

Abstract
This Lilliput explores the current epidemiological and virological arguments for a zoonotic origin of the COVID‐19 pandemic. While the role of bats, pangolins and racoon dogs as viral reservoirs has not yet been proven, a spill‐over of a coronavirus infection from animals into humans at the Huanan food market in Wuhan has a much greater plausibility than alternative hypotheses such as a laboratory virus escape, deliberate genetic engineering or introduction by cold chain food products. This Lilliput highlights the dynamic nature of the animal‐human interface for viral cross‐infections from humans into feral white tail deer or farmed minks (reverse zoonosis). Surveillance of viral infections at the animal‐human interface is an urgent task since live animal markets are not the only risks for future viral spill‐overs. Climate change will induce animal migration which leads to viral exchanges between animal species that have not met in the past. Environmental change and deforestation will also increase contact between animals and humans. Developing an early warning system for emerging viral infections becomes thus a societal necessity not only for human but also for animal and environmental health (One Health concept). Microbiologists have developed tools ranging from virome analysis in key suspects such as viral reservoirs (bats, wild game animals, bushmeat) and in humans exposed to wild animals, to wastewater analysis to detect known and unknown viruses circulating in the human population and sentinel studies in animal‐exposed patients with fever. Criteria need to be developed to assess the virulence and transmissibility of zoonotic viruses. An early virus warning system is costly and will need political lobbying. The accelerating number of viral infections with pandemic potential over the last decades should provide the public pressure to extend pandemic preparedness for the inclusion of early viral alert systems.
INTRODUCTION
“After the match is before the match” is a quote from a soccer trainer but it also applies to pandemics. One might doubt that we are already after the COVID‐19 pandemic since infections with SARS‐CoV‐2 are still ongoing and a new variant of concern (VOC) could still evolve that undermines the immunity induced in the population by natural infection and vaccination and thus restart a new epidemic wave. Historical evidence from the Spanish flu pandemic tells us that we should count with further, albeit smaller, waves of infection in the aftermath of the COVID‐19 pandemic (Brüssow, 2022). The Russian flu pandemic from 1889 which might have been a prior coronavirus pandemic (Brüssow & Brüssow, 2021) possibly caused a major resurge still 10 years after its onset (Brüssow, 2021). While the future of SARS‐CoV‐2 is still a matter of conjecture, it is likely that we are before and maybe not so far away from the next pandemic. Our generation already witnessed two major pandemics (AIDS and COVID‐19) and a number of epidemics with threatening pandemic potential by SARS, Middel East respiratory syndrome (MERS), Nipah, avian and swine flu, Zika, monkeypox and Ebola virus infections, just to quote the viral threats. Pandemic preparedness is therefore as important as ever. Experience has shown that even developed societies were rather unprepared when confronted by the beginning of the COVID‐19 pandemic. As The Economist, a British weekly, recently stated with respect to COVID‐19 containment strategies: “All governments make mistakes. What matters is whether they learn from them.” The scientific community has a particular societal duty to assist governments with their reflections on the lessons learnt so that politicians can take the right decisions in the future and act fast in case of new needs.
Pandemic preparedness has many facets. In this context, microbiologists can make an important contribution by developing an early warning system. It is a truism that most novel human viruses come from animals. Getting a better knowledge of pathogen characteristics favouring zoonosis (infections jumping from animals to humans) is thus of fundamental importance. Zoonosis can occur with any animal microbial pathogen, but in view of recent pandemics, concentrating on viruses might be a reasonable starting point.
RESEARCH ON THE ORIGIN OF SARS‐COV‐2
The wet market hypothesis
While the identity of the viral reservoir and intermediate animal host for SARS‐CoV‐2 has not yet been identified, important learning lessons can be gleaned from the COVID‐19 pandemic. A consortium of US, UK and Canadian researchers analysed the earliest 174 COVID‐19 cases which occurred in Wuhan in December 2019. There was a clear clustering of cases around the Huanan seafood wholesale market. The cases comprised people with direct links to the market and cases without direct links to the market lived at least close to the market. At the market seafood, poultry and other commodities were sold. More importantly, until at least November 2019 live wild‐caught or farmed mammals were on sale comprising foxes, badgers and racoon dogs which could thus serve as possible intermediate hosts for a SARS‐CoV‐2 spill‐over into the human population. The researchers evaluated social media check‐ins which showed that the Huanan market received much less visits than other markets in Wuhan. It is therefore not the sheer size of the visitors but something else which pushed the Huanan market into the prime suspect list for serving as origin of the pandemic. The researchers reconstructed a floor plan for the Huanan market with respect to the 585 environmental samples tested in early January 2020. Distance to the nearest vendor selling live mammals was predictive of the sample being positive for SARS‐CoV‐2. The southwest corner of the market was a positivity hotspot. In this area, the presence of cages with live racoon dogs on top of cages with live birds was photographically documented. The earliest COVID‐19 cases in food vendors all came from the western side of the Huanan market (Worobey et al., 2022). The Hubei province (for which Wuhan is the capital city) is home to wildlife farms with hundreds of thousands of racoon dogs and—notably—also of cave complexes inhabited by Rhinolophus bats. The Rhinolophus “horseshoe” bat genus carries the highest number of coronaviruses in China. A survey in Henan, the neighbour province to Hubei, revealed that about 6% of Rhinolophus bats harboured coronaviruses; some of which were—with the exception of the spike gene and orf 8—close relatives to SARS‐CoV‐1 virus, which caused the 2002–2003 SARS pandemic. The high contact rates between Rhinolophus bat species caused frequent host species switches and recombination between bat coronaviruses (Lin et al., 2017).
Genome analysis of early SARS‐CoV‐2 isolates provided additional information about the introduction of the virus into the human population. The earliest unambiguous COVID‐19 case in Wuhan is a seafood vendor at the Huanan market. He showed symptom onset on 10 December 2019 and was hospitalized on 16 December. An environmental sample taken from his shop showed a lineage B SARS‐CoV‐2 virus. A second viral lineage A SARS‐CoV‐2 was documented in a family cluster that showed symptom onset on 15 December 2019 and suffered hospitalization on 25 December. These people had not visited the Huanan market but lived close to the market. Lineage A and B SARS‐CoV‐2 differed by only two nucleotide substitutions. Bioinformatic analysis suggested that the pandemic most likely began with at least two separate zoonotic transmissions starting in November 2019. An earlier cryptic circulation of SARS‐CoV‐2 in the human population is excluded by the timing of phylogenetic trees constructed with the viral genome sequences (Pekar et al., 2022).
Between 2017 and November 2019 Chinese scientists in collaboration with colleagues from UK and Canada documented the sale of animals at Wuhan's wet markets (including the Huanan market). Vendors from 17 shops reported the sale of 36,000 animals from 38 species, including 31 protected species, but they recalled no selling of pangolins or bats. Most animals were sold alive as meat or as pet animals. Animals for meat were butchered in place. A third of the inspected animals showed wounds indicative of illegal wild hunting. With respect to mammals the most sold species were Amur hedgehogs and hares, followed by bamboo rats and racoon dogs (Xiao et al., 2021). For the farmed animals no certificate of origin could be presented making tracing efforts difficult since the Wuhan markets are served by a wide network of animal farms (Jiang & Wang, 2022).
Scientists had expressed their disappointment about the delayed—and some even suspected censored‐ release of virological data on animals traded at the Huanan market (Maxmen, 2022). With a delay of 3 years, such data are now published (Liu et al., 2023). From 1 January 2020, after the closure of the market, 923 samples were collected from the environment. Using Reverse Transcriptase‐quantitative PCR (RT‐qPCR) technique, SARS‐CoV‐2 was detected in 73 environmental samples, most were from the Western part of the market where live animals were sold. All four sewage wells of the market were positively tested. The detection of SARS‐CoV‐2 in multiple shops selling different product types suggested that SARS‐CoV‐2 may have been circulating in the market, especially the western zone already for a moment in December 2019. Half of the positive environmental sites were still positive when again sampled in February 2020, indicating a high initial viral load and persistence of the virus. Seven viral genomes could be reconstituted from the environmental samples, three genomes were absolutely identical with the Wuhan reference strain of SARS‐CoV‐2. On 18 January 2020, 457 samples were also collected from 18 species of animals, comprising unsold contents of refrigerators and freezers, swabs from stray animals and the contents of a fish tank. None tested positive for SARS‐CoV‐2. The Chinese researchers also conducted RNA‐seq analysis from 60 SARS‐CoV‐2 positive and 120 virus‐negative environmental samples. RNA sequences from humans, sheep, cattle, dogs, pigs and cats, but not from wildlife species were detected. Single spots of the market yielded racoon dog or hedgehog RNA and traces of bat RNA sequences. French researchers working with the Chinese data singled out the raccoon dog because its mtDNA was more abundant than that of other species in the SARS‐CoV‐2–laden samples (Cohen, 2023). In contrast, the Chinese researchers concluded that their data do not support racoon dogs as origin for the spread of SARS‐CoV‐2 and they did not rule out a human‐to‐animal transmission at the market or introduction by cold chain products. The late reporting of the animal data by the Chinese scientists and a preprint of French researchers conducting database mining on the Chinese data stirred a controversy in the scientific community (Mallapaty, 2023a, 2023b). However, this debate has created more heat than light for the origin of the COVID‐19 pandemic. Overall, a zoonosis at the Huanan food market remains the prime suspect for the origin of the COVID‐19 pandemic, but the available data clearly do not allow firm conclusions with respect to the animal species involved.
Zoonotic origin
Major knowledge gaps still exist concerning the zoonotic origin of SARS‐CoV‐2. We do not know the intermediate host transmitting the pandemic virus into the human population because all animals for sale at the Huanan market were culled without virological sampling when the market was sanitized on 1 January 2020. Nor do we know the original animal reservoir of SARS‐CoV‐2. Bats are commonly suspected as reservoirs for SARS‐CoV‐2, but so far the closest bat coronavirus with respect to genome sequence identity to SARS‐CoV‐2 were isolated in Laos (Temmam et al., 2022). If these viruses represent the source, SARS‐CoV‐2 must have diverged from them several decades ago (Boni et al., 2020). Either the true bat coronavirus has not yet been found or the precursor of SARS‐CoV‐2 might have circulated for several decades in animals other than bats.
A zoonotic origin of SARS‐CoV‐2 is likely because the known facts about its origin concur closely with epidemiological studies linking the SARS pandemic from 2003 caused by SARS‐CoV‐1 with cross‐infections from wild animals. The SARS epidemic started in 2002 in Guangdong province in Southern China before it experienced an international spread starting from Hong Kong in 2003. Early cases in 2002 were reported in restaurant workers handling exotic wildlife food. At that time Chinese researchers screened mammals sold for culinary purposes in a wildlife market in Shenzhen, Guangdong province. Indeed, four of six palm civets yielded a coronavirus that differed from the human SARS‐CoV‐1 by only 57 nucleotides (nt) (mostly in the S gene) and a 29‐nt deletion. A nearly identical coronavirus was also isolated from a racoon dog sold at this market. Animals that yielded this coronavirus also showed neutralizing antibodies to the virus, indicating a prior infection, but no clinical symptoms. Twenty per cent of the wild‐animal traders from this market but no local control subjects showed antibody to SARS‐CoV‐1. However, the food traders did not remember a respiratory disease. The researchers suspected that palm civets or racoon dogs served as intermediate hosts but did not represent the virus reservoir for SARS‐CoV‐1 (Guan et al., 2003). Coronaviruses that are relatively closely related to SARS‐CoV‐1 were isolated from Rhinolophus bats in China (Lin et al., 2017).
Laboratory escape?
An alternative hypothesis for the origin of SARS‐CoV‐2 is viral escape from a laboratory working with mammalian coronaviruses. While this is a possible scenario particularly since the Wuhan Institute of Virology (WIV) has been working with bat coronaviruses for many years, the epidemiological data do not support this hypothesis. Following the laboratory escape hypothesis, one would expect the first cases in a collaborator of the WIV or his or her family members or neighbours, which was not the case (Holmes et al., 2021). Serological evidence also points to the Huanan market and not WIV as likely point source for the start of the pandemic. A seroprevalence study conducted in April 2020 with 9500 subjects living in different districts of Wuhan showed the highest rate of seropositivity for SARS‐CoV‐2 antibodies in the districts of Qiaokuo (13% positive) and Jiang'an (11% positive) which flank the district where the Huanan market is located and not in the district Jiangxia (5% positive) where the Wuhan Institute of Virology (WIV) is situated (He et al., 2021). Also this data set is not in accordance with a laboratory virus escape hypothesis.
Molecular data exclude a longer period of silent viral transmission of SARS‐CoV‐2 with asymptomatic infections in the human population which could blur the location of the viral origin (Holmes et al., 2021). Laboratory infections have been documented in scientists working with live animals. Perhaps the most famous laboratory infections occurred in 1967 in Marburg, Frankfurt and Belgrade. At the time, all three virology institutes handled live monkeys imported from Uganda. Overall 24 lab workers were hospitalized and 7 died from the infection with Marburg virus, a member of the Filovirus group to which also Ebola virus belongs. Human‐to‐human transmission was not observed in this incident. There is also circumstantial evidence that the Russian flu epidemic from 1977 might have been initiated by an incompletely inactivated Influenza virus used in challenge trials (Brüssow, 2022). However, this incident does not provide any parallels to the origin of the COVID‐19 pandemic. Due to the increasing political antagonism between China and the US, the Chinese government has not allowed independent investigations by WHO experts at WIV. Due to political tensions, it seems unlikely that the current committee of the US House of Representatives will provide new scientific insights into the laboratory escape issue (Lenharo & Wolf, 2023).
Genetic engineering?
Finally, there are some arguments even in scientific journals suggesting that SARS‐CoV‐2 might have been deliberately engineered. The argument centres around an unusual fit of the viral spike protein to the human ACE2 receptor and on the polybasic furin cleavage site which in avian influenza viruses increases transmissibility and pathogenicity (Harrison & Sachs, 2022). The hypothesis of a deliberate construction has been repetitively rejected based on arguments that such traits are also found in some animal coronaviruses. In fact, coronaviruses from bats isolated in Laos share a closely related receptor recognizing domain (RBD) on the spike protein with SARS‐CoV‐2 (Temmam et al., 2022) and coronaviruses detected in trafficked pangolins caught by the Chinese customs showed an even closer match to RBD of SARS‐CoV‐2 (Lam et al., 2020; Xiao et al., 2020). In addition, a MERS‐like pangolin coronavirus showed a polybasic furin cleavage site in the spike protein enhancing the infection of human cells by magnitudes (Neil, 2023). The discrepancies in phylogenetic tree analyses when comparing different genes from bat and pangolin coronaviruses with SARS‐CoV‐2 are best explained by multiple genetic recombinations occurring naturally (Lam et al., 2020; Xiao et al., 2020). There is therefore no logical need for a genetic engineering hypothesis when Nature has shown with coronaviruses that she is the most formidable genetic engineer herself, as also demonstrated by the later appearance of variant viruses of concern, particularly the substantially mutated Omicron variant. Other researchers have pointed out that a plausible genetic backbone for construction of an engineered coronavirus yielding SARS‐CoV‐2 is entirely unknown, making a human construct a very unlikely hypothesis (Andersen et al., 2020; Garry, 2022; Holmes et al., 2021).
Biosafety issues
There is currently an ongoing discussion regarding the level of biosafety and regulatory surveillance needed when virologists transfer for example genes from the Omicron variant into earlier SARS‐CoV‐2 isolates to investigate the genetic basis for the pathogenicity attenuation of the Omicron variant. This research creates recombinant strains that potentially combine increased transmission properties of Omicron with the higher pathogenicity from earlier isolates. The US National Institute of Allergy and Infectious Diseases (NIAID) will approve such experiments under appropriate biosafety conditions for knowledge building on the genetic basis of viral pathogenicity, but wants to be immediately informed should unusual virulent viral strains be observed (Callaway & Kozlov, 2022).
Pangolins as transmitter of coronaviruses?
The argument with pangolins as intermediate hosts relies on the following observations. The pangolin is one the most trafficked animals, it is caught for consumption of meat which is appreciated as culinary delicacy and for its scales which are used in traditional medicine in China. The animal is threatened with extinction but illicit trade is booming. A batch of 25 smuggled pangolins was investigated in the summer 2019; 17 tested positive for coronavirus and developed signs of a respiratory disease with shortness of breath and 14 of them died within 2 months. One animal had developed an antiviral antibody response suggestive of a natural infection. Pathology showed alveolar damage, infiltration and bleeding in bronchi. Upon incubation of the coronavirus in Vero cell culture, cytopathic effects developed. Viral genome sequencing showed a coronavirus nearly identical between the pangolins and closely related to SARS‐CoV‐2, showing across the genome amino acid (aa) identity that varied from 91% to 100% with SARS‐CoV‐2. Notably, the RBD of the spike protein varied by only one aa from SARS‐CoV‐2 (Xiao et al., 2020). Another group of Chinese scientists detected coronavirus in lung and gut tissue and blood from pangolins obtained from an anti‐smuggle operation. The genomes of six sequenced pangolin viruses shared >99.8% identity and between 86% to 100% aa sequence identity with SARS‐CoV‐2 depending on the genome region analysed (Lam et al., 2020). Could pangolins thus represent a reservoir host for SARS‐CoV‐2? This is unlikely since the currently known pangolin viruses differ too much from SARS‐CoV‐2 to serve as a direct precursor, e.g., the Guangdong pangolin coronavirus differs by 1200 nucleotides (nt) from SARS‐CoV‐2 over the 21,300 nt‐long ORF1ab. Additionally, pangolins are nocturnal and solitary animals and their overall population size is small‐ these are all factors which do not favour the maintenance of viral infection chains. Their ecological interface with bats, the suspected virus reservoir, is small. It is therefore likely that the investigated pangolins got infected during mixed animal transport during smuggling. Pangolins developed under these conditions an acute lethal infection which is not compatible with a species serving as a virus reservoir. It is not known whether smugglers developed symptoms.
Bats as virus reservoirs for zoonosis?
Bats are a common suspect for viral zoonosis which is explained by their species richness (they represent 1400 species 20% of all mammalian species), mobility (the only flying mammal), longevity and a social lifestyle in dense colonies which favours viral infection. Ironically, except for species richness, humans share similar characteristics with bats. Notably, bats came to grips with massive virus exposure such that experimental infections with otherwise highly lethal viruses do not cause disease in bats. Some biologists think that the difference with humans is that bats evolved their lifestyle 60 million years ago leaving enough time to evolve tolerance to viral infection. Indeed, a bat genome sequencing project detected positive selection on several immunity‐related genes which include interleukins involved in immune regulation, activation of transcription factor NF‐κB and proteins involved in responses to pathogens as well as gene losses that potentiate cellular responses to multiple cytokines. In contrast, an expansion of cytidine deaminases displaying anti‐viral functions was observed (Jebb et al., 2020).
Bats have a robust interferon response to RNA viruses, constitutively expressing IFN‐alpha, but counteract the consequent inflammation by a dampened activation of the inflammasome. In addition, bats downregulate tumour necrosis factor‐alpha expression to suppress inflammation. Bats also exhibit dampened DNA sensing. The entire PYHIN gene family crucial for DNA sensing which binds microbial DNA and form caspase‐1‐activating inflammasomes or drive type I IFN gene transcription was found to be missing in bats. These immunological observations seem counterintuitive since they dampen the immune response rather than activating it. Apparently, controlling inflammation is more important than ramping up the immune system to combat the virus which is in line with pathogenesis models for severe COVID‐19 (Gorbunova et al., 2020).
Bats so far harbour the closest relatives of SARS‐CoV‐2 and the lack of pathology of coronavirus infections in bats are also a strong argument in favour of bat as infection source. Since coronaviruses show both genetic recombination and broad host ranges, SARS‐CoV‐2 could be the result of a genetic recombination event between coronaviruses circulating in the same or different species, which we might never identify.
REVERSE ZOONOSIS
The search for the animal origin of SARS‐CoV‐2 is complicated by reverse zoonosis, the phenomenon where humans pass the infection back to animals. Reverse zoonosis has important implication for animal species conservation and it will prevent eradication of SARS‐CoV‐2 by vaccination of the human population. Vaccination in many wild animal species is not feasible which means that we will have to live with SARS‐CoV‐2 in the future. The following paragraphs review recent data on coronaviral infections on the other side of the animal‐human interface.
White‐tailed deer
The situation is particularly intriguing in free‐ranging white‐tailed deer (WTD). When testing 600 deer serum samples from the northern US, wildlife scientists observed no neutralizing antibodies to SARS‐CoV‐2 in the ten years preceding the pandemic. This was followed by a rise to 3% seropositivity in 2020 and then a spectacular increase to 40% seropositivity in 2021. The animals displayed high virus neutralization titers. Seroprevalence differed highly between states (Illinois 7%, Michigan 67%) (Chandler et al., 2021). A subsequent study conducted in early 2021 by veterinarians in Ohio confirmed this observation with virus detection in nasal swabs of 36% WTD by RT‐PCR. Again, virus prevalence varied substantially between different geographical sites ranging from 14% to 70% and was higher in peri‐urban than in rural areas, suggesting human contact as risk factor for infection. Six independent human‐to‐WTD transmission events were identified by matching viral genome sequences, three were represented by a viral clade which was also detected in 50% of the human samples during the preceding winter peak in Ohio. An infectious virus was also recovered from WTD, suggesting the potential for deer‐to‐deer transmission. The WTD viruses showed uncommon aa substitutions not found in the human isolates indicating a potential adaptation of the virus to transmission between animals (Hale et al., 2022). In another study lymph nodes from 5000 WTD hunted in New York State were tested for SARS‐CoV‐2 by RT‐PCR. The prevalence of positive samples increased from 0.6% to 21% between the 2020 and 2021 hunting seasons suggesting widespread virus circulation in the deer population. Various variants of concern (Alpha, Gamma, Delta) were observed in WTD indicating that different variants of concern (VOCs) were transmitted to WTD and continued to co‐circulate. Transmission of Delta to WTD was observed during the peak of Delta circulation in humans. Notably, Alpha variants were still circulating in the WTD population when Alpha was not any longer seen circulating in humans; WTD might thus serve as a reservoir of virus lineages which became extinct in the human population. The mutational analysis provided some evidence for host adaptation (Caserta et al., 2023). In 2021 2% of nasal swabs and 6% of lymph nodes of 300 WTD from Ontario/Canada tested positive for SARS‐CoV‐2 by RT‐PCR. Sequencing revealed a highly divergent set of viral genomes with relatedness to human viral sequences from Michigan and from mink viruses. Analysis of the mutational signature suggested sustained viral transmission with minimal immune pressure in a susceptible animal population. All virus isolates from WTD were neutralized by sera from vaccinated or convalescent human subjects indicating that the accumulated mutations in deer have no impact on the spike protein antigenicity. One case of virus transmission from WTD to a human could be proven in a person who had a close contact with deer in the week before symptom onset in the person (Pickering et al., 2022).
Pet hamsters
Likely cases of “ping‐pong” infections where humans infected animals which then infected back humans have been documented for pet animals. A pet shop owner and his clients buying a hamster developed COVID‐19, and subsequently a cluster of people in contact with the buyers got COVID‐19. A direct epidemiological link to the pet animals was suspected. Indeed 50% of Syrian hamsters sold in a pet shop from Hong Kong and 58% of Syrian hamsters in a Hong Kong pet warehouse tested positive for SARS‐CoV‐2, while no infection was detected in dwarf hamsters or other pet animal species. The animals showed no disease symptoms. The infected hamsters were imported from The Netherlands. Notably the human cases from this cluster harboured a Delta variant which was at that time not circulating in Hong Kong but in The Netherlands. The sequences from the hamster and human viruses were closely related, but not identical differing by 1 to 13 nucleotides. Since several of the infected human subjects had no contact with the hamsters, the epidemiologists suspected human‐to‐human forward transmission of the virus transmitted by the hamsters (Chan et al., 2022; Yen et al., 2022).
Mink farms
Probable “ping‐pong” infections were also described in farmed fur animals from The Netherlands. In Spring 2020 16 farms reported outbreaks among minks. Epidemiological investigations revealed that in two farms SARS‐CoV‐2 infections occurred in farm workers shortly before respiratory symptoms were observed in minks. The sequence of virus from the farm workers and the minks differed by just 7 nucleotides but were quite distinct between the two farms. On another farm, workers became infected with highly related viruses after infections had occurred in minks. Contact tracing showed that the infection was transmitted to close contacts, but no forward infection into the community was observed. The viral genomes from infected farm workers differed clearly from the viral sequences circulating in unlinked human subjects from the same geographical area. Virus sequences from farm workers were nearly identical to mink virus sequences from the same farm while the sequences from different farms differed, suggesting frequent and independent cross‐species infection events. In some farms, a higher diversity in virus genome sequences was detected suggesting that the virus circulated in minks for some time before symptomatic infections were observed in animals (Oude Munnink et al., 2021). Infections on mink farms continued until November 2020 and ended when culling and strict hygiene measures on the farms were imposed. Overall, half of 120 mink farms were affected by infection. Back‐and‐forth infection between minks and farm workers occurred multiple times. Contacts between workers linked infection clusters from different farms. No infection spill‐over into the general population was seen while some infected cats and dogs and free‐ranging mustelids were identified around mink farms. Epidemiologically they did not spread the infection (Lu et al., 2021). Both WTD and minks can serve as secondary reservoirs of SARS‐CoV‐2 making eradication of the virus difficult, if not impossible. For farmed animals such as mink the European Food Safety Agency (EFSA) recommends regular testing of farm workers for SARS‐CoV‐2 by antigen test to suppress the introduction of SARS‐CoV‐2 into farms (EFSA Panel, 2023).
Wide animal host range
SARS‐CoV‐2 has a wide host range among mammals. Experimental infections have been achieved in hamsters, racoons, skunks, deer, racoon dogs, fruit bats, rabbits, shrews and non‐human primate species. Natural infections with SARS‐CoV‐2 have been described in cats, big zoo cats, gorillas, ferrets and minks. ACE2 receptor gene analysis indicated that also cetaceans could be susceptible to SARS‐CoV‐2 infection. With such a wide host range and the high environmental load of the virus during the COVID‐19 pandemic in the human population, SARS‐CoV‐2 could now be widespread in nature.
Genetic analysis showed that SARS‐CoV‐2 is a generalist virus with the potential to infect a wide range of mammalian hosts due to the conservation of the ACE2 receptor among many animal species. Pre‐adaptation of human SARS‐CoV‐2 for infection of many animal hosts is minimal and even in animal species where widespread transmission was observed (WTD, mink) only a few (one in deer, 5 in mink) aa changes have been associated with onward animal‐to‐animal transmission. No significantly increased mutation rate was observed in SARS‐CoV‐2 circulating in these two animal species and the resulting viral strains had not acquired a significantly higher transmission or pathogenicity in humans (Tan et al., 2022).
RISK ASSESSMENT AND MITIGATION: SOCIETAL ASPECTS
How to avert the next pandemic?
Public health specialists have developed a number of measures to contain an ongoing pandemic ranging from contact tracing, over social distancing, sanitization, the use of face masks, and vaccination, to lockdowns. Of course, their efficacy depends on the type of pandemic, what applies to respiratory infections such as influenza or coronavirus infection does not necessarily apply to a mainly sexually transmitted pandemic such as HIV. Some tools remain crucial with slight modifications, for example condom use instead of face masks, others simply do not apply, for example there is no vaccine against HIV. However, the quoted measures can curb the spread of an ongoing pandemic but they cannot diminish the risk of a future pandemic outbreak. To reduce the likelihood of viral spill‐over events, measures at the animal‐human interface are needed.
It is instructive to read what a prominent virologist wrote in 2003 on SARS: “Where next? Will SARS reappear? This question confronts public‐health officials worldwide, particularly infectious disease personnel in those regions of the world most affected by the disease and the economic burden of SARS” (Webster, 2004). The SARS epidemic was short‐lived, not really a pandemic with worldwide geographical spread, claimed a much small number of lives and had a modest economic cost compared with COVID‐19. Webster continued “Will the virus re‐emerge from wet markets or from laboratories working with SARS CoV, or are asymptomatic infections ongoing in human beings? Similar questions can be asked about a pandemic of influenza that is probably imminent.” Despite this warning published in The Lancet, COVID‐19 confronted a world that was largely unprepared for a new viral pandemic. It is now important for politicians and scientists to take up the challenge to design mitigation strategies against a new pandemic. Despite a death toll which goes into the 10 million confirmed deaths, SARS‐CoV‐2 remains of relatively low virulence. Indeed, there is worse in Pandora's box: the Spanish flu from 1918/9 claimed about 50 million lives and the black death pandemic of AD 1348 with Yersinia pestis wiped out more than 60% of the western Eurasian population over its 8 years course (Spyrou et al., 2022).
Should live animal markets be closed?
Some of Webster's questions from 2003 can be answered. There is no ongoing asymptomatic infection with SARS‐CoV‐1. In fact, the agent of SARS simply got extinct and SARS‐CoV‐2 is not a laboratory resurrection of SARS‐CoV‐1. Based on published research the most plausible scenario both for SARS and COVID‐19 are close viral encounters at the animal‐human interface on live animal markets. Should they be closed? In 2003 Webster was sceptical. Even at the height of the COVID‐19 pandemic, zoonosis researchers argued against a hasty conclusion. Wet markets are widespread in growing cities of low and middle‐income countries. Many people depend on these markets for their livelihoods and food supply. Particularly Asian people believe that fresh meat derived from live animals is safer, tastier and more natural, partly explained by the lack of cold chains. For poor people wet markets offer food security and equity since there they can there buy the quantities they can afford (Naguib et al., 2021). As Webster in 2003, these scientists are convinced that closing wet markets in face of persistent demand would not only put many people out of business (it is economically a large sector) and push their activity into the underground where it cannot be controlled. Webster reminds that even New York nearly doubled its live poultry markets which can—if carefully controlled—even serve as sentinels for detecting emerging influenza viruses. On the other hand, the argument remains that over 60% of human emerging infectious diseases are zoonotic and the large majority of these (72%) originate in wildlife (Naguib et al., 2021) and many have been traced to wet markets. How to solve this dilemma? Lin et al. (2021) propose to differentiate because the term “wet market” is not precisely defined. The name derives from the wet floors caused by the washing of stalls and the melting of the ice used to keep foods fresh. Lin and colleagues distinguished several types of wet markets, namely (1) those selling no live animals or only seafood; (2) those selling live domesticated animals; (3) those selling dead wild animals; and (4) those selling live wild animals (either captive‐reared or wild‐caught). Seafood‐even when sold live‐ can be neglected in a risk analysis because historically fish has not been linked to viral epidemics in humans. These researchers propose to base a political decision on an objective risk analysis. Risk increases with evolutionary relatedness of the traded animals to humans: mammals are thus more risky than birds (except for influenza virus where water fowls play an important ecological role in the emergence of new viruses) and birds are more risky than reptiles, amphibia, fish or even invertebrates. Within mammals bats pose a special risk (bats as food are popular in Indonesia), followed by rodents (bandicoots are popular food in China), carnivores (racoon dogs, civets are prestige food in China), and pangolins (as meat delicacy and scales for traditional medicine). There is an increasing risk gradient when going from live domesticated to non‐domesticated, captive‐reared animals to wild animals. Selling dead animals is clearly a lesser risk than selling live animals. Hygiene measures (vendor handwashing, stand cleaning, animal species separation and proper waste disposal) and the extent to which they are neglected are independent risk factors. The market size is a risk factor since it determines the extent of contact between animals and humans. The length and breadth of the supply chain is another risk factor since the stress of transport suppresses the animal's immune system activating latent infections and crowding of different species during transport favours cross‐species infections. From this analysis, it is clear that the political authorities should target wet markets selling live wild animals and impose strict controls if closure is politically not possible. Scientists should provide convincing data such that governments can motivate the stakeholders in a dialogue. This should not be impossible since food vendors are the primary victims because they are at the forefront of the animal‐human interface. Consumers should also be sensitized and educated about food hygiene and to species conservation issues since markets selling live and dead wild animals are a major threat to endangered species such as civets and pangolins. For example, Hong Kong has experience with live poultry markets. In Hong Kong, ducks and geese (the original source of influenza viruses) were ordered to be sold chilled and two obligatory cleaning days per month were imposed. With these two simple measures Hong Kong was able to reduce the spectrum of influenza virus isolation compared to mainland China (Webster, 2004).
In Africa bushmeat markets are suspected of origins for HIV, Ebola or monkeypox epidemics and similar targeted approaches should be considered, but might be more difficult to implement in resource‐poor countries. The situation in Europe is easier: EFSA has reviewed the SARS‐CoV‐2 situation in the EU. For companion animals risk of spill‐back infections to humans was considered as very low. Wild animals are not considered as a substantial risk because WTD is not widely distributed in the EU. EFSA recommends avoiding contact with diseased or dead wild animals (EFSA Panel, 2023).
One health initiative
However, reducing the risk of the next pandemic is more complex than just regulating wet markets and bushmeat markets. Numerous factors such as crowding, urbanization, increasing human populations with immunosuppression (AIDS or medically induced) or with comorbidities, deforestation, fragmentation of natural habitats, agricultural intensification and globalization have substantially enlarged the size and depth of the animal‐human viral interface. This expansion necessitates comprehensive and multidisciplinary approaches for risk management involving microbiologists, physicians, veterinarians, epidemiologists, sociologists and environmental scientists in a coordinated One Health approach. One Health was defined by WHO as “an integrated, unifying approach that aims to sustainably balance and optimize the health of people, animals, and ecosystems”. Of particular importance are sound statistical data able to reveal significant trends and associated risks. Researchers developed a database of emerging infectious diseases (EID) and compiled 335 events between 1940 and 2004, with a significant increase in EID over time and a peak in the 1990s with the onset of the HIV pandemic (Jones et al., 2008). Over this period, 60% of EID events were infections from animals and 72% of them had a wildlife origin. Vector‐borne diseases were responsible for 23% of EID events. The rise in EID was correlated with climate anomalies (rainfall, temperature, severe weather). The richness of human pathogens increased towards the Equator. Human population density and wildlife host species richness were predictors for EID. This database identified geographical hotspots for zoonotic EID from wildlife in South, East and South‐East Asia, sub‐Saharan Africa and Central America. Western Europe was another hotspot which the researchers explained by a reporting bias due to prominent zoonotic research centres in The Netherlands and the UK. Research on EID is thus concentrated on areas where the next pandemic is less likely to start (Jones et al., 2008). In an updated study from 2017 zoonotic EID risk was shown to be elevated in forested tropical regions experiencing land‐use changes and where wildlife biodiversity (mammalian species richness) is high. When accounting for a publication bias, India and China were the major hotspots for EID (Allen et al., 2017). Indeed proposals to limit zoonosis call for the protection of tropical and subtropical forests, a ban of wildlife animal trade, increased veterinary care for farmed animals and care for people suffering from immune suppression (Vora et al., 2022). The rationale for these interventions is straightforward. Clearance of tropical forests is perhaps the major driver of EID since it creates growing animal‐human interfaces in disturbed areas which also show a higher human population density than undisturbed ecosystems (Gibb et al., 2020). An illustrative example is the emergence of Nipah virus: clearance of forests in Malaysia diminished the food basis for fruit bats. Agronomists had recommended pig farmers to plant fruit trees on their farms such that they had an income from selling pigs and fruits. The starving fruit bats from deforested areas were attracted to these farms. Fruits from orchards were contaminated by bat secretions, dropped to the ground and were consumed by pigs. In this way, the first cross‐infection from bats to pigs occurred and subsequently pigs infected the pig farmers in Malaysia and slaughterhouse workers in Singapore, mediating the second cross‐infection. A comparable story can be told for Bangladesh: a forest decrease attracted fruit bats to the sampling pots of palm sap collectors which led to direct infections of humans with Nipah virus without an intermediate host. In general, bats when deprived of their natural habitat quickly adapt to live alongside people which also led to smaller infection chains of Hendra virus in Australia in horses and men (Brüssow, 2012). Immunosuppressed HIV patients developed susceptibilities to many previously rarely met pathogens and were unable to clear these infections allowing the pathogens to accumulate mutations. Similarly, medically immunosuppressed patients were unable to clear SARS‐CoV‐2 infections allowing the virus to accumulate many mutations possibly contributing to highly mutated variant viruses (Chaguza et al., 2023).
Climate change
Climate change will have a multitude of effects on human well‐being and ecosystem health in the future. While some effects are obvious, other consequences of temperature increase are at first glance perhaps surprising. A recent study predicted a marked increase in viral zoonosis over the next decades (Carlson et al., 2022). As the phylogenetic distance between animals and humans determines the likelihood of viral spillover, a recent study analysed the situation for mammalian viruses. The reasoning of their model calculation is straightforward: animals will respond to temperature increases by geographical host range extensions either to higher latitudes or higher altitudes to escape heat waves. This movement of both plants and animals has already been documented over the last decades. Their model stipulates that together with the animals also their viruses will move to new areas. This move leads not only to new overlaps between the investigated 3000 mammalian species but also to a multitude of new virus encounters with animals that had never seen these viruses in the past. These researchers predicted geographical hotspots for novel viral sharing between mammalian species. When accounting for geographical barriers to mammalian species dispersion as they track thermal optima for their survival, Southeast Asia will be the major hotspot. The majority of mammals will overlap with at least one unfamiliar mammalian species. Since bats are the only mammals which developed flight capacity, bats have a much higher dispersal capacity. As bats are also the most species‐rich mammalian group, have high population sizes and inhabit crowded rest places, they harbour a rich virus population. The model calculates that 90% of all new first viral encounters will be with bat viruses and that the majority of novel viral encounters will already occur with a global temperature increase of 1°C—which means that many new cross‐infections will occur before 2040. This novel virus sharing shortly will likely lead to some animal species extinction and novel human viral epidemics. Future potential greenhouse gas reduction will not affect this sharing of “new” viruses, hence prevention of future pandemics will need specific early detection and mitigation approaches. By extension, this argument also applies to parasitic and fungal infections which were not yet explored in their model. So what can microbiologists do to control the consequences of this increased viral sharing in the future? The final chapters will illustrate possible approaches described in recent publications.
RISK ASSESSMENT AND MITIGATION: MICROBIOLOGICAL ASPECTS
Animal virome analysis
Microbiologists can contribute to assessing, predict and possibly mitigating the risk of future viral spill‐overs from animals. A better knowledge of the virome of usual suspect animals will certainly help. In this vein, Chinese scientists sampled 1900 animals representing 18 mammalian species of game animals which were sold as delicacy food on wet markets. Respiratory and faecal samples were analysed by meta‐transcriptome sequencing and screened for viruses (He et al., 2022). The researchers identified 102 vertebrate viruses representing 13 virus families. Picornaviridae, Astroviridae and Parvovirdae were the most frequently identified viruses. Coronaviridae were identified in bamboo rats, civets, raccoon dogs and hedgehogs. Based on sequence matches, likely cases of cross‐species infections were identified for coronaviruses from bats to civets; from birds to porcupines; and from dogs to racoon dogs. In total 65 novel vertebrate viruses were detected. Putative pathogenic viruses were also observed in seemingly healthy animals. However, a subgroup of 21 game animals showed clinical symptoms associated with influenza‐, astro‐ and rotaviruses. SARS‐CoV‐2‐like sequences were not detected.
Somewhat narrower in scope was a survey of faecal, oral and urine samples from horseshoe bats living in and around a single botanical garden in southern China which was conducted between 2019 and 2020. By meta‐transcriptome sequencing 26 coronavirus genomes could be identified comprising 9 betacoronaviruses (the group which also includes SARS‐CoV‐2) and 17 alphacoronaviruses. One genome shared 94.5% nt sequence identity with SARS‐CoV‐2 but differed substantially over the spike gene. The researchers detected nearly 100% identical coronaviruses from multiple different bat species at nt level which suggested frequent host switching of coronaviruses across species barriers between bats (Zhou et al., 2021).
Wastewater analysis as sentinel
Virome analysis in animals identifies the virus reservoirs serving as potential viral “source” material. It is equally important to analyse which viruses circulate in the human population and which animal viruses actually crossed into the human population as viral “sink” material. Here the term sink is meant literally since wastewater turned out as suitable research material.
Swiss scientists conducted amplicon‐based sequencing on pooled wastewater from two cities. They used a bioinformatic method that searched for co‐occurrence of mutations on read pairs which allowed them to identify variant SARS‐CoV‐2 viruses with high confidence. The wastewater samples from Lausanne permitted to diagnose the 2020 arrival of the SARS‐CoV‐2 Alpha variant 13 days prior to its detection in local clinical samples. In 2021 longitudinal wastewater tests documented a low‐level circulation of the Delta variant even 118 days before it appeared in clinical samples from Lausanne. In other regions of Switzerland, e.g., in Zurich, the time prior to variant virus detection was shorter but nevertheless allowed to identify of the arrival of a new variant infection wave using the wastewater monitoring as a type of early warning system (Jahn et al., 2022). A US consortium analysed many wastewater samples from a single Californian university campus, achieving a high temporal and spatial resolution going down to single buildings. The analysis of both wastewater and clinical samples tracked waves of infections with different SARS‐CoV‐2 variants. Wastewater signals preceded that of clinical samples by about 10 days. The spread of virus on campus consisted of many separate, small outbreaks that clustered in nearby buildings. The researchers found matching viral sequences in wastewater collected from a single building over time representing either long shedding from a single person or an infection chain in the building (Karthikeyan et al., 2022). Wastewater sequencing has thus substantial potential for fine‐grained epidemiological analysis during an outbreak. For example, Austrian researchers developed a method to approximate the reproduction number of SARS‐CoV‐2 variants from the time of development of the virus load in wastewater. They demonstrated that the Alpha variant had a 1.4 higher reproduction number than preceding variants and the Omicron a 1.9 higher number than the Delta variant (Amman et al., 2022). Alternatively, a widespread geographical sampling across a country allowed to assess the regional spread of the infection or the effect of non‐pharmaceutical measures such as lockdowns on infection transmission (Morvan et al., 2022). However, all these SARS‐CoV‐2 targeted wastewater analyses track an ongoing pandemic. What is the potential to detect a cryptic epidemic with wastewater sequencing?
Metagenome/metatranscriptome sequencing
Untargeted metagenomic/metatranscriptomic shotgun sequencing can extend the search to an unsuspected circulation of viral pathogens but need a high enough concentration of pathogenic viruses for detection. This can be remedied by using probe‐based enrichment methods before shotgun sequencing (“semi‐targeted” approaches) (Levy et al., 2023). Wastewater sequencing demonstrated its value for advance warning in the UK where samples from February 2022 showed poliovirus genomes with shared mutations indicating an unsuspected person‐to‐person transmission in London (Guglielmi, 2022). This was a surprise since the last case of polio was seen in the UK in 1984 and the country was declared polio‐free in 2003. In response, the health authorities were able to initiate a polio vaccination campaign in children. Likewise in June 2022 an adult US patient with acute flaccid paralysis excreted poliovirus in stool. Retrospective wastewater investigation from his county and surrounding areas of New York City showed that 8% of local wastewater samples contained poliovirus genetically linked to the patient's poliovirus (Link‐Gelles et al., 2022). Wastewater analysis showed that poliovirus was widespread in New York City at this time (Ryerson et al., 2022). A local spread concomitant with the detection of a clinical case is expected since only 1 out of 2000 polio infections are associated with clinical symptoms (Pallansch, 2022). Wastewater will collect only viruses which are excreted by faeces and urine. Surprisingly, recent data from California indicate that a wide range of respiratory viruses (seasonal coronavirus, rhinovirus, influenza and parainfluenza virus, metapneumovirus and respiratory syncytial virus) can be detected in wastewater solids and correlate with clinical reporting of these viral infections in sentinel laboratories (Boehm et al., 2023). Other researchers tested whether air samplers from congregate settings (cafeterias, school classes) allowed the detection of respiratory pathogens by PCR. Several respiratory viruses and two herpesviruses were detected. In a longitudinal survey influenza virus A showed a distinct detection pattern from coronaviruses and bocavirus, detection was restricted to places frequented by young children (Ramuta et al., 2022). The quoted reports identified known viruses. It is less evident how to detect new viruses. One could conduct a virus particle purification procedure before starting the sequencing. Abundant bacterial virus genomes could be set aside from animal viruses by genome organization traits, but then the problem remains to differentiate potentially dangerous viral spill‐overs from relatively harmless viral “commensals” replicating in our bodies without causing any pathologies, a group of human viruses which remained heavily under‐investigated.
Risk ranking
Animal virome data are now created by a large, internationally coordinated global virus genomic surveillance network (Hill et al., 2023). In addition, such efforts need a curated viral genome sequence database annotated with a taxonomical classification as a reference tool. An advanced warning system also has to establish a ranking of the detected viruses into different risk categories. The most important risk criterion is the capacity of the detected virus to spread among humans.
Warren and Sawyer (2023) defined four properties that a viral pathogen must acquire to infect humans and cause an epidemic. First, such a virus must be able to enter a human cell (the need for a cellular receptor); second, it must be capable to replicate in a human cell (the need of fitting cellular cofactors for its cellular reproduction); third, the virus must bypass innate immunity and fourth, the virus must evade existing adaptive immunity in the human population. This set of four requirements is a useful start since it describes experimental approaches by which viruses can be classified into different risk categories.
Bypassing and evading the different arms of the immune system is certainly necessary for attributing a pandemic potential to a virus, but these are “defensive” viral traits. A zoonotic virus also needs “offensive” traits to develop a pandemic potential. A pandemic virus must achieve an efficient transmission from human to human to maintain infection chains (Figure 1 left). However, interhuman transmissibility is not easy to measure. The first, second and fourth Warren and Sawyer criteria can be evaluated in cell culture experiments, but the third criterium needs animal experiments. In contrast, an assessment of the transmission potential needs animal experiments as well as an evaluation of environmental and sociological conditions affecting virus transmission. The molecular genetics of viral transmission traits is less developed than those of the viral traits explored in the four Warren and Sawyer criteria. We clearly need further criteria for a pandemic risk assessment. The deeper we look into the human virome, the clearer it becomes that we harbour many viruses that infect us without causing detectable harm to our health. Allelovirus is such a case: most humans are either chronically infected or continuously re‐infected with Allelovirus (Tisza et al., 2020). So far no disease could be associated with Allelovirus infections (Kaczorowska & van der Hoek, 2020). Some data from chronic viral infections seem to indicate that it is not primarily the cytopathology of the viral infection at the cellular level which causes harm but an aggressive reaction of the immune system towards a “new, not yet adapted” virus that leads to severe clinical symptoms (Virgin et al., 2009). It is not obvious how to test the pathogenic potential of a virus since it frequently causes only mild or no disease in the reservoir species. Perhaps an antibody response to a spill‐over virus in humans could be used as a surrogate marker for a pathogenic potential.
FIGURE 1.
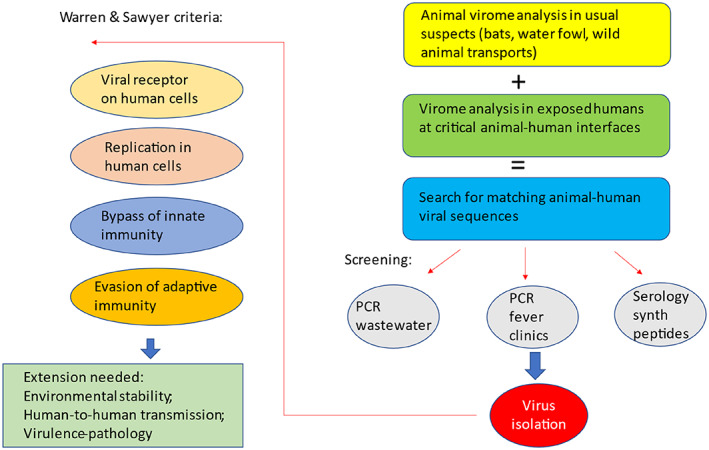
Criteria to assess the zoonotic risk of animal viruses to cause epidemics in the human population. The Warren and Sawyer (2023) criteria to assess the potential of an animal virus to enter and to be transmitted in the human population are illustrated on the left side of the diagram. These criteria need an extension to evaluate the potential of such viruses to be transmitted in the human population and to cause significant disease (left side, bottom). The Warren and Sawyer criteria do not specify how candidate animal viral viruses are chosen for investigation. The right side of the figure illustrates a possible flow scheme to identify candidate animal viruses for analysis. After virus isolation, such candidate viruses can be submitted to the modified Warren and Sawyer flow scheme to characterize their pandemic potential. See text for details.
Let us take recent publications to illustrate the Warren and Sawyer criteria for assessing the zoonotic potential of newly isolated animal viruses. Chinese researchers investigated anal swabs from 86 confiscated smuggled pangolins. Four pangolins tested positive for a coronavirus, sequencing identified a relative of a bat coronavirus and of MERS (87% and 68% genome identity). In fact, 13% of the pangolins showed antibodies to this virus. The virus was subsequently isolated and caused cytopathic effects in a human cell culture. The pangolin coronavirus used the human, bat and pangolin gene product DPP4 as cell entry receptor (as does MERS) and experiences proteolytic activation by furin. It can use DPP4 from a wide range of animals as cell entry receptor. The pangolin virus can replicate efficiently in human colon and primary human airway organoids. In mice transgenic with the human DPP4 gene, this pangolin virus replicates in the lungs and the brain. The lungs showed histologically a mild interstitial pneumonia but no weight loss was seen in the infected animals, indicating some pathogenicity and interferon‐suppressing anti‐innate immunity activity (Chen et al., 2023). Such a virus certainly represents a risk to infect humans but it needs more for a virus to cause a pandemic, most importantly the virus must acquire the capacity for efficient human‐to‐human spread. Predictions are that it needs at least critical changes in at least three viral proteins (polymerase, hemagglutinin, nucleocapdis protein) to confer such a phenotype to the avian influenza virus H5N1 (Kupferschmidt, 2023).
With 96% overall genome identity the Rhinolophus affinis bat coronavirus RaTG13 isolated in China back in 2013 is a closely related animal virus to SARS‐CoV‐2. However, its affinity for the human ACE2 receptor is very low. French virologists investigated 654 bats from Laos belonging to 46 species and sampled blood, saliva, faeces and urine; 24 animals yielded a positive signal for coronavirus by PCR. Genome sequencing placed the bat viruses between pangolin and human coronavirus from Wuhan. The RBD of the bat viruses resembled that of the spike protein from SARS‐CoV‐2 and human cell lines expressing ACE2 could be infected with faecal swabs from these Laotian bats. The cultures showed no cytopathic effects, but up to 106 pfu/ml infectious virus was produced in the cell supernatant. The kinetics of viral RNA synthesis by the bat coronaviruses in the human cells was slower than that by SARS‐CoV‐2. Orf8 which was associated with immune evasion in SARS‐CoV‐2 differed markedly in these bat coronaviruses from SARS‐CoV‐2 and the spike proteins of these coronaviruses lacked the furin cleavage site (Temmam et al., 2022).
Similar to the pangolin coronaviruses, the MERS coronaviruses use DPP4 as cellular receptor. A close relative of MERS (85% genome identity) isolated from African bats does not use DPP4, but bat ACE2 as receptor for cell entry. Use of human ACE2 is inefficient but a single amino acid change in the RBD of this bat coronavirus enhanced the affinity for human ACE2 and enabled this virus to enter human cells suggesting that these viruses hold a latent potential to infect humans when undergoing small mutational changes (Xiong et al., 2022).
Clinical sentinel studies
Applying the Warren and Sawyer criteria to these isolates indicate that many viruses are close to cross the animal‐human interface, whether they are imminent clinical threats is more difficult to judge. Attentive physicians and sentinel studies must therefore complement the efforts of microbiologists. A good example is the report by Zhang et al. (2022). These researchers conducted a sentinel surveillance study in eastern China among febrile patients with a history of recent animal exposure. The throat swabs of one patient yielded a new paramyxovirus which they called Langya virus, a new member of the Henipa virus group to which belongs the Hendra virus and the Nipah virus. Subsequently, they identified 35 further acute infections with Langya virus; in 26 patients Langya virus was the only identified pathogen. Symptoms were non‐specific and comprised fever, fatigue and cough. In 14 patients paired acute‐convalescent sera were obtained and 86% showed an IgG seroconversion to Langya virus. Further human‐to‐human transmission was not observed. Langya virus RNA was detected in a quarter of 262 investigated shrews identifying shrews as potential reservoirs for Langya virus.
OUTLOOK
There will be many viral spill‐overs of animal viruses into humans which are dead‐end infections since they are not further transmitted to contact persons. Waiting for this to occur to ring the alarm bell is a risky strategy. The identification of the SARS‐CoV‐2 virus plus its genome sequence (submitted on 20 January 2020 and published on 3 February 2020 just about 1 month after the first documented COVID‐19 case) was not quick enough (Zhou et al., 2020). When the authors wrote their report 2794 laboratory‐confirmed infections and 80 deaths had occurred already. The Chinese authorities closed and had cleaned the Huanan food market on 1 January 2020. Unfortunately, the worldwide spread of the infection could not any longer be prevented despite drastic control measures taken by the Chinese government such as taking the entire city of Wuhan under lockdown.
Early epidemic warning critically needs the involvement of microbiologists who must be supported by appropriate grants, necessitating the support of politicians. Politicians in democratic countries tend to deal with current problems and are less oriented towards problems that might occur when they will have already left their elected positions. Therefore, some push is needed from an informed public for the build‐up of a necessarily costly early virus warning system. However, the costs of such a system are small compared to the economic, societal and human costs of a further pandemic such as COVID‐19. This lobbying will however not be an easy task for science educators and science communicators since substantial parts of the public were even opposed to anti‐viral interventions in the middle of an unfolding pandemic.
Early warning of spill‐over infections from animals into humans is thus an uphill battle, but an essential one. The Warren and Sawyer scheme starts already with an isolated virus, but it is not obvious how to arrive to such a candidate for a spill‐over candidate. Alternative schemes could be imagined. For example, by starting with a virome analysis in usual suspects at the animal side of the animal‐human interface being it bats, water fowls or wild animals from trade and transport. Such investigations should be complemented by virome analyses in humans living or working at critical sites of the animal‐human interface such as wild animal traders and farmers, wild animal veterinarians or workers at deforestation zones. One could then compare both virome data sets and search for matching sequences pointing to viruses that crossed the animal‐human interface. These sequences could then be used to further substantiate the evidence for cross‐infections by screening wastewater for such putative viral sequences or clinical samples from patients in fever clinics from areas with disturbed ecosystems. One might also consider expressing predicted antigenic peptides from the matching virome sequences to use them in serological tests (Figure 1 right). Ultimately it needs an isolation of viruses with the identified sequences which can then be submitted to the Warren and Sawyer scheme to attribute a risk level to them.
When a new zoonotic infection has been identified, it needs measures to contain its spread so that it does not become an epidemic or a pandemic. To do so, we need a careful analysis of the efficacy of the different public health containment measures to curb a pandemic. COVID‐19 offers a lot of lessons learnt and scientists and politicians must take care to communicate the results of rigorous analyses objectively to a public which was deeply divided about the value of these measures mainly based on ideological prejudices. This task is important and goes beyond the skill of microbiologists and needs support from psychologists, sociologists and communication experts to overcome dangerous ideological splits that became apparent during the height of the COVID‐19 pandemic.
AUTHOR CONTRIBUTIONS
Harald Brüssow designed and wrote the Lilliput article without contributions from other scientists.
FUNDING INFORMATION
No funding information provided.
ACKNOWLEDGEMENTS
I thank Dr. Sophie Zuber for her critical reading of the manuscript.
Brüssow, H. (2023) Viral infections at the animal–human interface—Learning lessons from the SARS‐CoV‐2 pandemic. Microbial Biotechnology, 16, 1397–1411. Available from: 10.1111/1751-7915.14269
REFERENCES
- Allen, T. , Murray, K.A. , Zambrana‐Torrelio, C. , Morse, S.S. , Rondinini, C. , Di Marco, M. et al. (2017) Global hotspots and correlates of emerging zoonotic diseases. Nature Communications, 8(1), 1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amman, F. , Markt, R. , Endler, L. , Hupfauf, S. , Agerer, B. , Schedl, A. et al. (2022) Viral variant‐resolved wastewater surveillance of SARS‐CoV‐2 at national scale. Nature Biotechnology, 40(12), 1814–1822. [DOI] [PubMed] [Google Scholar]
- Andersen, K.G. , Rambaut, A. , Lipkin, W.I. , Holmes, E.C. & Garry, R.F. (2020) The proximal origin of SARS‐CoV‐2. Nature Medicine, 26(4), 450–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm, A.B. , Hughes, B. , Duong, D. , Chan‐Herur, V. , Buchman, A. , Wolfe, M.K. et al. (2023) Wastewater concentrations of human influenza, metapneumovirus, parainfluenza, respiratory syncytial virus, rhinovirus, and seasonal coronavirus nucleic‐acids during the COVID‐19 pandemic: a surveillance study. Lancet Microbe. Online ahead of print. Available from: 10.1016/S2666-5247(22)00386-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni, M.F. , Lemey, P. , Jiang, X. , Lam, T.T. , Perry, B.W. , Castoe, T.A. et al. (2020) Evolutionary origins of the SARS‐CoV‐2 sarbecovirus lineage responsible for the COVID‐19 pandemic. Nature Microbiology, 5(11), 1408–1417. [DOI] [PubMed] [Google Scholar]
- Brüssow, H. (2012) On viruses, bats and men: a natural history of food‐borne viral infections. In: Witzany, G. (Ed.) Viruses: essential agents of life. Dordrecht: Springer Publisher. [Google Scholar]
- Brüssow, H. (2021) What we can learn from the dynamics of the 1889 ‘Russian flu’ pandemic for the future trajectory of COVID‐19. Microbial Biotechnology, 14(6), 2244–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüssow, H. (2022) The beginning and ending of a respiratory viral pandemic‐lessons from the Spanish flu. Microbial Biotechnology, 15(5), 1301–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüssow, H. & Brüssow, L. (2021) Clinical evidence that the pandemic from 1889 to 1891 commonly called the Russian flu might have been an earlier coronavirus pandemic. Microbial Biotechnology, 14(5), 1860–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway, E. & Kozlov, M. (2022) Which COVID studies pose a biohazard? Lack of clarity hampers research. Nature, 611(7934), 17–18. [DOI] [PubMed] [Google Scholar]
- Carlson, C.J. , Albery, G.F. , Merow, C. , Trisos, C.H. , Zipfel, C.M. , Eskew, E.A. et al. (2022) Climate change increases cross‐species viral transmission risk. Nature, 607(7919), 555–562. [DOI] [PubMed] [Google Scholar]
- Caserta, L.C. , Martins, M. , Butt, S.L. , Hollingshead, N.A. , Covaleda, L.M. , Ahmed, S. et al. (2023) White‐tailed deer (Odocoileus virginianus) may serve as a wildlife reservoir for nearly extinct SARS‐CoV‐2 variants of concern. Proceedings of the National Academy of Sciences of the United States of America, 120(6), e2215067120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaguza, C. , Hahn, A.M. , Petrone, M.E. , Zhou, S. , Ferguson, D. , Breban, M.I. et al. (2023) Accelerated SARS‐CoV‐2 intrahost evolution leading to distinct genotypes during chronic infection. Cell Reports Medicine, 4(2), 100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, J.F.W. , Siu, G.K.H. , Yuan, S. , Ip, J.D. , Cai, J.P. , Chu, A.W.H. et al. (2022) Probable animal‐to‐human transmission of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) Delta variant AY.127 causing a pet shop‐related coronavirus disease 2019 (COVID‐19) outbreak in Hong Kong. Clinical Infectious Diseases, 75(1), e76–e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler, J.C. , Bevins, S.N. , Ellis, J.W. , Linder, T.J. , Tell, R.M. , Jenkins‐Moore, M. et al. (2021) SARS‐CoV‐2 exposure in wild white‐tailed deer (Odocoileus virginianus). Proceedings of the National Academy of Sciences of the United States of America, 118(47), e2114828118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Yang, X. , Si, H. , Gong, Q. , Que, T. , Li, J. et al. (2023) A bat MERS‐like coronavirus circulates in pangolins and utilizes human DPP4 and host proteases for cell entry. Cell, 186(4), 850–863.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, J. (2023) New clues to pandemic's origin surface, causing uproar. Science, 379(6638), 1175–1176. [DOI] [PubMed] [Google Scholar]
- EFSA Panel on Animal Health and Welfare (AHAW) , Nielsen, S.S. , Alvarez, J. , Bicout, D.J. , Calistri, P. , Canali, E. et al. (2023) SARS‐CoV‐2 in animals: susceptibility of animal species, risk for animal and public health, monitoring, prevention and control. EFSA Journal, 21(2), e07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garry, R.F. (2022) The evidence remains clear: SARS‐CoV‐2 emerged via the wildlife trade. Proceedings of the National Academy of Sciences of the United States of America, 119(47), e2214427119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb, R. , Redding, D.W. , Chin, K.Q. , Donnelly, C.A. , Blackburn, T.M. , Newbold, T. et al. (2020) Zoonotic host diversity increases in human‐dominated ecosystems. Nature, 584(7821), 398–402. [DOI] [PubMed] [Google Scholar]
- Gorbunova, V. , Seluanov, A. & Kennedy, B.K. (2020) The world goes bats: living longer and tolerating viruses. Cell Metabolism, 32(1), 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, Y. , Zheng, B.J. , He, Y.Q. , Liu, X.L. , Zhuang, Z.X. , Cheung, C.L. et al. (2003) Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science, 302(5643), 276–278. [DOI] [PubMed] [Google Scholar]
- Guglielmi, G. (2022) What polio's UK presence means for global health. Nature, 607(7918), 225. [DOI] [PubMed] [Google Scholar]
- Hale, V.L. , Dennis, P.M. , McBride, D.S. , Nolting, J.M. , Madden, C. , Huey, D. et al. (2022) SARS‐CoV‐2 infection in free‐ranging white‐tailed deer. Nature, 602(7897), 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, N.L. & Sachs, J.D. (2022) A call for an independent inquiry into the origin of the SARS‐CoV‐2 virus. Proceedings of the National Academy of Sciences of the United States of America, 119(21), e2202769119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, W.T. , Hou, X. , Zhao, J. , Sun, J. , He, H. , Si, W. et al. (2022) Virome characterization of game animals in China reveals a spectrum of emerging pathogens. Cell, 185(7), 1117–1129.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Z. , Ren, L. , Yang, J. , Guo, L. , Feng, L. , Ma, C. et al. (2021) Seroprevalence and humoral immune durability of anti‐SARS‐CoV‐2 antibodies in Wuhan, China: a longitudinal, population‐level, cross‐sectional study. Lancet, 397(10279), 1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, V. , Githinji, G. , Vogels, C.B.F. , Bento, A.I. , Chaguza, C. , Carrington, C.V.F. et al. (2023) Toward a global virus genomic surveillance network. Cell Host & Microbe. S1931‐3128(23)00107‐5. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, E.C. , Goldstein, S.A. , Rasmussen, A.L. , Robertson, D.L. , Crits‐Christoph, A. , Wertheim, J.O. et al. (2021) The origins of SARS‐CoV‐2: a critical review. Cell, 184(19), 4848–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn, K. , Dreifuss, D. , Topolsky, I. , Kull, A. , Ganesanandamoorthy, P. , Fernandez‐Cassi, X. et al. (2022) Early detection and surveillance of SARS‐CoV‐2 genomic variants in wastewater using COJAC. Nature Microbiology, 7(8), 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jebb, D. , Huang, Z. , Pippel, M. , Hughes, G.M. , Lavrichenko, K. , Devanna, P. et al. (2020) Six reference‐quality genomes reveal evolution of bat adaptations. Nature, 583(7817), 578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, X. & Wang, R. (2022) Wildlife trade is likely the source of SARS‐CoV‐2. Science, 377(6609), 925–926. [DOI] [PubMed] [Google Scholar]
- Jones, K.E. , Patel, N.G. , Levy, M.A. , Storeygard, A. , Balk, D. , Gittleman, J.L. et al. (2008) Global trends in emerging infectious diseases. Nature, 451(7181), 990–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowska, J. & van der Hoek, L. (2020) Human anelloviruses: diverse, omnipresent and commensal members of the virome. FEMS Microbiology Reviews, 44(3), 305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthikeyan, S. , Levy, J.I. , De Hoff, P. , Humphrey, G. , Birmingham, A. , Jepsen, K. et al. (2022) Wastewater sequencing reveals early cryptic SARS‐CoV‐2 variant transmission. Nature, 609(7925), 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt, K. (2023) From bad to worse. Science, 380(6640), 28–29. [DOI] [PubMed] [Google Scholar]
- Lam, T.T. , Jia, N. , Zhang, Y.W. , Shum, M.H. , Jiang, J.F. , Zhu, H.C. et al. (2020) Identifying SARS‐CoV‐2‐related coronaviruses in Malayan pangolins. Nature, 583(7815), 282–285. [DOI] [PubMed] [Google Scholar]
- Lenharo, M. & Wolf, L. (2023) US COVID‐origins hearing renews debate over lab‐leak hypothesis. Nature, 615(7952), 380–381. [DOI] [PubMed] [Google Scholar]
- Levy, J.I. , Andersen, K.G. , Knight, R. & Karthikeyan, S. (2023) Wastewater surveillance for public health. Science, 379(6627), 26–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, B. , Dietrich, M.L. , Senior, R.A. & Wilcove, D.S. (2021) A better classification of wet markets is key to safeguarding human health and biodiversity. Lancet Planet Health, 5(6), e386–e394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, X.D. , Wang, W. , Hao, Z.Y. , Wang, Z.X. , Guo, W.P. , Guan, X.Q. et al. (2017) Extensive diversity of coronaviruses in bats from China. Virology, 507, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link‐Gelles, R. , Lutterloh, E. , Schnabel Ruppert, P. , Backenson, P.B. , St George, K. , Rosenberg, E.S. et al. (2022) Public health response to a case of paralytic poliomyelitis in an unvaccinated person and detection of poliovirus in wastewater—New York, June‐August 2022. Morbidity and Mortality Weekly Report, 71(33), 1065–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W.J. , Liu, P. , Lei, W. , Jia, Z. , He, X. , Shi, W. et al. (2023) Surveillance of SARS‐CoV‐2 at the Huanan seafood market. Nature. Online ahead of print. Available from: 10.1038/s41586-023-06043-2 [DOI] [PubMed] [Google Scholar]
- Lu, L. , Sikkema, R.S. , Velkers, F.C. , Nieuwenhuijse, D.F. , Fischer, E.A.J. , Meijer, P.A. et al. (2021) Adaptation, spread and transmission of SARS‐CoV‐2 in farmed minks and associated humans in The Netherlands. Nature Communications, 12(1), 6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallapaty, S. (2023a) COVID‐origins study links raccoon dogs to Wuhan market: what scientists think. Nature, 615(7954), 771–772. [DOI] [PubMed] [Google Scholar]
- Mallapaty, S. (2023b) COVID‐origins report sparks debate over major genome hub GISAID. Nature, 616(7955), 13–14. [DOI] [PubMed] [Google Scholar]
- Maxmen, A. (2022) Scientists struggle to probe COVID's origins amid sparse data from China. Nature, 603(7903), 773–775. [DOI] [PubMed] [Google Scholar]
- Morvan, M. , Jacomo, A.L. , Souque, C. , Wade, M.J. , Hoffmann, T. , Pouwels, K. et al. (2022) An analysis of 45 large‐scale wastewater sites in England to estimate SARS‐CoV‐2 community prevalence. Nature Communications, 13(1), 4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naguib, M.M. , Li, R. , Ling, J. , Grace, D. , Nguyen‐Viet, H. & Lindahl, J.F. (2021) Live and wet markets: food access versus the risk of disease emergence. Trends in Microbiology, 29(7), 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil, S.J.D. (2023) Pangolin merbecovirus gets down to (poly)basics. Cell, 186(4), 688–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oude Munnink, B.B. , Sikkema, R.S. , Nieuwenhuijse, D.F. , Molenaar, R.J. , Munger, E. , Molenkamp, R. et al. (2021) Transmission of SARS‐CoV‐2 on mink farms between humans and mink and back to humans. Science, 371(6525), 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallansch, M.A. (2022) Circulating poliovirus in New York—new instance of an old problem. The New England Journal of Medicine, 387(19), 1725–1728. [DOI] [PubMed] [Google Scholar]
- Pekar, J.E. , Magee, A. , Parker, E. , Moshiri, N. , Izhikevich, K. , Havens, J.L. et al. (2022) The molecular epidemiology of multiple zoonotic origins of SARS‐CoV‐2. Science, 377(6609), 960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering, B. , Lung, O. , Maguire, F. , Kruczkiewicz, P. , Kotwa, J.D. , Buchanan, T. et al. (2022) Divergent SARS‐CoV‐2 variant emerges in white‐tailed deer with deer‐to‐human transmission. Nature Microbiology, 7(12), 2011–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramuta, M.D. , Newman, C.M. , Brakefield, S.F. , Stauss, M.R. , Wiseman, R.W. , Kita‐Yarbro, A. et al. (2022) SARS‐CoV‐2 and other respiratory pathogens are detected in continuous air samples from congregate settings. Nature Communications, 13(1), 4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryerson, A.B. , Lang, D. , Alazawi, M.A. , Neyra, M. , Hill, D.T. , St George, K. et al. (2022) Wastewater testing and detection of poliovirus type 2 genetically linked to virus isolated from a paralytic polio case—New York, march 9‐October 11, 2022. Morbidity and Mortality Weekly Report, 71(44), 1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyrou, M.A. , Musralina, L. , Gnecchi Ruscone, G.A. , Kocher, A. , Borbone, P.G. , Khartanovich, V.I. et al. (2022) The source of the black death in fourteenth‐century Central Eurasia. Nature, 606(7915), 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, C.C.S. , Lam, S.D. , Richard, D. , Owen, C.J. , Berchtold, D. , Orengo, C. et al. (2022) Transmission of SARS‐CoV‐2 from humans to animals and potential host adaptation. Nature Communications, 13(1), 2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temmam, S. , Vongphayloth, K. , Baquero, E. , Munier, S. , Bonomi, M. , Regnault, B. et al. (2022) Bat coronaviruses related to SARS‐CoV‐2 and infectious for human cells. Nature, 604(7905), 330–336. [DOI] [PubMed] [Google Scholar]
- Tisza, M.J. , Pastrana, D.V. , Welch, N.L. , Stewart, B. , Peretti, A. , Starrett, G.J. et al. (2020) Discovery of several thousand highly diverse circular DNA viruses. eLife, 9, e51971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin, H.W. , Wherry, E.J. & Ahmed, R. (2009) Redefining chronic viral infection. Cell, 138(1), 30–50. [DOI] [PubMed] [Google Scholar]
- Vora, N.M. , Hannah, L. , Lieberman, S. , Vale, M.M. , Plowright, R.K. & Bernstein, A.S. (2022) Want to prevent pandemics? Stop spillovers. Nature, 605(7910), 419–422. [DOI] [PubMed] [Google Scholar]
- Warren, C.J. & Sawyer, S.L. (2023) Identifying animal viruses in humans. Science, 379(6636), 982–983. [DOI] [PubMed] [Google Scholar]
- Webster, R.G. (2004) Wet markets—a continuing source of severe acute respiratory syndrome and influenza? Lancet, 363(9404), 234–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey, M. , Levy, J.I. , Malpica Serrano, L. , Crits‐Christoph, A. , Pekar, J.E. , Goldstein, S.A. et al. (2022) The Huanan seafood wholesale market in Wuhan was the early epicenter of the COVID‐19 pandemic. Science, 377(6609), 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, K. , Zhai, J. , Feng, Y. , Zhou, N. , Zhang, X. , Zou, J.J. et al. (2020) Isolation of SARS‐CoV‐2‐related coronavirus from Malayan pangolins. Nature, 583(7815), 286–289. [DOI] [PubMed] [Google Scholar]
- Xiao, X. , Newman, C. , Buesching, C.D. , Macdonald, D.W. & Zhou, Z.M. (2021) Animal sales from Wuhan wet markets immediately prior to the COVID‐19 pandemic. Scientific Reports, 11(1), 11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, Q. , Cao, L. , Ma, C. , Tortorici, M.A. , Liu, C. , Si, J. et al. (2022) Close relatives of MERS‐CoV in bats use ACE2 as their functional receptors. Nature, 612(7941), 748–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen, H.L. , Sit, T.H.C. , Brackman, C.J. , Chuk, S.S.Y. , Gu, H. , Tam, K.W.S. et al. (2022) Transmission of SARS‐CoV‐2 delta variant (AY.127) from pet hamsters to humans, leading to onward human‐to‐human transmission: a case study. Lancet, 399(10329), 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.A. , Li, H. , Jiang, F.C. , Zhu, F. , Zhang, Y.F. , Chen, J.J. et al. (2022) A zoonotic Henipavirus in febrile patients in China. The New England Journal of Medicine, 387(5), 470–472. [DOI] [PubMed] [Google Scholar]
- Zhou, H. , Ji, J. , Chen, X. , Bi, Y. , Li, J. , Wang, Q. et al. (2021) Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS‐CoV‐2 and related viruses. Cell, 184(17), 4380–4391.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Yang, X.L. , Wang, X.G. , Hu, B. , Zhang, L. , Zhang, W. et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]