

Abstract
l-2-Haloacid dehalogenases, industrially and environmentally important enzymes that catalyze cleavage of the carbon-halogen bond in S-2-halocarboxylic acids, were known to hydrolyze chlorinated, brominated and iodinated substrates but no activity towards fluorinated compounds had been reported. A screen for novel dehalogenase activities revealed four l-2-haloacid dehalogenases capable of defluorination. We now report crystal structures for two of these enzymes, Bpro0530 and Rha0230, as well as for the related proteins PA0810 and RSc1362, which hydrolyze chloroacetate but not fluoroacetate, all at ~2.2 Å resolution. Overall structure and active sites of these enzymes are highly similar. In Molecular Dynamics (MD) calculations, only the defluorinating enzymes sample more compact conformations, which in turn allow more effective interactions with the small fluorine atom. Structural constraints, based on X-ray structures and MD calculations, correctly predict the defluorination activity of the homologous enzyme ST2570.
Keywords: L-2-haloacid dehalogenases, enzymic defluorination, protein structures, enzyme catalysis, molecular dynamics
Graphical Abstract
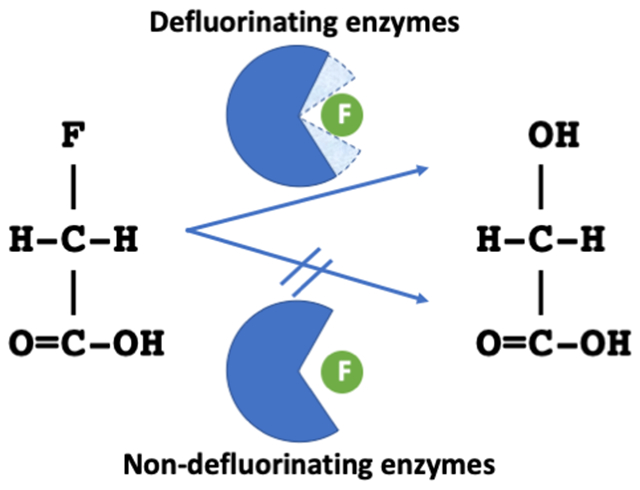
You have to come close if you want a reaction. Several members of the haloacid dehalogenase-like hydrolases superfamily were found to break carbon-fluorine bonds. The only features that differentiate between them and other family members unable to achieve this feat were small changes in distance between selective active site residues and protein dynamics leading to tighter interactions between protein and halogen.
Introduction
l-2-Haloacid dehalogenases (HADs, EC 3.8.1.2) are enzymes of industrial and environmental importance. They catalyze the hydrolysis of S-2-haloalkanoic acids into hydrogen halide and R-2-hydroxyalkanoic acids. Since the 1980s, numerous HADs have been identified and over twenty homologues have been characterized in more detail.[1] Many HADs were isolated from microbes capable of metabolizing toxic halogenated waste as the sole source of carbon and energy.[2] For example, the well-studied DhlB enzyme participates in the degradation of the underground water contaminant 1,2-dichloroethane by Xanthobacter autotrophicus GJ10.[3] HADs belong to the very large and ubiquitous haloacid dehalogenase-like hydrolases superfamily (HADSF), most of whose members, however, handle phosphoryl transfer chemistry.[4–6] HADs are regio- and stereospecific, acting only on the S-enantiomer of their substrates, and can collectively process 2-haloacids of 2 to 16 carbon atoms in length.[7] For these reasons, HADs have long been of interest in biotechnology for both the production of optically active compounds as well as the bioremediation of environmental contaminants.[2,8]
Despite the broad substrate specificity of HADs, which cover a variety of chlorinated, brominated and iodinated carboxylic acids, none of the initially characterized members are active against 2-fluorooacids.[2] The large dissociation energy of the C-F bond (up to 130 kcal/mol)[9] renders it significantly less susceptible to cleavage than other carbon-halogen bonds. For comparison, the C-X bond dissociation energies in the series of halomethanes are 115, 83.7, 72.1 and 57.6 kcal/mol for F, Cl, Br and I, respectively.[10] In an effort to identify dehalogenases with novel activities through a functional genomics screen, we discovered four l-2-haloacid dehalogenases capable of defluorination. Bpro0530 and Bpro4516 from Polaromonas sp. JS666, RHA1_ro00230 (referred to as Rha0230 hereafter) from Rhodococcus sp. RHA1, and Adeh3811 from Anaeromyxobacter dehalogenans 2CP-C are the first HAD-like family members to exhibit such a defluorinating activity.[1]
The reaction mechanism of HADs has been established based on functional and structural investigations of three model enzymes: L-Dex YL from Pseudomonas sp. YL,[11–13] DhlB from Xanthobacter autotrophicus GJ10[14,15] and DehIVa from Burkholderia cepacia MBA4.[16,17] HADs catalyze a two-step dehalogenation reaction analogous to that of the haloalkane[18,19] and fluoroacetate dehalogenases,[20–22] both of which belong to the α/β hydrolase superfamily (ABHSF) of proteins.[23] Following substrate binding, the carbon-halogen bond is cleaved in an SN2-like fashion, where an aspartate nucleophile attacks the halogen-bearing carbon atom (C2) to directly expel the halide. Simultaneously, the covalent ester intermediate is formed, which in turn is hydrolyzed by an activated water molecule to release the alcohol product and regenerate the enzyme (Figure 1).
Figure 1.
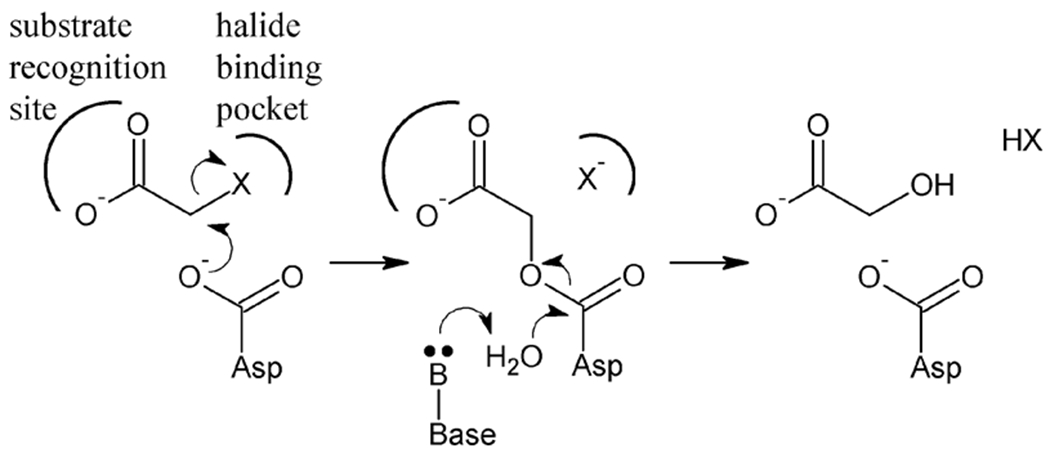
Two-step reaction mechanism of HADs. An aspartate nucleophile first attacks the halide-bearing carbon atom in an SN2-like manner, which directly displaces the halide anion into the halide-binding pocket. Simultaneously, the covalent ester intermediate forms, which is subsequently hydrolyzed using an activated water molecule.
Although organofluorine compounds include most useful products with diverse applications[24] their production and application continue to be controversial because of their adverse global impact and environmental persistence.[25–27] Under-standing the structural basis of enzymatic defluorination is therefore fundamental to the development of improved enzymes for biotechnological remediation.
Here, we present the crystal structures, together with kinetic data and repeated molecular dynamics (MD) simulations, of two defluorinating HADs, Bpro0530 and Rha0230, and of two non-defluorinating HADs, PA0810 from Pseudomonas aeruginosa PAO1 and RSc1362 from Ralstonia solanacearum. We also include sequence comparisons for the two defluorinating but so far crystallization-resistant HADs Bpro4516 and Adeh3811. In addition, we report kinetic measurements and MD calculations for ST2570, an L-haloacid dehalogenase from the thermophilic archaeon Sulfolobus tokodaii, whose atomic coordinates had been deposited in the Protein Data Bank (PDB code 2W43).[28]
We explore various functional and biochemical aspects of these enzymes and relate them to the mechanistically similar dehalogenases in the ABHSF.[20,29–32] Subtle modifications in the halide binding site, like changes in protein dynamics, can tune enzyme selectivity. Our results would suggest that among HADs the ability to place the halide interacting residues closer to the smaller fluorine atom is essential to conferring defluorination activity. This structural adaptation appears to be a common evolutionary theme shared by other enzymes involved with fluorine chemistry.[33–36]
Results and Discussion
Overall structure
The four HADs Bpro0530, Rha0230, PA0810, and RSc1362 were crystallized and their crystal structures determined at resolutions of ca. 2.2 Å. Data collection and refinement statistics are listed in Table S1. The HAD structures belong to the C1 subfamily of the HADSF protein fold,[5] comprising an α/β/α core domain, an α-helical cap domain inserted at a conserved location, and an active site located at the domain interface. The core domain consists of six parallel β-strands arranged in the order of β5-β4-β1-β6-β7-β8 (or 3-2-1-4-5-6 when disregarding the two short interdomain strands β2 and β3), which are flanked by α-helices. The cap domain comprises a bundle of four α-helices and is inserted immediately after β1 of the core domain (Figures 2 and S1).
Figure 2.
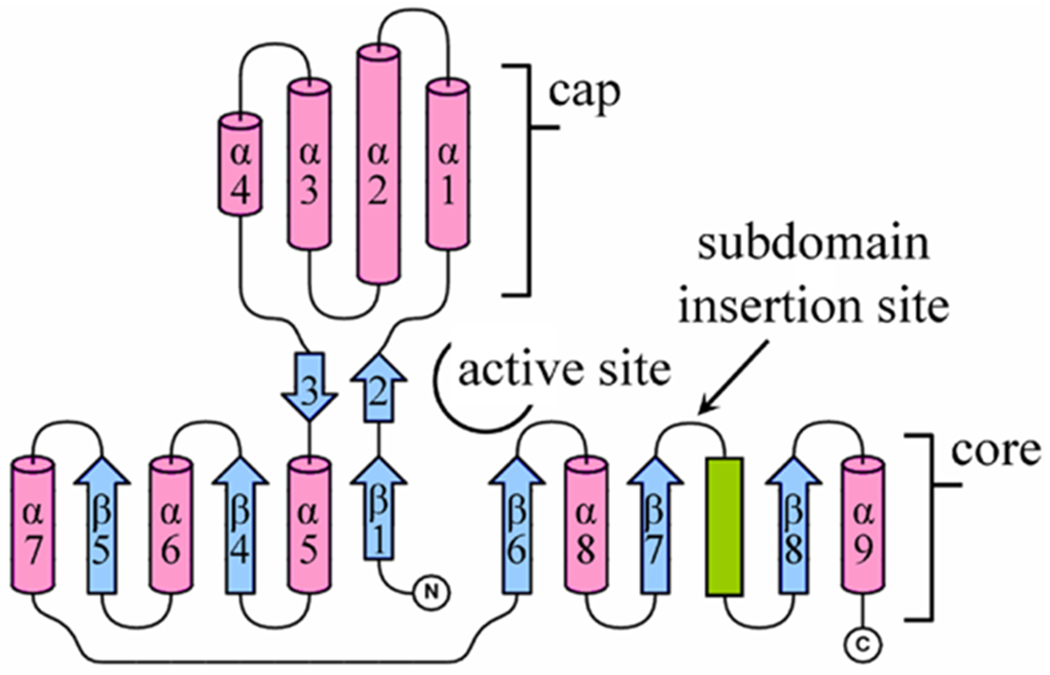
Topology of secondary structure elements of HADs. The HADs share a common fold comprising a core and cap domain, creating the active site at the domain interface. The core domain consists of a six-stranded parallel β-sheet (blue arrows) sandwiched between α-helices (pink cylinders) and a coil (green bar). The cap domain comprises a bundle of four α-helices, inserted between β1 and α5. The insert between β7 and the coil varies greatly in size. In DhlB, there are 23 residues folded into two α-helices as an ‘atrium’ subdomain, which partly covers the active site opening. In Rha0230 and PA0810, the subdomain comprises a 10-residue coil-structured ‘flap’. All other HADs possess only a 4-residue loop at this location. The figure is prepared with the help of TopDraw.[48]
All HADs share a common fold; the degree of structural similarity can be judged from Table S2, which lists RMSD values for Cα atoms with the cap and core domains superposed independently to account for variations in the interdomain angle. While reasonably similar overall, slightly elevated numbers set the three enzymes Rha0230, PA0810 and ST2570 somewhat apart from the remaining HADs. There are also some clearly distinct structural features. First, the insert between β7 and the downstream coil structure varies considerably in length. DhlB has the longest insert known; the stretch of 23 amino acids folds into an extra double-helical ‘atrium’ subdomain, partly obstructing access to the active site.[14] Rha0230 and PA0810 present a similar, but miniaturized, 10-residue coil-structured ‘flap’ subdomain. All other HADs have only a loop of four residues bridging β7 and the downstream coil region (Figures 3 and S1).
Figure 3.
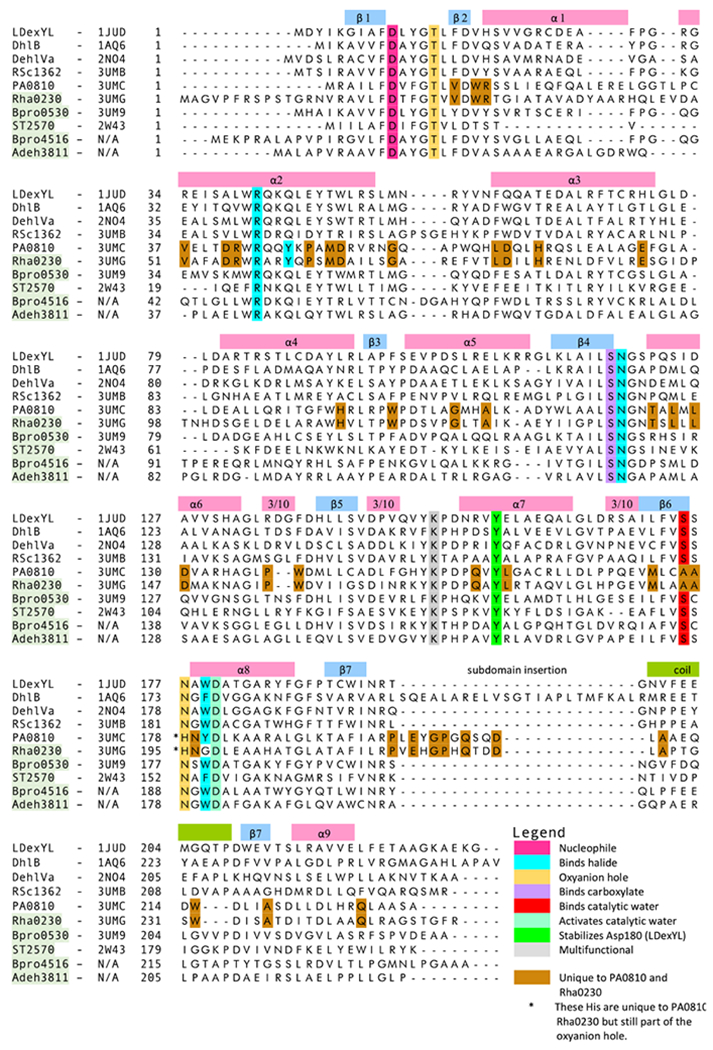
Sequence alignment of HADs. The sequences of three previously structurally characterized and confirmed HADs (L-Dex YL, DhlB, DehIVa) are aligned with those of the five HADs discussed in depth in this study (RSc1362, PA0810, Rha0230, Bpro0530, ST2570) and two additional defluorinases of unknown structure (Bpro4516, Adeh3811). The names of defluorinating enzymes have a green background. The position of the leading residue in each row is numbered. The conserved multi-function lysine residue (Lys151 of L-Dex YL) mediates substrate binding, positioning of the aspartate nucleophile and stabilization of the conserved aspartate (Asp180 of L-Dex YL), which activates the catalytic water molecule. The alignment is generated using the CLUSTALW algorithm.[49] The alignment of α1, α2, subdomain insertion, and coil was adjusted manually based on the crystal structures. The secondary structural elements are derived from the crystal structure of L-Dex YL using DSSP.[50]
Many HADs identified to date are dimeric, and employ a conserved dimerization mode (Figure 4A). The interface is primarily formed between the second half of α2 and α3 of one subunit, and α8 and the following coil of the other subunit. In DhlB, dimerization further involves the atrium subdomain.[14] This arrangement results in an active site that is partly covered by the partner subunit. However, in Rha0230 and PA0810, which are members of a new HAD subfamily, dimerization involves the edge of β5 and the surrounding helices, leaving the active site cavity much more exposed to solvent (Figure 4B).
Figure 4.
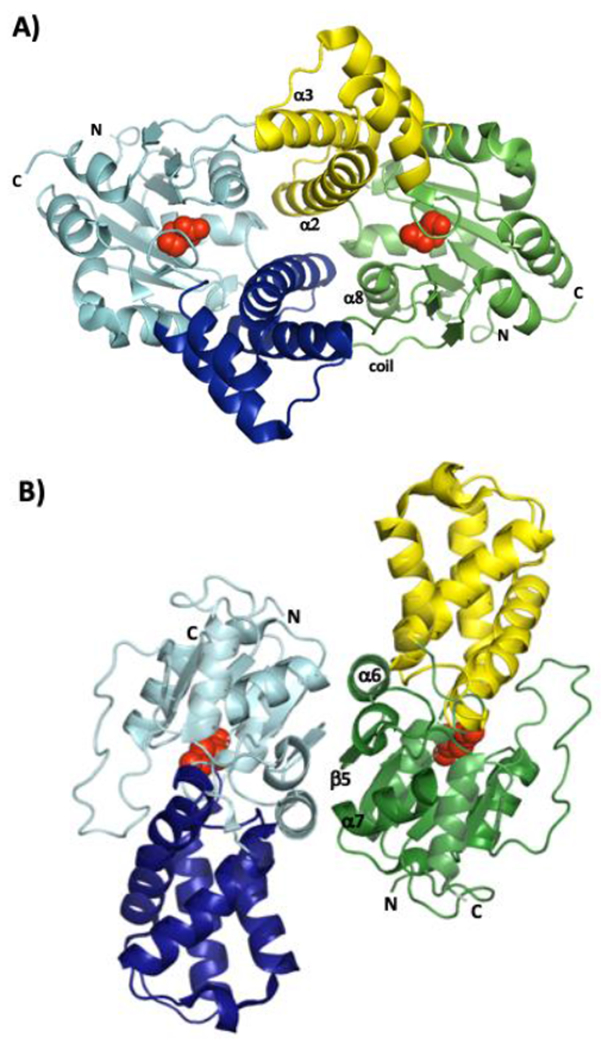
Cartoon representations of the A) Bpro0830 and B) Rha0230 dimers as seen along their respective twofold symmetry axes. Core domains are in aquamarine and green whereas the cap domains are in dark blue and yellow, respectively. Structural elements that form most of the interface are labelled together with the N- and C-termini. The side chains of the covalent intermediate-forming aspartate residues, are shown as red spheres to indicate the location of the active sites.
Protein flexibility
Protein dynamics are essential in catalysis although they can differ greatly in mode, magnitude and timescale. To date, two types of dynamics have been described in HADs.[17] First, the halide-binding arginine side chain adopts considerably different conformations among the HADs, which either permit or block access to the buried active site cavity. This arginine is thus proposed to traffic substrates and products in and out of the active site.
Second, a normal mode analysis performed on DehIVa predicted that up to 20° of hinge-like interdomain motion is possible. This feature is supported by the structural superposition of all HADs (Figure S2). It is believed to contribute to regulation of entry and exit of substrates and products, as well as to exclude solvent from the active site during catalysis.[17] Similar interdomain dynamics were also reported in several phosphoryl transferases structurally related to the HADSF.[4]
Sequence comparison
In general, when referring to residues, Bpro0530 numbering is applied but when selected enzymes are discussed their respective residue numbers are used. Despite their moderate to low overall sequence identities, structure-based alignments highlight the highly conserved active sites of HADs. The amino acid sequences of the six newly identified HADs, Bpro0530, Bpro4516, Rha0230, Adeh3811, PA0810, and RSc1362, are compared with those of HADs for which crystal structures were previously available, L-Dex YLL,37] DhlB,[14] DehIVa[17] and ST2570 from Sulfolobus tokodaii[28] (Figure 3). PH0459 from Pyrococcus horikoshii OT3[38] (PDB CODE 1X42) and ST2620 from Sulfolobus tokodaii (PDB CODE 2ZG6), despite being annotated as probable HADs, are excluded here because they harbor sequence motifs indicative of phosphoryl transferases and also did not show dehalogenase activity in our assays.
The sequence alignment reveals a low to moderate level of identity (20% to 54%) but several key HADSF sequence motifs are fully conserved although none could be identified that would correlate with defluorination activity. Strict conservation is also found for almost all active site residues with assigned catalytic roles (Figure 3 and Table 1).[1,5,39] The exceptions are the aromatic halide-interacting amino acid (W179 in Bpro0530) and the serine that binds the catalytic water (S175 in Bpro0530). The former varies from tryptophan to tyrosine or phenylalanine, and even to glycine in Rha0230, while the latter is replaced by an alanine in both the defluorinating Rha0230 and the non-defluorinating PA0810. Along their whole chains, there are residues that are common to the two last enzymes but different from amino acids placed at corresponding spots in other HADs identifying Rha0230 and PA0810 as members of a new HAD subfamily.
Table 1.
Listing and residue number correlation of amino acids with primary catalytic roles as inferred from model HADs L-Dex YL (PDB ID 1JUD), DhlB (1QQ5) and DehIVa (2NO5).
| Enzyme | L-Dex YL | DhlB | DehIVa | RSc1362 | PA0810 | Rha0230 | Bpro0530 | ST2570 |
|---|---|---|---|---|---|---|---|---|
| SwissProt ID | Q53464 | Q60099 | Q51645 | Q8XZN3 | Q9I5C9 | Q0SK70 | Q12G50 | Q96XE7 |
|
| ||||||||
| Nucleophile | D10 | D8 | D11 | D10 | D7 | D21 | D10 | D7 |
|
| ||||||||
| Halide-binding | R41a | R39 | R42 | R41 | R44 | R58 | R41 | R23 |
| q44 b | q42 | q45 | q44 | Y47 | Y61 | q44 | q26 | |
| N119 | N115 | q45 | N123 | N122 | N139 | N119 | N96 | |
| W179 c | F175 | W180 | W183 | Y180 | G197d | W179 | F154 | |
|
| ||||||||
| Carboxylate binding | S118 | S114 | S119 | S122 | S121 | S138 | S118 | S95 |
|
| ||||||||
| Hydrolysis of covalent ester intermediate | T14e | T12 | T15 | T14 | T11 | T25 | T14 | T11 |
| S175ef | S171 | S176 | S179 | A176g | A193g | S175 | S150 | |
| N177ef | N173 | N178 | N181 | H178 | H195 | N177 | N152 | |
| D180fgh | D176 | D181 | D184 | D181 | D198 | D180 | D155 | |
|
| ||||||||
| Multifunction | K151i | K147 | K152 | K155 | K152 | K169 | K151 | K128 |
|
| ||||||||
| Stabilizes Asp180 | Y157 | Y153 | Y158 | Y161 | Y158 | Y175 | Y157 | Y134 |
The conserved arginine is also proposed to recruit the substrate in a ‘lockdown’ mechanism based on its highly dynamic nature.
The lower case letter q is meant to indicate the glutamine amino acid found in most HADs at this position. Its side chain invariably points away from the active site and is not expected to support catalysis. A grey background indicates residues that are unique to the new PA0810/Rha0230 subfamily; Y47 and Y61, respectively, are in positions to interact with the halide atom.
Although the chemical identity of the residues in this position changes their aromatic character is conserved with the exception of
Rha0230, which lacks the conserved aromatic side chain that interacts with the halide.
Forms the oxyanion hole.
Binds the catalytic water.
Activates the catalytic water.
Lacks the hydroxyl group of the conserved serine, binding of catalytic water may be taken over by the histidine found two residues downstream.
Orients the aspartate nucleophile, binds substrate and stabilizes Asp180 (L-Dex YL). A green background indicates the three defluorinating enzymes.
Active site comparison
In general, the active sites of HADs are highly similar in both amino acid composition and spatial organization. This includes the aspartate nucleophile, the substrate binding serine and lysine, and the halide-binding arginine, asparagine and aromatic residues. Figure 5 provides a visual guide to the general arrangement of residue side chains around the product of the first half reaction, the covalent intermediate and the halide ion. The DhlB complex is chosen because it represents the only crystal structure that contains both covalent intermediate and halide ion close together and therefore can be seen as a good approximation of the Michaelis-Menten complex. Superposition and detailed analysis of these active sites, however, does reveal distinct structural characteristics in the novel HADs (Figure 6 and Table 1).
Figure 5.
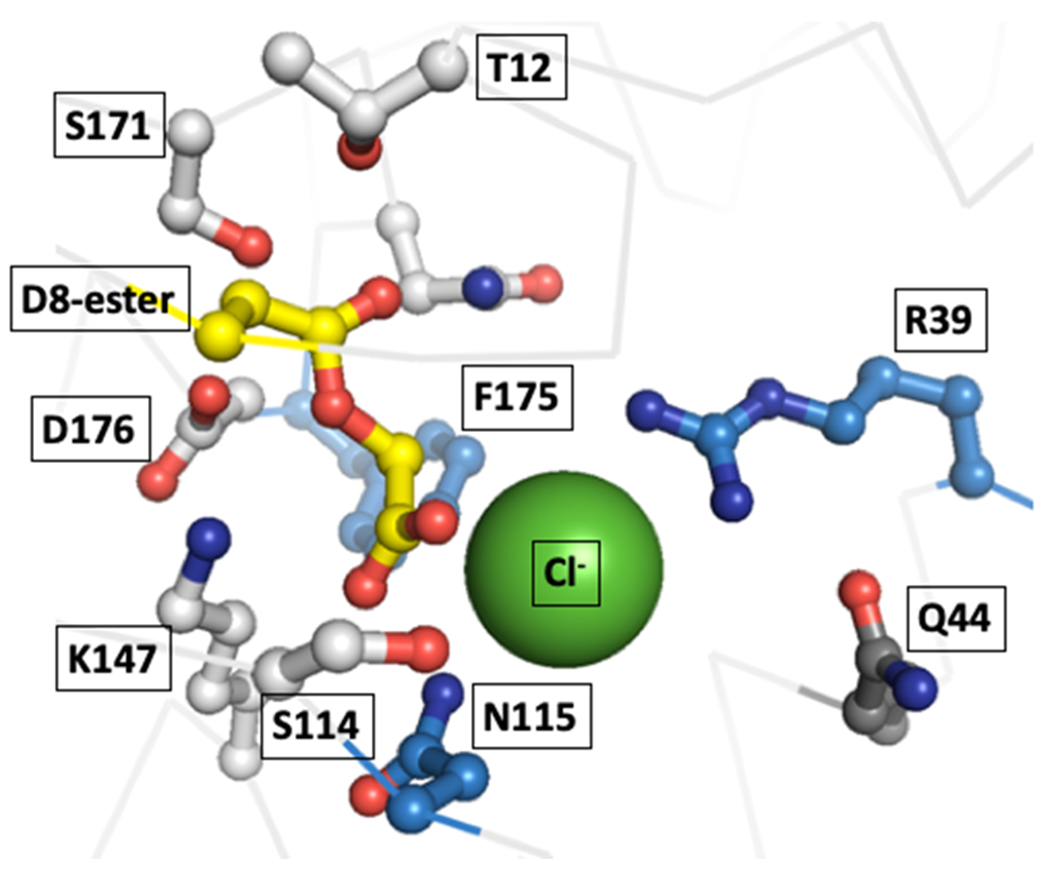
Active site of the enzyme DhlB (PDB CODE 1QQ7) whose structure is the only one that features the covalent aspartate-acetyl intermediate and a bound halide (Cl−, shown as a green sphere) in the active site, making it the best model of the catalytically important interactions available. Residues close to the halide ion are shown as blue and those contacting the covalent intermediate as white sticks. Although Q44 points away into solvent it is shown to indicate the spot where members of the PA0810/Rha0230 subfamily place a tyrosine residue in position to interact with the halide ion (see also Figure 9).
Figure 6.
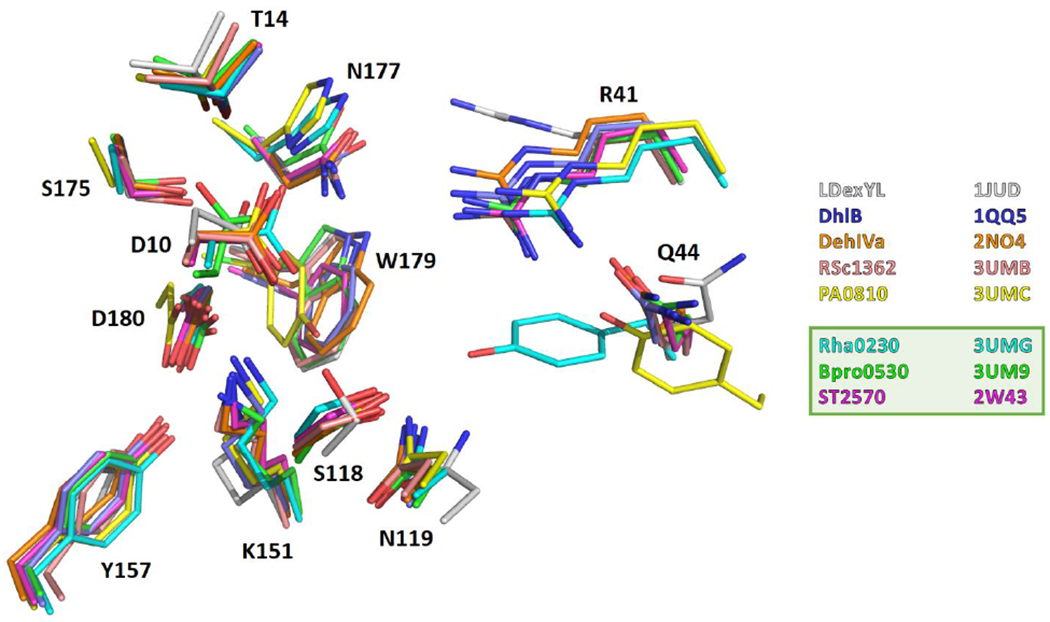
Superposition of HAD active sites. The amino acid side chains forming the active sites of Bpro0530 (PDB code 3UM9; chain A), Rha0230 (3UMG; chain A), ST2570 (2W43; chain A), PA0810 (3UMC; chain B), RSc1362 (3UMB; chain A), L-Dex YL (1JUD; chain A), DhlB (1QQ5; chain A), and DehIVa (2NO4; chain A) are superimposed using only the Cα atoms of those active site residues that do not interact with halide (i.e. Asp10, Thr14, Ser118, Lys151, Tyr157, Ser175, Asn177 and Asp180 of Bpro0530 and the corresponding residues of the other HADs; see also ). All these residues belong to the respective core domains. This course of action should enable a comparison of the arrangement of the halide pocket relative to the rest of the active site.
For example, in the non-defluorinating enzyme RSc1362, the arrangement of active site residues is largely similar to that of L-Dex YL. The most notable difference is the out-of-plane shift (relative to the guanidinium group) of the halide-binding Arg41 by ~1.7 Å. Compared to L-Dex YL, RSc1362’s Arg41 side chain is found closer to the halide-binding Asn123 [Asn(Nδ2)--Arg(Nη1) distance of 6.2 Å and Asn(Nδ2)--Arg(Nη2) distance of 6.3 Å], but not to the Asp10 nucleophile [Asp(Oδ1)--Arg(Nη2) distance of 6.5 Å] (Figure 7).
Figure 7.
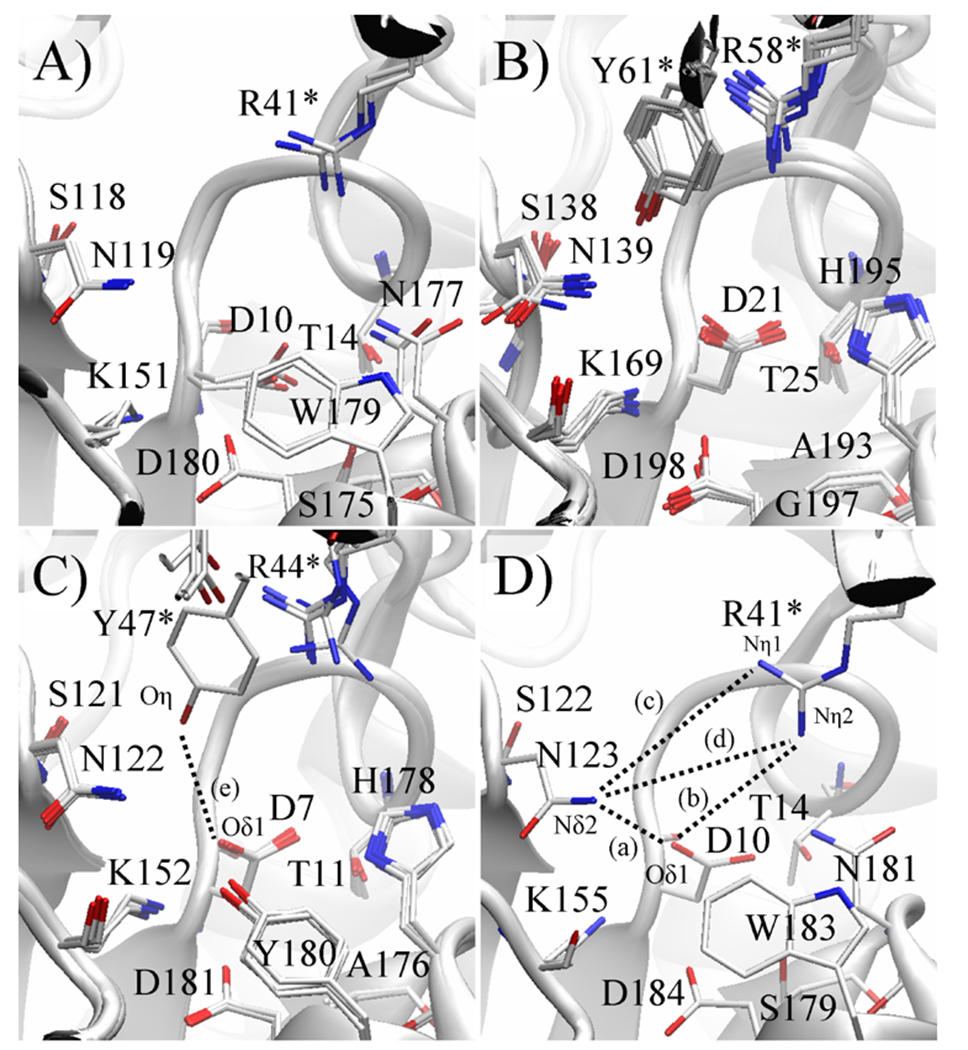
Conformational variability of key active site residues of four novel HADs. Active sites of crystallographically independent protein subunits of the same enzyme are superimposed, superpositioning only core domain Cα atoms to highlight any potential differences due to interdomain motion. The conserved arginine and the partly conserved tyrosine three residues downstream belong to the cap domain (marked by *) while the remaining labeled residues belong to the core domain. A) In Bpro0530, both active sites are similar. However, the Asp10 nucleophile has poorly defined electron density for the carboxylate group and is thus likely disordered. The conformation with the best geometry, which differs from that in all other HADs, is refined. B) In Rha0230, all 8 active sites closely resemble one another. The cap domain’s α2 contains a unique tyrosine, which protrudes towards the halide-binding cavity. C) In PA0810, the cap domain also harbours the unique tyrosine residue, which in one subunit (subunit B) adopts the conformation observed in Rha0230, placing closer to the halide. The other three active sites are practically superimposable with the tyrosine ring shifted towards the protein surface. D) In RSc1362, the composition of the active site is identical to that in L-Dex YL, DehIVa and Bpro0530 and probably also in the defluorinating enzymes Bpro4516 and Adeh3811. The dashed lines represent the key active site measurements, and correspond to the labels A) to E) in Figure 10.
In the defluorinating enzyme Bpro0530, a typical HAD, one finds two unique structural features. First, the carboxylate group of the Asp10 nucleophile appears disordered judged by its relatively poorly defined electron density. Second, Arg41 of the halide pocket surveys a conformation tucking it slightly deeper into the enzyme. This places the guanidinium group closer to the halide-binding asparagine [Arg(Nη1)--Asn(Nδ2) distance as short as 6.0 Å; Table S3], which effectively reduces the halide-binding cavity. Arg41 also appears closer to the reactive oxygen atom of the aspartate nucleophile to again create a slightly more compact active site, because of its inherent disorder, the Asp(Od1)—Arg(Nh2) distance in Bpro0530 can only be estimated as 5.7 Å from the position of D10 in the superposed structure of L-Dex YL (PDB code 1ZRM).
Not too surprisingly, the members of the Rha0230/PA0810 subfamily display a few more changes at their active sites. In the defluorinating enzyme Rha0230, the average position of the halide-binding Arg58 (corresponding to Arg41 in Bpro0530) is shifted slightly, in-plane of the guanidinium group, which moves closer to the aspartate nucleophile [Asp(Oδ1)--Arg(Nη2) distance as little as 5.5 Å] but does not come particularly close to the halide-binding Asn139 (Asn119 in Bpro0530; Figure 7). Also, the enzyme is so far the only HAD with a glycine residue, Gly197, at the position of the generally conserved aromatic halide-binding residue (Table 1). Meanwhile, Tyr61 projects from α2 of the cap domain towards the active site (Figures 6, 8. 9), placing its side chain hydroxyl only ~5.4 Å from Oδ1 of the Asp21 (Asp10 in Bpro0530) nucleophile. This hydroxyl group is located approximately midway between Arg58 and Asn139 and apparently contributes to the tightening of the halide pocket. Lastly, the conserved Ser175-Ser/Cys176-Asn177 motif (Bpro0530 numbering) required for the turnover of the covalent ester intermediate has been substituted by Ala193-Ala194-His195 in Rha0230.
Figure 8.
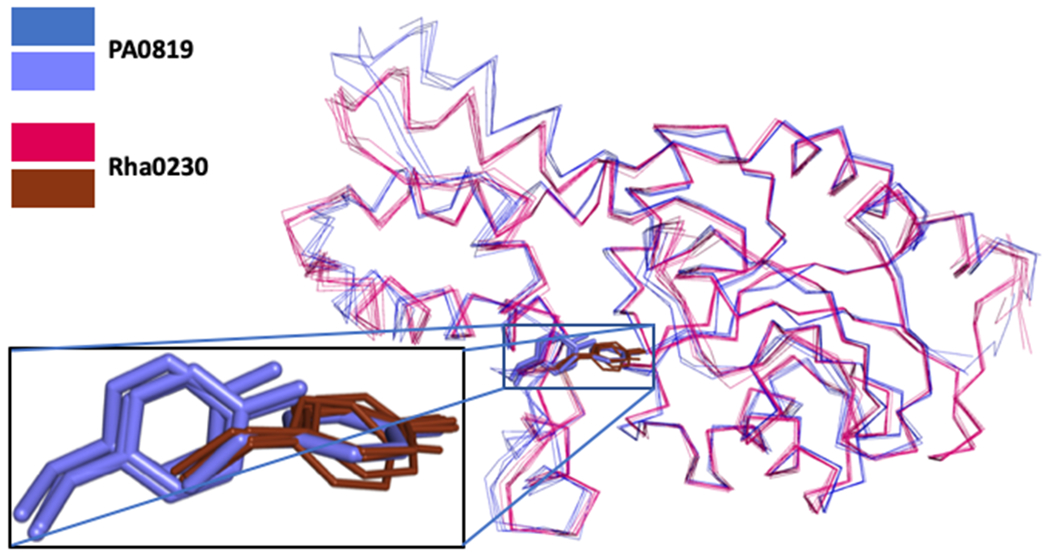
Superposition of Cα-positions of the eight crystallographically independent subunits of the defluorinating enzyme Rha0230 (red) and the four subunits (two dimers) of the non-defluorinating enzyme PA0810 (blue). The zoom box displays the relative positions of Y47 (Rha0230; thin brown) and Y61 (PA0810 in thick blue sticks) side chains. In all four subunits of Rha0230, Y47 residues overlap very closely. In the four subunits of PA0810, however, only one of them (subunit B) adopts a similar orientation. In the remaining subunits, the Y61 side chains still overlap well but retreat from the active site by a change of the backbone angles of a short stretch of amino acids. This conformational change slightly enlarges the halide-binding part of the active site. Figure 9 depicts the two conformations of Y47 in PA0810 and its interactions with neighbouring side chains in more detail.
Figure 9.
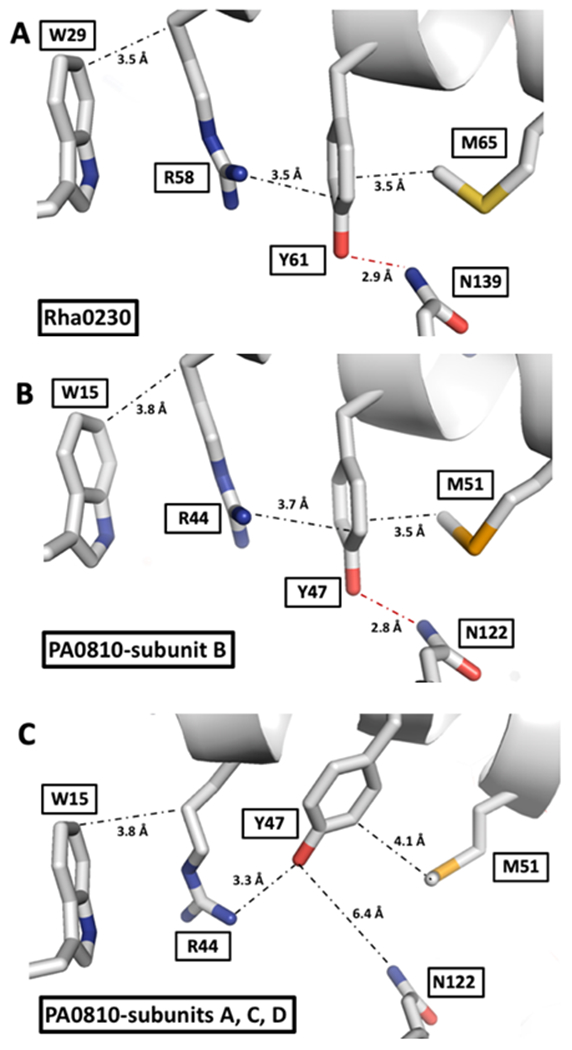
Interactions stabilizing the positions of the tyrosine residues unique to the Rha0230/PA0810 subfamily. Distances in A, B and C are given in Ångstrom and represent the average of all crystallographically independent subunits. The side chains of the residues surrounding the unique tyrosines in A) Rha0230 and B) in subunit B of PA0810 are tightly packed at van der Waals distances and Tyr-61/Tyr-47 forms a short hydrogen bond to an Asn residue. C) In the majority of PA0810 subunits, however, this H-bond is broken; Tyr-47 now engages in an ionic interaction with Arg-44 while the distances to the bracketing tryptophan and methionine residues are also looser. This rather localized conformational change affects stable packing in Rha0230 and one subunit of PA0810 and looser interactions in the majority of PA0810 subunits.
The other member of the new subfamily, the non-defluorinating enzyme PA0810, has not lost the halide-interacting aromatic residue Tyr180. Its halide-binding Arg44 is shifted in the same direction as Arg58 in Rha0230, although it is slightly further away from Oδ1 of the Asp7 nucleophile [Asp(Oδ1)--Arg(Nη2) distance as small as 6.5 Å; Figure 10 and Table S3]. PA0810 employs an identical Ala176-Ala177-His178 motif for the hydrolysis of the ester intermediate. Its cap domain also contributes a corresponding tyrosine, Tyr47, to the active site. However, of the four crystallographically independent subunits, only one shows the tyrosine ring in the same position as Tyr61 in Rha0230 with an Asp(Oδ1)--Tyr(Oη) distance of 5.7 Å.
FIGURE 10.
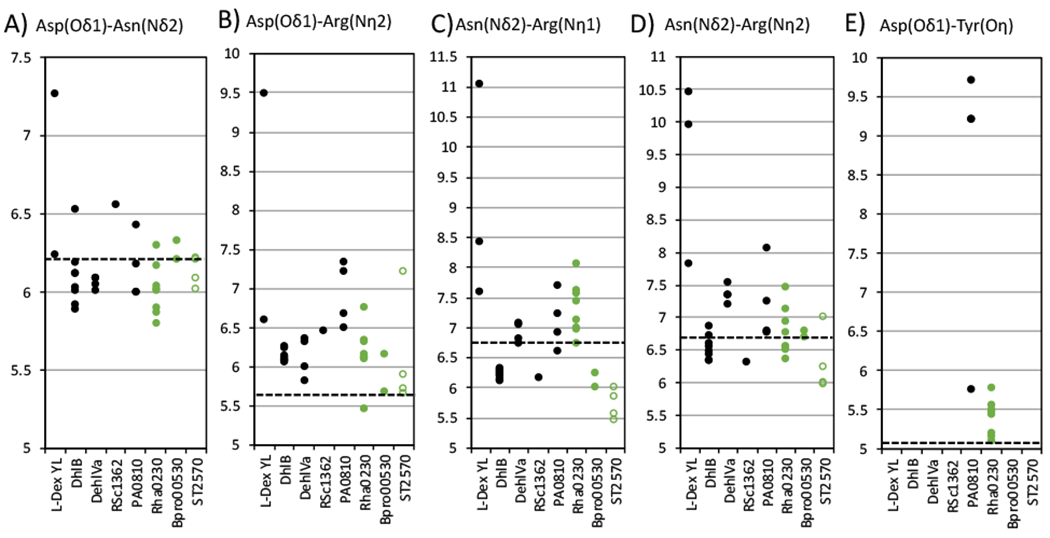
Analysis of the geometry of active sites of HADs. In columns A) to D), the interatomic distances (in Å)are given between the reactive Oδ1 atom of the Asp10 nucleophile, the halide-binding atoms in Arg41 (Nη1 and Nη2) and Asn119 (Nδ2) residues (Bpro0530 numbering; for equivalent residues in other enzymes see Tables 1 and S2). In column E, there are only two entries for the distance between the covalent intermediate-forming aspartate and the hydroxyl group of the unique cap domain tyrosine (Oη), which is unique to PA0810 (Y47) and Rha0230 (Y61). In the equivalent position, all other known HADs carry a glutamine residue, which without exception points into solvent. These measurements are also listed in supplemental Table S4. In essence, A) B) and E) compare how close the halide pocket is to the aspartate nucleophile, and C) and D) correlate with the size of the halide pocket. Black filled circles show distances in the non-defluorinating HADs L-Dex YL, DhlB, DehIVa, PA0810 and RSc1362. Green filled circles mark measurements in the defluorinating HADs Bpro0530 and Rha0230. Open green circles correspond to distances in ST2570, whose structure was previously known (PDB CODE 2W11) but whose defluorination capability was suggested by the values measured here and subsequently confirmed by kinetic measurements. Multiple plots for each HAD represent measurements from different crystal structures or different active sites in a single crystal structure, and the distribution of the plot correlates with the dynamic nature of the active site structures. The dashed horizontal line in each panel represents the apparent cutoff required for defluorination; it is set to the larger of the minimum distances found in either Bpro0530 or Rha0230, the two defluorinating enzymes. No other HAD besides ST2570 exhibits distances at or below every measurement cutoff. Distances to the aspartate nucleophile of Bpro0530 are estimated from the superposed aspartate in L-Dex YL (PDB code 1ZRM).
Local backbone flexibility in α2 of the cap domain favours an alternate conformation, which retracts Tyr47 away from the active sites of the remaining three subunits in the crystal’s unit cells (Figure 8). This distribution would indicate that for PA0810 the location of the tyrosine ring away from the active site represents a lower energy conformation and therefore the prevalent location of this aromatic side chain leading to more space around the halide-binding site. Figure 9 shows in more detail the interactions between the various side chains that stabilize the location and orientation of Tyr47 in its two conformations found in PA0810.
Catalytic roles of active site residues
The two-step hydrolytic dehalogenation mechanism catalyzed by HADs includes: recruitment and binding of the substrate, SN2-attack, binding and activation of the catalytic water, hydrolysis of the covalent ester intermediate, and product release (Figure 1). Numerous structural and functional studies have elucidated the residues that mediate these steps. We attempt a detailed description of the mechanism that combines biochemical and structural findings and apply it to the enzymes investigated in this study. For this purpose, residues, conserved in sequence and structure among homologues, are assumed to share identical roles (Table 1).
Recruitment of the substrate is facilitated by the dipole moment created by dimerization, drawing the substrate’s negatively charged carboxylate towards the positively charged active site.[14] The highly mobile Arg41 (Bpro-0530 numbering) at the active site entrance assists in this process using a “lockdown” mechanism, latching onto the substrate’s halogen atom upon entry to the active site.[17] The halogen atom is further held by Asn119 and Trp179,[13,15,17] while the substrate’s carboxylate group is bound by Ser118 and Lys151. [15,17,40]
The bond-breaking SN2 attack is initiated by the Asp10 nucleophile, which is maintained in a productive conformation through hydrogen bonding to Lys151 and Asp180.[40] Additionally, Lys151 is believed to reduce the pKa of Asp10 thereby keeping it in the deprotonated (i.e. reactive) state.[15] Since Lys151’s positive charge also interacts with the substrate’s carboxylate, we propose that substrate binding automatically releases Asp10 for the nucleophilic attack. It is interesting to note that in the crystal structure of Bpro0530, the Asp10 side chain is disordered. This may be attributed to the binding of a negatively charged sulfate, which could repel Asp10 from its anticipated orientation, although the binding of sulfate or phosphate does not cause such perturbation in other HADs. Alternatively, Asp10 in Bpro0530 may be inherently dynamic. Independent of which of these interpretations is correct, the mobility shows Asp10’s potential for bond-forming attack.
As Oδ1 of Asp10 approaches the halide-bearing carbon atom (C2) of the haloacid, the C-X bond is stretched and eventually broken, after which the halogen ion is displaced into the halide-binding pocket (Arg41, Asn119 and Trp179).[13,15,17] This SN2 mechanism likely proceeds via a pentavalent trigonal bypiramidal transition state with Walden inversion at the electrophilic stereocenter (C2).[13] This yields the covalent ester intermediate that contains a new C-O bond formed between the dehalogenated substrate moiety and Asp10.
Hydrolysis of the ester intermediate is better understood following the identification of the catalytic water molecule that had been missing from earlier HAD crystal structures [17] this water molecule is also observed in the crystal structures of Rha0230, PA0810 and RSc1362. The hydrolysis step involves the binding and activation of the catalytic water, as well as the stabilization of the oxyanion of the tetrahedral intermediate. The catalytic water is held in position by Ser175, Asn177 and Asp180, and deprotonated (i.e. activated) by Asp180, which in turn is stabilized by Lys151 and Tyr157.[40] The attack of the hydroxide ion on the Cγ of Asp10 results in the formation of the tetrahedral intermediate and the accumulation of negative charge on the (former) carbonyl oxygen atom of Asp10. This negative charge is stabilized by an oxyanion hole composed of the side chains of Thr14, Ser175 and Asn177.[13,15] The tetrahedral intermediate then collapses to liberate the alcohol product, probably with assistance from Lys151 and/or Asp180 in protonating the leaving group. Finally, the catalytic cycle completes with product dissociation from the active site.
Catalytic residues in PA0810 and Rha0230 that differ from consensus amino acids
Our previous dehalogenase screens identified the first members of a new subfamily of HADs, Rha0230 and PA0810, which despite distinct overall amino acid sequence similarity to HADs such as L-Dex YL, DhlB and DehIVa also show their own unique features, e.g. amino acid sequence (Figure 3) and dimerization mode (Figure 4). Members of this new HAD subfamily generally have a tyrosine (Tyr61 in Rha0230; Tyr47 in PA0810) three residues downstream of the halide-binding arginine. This tyrosine replaces the conserved glutamine (Gln44 in L-Dex YL), which does not seem to interact with halide at all. PA0810 still possesses the common aromatic halide-binding residue Tyr180 and its Tyr47 preferentially occupies the space of the common glutamine it replaces. Rha0230, however, puts the corresponding Tyr61 significantly deeper into the active site (Figures 6 and 8) as a member of a 4-member stack of side chains encompassing Trp29, Arg58, Tyr61, and Met65 (Figure 9). This places the side chain closer to the halide and, by reducing its mobility, should keep it there longer. This could counter the loss of halide interaction caused by the replacement of the aromatic residues common in this position in all other HADs with a glycine in Rha0230 (Figure 3 and Table 1).
In addition, these subfamily members use different residues to hydrolyze the ester intermediate. Instead of the Ser175-Ser176-Asn177 motif in L-Dex YL or Ser175-Cys176-Asn177 in Bpro0530, they employ Ala193-Ala194-His195 (Rha0230 numbering); the catalytic residues are underlined. To find an alanine residue in position 193 of Rha0230 is rather surprising because the corresponding Ser175Ala mutation in L-Dex YL prevents hydrolysis of the covalent ester intermediate, because it can no longer efficiently bind the catalytic water nor stabilize the oxyanion of the tetrahedral intermediate. [13] Since Rha0230 is active, however, it is very likely that His195 provides compensatory effects to sustain catalytic function. Notably, this residue is hydrogen bonded to Glu217 in the unique flap subdomain, which is also conserved in PA0810. This water-His195-Glu217 hydrogen-bonding network resembles that of the ABHSF hydrolases, in which a conserved histidine side chain activates the catalytic water, too.
It is tempting to look to those amino acid changes for an explanation as to which HADs will display defluorinating activity. None of the differences, however, correlate with an enzyme’s capability to hydrolyze fluoroacetate. The amino acid sequences of the recently identified defluorinases Bpro0530, Bpro4516, Adeh3811 and ST2570 align well with those of the previously characterized non-defluorinating enzymes L-DexYL, DhlB and DehlVa as well as that of the new addition RSc1362. All amino acids that have been identified as critical for catalysis are fully conserved. The new subfamily also includes one non-defluorinating, PA0810, as well as a defluorinating member, Rha0230.
Enzyme kinetics
The dechlorination activities of the five enzymes investigated here are generally comparable to those of the established HADs documented previously.[1] Steady-state kinetic parameters of the novel HADs for fluoroacetate and chloroacetate hydrolysis were determined near their optimal pH values (Table 2). The kcat values of dechlorination span one order of magnitude and are significantly larger than the kcat values of defluorination. The KM values for the tested substrates cover several orders of magnitude. We have, however, identified KM values at new extremes for chloro-acetate as the substrate; 5 μM and 3.5 μM measured for Bpro0530 and ST2570, respectively, are the lowest KM values known, whereas the 18 mM KM of Rha0230 is unusually high. Defluorination activity was also identified in ST2570 and is comparable to that of the other defluorinating HADs Bpro0530 and Rha0230. The Y61F mutant of Rha0230 was engineered to probe the role of Tyr61’s side chain hydroxyl group in catalysis. This mutation decreased the KM slightly for both fluoroacetate and chloroacetate hydrolysis when compared to WT. In contrast, kcat is reduced 50-fold for defluorination but increased 1.5-fold for dechlorination.
Table 2.
Steady-state defluorination kinetics of various HADs
| Enzyme | kcat (min−1) | K M (mM) | Kcat/KM (s−1M−1) |
|---|---|---|---|
| Bpro0530 | 13 ± 1 | 20.3 ± 0.3 | 11 ± 0.8 |
| Rha0230 | 17 ± 3 | 18 ± 3 | 16 ± 4 |
| Rha0230-Y61 | 0.36 ± 0.06 | 15 ± 2 | 0.41 ± 0.09 |
| PA0810 | No activity | detected | |
| RSc1362 | No activity | detected | |
| ST2570 | 7.0 ± 0.4 | 2.4 ± 0.7 | 49 ± 15 |
| Steady-state dechlorination
kinetics of various HADs | |||
| Enzyme | kcat (min−1) | K M (mM) | Kcat/KM (s−1M−1) |
|
| |||
| Bpro0530 | 320 ± 20 | 0.005 ± 0.001 | 1.1 × 106 ± 2.2 × 105 |
| Rha0230 | 40 ± 4 | 18 ± 2 | 37 ± 6 |
| Rha0230-Y61 | 61 ± 6 | 14 ± 2 | 73 ± 13 |
| PA0810 | 4 ± 2 | 0.6 ± 0.2 | 111 ± 67 |
| RSc1362 | 500 ± 100 | 0.06 ± 0.02 | 1.4 × 105 ± 5.4 × 104 |
| ST2570 | 15.4 ± 0.2 | 3.53 ×
10−3 ± 3 × 10−5 |
7.3 × 104 ± 1.1 × 103 |
All kinetic measurements were done at the enzymes’ pH optima. Bpro0530, Rha0230 and ST2570 were assayed in 100 mM Tris-SO4, pH 8.5, 30°C. PA0810 and RSc1362 were assayed in 50 and 25 mM CHES pH 9.5, 25°C, respectively.
Defluorination in HADs is significantly slower than dechlorination,[1] in line with the increased stability of the C-F bond. This contrasts with the fluoroacetate dehalogenases of the ABHSF, which are significantly better at defluorination and release fluorine faster than chlorine ions.[20–22] Potential explanations for this difference would regard the defluorination capability in HADs as either a novel enzymatic activity that is still undergoing evolutionary improvements or merely a fortuitous activity; their much higher KM towards fluoroacetate compared to chloroacetate is consistent with both of these possibilities.
We also suggest a catalytic role for Tyr61 in the halide pocket of Rha0230. The Tyr61Phe mutation enlarges the halide pocket at the cost of a polar contact from the hydroxyl group. It slows defluorination but enhances dechlorination. This suggests that Tyr61’s hydroxyl group only serves to assist defluorination, since it seems to interfere with dechlorination through steric hindrance.
Structural and dynamic adaptations for defluorination
Defluorination is a rare activity identified in only a few enzymes to date.[41] For the fluoroacetate dehalogenases of the ABHSF,[20–22] it was shown that the halide pocket plays an essential role in efficiently breaking the C-F bond.[20,22,42] First, the amino acids contributing to the pocket must sufficiently destabilize the carbon-fluorine bond before providing stabilization to the displaced fluoride ion. This is achieved through three residues (a histidine, a tryptophan and a tyrosine) that form dipole-dipole interactions or hydrogen bonds with the fluorine. Second, to ensure that these interactions are effective as well as to impose selectivity for fluoride, these residues must be placed so they tightly surround the small fluorine atom, allowing for stronger interactions. In a His→Asn fluoroacetate dehalogenase mutant whose halide pocket is enlarged by ~1.4 Å, defluorination activity is reduced 10-fold while dechlorination is increased 4-fold when compared to WT.[20] In a Tyr→Phe mutant whose halide pocket is also enlarged but has lost one halide-binding contact, defluorination is reduced 190-fold and dechlorination is reduced 3-fold. Since both mutants lost the WT’s original fluoride selectivity and the mutant with fewer halide interactions became less efficient, it was concluded that steric interactions control halide selectivity while electrostatics maintain the catalytic efficiency of the fluoroacetate dehalogenases.[42]
Since all HADs employ multiple side chains to activate the halogen atom (primarily arginines and asparagines, but also aromatic side chains), one could argue that they intrinsically possess the electrostatic interactions required for defluorination. Yet, the inability of most HADs to defluorinate suggests that certain, probably structural, criteria have not been met. The comparison of structurally known HADs reveals geometric arrangements in the active sites that correlate well with the presence or absence of defluorination activity (Figure 10 and Table S3). Although each subunit represents a static picture, their sum provides a reasonable sampling of the range of low-energy conformations accessible to these HADs. We found that pairwise distances between key catalytic residues can be used to identify defluorination activity. Despite the expected partial overlaps between these measurements, only the active sites of Bpro0530 and Rha0230, the two newly identified defluorinating enzymes, and the previously structurally characterized ST2570 can sample the shortest distances, which allows them to assume a conformation suitable for forming the stronger interactions with the smaller fluorine atom that seem necessary for defluorination to occur.
Adaptive changes include a smaller halide pocket which apparently requires Asn(Nδ2)--Arg(Nη1) and Asn(Nδ2)--Arg(Nη2) distances shorter than 6.8 Å (for definition of these distances see legends of Figures 7 and 10). This condition alone, however, is not sufficient to enable defluorination as neither DhlB nor RSc1362 can hydrolyze fluoroacetate despite satisfying this criterion. A second requirement appears to be concomitant placement of the halide pocket closer to the aspartate nucleophile, creating a more compact active site that can better stabilize the transition state of the SN2 cleavage of the C-F bond. The transition state of defluorination is expected to be more compact than its counterparts in the reactions involving larger halides because of the shorter scissile bond and the smaller atomic radius of the fluorine atom. It seems that Asp(Oδ1)--Asn(Nδ2) and Asp(Oδ1)--Arg(Nη2) distances below 6.2 Å and 5.7 Å, respectively, fulfill this requirement (Figure 10).
It is obvious that the differences in the distances we identified above as separating defluorinating from non-defluorinating HADs can be very small, separating the distinguishing values by only fractions of an Å (Figure 10 and Table S3). Such a narrow margin, however, should not surprise as there is only a 0.28 Å difference in the van der Waals radii of fluorine (1.47 Å) and chlorine (1.75 Å) atoms.
Keeping the closeness of the distinguishing features in mind and to overcome the disadvantage of crystallography, which deals with static structures, we added time-dependent dynamic aspects to the comparisons. We performed ten 300 ns MD simulations for each of the five enzymes discussed in more depth, RSc1362, PA0810, Rha0230, Bpro0530, and ST2570. As approximations of the size of the respective active sites, the probability and cumulative probability distributions of the Euclidian distance of the Cα-Cα backbone atoms of the same residues analyzed in the crystal structures, i.e. Asp10, Arg41, and Asn123 in Bpro0530 numbering, are shown in Figure 11. Distance distributions of individual simulation repeats are shown in Figure S3.
Figure 11.
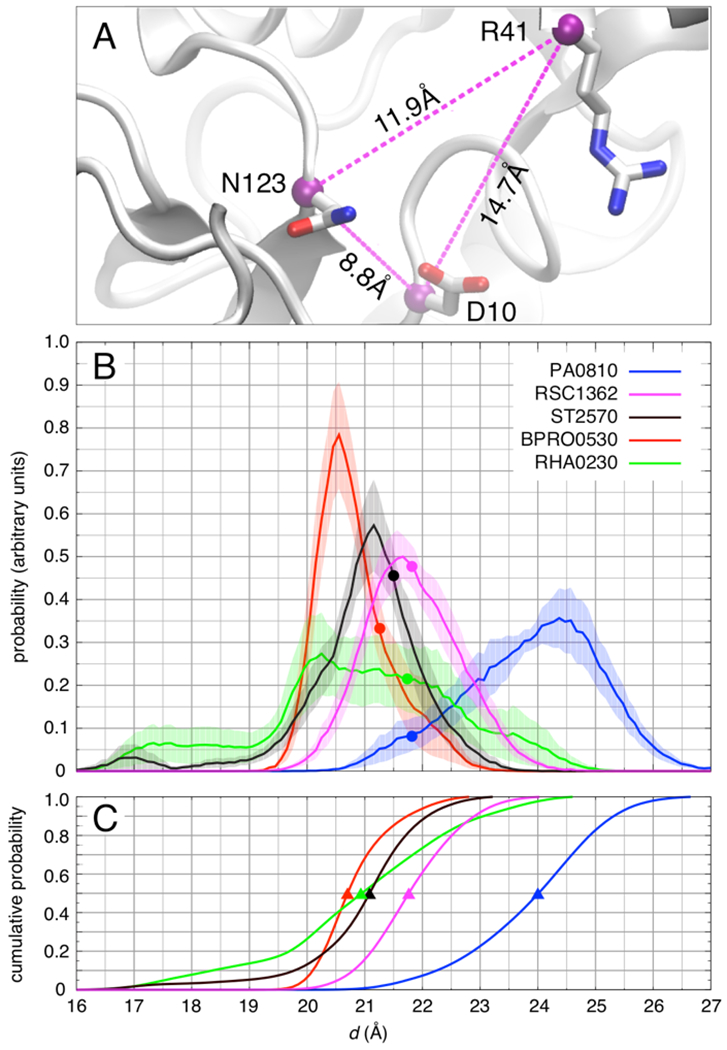
Structural fluctuations of the binding pocket in five dehalogenase enzymes obtained from MD simulations at 300 K. A) Molecular rendering from a simulation of RSc1362 with three selected Cα-Cα distances shown. The three pairwise distances, dDR, dRN, and dND are used to define the Euclidian distance d = (dDR2 + dRN2 + dND2)1/2 as a measure of the size of the binding pocket. B) Probability distribution of d. Shading represents the standard error of the mean estimated over ten independent 300-ns simulations of each enzyme. Filled circles represent the value of d in the crystallographic structure of each enzyme. C) Cumulative distribution of d. The triangles highlight the median (50% cumulative population) of d in each enzyme.
PA0810 and especially Rha0230 show a broader distribution of d values compared to the other dehalogenases. In the latter enzyme, this mostly occurs as a result of loss of secondary structure in the Arg58 containing helix in some but not all simulation repeats. It should be noted, however, that for PA0810 the probability distribution of the Euclidian distance nevertheless does not reach the lower values correlated with defluorination activity but extends to larger numbers, i.e. a less compact active site. In Rha0230, however, the Euclidian distance distribution peaks at 20 Å and even extends to significantly shorter values. According to sequence alignments (Figure 3), PA0810 and Rha0230 belong to a distinct subfamily. A looser dimerization mode (Figure 4) and increased Cα RMSD values against the remaining HADs (Table S2) support this interpretation leading us to speculate that the increased conformational mobility might be a property of all members of this protein subfamily.
As stated before, these distances differ only slightly in the crystallographic structures. The simulations, however, indicate that significant structural fluctuations can occur in all five enzymes at physiologically relevant temperatures in the absence of substrate. Furthermore, the size of the active site in the three defluorinating enzymes is more compact and the catalytic residues can come closer together than in the dechlorinating enzymes (Figure 11B). Accordingly, the median Euclidian distances of Bpro0530, Rha0230, and ST2570 are tightly clustered around 21 Å, whereas those of RSc1362 and PA0810 accumulate in their more expanded conformation (Figure 11C).
In an effort to test whether the criteria we identified had any predictive value, we searched the Protein Data Bank for further coordinate sets of HADs. The only other structurally known HAD is the enzyme ST2570. Its static active site distance measurements and the computations of both its probability and cumulative probability distributions of the Euclidian distance of the Cα-Cα backbone atoms conform to the previously established structural criteria for defluorinating HADs. Assays experimentally confirm the predicted catalytic activity, validating our structure-based assignments (Table 2).
From a structural point of view, the evolutionary steps necessary to enhance defluorination seem to involve optimizing the interaction geometries, which most likely requires trade-offs in dechlorination activity. Nevertheless, defluorination by Bpro0530, Rha0230, and ST2570 still is much slower than dechlorination. There could be, however, valid reasons for this discrepancy: fluoroacetate, which we used in our activity assays, probably does not represent the natural substrate of most if not all of these enzymes and, given the rather recent introduction of most fluorinated compounds into the environment, these enzymes might be rather new in evolutionary terms and therefore not yet fully optimized. Active enough to enable the source organisms’ survival in a contaminated environment, they have developed the capability to form tighter contacts to the halogen atom, a seemingly crucial property, but have not sufficiently refined other parameters critical for efficient catalysis.
Among the halogenating enzymes, which also use nucleophilic substitution to catalyze the reverse reaction (i.e. formation of C-X bonds), the S. cattleya enzyme is the only one known to be capable of fluorination.[33,34] Its halide pocket is distinct in that it is smaller than those of related non-fluorinating chlorinases.[35,36] It is tempting to speculate that halogen-manipulating enzymes have evolved around this common structural theme to successfully engage in fluorine biochemistry.
Conclusion
Here, we present the crystal structures of defluorinating enzymes Bpro0530 and Rha0230, determined at resolutions of 2.2 Å and 2.25 Å, respectively. Crystallization efforts for defluorinases Bpro4516 and Adeh3811 unfortunately were unsuccessful. We also describe the structures of two homologous dehalogenases, PA0810 (2.15 Å) and RSc1362 (2.20 Å), which hydrolyze chloroacetate but not fluoroacetate. Structural comparison of these enzymes reveals that their active sites display highly similar architectures and molecular dynamics simulations show considerable structural fluctuations. The defluorinating enzymes alone, however, have evolved the ability to sample more compact active site conformations. These features can provide more effective interactions to the substrate’s smaller fluorine halogen atom, which seem to be required for activation and cleavage of the C-F bond. We condense these structural constraints into a few parameters, based on X-ray crystallography and MD simulations, and demonstrate their usefulness through correctly predicting and then confirming the defluorination activity of the homologous enzyme ST2570. Illuminating the structure-function relationship of this rare enzymatic activity should provide valuable insights for the development of efficient biocatalytic processes to detoxify fluorinated organic compounds.
Experimental Section
DNA manipulation:
The dehalogenase genes were cloned into plasmid p15TV-L (GenBank Accession Number EF456736) as described.[1] The gene for enzyme ST2570 was optimized for expression in E. coli, synthesized, and cloned by Bio Basic Inc. (Markham, ON, Canada). Site-directed mutagenesis was performed using the QuikChange kit as directed by Stratagene (La Jolla, CA, USA).
Protein purification:
Protein purification was performed as described.[1] In brief, the protein was overexpressed in E. coli BL21(DE3) LB cultures overnight at 16-24 °C using 0.3-1 mM IPTG. For Se-Met labeled proteins, the cells were grown in M9 selenomethionine growth media instead, as recommended by Medicilon (Shanghai, PRC). The cells were harvested by centrifugation, resuspended and sonicated to produce the cell-free extract. After another centrifugation, the soluble HADs were purified from the supernatant by Ni-affinity chromatography, followed by cleavage of the His6-tag using TEV protease and a second round of Ni-affinity chromatography. The samples were further purified by size exclusion chromatography, concentrated, flash-frozen in liquid nitrogen and stored at −80 °C. The various buffers employed are presented in the relevant sections below. Protein concentrations were determined from their absorbance at 280 nm using extinction coefficients deduced by ProtPARAM.[43]
Enzymatic kinetics:
The steady-state kinetic parameters (kcat and KM) and apparent reaction enthalpy (ΔHapp) were determined by isothermal calorimetry using the VP-ITC microcalorimeter (MicroCal, LLC, Northampton, MA, USA) or the TAM III microcalorimeter (TA Instruments, New Castle, DE, USA) following published procedures;[1,44] buffers employed were 100 mM Tris-SO4, pH 8.5, at 30 °C for Bpro0530, Rha0230 and ST2570 as well as 25 or 50 mM CHES, pH 9.5 at 25°C for RSc1362 and PA0810, respectively. They were selected to allow each enzyme to be investigated at its pH-dependent peak activity.
In brief, the nature of each enzyme reaction (i.e. product inhibition and irreversibility) was first characterized by consecutive single-injection experiments, using saturating substrate concentrations and sufficient enzyme for complete turnover within 30 min. Since hydrolytic dehalogenation was practically irreversible, the kinetic parameters were directly extracted from the peak shape of the thermograms when product inhibition was absent. Otherwise, multiple-injection experiments were performed using minimal amounts of enzyme to ensure negligible product formation. Substrate was injected successively as 6×3 μl, 6×10 μl and 6×32 μl to 1.4 ml of reaction solution at 5 min intervals and the final concentrations accumulated from KM/10 to 10×KM whenever achievable. The kinetic parameters were obtained by non-linear regression fitting to the Michaelis-Menten equation in the software package GraphPad Prism (La Jolla, CA, USA) and averaged from triplicate runs.
X-ray crystallography:
Bpro0530 was crystallized in 1 M Li2SO4 and 0.1 M Tris-HCl, pH 8.5; Rha0230 in 0.6 M trisodium citrate and 0.1 M Na-HEPES, pH 7.5; PA0810 in 20% PEG3350 and 0.2 M diammonium citrate, and RSc1362 in 15% isopropanol, 0.1 M KCl, 25 mM MgCl2, 2% 1,4-dioxane and 50 mM sodium cacodylate, pH 6.0. The details on crystallization, data collection and structure determination for each protein are described in the Supporting Information. Structural biology software applications used in this project were compiled and configured by SBGrid.[45] All figures presenting protein structure were generated with the program Pymol.[46] The coordinates and structure factors with ID-codes 3UMC (PA0810), 3UMB (RSc1362), 3UM9 (Bpro0530) and 3UMG (Rha0230) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Molecular Dynamics Simulations:
Using the molecular simulation program GROMACS[47] all-atom molecular dynamics simulations were accomplished for five dehalogenases, Bpro0530, Rha0230, PA0810, RSc1362, and ST2570. A detailed description of the computational procedures is given in the Supporting Information. In short, a total of 3 μs of unrestrained simulations of each enzyme in explicit water at 300 K were performed (see Supporting Information for details). Structural fluctuations in the arrangement of residues making up the respective halide-binding pockets were analyzed. To characterize the size of the binding pocket, the Cα-Cα distances for the following pairs of residues were monitored: Asp10-Arg41, Arg41-Asn119, and Asn119-Asp10 (residue numbering according to Bpro0530; corresponding residues were monitored in the other enzymes) over of ten independent MD trajectories. These conserved residues were chosen because they are involved in halide binding and correspond to the ones for which relevant crystallographically-derived side-chain distances can be identified.
Supplementary Material
Acknowledgements
We thank Julia Barette, Aiping Dong, Yan Liu, Max Wong, Xiaohui Xu and Hong Zheng for assistance with experiments. We are also grateful to the staff at beamline X8C at the Brookhaven National Laboratory and at BioCARS and the Structural Biology Center - CAT, both at the Advanced Photon Source at Argonne National Laboratory, for their help during data collection. This work was supported by the Natural Sciences and Engineering Research Council of Canada through a graduate scholarship (PWYC) and operating grants to EAE (RGPIN-2015-06663) and EFP (RGPIN-2015-04877 and RGPIN-2020-068), by the Canada Research Chairs Program (EAE, EFP and RP), by a grant (MOP-130461) from the Canadian Institutes for Health Research (RP), by the Government of Canada through Genome Canada and the Ontario Genomics Institute (2009-OGI-ABC-1405; EAE, AS, AFY), and by the Protein Structure Initiative of the National Institutes of Health (Midwest Center for Structural Genomics, NIH grant GM074942; AS). Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. SBC-CAT is operated by the University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant No. RR007707.
Footnotes
Dedicated to the memory of an exceptional scientist, Prof. Francois Diederich.
Supporting information for this article is given via a link at the end of the document.
References
- [1].Chan WY, Wong M, Guthrie J, Savchenko AV, Yakunin AF, Pai EF, Edwards EA, Microb. Biotechnol 2010, 3, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kurihara T, Esaki N, N. (2008) Chem. Rec. (New York, N.Y) 2008, 8, 67–74. [DOI] [PubMed] [Google Scholar]
- [3].van der Ploeg J, van Hall G, Janssen DB J. Bacteriol 1991, 173, 7925–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Allen KN, Dunaway-Mariano D Curr. Op. Struct. Biol 2009, 19, 658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L, J. Mol. Biol 2006, 361, 1003–1034. [DOI] [PubMed] [Google Scholar]
- [6].Kuznetsova E, Proudfoot M, Gonzalez CF, Brown G, Omelchenko MV, Borozan I, Carmel L, Wolf YI, Mori H, Savchenko AV, Arrowsmith CH, Koonin EV, Edwards AM, Yakunin AF, J. Biol. Chem 2006, 281, 36149–36161. [DOI] [PubMed] [Google Scholar]
- [7].Kurihara T, Esaki N, Soda K, J. Mol. Catal. B: Enzymatic 2000, 10, 57–65. [Google Scholar]
- [8].Swanson PE, Curr. Opin. Biotechnol 1999, 10, 365–369. [DOI] [PubMed] [Google Scholar]
- [9].Lemal DM, J. Org. Chem 2004, 69, 1–11. [DOI] [PubMed] [Google Scholar]
- [10].Blanksby SJ, Ellison GB, Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- [11].Kurihara T, Liu JQ, Nardi-Dei V, Koshikawa H, Esaki N, Soda K, J. Biochem 1995, 117, 1317–1322. [DOI] [PubMed] [Google Scholar]
- [12].Li YF, Hata Y, Fujii T, Kurihara T, Esaki N, J. Biochem 1998, 124, 20–22. [DOI] [PubMed] [Google Scholar]
- [13].Li YF, Hata Y, Fujii T, Hisano T, Nishihara M, Kurihara T, Esaki N, J. Biol. Chem 1998, 273, 15035–15044. [DOI] [PubMed] [Google Scholar]
- [14].Ridder IS, Rozeboom HJ, Kalk KH, Janssen DB, Dijkstra BW, J. Biol. Chem 1997, 272, 33015–33022. [DOI] [PubMed] [Google Scholar]
- [15].Ridder IS, Rozeboom HJ, Kalk KH, Dijkstra BW, J Biol Chem 1999, 274, 30672–30678. [DOI] [PubMed] [Google Scholar]
- [16].Pang BC, Tsang JS, FEMS Microbiol. Lett 2001, 204, 135–140. [DOI] [PubMed] [Google Scholar]
- [17].Schmidberger JW, Wilce JA, Tsang JS, Wilce MC, J. Mol. Biol 2007, 368, 706–717. [DOI] [PubMed] [Google Scholar]
- [18].Verschueren KH, Seljee F, Rozeboom HJ, Kalk KH, Dijkstra BW, Nature 1993, 363, 693–698. [DOI] [PubMed] [Google Scholar]
- [19].Prokop Z, Monincova M, Chaloupkova R, Klvana M, Nagata Y, Janssen DB, Damborsky J, J. Biol. Chem 203, 278, 45094–45100. [DOI] [PubMed] [Google Scholar]
- [20].Chan PWY, Yakunin AF, Edwards EA, Pai EF, J. Am. Chem. Soc 2011, 133, 7461–7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu JQ, Kurihara T, Ichiyama S, Miyagi M, Tsunasawa S, Kawasaki H, Soda K, Esaki N, J. Biol. Chem 1998, 273, 1998, 30897–30902. [DOI] [PubMed] [Google Scholar]
- [22].Jitsumori K, Omi R, Kurihara T, Kurata A, Mihara H, Miyahara I, Hirotsu K, Esaki N, J. Bacteriol 2009, 191, 2630–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Holmquist M, M. Curr. Prot. Pept. Sci 2000, 1, 209–235. [DOI] [PubMed] [Google Scholar]
- [24].Kirsch P, in P Modern Fluoroorganic Chemistry – Synthesis, Reactivity, Applications. WILEY-VCH, Weinheim. 2004. [Google Scholar]
- [25].Key BD, Howell RD, Criddle CS, Env. Sci. Technol 1997, 31, 2445–2454. [Google Scholar]
- [26].Houde M, Martin JW, Letcher RJ, Solomon KR, Muir DC, Env. Sci Technol 2006, 40, 3463–3473. [DOI] [PubMed] [Google Scholar]
- [27].Ritter SK, Chem SK. Eng. News 2010, 88, 12–17. [Google Scholar]
- [28].Rye CA, Isupov MN, Lebedev AA, Littlechild JA, Extremophiles 2009, 13, 179–190. [DOI] [PubMed] [Google Scholar]
- [29].Kim TH, Mehrabi P , Ren Z, Sljoka A, Ing C, Bezginov A, Ye L, Pomès R, Prosser RS, and Pai EF, Science 217, 355, eaag2355. [DOI] [PubMed] [Google Scholar]
- [30].Mehrabi P, Di Pietrantonio C, Kim TH, Sljoka A, Taverner K, Ing C, Kruglyak N, Pomès R, Pai EF, and Prosser RS, J. Amer. Chem. Soc 2019, 141, 11540–11556. [DOI] [PubMed] [Google Scholar]
- [31].Mehrabi P, Schulz EC, Dsouza R, Müller-Werkmeister HM, Tellkamp F, Miller RJD, and Pai EF, Science 2019, 365, 1167–1170. [DOI] [PubMed] [Google Scholar]
- [32].Yue Y, Fan J, Xin G, Huang Q, Wang J-b., Li Y, Zhang Q, and Wang W, (2021) Environ. Sci. Technol 2021, 55, 9817–9825. [DOI] [PubMed] [Google Scholar]
- [33].Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH, H J Nature 2004, 427, 561–565. [DOI] [PubMed] [Google Scholar]
- [34].O’Hagan D, Schmidberger JW, Nat JW. Prod. Rep 2010, 27, 900–918. [DOI] [PubMed] [Google Scholar]
- [35].Eustaquio AS, Pojer F, Noel JP, Moore BS, Nat. Chem. Biol 2008, 4, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Blasiak LC, Drennan CL, Acc. Chem. Res 2009, 42, 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hisano T, Hata Y, Fujii T, Liu J-Q, Kurihara T, Esaki N, Soda K, J. Biol. Chem 1996, 271, 20322–20330. [DOI] [PubMed] [Google Scholar]
- [38].Arai R, Kukimoto-Niino M, Kuroishi C, Bessho Y, Shirouzu M, Yokoyama S, Protein Sci. 2006, 15, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Koonin EV, Tatusov RL, J. Mol. Biol 1994, 244, 125–132. [DOI] [PubMed] [Google Scholar]
- [40].Nakamura T, Yamaguchi A, Kondo H, Watanabe H, Kurihara T, Esaki N, Hirono S, Tanaka S, J. Comput. Chem 2009, 30, 2625–2634. [DOI] [PubMed] [Google Scholar]
- [41].Natarajan R, Azerad R, Badet B, Copin E, J. Fluor. Chem 2005, 126, 424–435. [Google Scholar]
- [42].Kamachi T, Nakayama T, Shitamichi O, Jitsumori K, Kurihara T, Esaki N, Yoshizawa K, Chemistry 2009, 15, 7394–7403. [DOI] [PubMed] [Google Scholar]
- [43].Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF, Methods Mol. Biol. (Clifton, N.J) 1999, 112, 531–552. [DOI] [PubMed] [Google Scholar]
- [44].Tran C in Characterization of hydrolytic dehalogenases: Substrate specificity and carbon isotope fractionation. 2013, M.Sc. thesis, University of Toronto [Google Scholar]
- [45].Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P, Elife, 2013, 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schrödinger LLC. The PyMOL Molecular Graphics System. Version 1.6.0.0 2010. [Google Scholar]
- [47].Hess B, Kutzner C, van der Spoel D, E. Lindhal E, J. Chem. Theory Comput 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- [48].Bond CS, Bioinformatics (Oxford) 2003, 19, 311–312. [DOI] [PubMed] [Google Scholar]
- [49].Thompson JD, Higgins DG, Gibson TJ, Nucl. Acids Res 1994, 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kabsch W, Sander C, Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.