

Abstract
Solar ultraviolet (UV) radiation induces DNA photoproducts in skin cells and is the predominant cause of human skin cancers. To understand human susceptibility to skin cancer and to facilitate the development of prevention measures, highly specific reagents to detect and quantitate UV-induced DNA adducts in human skin will be needed. One approach towards this end is the use of monoclonal antibody-based molecular dosimetry methods. To facilitate the development of photoproduct-specific antibody reagents we have: (i) cloned and sequenced a single chain variable fragment (ScFv) gene coding for one such high affinity monoclonal antibody, αUVssDNA-1 (mAb C3B6), recognizing the thymidine(6–4)thymidine photoproduct; (ii) expressed and displayed the cloned ScFv gene on the surface of phage; (iii) selected functional recombinant phage by panning; (iv) purified the ScFv peptide; (v) shown that the purified ScFv peptide binds to UV-irradiated polythymidylic acid but not unirradiated polythymidylic acid. This is the first demonstration of the use of phage display to select a ScFv recognizing DNA damage. In addition, this is the initial step towards immortalizing the antibody gene for genetic manipulation, structure–function studies and application to human investigations.
INTRODUCTION
Exposure to carcinogenic agents, both chemical and physical in nature, is responsible for the initiation and promotion of the majority of human cancers. DNA modification is a requisite event for mutation and heritable changes in cells and the measurement of DNA adducts in human tissues is important for both biomonitoring and mechanistic investigations in carcinogenesis. In recent years, the study of DNA adducts in human samples and mutations resulting from agent-specific DNA adducts have provided valuable tools in the field of molecular epidemiology (1–3).
Ultraviolet (UV) radiation is the most common physical carcinogen in our environment and the major cause of skin cancer in humans. The incidence of skin cancer among light-skinned individuals in the US population is increasing at 3–5% per year (4–6). It is estimated that >600 000 individuals will develop new basal cell carcinoma or squamous cell carcinoma of the skin each year in the US (5). Thus, non-melanoma skin cancer is a common environmentally caused cancer and of considerable clinical importance, by virtue of its relatively frequent occurrence (6,7). In addition, concern about the depletion of stratospheric ozone due to environmental pollution with chlorofluorocarbons, resulting in an increased intensity of UV radiation at the Earth’s surface (8), has emphasized the need for assessment of potential risk and susceptibility determinants for skin cancers in humans. Evaluating the impact of individual susceptibility to skin cancer on overall skin cancer burden requires an understanding of the capacity for DNA to sustain and repair UV-induced DNA damage.
Several immunological assays have been developed for quantitation of photoproducts in DNA extracted from UV-exposed tissues. For 15 years we have used a monoclonal antibody, αUVssDNA-1 or mAb C3B6, produced by hybridoma cell line 25JF.C3B6. This was originally selected from cell fusions using spleen cells from mice immunized with UV-irradiated polydeoxynucleotides (9). mAb C3B6 is specific for UV-irradiated polynucleotides and oligonucleotides that contain the (6–4)-dipyrimidine photoproduct (10). However, there is a need to improve and alter the specificities and affinities of such reagents to expand their usefulness as analytical reagents.
Here we report the cloning and sequencing of a ScFv gene coding for the high affinity monoclonal antibody mAb C3B6, recognizing the thymidine(6–4)thymidine [T(6–4)T] photoproduct (Fig. 1). ScFv polypeptides represent the antigen-binding domains of antibodies and they are useful tools for molecular recognition and structure–activity investigations. We have purified the ScFv fragment to homogeneity and analyzed its binding characteristics to UV-irradiated polythymidylic acid [UV-poly(dT)]. Recombinant C3B6 ScFv binds specifically to UV-poly(dT) and does not cross-react with unirradiated polythymidylic acid [poly(dT)]. Recombinant ScFv offers a tool to understand protein–DNA recognition by antibody molecules. Furthermore, we have expressed the ScFv peptide recognizing the T(6–4)T photoproduct on the surface of phage. The recombinant phage binds specifically to UV-poly(dT).
Figure 1.

Structure of the T(6–4)T photoproduct.
MATERIALS AND METHODS
Production of UV-poly(dT)
A mercury vapor lamp was used to irradiate 6.25 ml of phosphate-buffered saline, pH 7.4, containing 100 µg/ml poly(dT) (Sigma) with 254 nm radiation. The UV lamp was placed 5 cm from the surface of the liquid, which was irradiated with constant stirring for 1000 s at 5 J/m2/s to generate T(6–4)T photoproducts. UV fluorescence was measured with an IL700 radiometer.
Cloning and sequencing of the ScFv gene coding for the T(6–4)T antibody
Poly(A)+-rich mRNA was isolated from the 25JF.C3B6 hybridoma clone, which produces the mAb C3B6, which is specific for photoproducts with the characteristics of (6–4)-dipyrimidine adducts, using the Mini Ribosep™ mRNA Isolation Kit from Collaborative Biomedical Products. Approximately 107 cells were thawed in 10 ml of lysis buffer containing 200 µg/ml proteinase K warmed to 37°C. Cells were sheared by forcing the lysate through an 18 gauge needle 20 times until the viscosity was reduced to that of the lysis buffer. Following incubation at 45°C for 2 h, the concentration of NaCl was adjusted to 0.5 M and the mRNA purified over oligo(dT)–cellulose affinity chromatography medium. First-strand cDNA synthesis was performed using the isolated mRNA, random hexanucleotides p(dN)6 and reverse transcriptase. The variable heavy chain (VH) and variable light chain (VL) regions were amplified in separate reactions using mouse PCR primers (11) supplied with the Pharmacia Recombinant Phage Antibody System (catalog no. 27-9400-01) and Taq DNA polymerase (AmpliTaq) for 30 cycles consisting of melting at 94°C for 1 min, annealing at 55°C for 2 min and extension at 72°C for 2 min. Following gel purification, the VH and VL regions were joined through a linker segment encoding amino acid residues GGGGSGGGGSGGGGS. The assembled ScFv fragment was amplified with PCR primers containing SfiI and NotI sites to aid ligation into the pCANTAB5E phagemid vector (Pharmacia Biotech).
For sequencing, the ScFv gene was inserted as a blunt-ended fragment at the SmaI site of the pUC18 vector. Double-strand sequencing reactions were performed using Sequenase, forward/reverse sequencing primers and deoxyadenosine 5′-[α-35S]thiotriphosphate (1300 Ci/mmol) for labeling. The products of the sequencing reactions were separated on 8% polyacrylamide gels (12).
Recombinant phage expressing the specific ScFv fragment on the surface
After digestion with SfiI and NotI, the PCR-amplified ScFv fragments were ligated into the pCANTAB 5E phagemid vector. The recombinants were used to transfect competent XL-1 Blue cells. The transformation mix (200 µl) was spread on five SOB plates (20 g Bacto-tryptone, 5 g Bacto-yeast extract, 0.5 g NaCl, 0.01 M MgCl2 and 15 g Bacto-agar per liter of medim) supplemented with 2% glucose and 100 µg/ml ampicillin and incubated at 30°C overnight. Each plate was flooded with 5 ml of 2× YT (17 g Bacto-tryptone, 10 g Bacto-yeast extract, 5 g NaCl per liter of medium) containing 100 µg/ml ampicillin and 2% glucose and the colonies were scraped together. An aliquot of 300 µl of the scraped culture was used to inoculate 25 ml of 2× YT containing 100 µg/ml ampicillin and 2% glucose. The cells were grown at 37°C to an A600 of 0.5. Recombinant phage were packaged using M13K07 helper phage. M13K07 phage (3 × 1010 p.f.u.) were added for each 15 ml of cells and the culture grown for 1 h at 37°C. The cells were spun down at 1000 g for 10 min at room temperature and the pellet resuspended in 10 ml of 2× YT containing 100 µg/ml ampicillin and 50 µg/ml kanamycin and the culture grown overnight at 37°C. After overnight growth the cells were pelleted and the phage recovered from the supernatant by PEG precipitation. Two milliliters of PEG/NaCl (200 g polyethylene glycol 6000 and 146.1 g NaCl in 1 l final volume) was added for each 10 ml of supernatant and the mixture incubated on ice for 45 min. After centrifugation at 10 000 g for 20 min at 4°C, the phage were resuspended in 3 ml of 0.2× PBS and an aliquot (30 ml) was used to determine the phage titer. Non-fat dry milk (Richfood brand) was added to a concentration of 2% (w/v) and sodium azide (NaN3) to a concentration of 0.01% to the resuspended phage prior to panning.
Phage panning protocol
UV-poly(dT) was diluted to 0.5 µg/ml in 0.1× PBS and 100 µl of this solution was added to each well of microtiter plates (Costar). The plates were completely dried at 32°C in a convection oven and blocked with 5% non-fat dry milk in 0.2× PBS. The plates were washed three times with 0.2× PBS and 100 µl of preblocked phage were added to each well. Plates were incubated at 37°C for 2 h with mild shaking, washed 20 times with 0.2× PBS, 20 times with 0.2× PBS containing 0.05% Tween 20 and three times with 0.2× PBS. Each well was eluted with 100 µl of 1 M triethylamine at 37°C for 15 min and neutralized with 50 µl of 1 M Tris–HCl, pH 7.4. The eluted phage were added to 10 ml of log phase XL-1 Blue cells in 2× YT medium and grown at 37°C for 1 h. Ampicillin was added to a final concentration of 100 µg/ml and glucose to a final concentration of 2% (w/v). M13K07 phage (2 × 1010 p.f.u.) was added and the culture was incubated at 37°C for 1 h before pelleting the cells at 1000 g for 10 min at room temperature. The cells were then resuspended in 10 ml 2× YT containing 100 µg/ml ampicillin and 50 µg/ml kanamycin and grown overnight at 37°C. The phage panning protocol was repeated twice more.
ELISA of isolated clones
Following the final round of panning, eluted phage were used to infect log phase XL-1 Blue cells and the cells were plated on SOB medium containing 100 µg/ml ampicillin and 2% glucose and incubated overnight at 30°C. Individual colonies were used to inoculate 100 µl of 2× YT supplemented with 100 µg/ml ampicillin, 15 µg/ml tetracycline and 2% glucose and incubated overnight at 30°C with shaking. Twenty microliters of the saturated overnight culture were used to inoculate 200 µl of 2× YT containing 1.2 × 108 p.f.u. M13KO7 helper phage and incubated at 37°C for 2 h with shaking. The cultures were centrifuged at 1500 g for 20 min at room temperature and the pellets resuspended in 200 µl of 2× YT containing 100 µg/ml ampicillin and 50 µg/ml kanamycin and grown overnight at 37°C. Cultures were spun down at 1500 g for 20 min at room temperature and the supernatant removed and blocked in an equal volume of 5% non-fat dry milk in 0.2× PBS and 0.01% sodium azide for use in direct ELISA analyses of recombinant phage binding.
The overnight phage cultures grown under conditions of adequate aeration (10 ml) were centrifuged at 1500 g for 20 min at room temperature. The phage in the supernatant were precipitated by adding 2 ml of PEG/NaCl solution (200 g polyethylene glycol 6000 and 146 g NaCl was dissolved in water to 1 l). After centrifugation at 10 000 g for 20 min at 4°C, the recombinant phage were resuspended in 3 ml of 0.2× PBS containing 2% non-fat dry milk and 0.01% sodium azide (NaN3).
The microtiter plates containing UV-poly(dT) were blocked with 5% non-fat dry milk in 0.2× PBS for 1 h at room temperature. The ELISA plates were washed four times with 0.2× PBS and 120 µl of preblocked phage were transferred to each well and incubated at 37°C with shaking. The plates were washed six times with 0.2× PBS containing 0.05% Tween 20. A 100 µl aliquot of the diluted secondary antibody (αM13 polyclonal, 1:10 000 dilution in 0.2× PBS containing 2.5% milk and 0.05% Tween 20) was added to each well and the plates incubated at 37°C for 45 min. The plates were washed six times with 0.2× PBS containing 0.05% Tween 20, and 100 µl of 2′,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid)diammonium salt (ABTS) substrate containing 0.09% H2O2 was added to each well. Absorbance at 405 nm was monitored at regular intervals for each clone. Control ELISA plate wells containing no antigen or unirradiated poly(dT) were processed at the same time as described above.
Purification of T(6–4)T-specific ScFv peptides
Production of soluble ScFv peptides specific for the T(6–4)T photoproduct was achieved by transfecting recombinant phage (220-F5) into Escherichia coli HB2151. This strain does not carry the suppressor that is needed for production of ScFv–gIIIp fusion proteins, resulting in the production of only ScFv peptides. The procedures for infecting E.coli HB2151 cells and production of soluble antibodies are described in detail in the instructions for the expression module of the RPAS system of the RPAS system (Pharmacia Biotech).
RESULTS
Cloning and sequencing of the ScFv gene coding for the T(6–4)T-specific antibody
The nucleotide sequence and the predicted amino acid sequence of the ScFv gene (VH and VL) coding for the T(6–4)T-specific antibody were determined. Comparison of the protein sequence with GenBank sequences showed >90% homology (85% amino acid sequence identity) with mouse rearranged heavy chains for VH and >98% homology (94% amino acid sequence identity) with the mouse immunoglobulin κ chain variable region for VL. All the predicted highly conserved residues of antibody protein sequences are present. The complementarity determining regions (CDRs) of VH and VL have been identified and are indicated in Figure 2.
Figure 2.

Alignment of the amino acid sequence of the VH and VL regions of the C3B6 ScFv peptide (bottom) with that of 64M5 ScFv (top), which also recognizes the T(6–4)T photoproduct (21). The conserved amino acids between the two sequences are indicated below and the non-conserved residues are shown by dashes. CDR1, CDR2 and CDR3 and the glycine-serine linker (-G4S-)3 are shown in bold. Deletions are indicated by closed circles.
Recently, the amino acid sequences of four other monoclonal antibodies specific for DNA photoproducts (64M2, 64M3, 64M4 and 64M5) were reported by Morioka et al. (13,14). 64M4 apparently bound all four possible pyrimidine–pyrimidone photoproducts, whereas others were selective for T(6–4)C and T(6–4)T sequences. In addition, 64M5 was reported to have the highest binding affinity for photodamaged DNA. Figure 2 shows the alignment of the amino acid sequence of C3B6 ScFv peptide compared to that of 64M5. A close examination of the two sequences indicates that while most of the amino acid residues of the two sequences are highly conserved, the CDR regions of the two ScFv peptdies are quite dissimilar. The CDR3 loop of VH and CDR1 loop of VL of C3B6 are much shorter than the corresponding loops in 64M5.
Recombinant phage expressing photoproduct-specific ScFv on the surface
Our initial attempts to use TG1 cells as host for producing recombinant phage expressing the ScFv peptide on the surface were not successful. Therefore, the recombinant phagemids carrying the ScFv gene was used to transfect different E.coli strains. These included SURE, XL-1 Blue, NM522, JM109 and DH5α cells. The recombinant phage were packaged using M13K07 helper phage. Figure 3 shows expression of these phage in various E.coli strains. Three strains, SURE, XL-1 Blue and DH5α, appear to express the ScFv–gIIIp fusion well. Of these, XL-1 Blue was chosen as the best strain for production of the recombinant phage based on maximal expression of the ScFv–gIIIp fusion protein.
Figure 3.
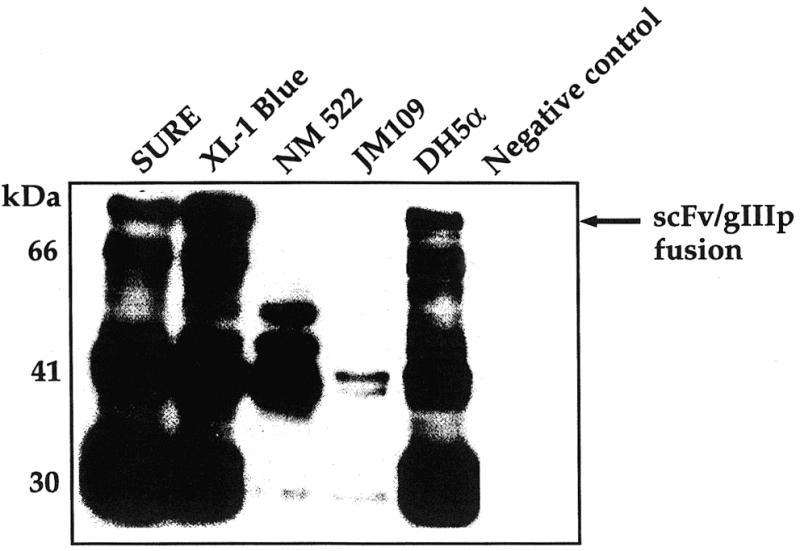
Western blot profile showing expression of the ScFv–gIIIp fusion protein in various amber suppressor bacterial strains. The pCANTAB 5E C3B6 ScFv phagemid construct was transfected into different suppressor strains, namely SURE, XL-1 Blue, NM522, JM109 and DH5α. One milliliter cultures of each were treated with 1 mM IPTG to induce expression of ScFv–gIIIp fusion proteins. After sonication of the cells, the protein species in 30 µl of the supernatant were separated by PAGE and transferred to a nitrocellulose membrane. The western blot was probed with an αE tag–HRP monoclonal antibody purchased from Pharmacia Biotech. Location of the ScFv–gIIIp fusion is indicated by an arrow. Negative control, SURE cells without the recombinant phagemid.
After three rounds of selective panning, the recombinant phage were analyzed by ELISA using microtiter plates coated with UV-poly(dT). Several clones were isolated that yielded A405 readings in direct ELISA analyses showing 10- to 15-fold higher binding to UV-poly(dT) than to poly(dT) (Fig. 4). However, these results from the microtiter plates were not reproducible. We reasoned that this was due to variations in cell growth and production of recombinant phage under the minimally aerated conditions that exist within the microtiter plate wells. Selected clones from the microtiter plates were grown under adequately aerated conditions (in flasks) as well as under minimally aerated conditions (microtiter plate wells) for comparison by specific binding in an ELISA. The results indicated that the phage clones should be grown under adequate aeration for efficient production of recombinant phage titers expressing ScFv–gIIIp fusion protein on the surface.
Figure 4.

ELISA on C3B6 ScFv recombinant phage after three rounds of panning. Graphic depiction of microtiter plate readings of selected C3B6 ScFv recombinant phage clones. Phage grown under adequately aerated conditions (in flasks) were PEG precipitated and concentrated 3-fold before use in the ELISA. The absorbance readings are an average of five replicate wells. The range of absorbance is indicated by error bars.
Characterization of ScFv peptides
A T(6–4)T-specific recombinant phage (220-F5) was used to infect a non-suppressor strain of E.coli (HB2151) to produce soluble ScFv fragments. The UV-poly(dT)-specific ScFv peptide was purified to homogeneity using an anti-E tag/protein G affinity column. The size of the ScFv peptide was ~31 kDa, which agrees well with that predicted from the ScFv gene. The western blot profiles of the protein species in each fraction eluted from the affinity column are shown in Figure 5. Competitive ELISA analyses were performed to compare the C3B6 ScFv peptide binding characteristics to that of the original monoclonal antibody, mAb C3B6 (Fig. 6). Antigen-specific binding inhibition was determined using both unirradiated poly(dT) and UV-poly(dT) as competitors. ScFv specifically recognized UV-poly(dT) with no evidence of cross-reactivity with unirradiated poly(dT) up to 1000 ng of inhibitor. It appears that while the specificity of the C3B6 ScFv peptide is similar to that of the original antibody, its apparent binding affinity appears to be 60-fold lower than that of the parent monoclonal antibody.
Figure 5.
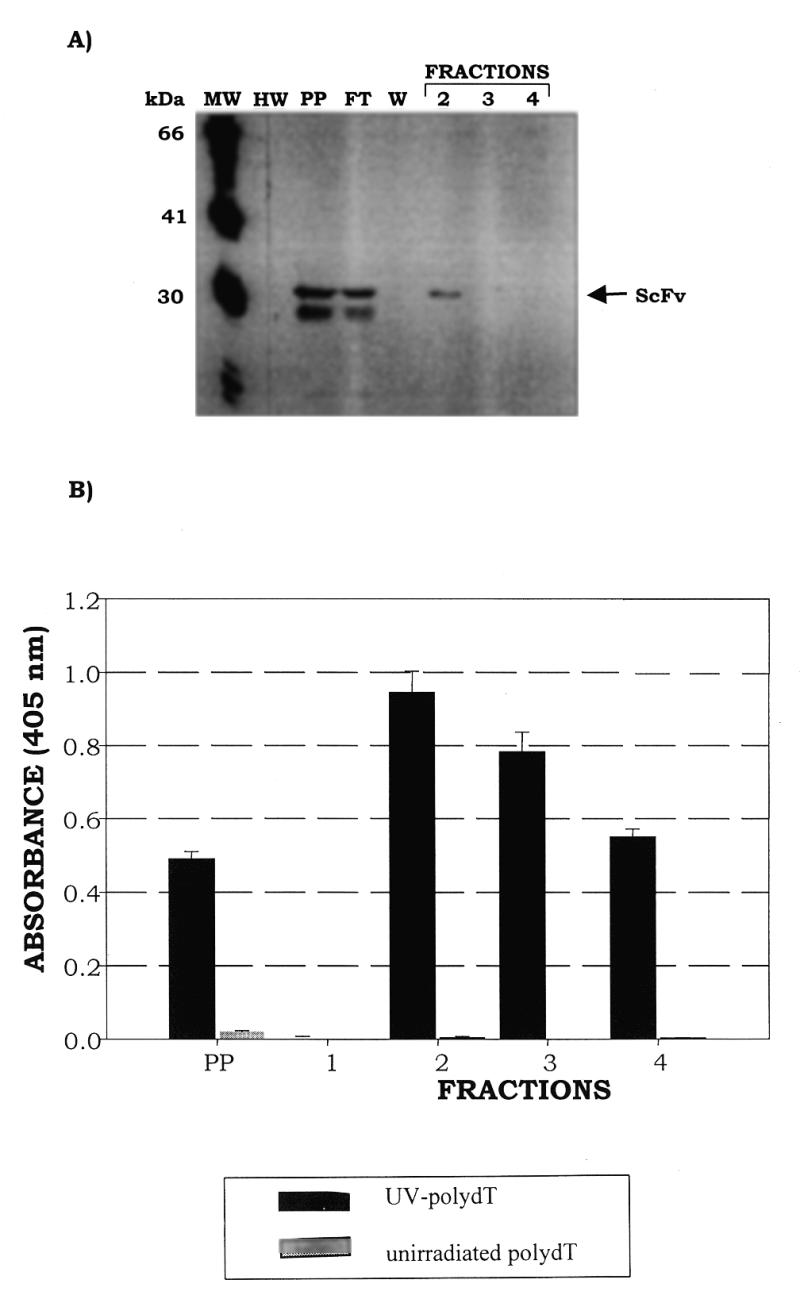
Purification of soluble C3B6 ScFv from recombinant phage clone 220-F5 using an anti-E tag/protein G affinity column. (A) Western blot of fractions eluted from the anti-E tag column; the blot was probed with anti-F(ab′)2 antibody (Jackson ImmunoResearch Laboratories). C3B6 ScFv is indicated by an arrow. Lane MW, FokI molecular weight markers; lane HW, prestained high molecular weight markers; lane PP, periplasmic extract; lane FT, flow-through; lane W, wash; lanes 2–4, fractions eluted with 1 M glycine, pH 3.0. (B) Binding activity, assessed by ELISA, of C3B6 ScFv fractions eluted from the anti-E tag column. Soluble ScFv, produced in the non-amber suppressor HB2151 strain, was passed over a protein G column to which the monoclonal anti-E tag antibody (Pharmacia Biotech) had been covalently bound. After washing with 6 column vol of 0.2 M phosphate buffer containing 0.05% NaN3 the bound protein was eluted in 1.0 M glycine, pH 3.0. ELISA plates were prepared as described in Materials and Methods and blocked for 1 h at room temperature in a solution of 5% non-fat dry milk in 0.2× PBS. Plates were then washed four times with 0.2× PBS. ScFv fractions used were diluted 1:5 to a final concentration of 2% non-fat dry milk in 0.2× PBS containing 0.02% NaN3 and 100 µl were added to four wells containing 50 ng UV-poly(dT) and four wells containing unirradiated poly(dT). Plates were incubated at 37°C with moderate shaking for 1.5 h and then washed six times with 0.2× PBS containing 0.05% Tween 20 (TPBS). Peroxidase-conjugated rabbit anti-mouse polyclonal sera with F(ab′)2 specificity (Jackson ImmunoResearch Laboratories) was diluted 1:12 000 in 2.5% non-fat dry milk in 0.2× PBS and 100 µl were added per well. Plates were incubated at 37°C with moderate shaking for 1 h and then washed six times in TPBS. Aliquots of 100 µl of ABTS substrate were added to each well and readings were taken at 405 nm after 30 min development.
Figure 6.

Competitive ELISA comparing C3B6 ScFv fragment affinity for the T(6–4)T photoproduct to that of the parent antibody, mAb C3B6. Antigen-specific binding inhibition was determined using both unirradiated poly(dT) and UV-poly(dT) as competitors. Initial competitor concentrations were 10 and 1.1 µg/ml for the C3B6 ScFv peptide and mAb C3B6, respectively. Competitor concentrations were reduced by 3-fold each time by serial dilution. Binding activity was determined using the ABTS substrate. The experiments were performed in triplicate and absorbance readings from the three wells were averaged and used to determine percent inhibition. Non-linear regression was used to fit the data shown in figure 7 of Gange et al. (22). The data was then plotted using SigmaPlot v.4.0 (SPSS, Chicago, IL).
DISCUSSION
All chemical-specific DNA adduct measurements are dependent upon the specificity and affinity of the analytical reagents and the methods used in their determination. Monoclonal antibodies and polyclonal antisera have become essential tools for the isolation and measurement of DNA adducts. Since ScFv peptides represent the antigen-binding domains of antibodies, they have become useful tools for molecular recognition and structure–activity investigations.
Phage display systems have provided powerful selection methods for isolating and studying proteins and peptides (15–20). This is made possible by making the chemistry (or function) selectable by linking it to infectivity. Thus, the recombinant phage antibody technology mimics the crucial step between recognition and replication found in the humoral immune reponse. In order to explore the usefulness of phage display for studying antibody–DNA adduct interactions, we have expressed a ScFv peptide recognizing UV-poly(dT) on the surface of filamentous phage. The starting point for these experiments was the monocloncal antibody αUVssDNA-1 (mAb C3B6) produced by hybridoma cell line 25JF.C3B6, which was originally selected from cell fusions using spleen cells from mice immunized with UV-irradiated polydeoxyribonucleotides. mAb C3B6 is specific for UV-irradiated polynucleotides and oligonucleotides that contain the T(6–4)T dipyrimidine photoproduct. We were successful in producing useful titers of the recombinant phage that bound specifically to UV-poly(dT).
Isolation of a number of recombinant phage clones clearly demonstrates the feasibility of generating and expressing functional anti-DNA adduct ScFv antibody fragments on the surface of filamentous phage (Fig. 4). A viable filamentous phage expresses three or five copies of the gene –3-encoded adsorption protein (gIIIp) on its tip. These are required for infection by the bacterium. In fusion phage about one or two copies of the recombinant ScFv–gIIIp protein were packaged into the phage coat, allowing the recombinant phage to retain infectivity. We encountered several technical difficulties during the course of these experiments. The ScFv–gIIIp fusion protein appears to be toxic to several bacterial strains. For example, NM522 and JM109 do not produce the fusion protein at all. Only cleavage products of the ScFv–gIIIp fusion protein were observed in the cell extracts (Fig. 3). SURE, XL-1 Blue and DH5α appear to tolerate the toxic ScFv–gIIIp fusion protein. Recombinant phage were packaged successfully in these cultures. Another technical difficulty that we encountered during the course of these experiments relates to adequate aeration of the medium for optimal production of recombinant phage titers. We reproducibly obtained high phage titers in well-aerated flasks as compared to microtiter plates.
As part of this study, we have also determined the nucleotide sequence of the genes (VH and VL) coding for the monoclonal antibody recognizing the T(6–4)T photoproduct. The inferred amino acid sequence was compared with the sequences of four monoclonal antibodies (64M2, 64M3, 64M4 and 64M5) reported earlier this year by Morioka et al. (13,14). Apparently, 64M4 bound all four possible pyrimidine–pyrimidone photoproducts with equal affinities, whereas the others were selective for T(6–4)C and T(6–4)T sequences. In addition, 64M5 had the highest binding affinity for photodamaged DNA. Unlike mAb C3B6, which preferentially recognizes T(6–4)T in single-stranded DNA, 64M5 has been shown to be selective for T(6–4)C and T(6–4)T sequences in both single-stranded and double-stranded DNA. Alignment of the amino acid sequences of the variable regions of the C3B6 ScFv with that of 64M5 indicates striking dissimilarity in the CDR regions of the two ScFv peptides, suggesting that recognition of the T(6–4)T adduct could be achieved in more than one way. The CDRs form the protein structural motifs, namely loops, through which the critical interaction between the antibody and ligand occurs. The CDR3 loop of VH and CDR1 loop of VL of C3B6 are much shorter than the corresponding loops in 64M5 (Fig. 2). In fact, a careful inspection of the CDRs of various antibodies reveals that these short loops more closely resemble the corresponding short loops in 64M4. The significance of this is not yet clear. It may be a reflection of the immense potential of biological systems to evolve more than one complex biological solution to the problem of recognizing photodamaged DNA. Crystal structures of ScFv peptides complexed with the T(6–4)T antigen may shed some light on this observation.
We have also purified the ScFv antibody fragment which contains the antigen recognition domain of mAb C3B6 to homogeneity (Fig. 5). We have compared the specificity of the ScFv peptide to that of the original monoclonal antibody using competitive ELISA (Fig. 6). Like the original monoclonal antibody, the ScFv peptide binds specifically to UV-poly(dT). While the binding affinity of ScFv appears to be ~60-fold lower than that of the original antibody, this could be due to differential affinity of the anti-(Fab′)2 secondary antibody for ScFv and the parent antibody. Other ScFv peptides with lower affinity as compared to the original antibody have been reported in the literature (21). The lower affinity could also be attributed to lower stability of the ScFv peptide. Increasing the stability of the ScFv peptide is likely to increase its binding affinity for the ligand. This could be examined by exploring the use of alternative linkers to connect the VH and VL regions of the ScFv peptide.
This work is the first demonstration of the use of phage display to select ScFv proteins that recognize DNA damage. Also, it represents the initial step towards future genetic manipulation and structure–function studies. It is quite clear that we can easily produce large quantities of ScFv peptide in bacteria, avoiding the use of animals. Successful generation and expression of functional DNA adduct-specific ScFv antibody fragments on the surface of filamentous phage and production of useful titers of the recombinant phage that bind specifically to UV-poly(dT) increases the possibility of altering the affinity and specificity of ScFv peptides. In future experiments, we will attempt to alter the affinity and specificity of the ScFv fragment by region-specific mutagenesis of the loops that constitute the CDRs, followed by affinity selection with appropriate DNA adducts using antibody phage display. The power of this technique lies in the repeated cyclic amplification of the phage followed by screening of these phage with the appropriate DNA adduct. Using this technique, we propose to identify ScFv peptides with altered affinity and specificity.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Prof. Hamilton O. Smith for encouragement and helpful suggestions. We thank K. Castleberry for typing the manuscript. This work was supported by a Shannon Award (ES 07568) from NIEHS. The work is currently funded by grants from the NIEHS (ES07568 and ES06052). We are grateful to the Environmental Health Sciences Center Core Facility (supported by grant ES03819) for synthesis of oligonucleotides. A.G.Z. was supported by a pre-doctoral Ford Foundation fellowship and NIEHS grant PO1 ES06052.
REFERENCES
- 1.Strickland P.T., Routledge,M.N. and Dipple,A. (1993) Cancer Epidemiol. Biomarkers Prev., 2, 607–619. [PubMed] [Google Scholar]
- 2.Groopman J.D., Donahue,P.R., Zhu,J., Chen,J. and Wogan,G.N. (1985) Proc. Natl Acad. Sci. USA, 82, 6492–6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schulte P.A. and Pereva,E.P. (1993) Molecular Epidemiology: Principles and Practices. Academic Press, San Diego, CA.
- 4. IARC (1992) Solar and Ultraviolet Radiation, IARC Monograph. IARC, Lyon. [PMC free article] [PubMed]
- 5.Scotto J., Fears,T.R. and Fraumeni,J.F. (1981) Incidence of Nonmelanoma Skin Cancers in the United States. NCI Publication no. (NIH) 82-2433.
- 6.Glass A.G. and Hoover,R.N. (1989) J. Am. Med. Assoc., 262, 2097–2100. [Google Scholar]
- 7.Weinstock M.A. (1989) J. Am. Med. Assoc., 262, 2138–2140. [Google Scholar]
- 8.Kerr J.B. and McElroy,C.T. (1993) Science, 262, 1032–1034. [DOI] [PubMed] [Google Scholar]
- 9.Strickland P.T. and Boyle,J.M. (1981) Photochem. Photobiol., 34, 595–601. [PubMed] [Google Scholar]
- 10.Strickland P.T., Nikaido,O., Matsunaga,T. and Boyle,J.M. (1992) Photochem. Photobiol., 55, 723–727. [PubMed] [Google Scholar]
- 11.Kettleborough C.A., Saldanhe,J., Ansell,K.H. and Bendig,M.M. (1993) Eur. J. Immunol. 23, 206–211. [DOI] [PubMed] [Google Scholar]
- 12.Sanger F., Nicklen,S. and Coulson,A.R. (1977) Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morioka H., Miura,H., Koabyashi,H., Koizumi,T., Fujii,K., Asano,K., Matsunaga,T., Nikaido,O., Stewart,J.D. and Ohtsuka,E. (1998) Biochim. Biophys. Acta, 1385, 17–32. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi H., Morioka,H., Torizawa,T., Kat,K., Shimada,I., Nikaido,O. and Ohtsuka,E. (1998) J. Biochem. Soc., 124, 182–188. [DOI] [PubMed] [Google Scholar]
- 15.Parmley S.F. and Smith,G.P. (1988) Gene, 73, 305–318. [DOI] [PubMed] [Google Scholar]
- 16.Smith G.P. and Scott,J.K. (1993) Methods Enzymol., 217, 228–253. [DOI] [PubMed] [Google Scholar]
- 17.McCafferty J., Griffiths,A.D., Winter,G. and Chriswell,J. (1990) Nature, 348, 552–554. [DOI] [PubMed] [Google Scholar]
- 18.Barbas C.F., Rosenblum,J.S. and Lerner,R.A. (1993) Proc. Natl Acad. Sci. USA, 90, 6385–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winter G. and Milstein,C. (1991) Nature, 349, 293–299. [DOI] [PubMed] [Google Scholar]
- 20.Raag R. and Whitlow,M. (1995) FASEB J., 9, 73–80. [DOI] [PubMed] [Google Scholar]
- 21.Kipriyanov S.M., Moldenhauser,G. and Little,M. (1997) J. Immunol. Methods, 200, 69–77. [DOI] [PubMed] [Google Scholar]
- 22.Gange S.J., Muñoz,A., Wang,J.-S. and Groopman,J.D. (1996) Cancer Epidemiol. Biomarkers Prev., 5, 57–62. [PubMed] [Google Scholar]