultaneously, digested with a restriction enzyme, separated by electrophoresis, blotted and labeled with the cloned insert of a target DNA probe (to minimize hybridization to the BAC vector, the 7.1 kb band common to all pools is indicated by an arrow on the left). Differences in the size of restriction fragments are the basis for assigning BACs to individual loci in the source genome (second lane). Multiple restriction fragments that correspond to a single genetic locus co-occur in the same pools (2). Pools that lack a positive BAC show only the vector band (1, 3, 4 and 6). If robotic gridding of BACs is available, it is practical to probe dot blots and digests simultaneously. Lacking robotic gridding, by first screening digests, one need only screen the subset of dot blots for the pools that contain a positive clone.
Figure 2.
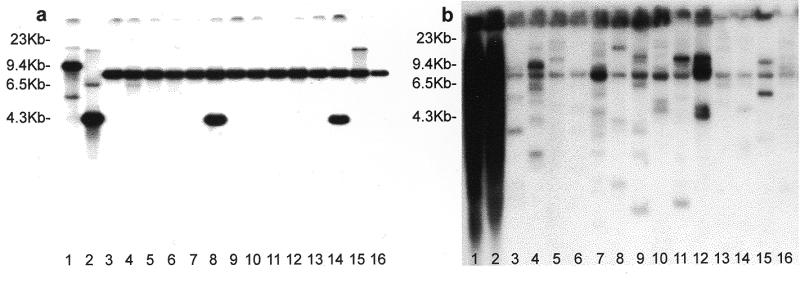
Screening of BAC-RF blots for two DNA probes. Sorghum genomic DNA probes SHO74 (a) and pSB1172 (b) were applied to S.bicolor (genotype BTx623; lane 1), S.propinquum (unnamed accession; lane 2), and 14 BAC pools each comprised of 384 clones, collectively representing about one-genome coverage of S.propinquum. Previous RFLP mapping of SHO74 showed that the 9.4 kb S.bicolor band was allelic to the 4.3 kb S.propinquum band, and that the locus maps to linkage group D. While the additional bands could be faintly discerned, they could not be mapped. Pools 2 and 13 (lanes 4 and 15) clearly contain BACs that harbor the additional loci, while pools 6 and 12 (lanes 8 and 14) each contain BACs that correspond to the mapped locus. pSB1172 shows a smear in the parental genotypes (even with shorter exposures), which is dissected into individual constituent loci in the BAC pools.
Only those BAC pools (and underlying BACs) that contain a restriction fragment that is indistinguishable in size from a genomic fragment are considered to be true positives. The most common false-positives (artifacts) we have found are additional bands that correspond to supercoiled or closed-circular BAC vector, migrating at apparent mobilities of ~4.2 and 8 kb, respectively. These can be minimized by good quality control during BAC library development as they are especially prominent in batches of BACs that contain a high frequency of empty clones. Restricting the BAC pools with the enzyme used for making the BAC library is a convenience in high-throughput applications, as it cleanly separates BAC DNA from vector DNA, leaving a 7.4 kb vector band common to all pools (which cross-hybridizes with many other common cloning vectors). Direct correspondence in size of BAC restriction fragments and previously mapped genomic restriction fragments permits direct identification of BACs containing the mapped locus (Fig. 2a). When prior information (such as RFLPs) impels the use of other restriction enzymes to digest the BAC pools, prehybridization to cold vector DNA may be necessary to block out false-positive bands resulting from restriction sites within the BAC vector. Methylation-insensitive enzymes are recommended to avoid artifacts due to differential methylation in bacteria versus host cells.
A limitation to the number of BACs that can be used in each pool is the extent to which vector hybridization signal can be quenched. We subdivided our sorghum library of 38 016 clones into 99 pools, each comprised of a single plate of 384 BACs representing ~6% of the genome and 1% of the library. In such a pool, the molar ratio of vector DNA to target DNA is 384:1. Without exception, residual vector DNA was present in sufficient quantity to detect the 7.4 kb band. The ratio of vector to target signal rises linearly with increasing number of BACs per pool: by our methods, a practical limit to reliable detection of positive bands is about three to four 384-well plates per pool. By probing with a labeled synthetic oligonucleotide internal to the target sequence (21), vector signal may be virtually eliminated, although at additional cost.
If robotic gridding of BACs is available, it is practical to probe pools and ‘dot blots (grids)’ simultaneously. Lacking robotic gridding, a two-step approach provides an alternative that economizes both labor and materials—first pre-selecting the subset of pools that contain positive BACs, then screening dot blots only for the subset of pools that contain a positive clone. The economies of pooling for PCR-based screens have previously been recognized (16), but hybridization-based screens have relied on dot blots.
Use of DNA fingerprint data to assemble locus-specific contigs for duplicated chromosomal regions requires a means to distinguish between differences that have accumulated as a result of divergence between the loci subsequently to duplication, and differences in the genomic DNA content of the underlying clones. When the duplication event is ancient and the derived loci have undergone substantial divergence, fingerprint data alone may be sufficient information to resolve locus-specific contigs. When the duplication event is recent or homogenizing forces are acting, such as in autopolyploids that contain many homologous chromosomes that regularly recombine with one another, additional data are likely to be necessary. The specific need for BAC-RF to help resolve locus-specific contigs will certainly vary among taxa and may also vary for different duplication events within a taxon.
Regardless of the antiquity of duplication, subsets of BACs containing a common RFLP allele should be much more similar to each other than to those containing different RFLP alleles detected by the same probe. Two test cases are presented: one is in sorghum, which is thought to be an ancient polyploid (3,4,22), and the other is in sugarcane, which is a polyploid of very recent origin (14). Two sorghum DNA probes, one detecting three loci (pSB1140a, -b, -c), and one detecting four loci (pSB1698a, -c, -d, -e) hybridized to 9 and 21 BACs respectively, in the S.propinquum library. A minimum of three BACs putatively corresponded to each locus, based on the BAC-RF method (Table 1). For each clone, the lowest probability of coincidence (see Materials and Methods) obtained in comparing the clone with the other clones assigned to the same locus by BAC-RF (i.e. containing common RFLP alleles) was noted as the best ‘internal match,’ and the lowest probability of coincidence obtained in comparing the clone with those assigned to other loci was noted as the best ‘cross-match.’ The log of the ratio of the best internal match divided by the best cross-match provides a convenient summary statistic expressing the extent of similarity of BACs tenatively assigned to the same locus. Two of the 30 BACs (31L13 and 72N14, both detected by pSB1698) were excluded from consideration due to contamination (simultaneous presence of bands with non-stoichiometric intensities in the gel). For 25 (89%) of the 28 remaining BACs, internal matches were all higher than 1e-15 while cross-matches were all lower than 2e-11, indicating a high level of reliability of the BAC-RF assembled contigs. Fingerprint-based contig assembly for these groups of clones using a cutoff (level of coincidence required for inclusion) of 1e-14 was straightforward. For the other three (11%) clones, internal and cross-matches were similar and poor. 67E04 and 96D19 RFLP corresponded to a common unmapped genomic restriction fragment (designated pSB1698e); their low correspondence may simply reflect a small region of overlap. Finally, 34N18 (pSB1140b) showed an ambiguous result with a best internal match of 4e-14 and a best cross-match of 7e-13 with 32J21, a BAC detected by pSB1698a which is on a different linkage group. Increasing the depth (redundancy) of the BAC library may help to determine if these few ambiguous BACs are assigned to their proper locus.
Table 1. DNA fingerprint analysis of locus-specific groups of BACs as determined by the BAC-RF method. (A) Sorghum BACs. (B) Sugarcane BACs.
| A. Sorghum BACs | ||||||
|---|---|---|---|---|---|---|
| |
|
|
|
|
|
|
| Locus | Clone | Best internal match | Best cross-match | |||
| Clone | Score (a) | Clone | Score (b) | Log (a/b) | ||
| pSB1140a | 10A14 | 42F20 | 2e-24 | 04A14 | 2e-08 | –16.0 |
| pSB1140a | 42F20 | 10A14 | 2e-24 | 04A14 | 6e-11 | –13.5 |
| pSB1140a | 42L10 | 42F20 | 2e-21 | 04A14 | 1e-10 | –10.7 |
| |
|
|
|
|
Average |
–13.4 |
| pSB1140b | 34N18 | 64A06 | 4e-14 | 32J21 | 7e-13 | –1.2 |
| pSB1140b | 55E08 | 64A06 | 5e-20 | 98E20 | 2e-07 | –12.6 |
| pSB1140b | 64A06 | 55E08 | 5e-20 | 32J21 | 3e-11 | –8.8 |
| Average | –7.5 | |||||
| pSB1140c | 62L04 | 83N10 | 1e-22 | 56H18 | 2e-11 | –11.3 |
| pSB1140c | 83N10 | 92A13 | 8e-24 | 41E03 | 6e-09 | –14.9 |
| pSB1140c | 92A13 | 83N10 | 8e-24 | 10K22 | 2e-10 | –13.4 |
| |
|
|
|
|
Average |
–13.2 |
| pSB1698a | 10K22 | 32K17 | 1e-15 | 92A13 | 2e-10 | –5.3 |
| pSB1698a | 32J21 | 32K17 | 5e-19 | 34N18 | 7e-13 | –6.1 |
| pSB1698a | 32K17 | 32J21 | 5e-19 | 12O13 | 7e-10 | –9.1 |
| pSB1698a | 41E03 | 62G07 | 8e-28 | 04A14 | 7e-10 | –17.9 |
| pSB1698a | 41G18 | 41E03 | 1e-22 | 34N18 | 5e-11 | –11.7 |
| pSB1698a | 62G07 | 41E03 | 8e-28 | 04A14 | 2e-11 | –16.4 |
| pSB1698a | 74O08 | 85O16 | 1e-25 | 70I16 | 8e-09 | –16.9 |
| pSB1698a | 85O16 | 74O08 | 1e-25 | 42L10 | 2e-07 | –18.3 |
| pSB1698a | 89J06 | 85O16 | 3e-18 | 62L04 | 2e-08 | –9.8 |
| |
|
|
|
|
Average |
–12.4 |
| pSB1698c | 04A14 | 56H18 | 1e-32 | 62G07 | 2e-11 | –21.3 |
| pSB1698c | 56H18 | 04A14 | 1e-32 | 62L04 | 2e-11 | –21.3 |
| pSB1698c | 70I16 | 56H18 | 2e-17 | 41G18 | 4e-10 | –7.3 |
| pSB1698c | 92O11 | 04A14 | 3e-30 | 42L10 | 2e-10 | –19.8 |
| Average | –17.4 | |||||
| pSB1698d | 12O13 | 98E20 | 6e-25 | 04A14 | 6e-10 | –15.0 |
| pSB1698d | 48D01 | 98E20 | 1e-16 | 04A14 | 2e-09 | –7.3 |
| pSB1698d | 60B15 | 98E20 | 9e-24 | 32K17 | 3e-08 | –15.5 |
| pSB1698d | 98E20 | 12O13 | 6e-25 | 04A14 | 4e-09 | –15.8 |
| |
|
|
|
|
Average |
–13.4 |
| pSB1698e | 67E04 | 96D19 | 4e-10 | 92O11 | 3e-10 | 0.1 |
| pSB1698e | 96D19 | 67E04 | 4e-10 | 70I16 | 5e-10 | –0.1 |
| |
|
|
|
|
Average |
0.0 |
| Per group average Log (a/b) | –11.1 | |||||
| Per BAC average Log (a/b) | –12.0 | |||||
| Average no. BACs per locus | 3.1 | |||||
| B. Sugarcane BACs | ||||||
|---|---|---|---|---|---|---|
| |
|
|
|
|
|
|
| Locus | Clone | Best internal match | Best cross-match | |||
| Clone | Score (a) | Clone | Score (b) | Log (a/b) | ||
| SH2a | 13A18 | 161B11 | 7E-05 | 87I1 | 2E-06 | 1.5 |
| SH2a | 161B11 | 199A10 | 2E-09 | 38M8 | 1E-06 | –2.7 |
| SH2a | 199A10 | 238N13 | 6E-13 | 130F11 | 7E-07 | –6.1 |
| SH2a | 238M13 | 199A10 | 6E-13 | 12A1 | 9E-06 | –7.2 |
| |
|
|
|
|
Average |
–3.6 |
| SH2b | 12A1 | 130F11 | 8E-12 | 9B1 | 5E-13 | 1.2 |
| SH2b | 54I3 | 130F11 | 1E-17 | 9B1 | 5E-08 | –9.7 |
| SH2b | 130F11 | 54I3 | 1E-17 | 9B1 | 3E-08 | –9.5 |
| |
|
|
|
|
Average |
–6.0 |
| SH2c | 133J7 | 235B15 | 3E-17 | 87I1 | 6E-07 | –10.3 |
| SH2c | 174G16 | 133J7 | 2E-07 | 87I1,166O17 | 1E-04 | –2.7 |
| SH2c | 235B15 | 133J7 | 3E-17 | 51N2 | 3E-06 | –11.0 |
| |
|
|
|
|
Average |
–8.0 |
| SH2d | 54N6 | 52I37 | 7E-07 | N.A. | ||
| SH2e | 3M6 | 182N4 | 4E-06 | 235B15 | 1E-04 | –1.4 |
| SH2e | 7B15 | 91D11 | 2E-07 | 179N14 | 2E-05 | –2.0 |
| SH2e | 7B16 | 7B15 | 4E-05 | 38K14,235B15 | 2E-04 | –0.7 |
| SH2e | 51N2 | 52I3 | 1E-19 | 143N19 | 6E-14 | –5.8 |
| SH2e | 52I3 | 51N2 | 1E-19 | 143N19 | 6E-14 | –5.8 |
| SH2e | 91D11 | 7B15 | 2E-07 | 38K14 | 2E-06 | –1.0 |
| SH2e | 143N19 | 177O18 | 2E-19 | 63G10 | 2E-06 | –13.0 |
| SH2e | 146B3 | 52I3 | 4E-06 | 26K6,54N6 | 2E-06 | 0.3 |
| SH2e | 152G10 | 51N2,52I3 | 5E-04 | 68H8 | 3E-05 | 1.2 |
| SH2e | 160O6 | 177O18 | 8E-08 | 87I1 | 6E-07 | –0.9 |
| SH2e | 143N19 | 182N4 | 1E-21 | 143N19 | 2E-19 | –2.3 |
| SH2e | 182N4 | 177O18 | 1E-21 | 143N19 | 1E-14 | –7.0 |
| SH2e | 198L13 | 216J5 | 6E-12 | 133J17 | 8E-06 | –6.1 |
| SH2e | 216J5 | 198L13 | 6E-12 | 94E5′ | 6E-06 | –6.0 |
| SH2e | 230F15 | 240M3 | 6E-10 | 63G10 | 6E-07 | –3.0 |
| SH2e | 240M3 | 230F15 | 6E-10 | 63G10 | 2E-07 | –2.5 |
| |
|
|
|
|
Average |
–3.5 |
| SH2f | 22D5 | 22M20 | 4E-23 | 54I3 | 6E-05 | –18.2 |
| SH2f | 22M20 | 22D5 | 4E-23 | 54I3 | 6E-06 | –17.2 |
| SH2f | 26K6 | 65G1 | 3E-02 | 146B3 | 2E-06 | 4.2 |
| SH2f | 29G4 | 148B7 | 6E-14 | 7B15,54I3,4O5 | 1E-03 | –10.2 |
| SH2f | 38I8 | 22D5 | 2E-11 | 161B11 | 5E-06 | –5.4 |
| SH2f | 38K14 | 38I8 | 5E-08 | 91D11 | 2E-06 | –1.6 |
| SH2f | 38M8 | 38I8 | 2E-08 | 179N14 | 3E-09 | 0.8 |
| SH2f | 63G10 | 38K14 | 7E-04 | 240M3 | 2E-07 | 3.5 |
| SH2f | 65G1 | 148B7 | 4E-05 | 143N19 | 3E-05 | 0.1 |
| SH2f | 94E5′ | 148B7 | 7E-11 | 216J5 | 6E-06 | –4.9 |
| SH2f | 148B7 | 29G4 | 6E-14 | 143N19 | 1E-06 | –7.2 |
| |
|
|
|
|
Average |
–5.1 |
| SH2g | 7C7 | 87I1 | 4E-04 | 5C19,19K12 | 1E-05 | 1.6 |
| SH2g | 66E14′ | 87I1 | 5E-03 | 13A18 | 5E-05 | 2.0 |
| SH2g | 87I1 | 7C7,99D20 | 7E-04 | 133J7,160O6 | 6E-07 | 3.1 |
| SH2g | 99D20 | 87I1 | 7E-04 | 179N14 | 1E-06 | 2.8 |
| |
|
|
|
|
Average |
2.4 |
| SH2h | 9B1 | 240H12 | 4E-03 | 12A1 | 5E-13 | 9.9 |
| SH2h | 212D23 | 240H12 | 1E-05 | 7B16 | 3E-03 | –2.5 |
| SH2h | 240H12 | 212D23 | 1E-05 | 62L4 | 2E-02 | –3.3 |
| |
|
|
|
|
Average |
1.4 |
| CDSR029a | 17P9 | 62L4 | 5E-11 | 68H8 | 2E-10 | –0.6 |
| CDSR029a | 62L4 | 17P9 | 5E-11 | 68H8 | 1E-15 | 4.7 |
| |
|
|
|
|
Average |
2.0 |
| CDSR029b | 5C19 | 5C20 | 2E-23 | 4O5 | 5E-06 | –17.4 |
| CDSR029b | 5C20 | 5C19 | 2E-23 | 146B3 | 3E-06 | –17.2 |
| CDSR029b | 19K12 | 5C20 | 2E-11 | 4O5 | 2E-06 | –5.0 |
| CDSR029b | 161B16 | 5C19 | 7E-09 | 187M21 | 5E-06 | –2.9 |
| |
|
|
|
|
Average |
–10.6 |
| CDSR029c |
166O17 |
|
|
140E14′ |
1E-05 |
N.A. |
| CDSR029d | 4O5 | 440E14′ | 1E-11 | 19K12 | 2E-06 | –5.3 |
| CDSR029d | 107M24 | 440E14′ | 1E-03 | 199A10 | 1E-05 | 2.0 |
| CDSR029d | 140E14′ | 4O5 | 1E-11 | 187M21 | 2E-07 | –4.3 |
| |
|
|
|
|
Average |
–2.5 |
| CDSR029e | 68H8 | 179N14 | 3E-12 | 62L4 | 1E-15 | 3.5 |
| CDSR029e | 179N14 | 68H8 | 3E-17 | 62L4 | 1E-11 | –5.5 |
| |
|
|
|
|
Average |
–1.0 |
| CDSR029f |
187M21 |
|
|
52I3 |
7E-07 |
N.A. |
| Per group average Log a/b | –3.1 | |||||
| Per BAC average Log a/b | –3.7 | |||||
| Average no. BACs per locus | 4.1 | |||||
Analysis of sugarcane BACs reinforced the validity of the BAC-RF technique for clustering BACs into locus-specific groups, and highlighted the special problems that will be faced in physical mapping of autopolyploids. Table 1 illustrates the results of BAC-RF and fingerprint analysis of sugarcane BACs detected by the shrunken-2 and CDSR029 probes, respectively. As for sorghum, BACs within a BAC-RF assembled group are much more similar to one another than to BACs in other groups: on average, the best internal match is nearly 104 more likely than the best cross-match. However, this is not nearly so clear a distinction as the 1012 improvement realized in sorghum. Further, a high level of heterogeneity is evident within BAC-RF groupings in sugarcane. For example, the SH2-B grouping included four BACs that had >107 better internal matches than cross-matches, and also four that had better matches outside the group than within it. This curious result is consistent with the molecular genetics of sugarcane (and other autopolyploids), where genetic mapping is based on DNA polymorphisms that segregate according to simplex ratios to avoid the possibility that DNA marker ‘alleles’ of the same size may occur at several independently segregating loci. The BAC-RF method has grouped clones based only on fragment size, as many of the DNA polymorphisms do not show simplex segregation: therefore, it may provide necessary, but insufficient, information to group all BACs into locus-specific groups in autopolyploids. The comparison of internal matches to cross-matches should help to highlight which BACs do belong in a grouping and which do not.
Dissection of a multigene family is illustrated in Figure 2b. For the probe pSB1172, the S.propinquum autoradiogram shows a ‘smear’. Across the 14 BAC pools shown (lanes 3–16: approximately one-genome coverage of sorghum), at least 21 different restriction fragments are clearly discernible in the 15 pools shown, with two to eight different fragments per pool. Because virtually all pools contain multiple positive BACs, ‘dot blots’ will need to be supplemented by further analysis of the individual BACs to identify a non-redundant set of individual family members. Nonetheless, by this means one can obtain all members of a multigene family in a reasonably short time.
DISCUSSION
The combination of polyploidy and heterozygosity that is inherent to many crop genomes, but rare in models such as Caenorhabditis and Arabidopsis, or mammals such as human and mouse, will be a new challenge to structural genomics. The combination of qualitative (dot or positive PCR) and quantitative (fragment size) data about positive BACs will complement high-efficiency methods for fingerprinting large-insert clones (10), toward robust contig assembly in the highly-duplicated genomes of major crops such as sugarcane, autopolyploids with 10 or more homologous chromosomes that are highly heterozygous. Integration with high-density RFLP maps aligns BAC contigs with a host of important genes and QTLs that have been located over two decades of research, fostering accelerated identification of the underlying genes by low-coverage sequencing (23).
BAC-RF pools offer a facile alternative to radiation hybrids for constructing high-resolution chromosome maps. BAC pools and radiation hybrids differ primarily in that (i) individual chromosome segments in BAC pools tend to be smaller, affording finer map resolution, and (ii) individual BAC pools tend to contain a larger number of unlinked chromosome segments than individual radiation hybrids, increasing the likelihood of false positive associations. BAC pools afford a level of resolution that is determined by the insert size and depth of genome coverage of the underlying BAC library. In principle, well-developed algorithms for mapping radiation hybrids (cf. 24) may be applied to BAC pools by substituting the average frequency of restriction sites represented in the BACs for the X-ray dose (‘centiRays’). ‘Deep’ BAC libraries in which individual genetic loci occur 10 or more times on average, and terminate at a larger number of different restriction sites, will improve resolution accordingly.
Direct hybridization of labeled DNA probes to pools of BAC DNA offers an economical alternative to robotic gridding, eliminating the need for sequence information or primer synthesis. In well-mapped taxa, physical maps will be quickly anchored to genetic maps by application of previously mapped DNA probes. In taxa for which maps are not yet established, it may be practical to assemble BAC-RF-based physical maps as an alternative to recombination-based or radiation hybrid-based genetic maps.
Acknowledgments
ACKNOWLEDGEMENTS
We appreciate the support of the NSF Plant Genome Research Program, International Consortium for Sugarcane Biotechnology (A.H.P., R.A.W); USDA Plant Genome Research Program, Texas Higher Education Coordinating Board, Texas and Georgia Agricultural Experiment Stations (A.H.P.); Belgian-American Educational Foundation (X.D.), and Rockefeller Foundation (Q.X., A.H.P.).
REFERENCES
- 1.Wendel J., Stuber,C., Edwards,M. and Goodman,M. (1986) Theor. Appl. Genet., 72, 178–185. [DOI] [PubMed] [Google Scholar]
- 2.Helentjaris T., Weber,D. and Wright,S. (1988) Genetics, 118, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chittenden L., Schertz,K., Lin,Y.-R., Wing,R. and Paterson,A. (1994) Theor. Appl. Genet., 87, 925. [DOI] [PubMed] [Google Scholar]
- 4.Pereira M., Lee,M., Bramel-Cox,P., Woodman,W., Doebley,J. and Whitkus,R. (1994) Genome, 37, 236–243. [DOI] [PubMed] [Google Scholar]
- 5.Kishimoto N., Higo,H., Abe,K., Arai,S., Saito,A. and Higo,K. (1994) Theor. Appl. Genet., 88, 722–726. [DOI] [PubMed] [Google Scholar]
- 6.Nagamura Y., Inoue,T., Antonio,B., Shimano,T., Kajiya,H., Shomura,A., Lin,S., Kuboki,Y., Harushima,Y., Kurata,N., Minobe,Y., Yano,M. and Sasaki,T. (1995) Breeding Sci., 45, 373–376. [Google Scholar]
- 7.Kowalski S., Lan,T.-H., Feldmann,K. and Paterson,A. (1994) Genetics, 138, 499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paterson A. (1996) Genome Mapping in Plants. Academic Press/Landes Bioscience, Austin, TX, USA.
- 9.Shizuya H., Birren,B., Kim,U.-J., Mancino,V., Slepak,T., Tachiiri,Y. and Simon,M. (1992) Proc. Natl Acad. Sci. USA, 89, 8794–8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marra M., Kucaba,T., Dietrich,N., Green,E., Brownstein,B., Wilson,R., McDonald,K., Hillier,L., McPherson,J. and Waterston,R. (1997) Genome Res., 7, 1072–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gillett W., Hanks,L., Wong,G., Yu,J., Lim,R. and Olson,M. (1996) Genomics, 33, 389–408. [DOI] [PubMed] [Google Scholar]
- 12.Soderlund C., Longden,I. and Mott,R. (1997) Comp. Appl. Biosci., 13, 523–535. [DOI] [PubMed] [Google Scholar]
- 13.Stebbins G. (1966) Science, 152, 1463–1469. [DOI] [PubMed] [Google Scholar]
- 14.Ming R., Liu,S., Lin,Y., Silva,J.D., Wilson,W., Braga,D., Deynze,A.V., Wenslaff,T., Wu,K., Moore,P., Burnquist,W., Irvine,J., Sorrells,M. and Paterson,A. (1998) Genetics, 150, 1663–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reinisch A., Dong,J.-M., Brubaker,C., Stelly,D., Wendel,J. and Paterson,A. (1994) Genetics, 138, 829–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green E. and Olson,M. (1990) Proc. Natl Acad. Sci. USA, 87, 1213–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goss S. and Harris,H. (1975) Nature, 255, 680–684. [DOI] [PubMed] [Google Scholar]
- 18.Cox D., Burmeister,M., Price,E., Kim,S. and Myers,R. (1990) Science, 250, 245–250. [DOI] [PubMed] [Google Scholar]
- 19.Lin Y., Zhu,L., Ren,S., Yang,J., Schertz,K. and Paterson,A. (1999) Mol. Breeding, 5, 511–520. [Google Scholar]
- 20.Sambrook J., Fritsch,E. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, second edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 21.Cai W., Reneker,J., Chow,C., Vaishnav,M. and Bradley,A. (1998) Genomics, 54, 387–397. [DOI] [PubMed] [Google Scholar]
- 22.Whitkus R., Doebley,J. and Lee,M. (1992) Genetics, 132, 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouck J., Miller,W., Gorrell,J., Muzny,D. and Gibbs,R. (1998) Genome Res., 8, 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boehnke M., Lange,K. and Cox,D. (1991) Am. J. Hum. Genet., 49, 1174–1188. [PMC free article] [PubMed] [Google Scholar]