

Summary
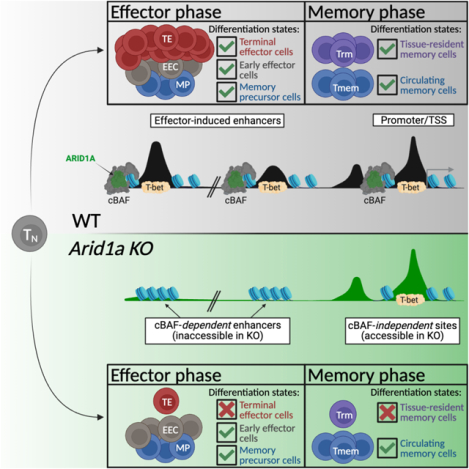
CD8+ T cells provide host protection against pathogens by differentiating into distinct effector and memory cell subsets, but how chromatin is site-specifically remodeled during their differentiation is unclear. Due to its critical role in regulating chromatin and enhancer accessibility through its nucleosome remodeling activities, we investigated the role of the canonical BAF (cBAF) chromatin remodeling complex in antiviral CD8+ T cells during infection. ARID1A, a subunit of cBAF, was recruited early after activation and established de novo open chromatin regions at enhancers. Arid1a deficiency impaired the opening of thousands of activation-induced enhancers, leading to loss of TF binding, dysregulated proliferation, gene expression, and failure to undergo terminal effector differentiation. While Arid1a was dispensable for circulating memory cell formation, tissue-resident memory formation was strongly impaired. Thus, cBAF governs the enhancer landscape of activated CD8+ T cells that orchestrates TF recruitment and activity and the acquisition of specific effector and memory differentiation states.
Keywords: Antiviral immunity, ARID1A, BAF Complex, Chromatin Remodeling, Epigenetics, CD8+ T cells, effector T cell, memory T cell, immunotherapy
eTOC Blurb
Chromatin remodeling is a critical step for cellular differentiation. McDonald, Chick et al. show that the canonical BAF complex mediates chromatin remodeling to establish de novo enhancers in recently activated virus-specific CD8+ T cells, which in turn allows the activated cells to acquire specialized effector and memory cell fates.
Graphical Abstract
Introduction
CD8+ T cells play an essential role in host protection against intracellular pathogens and cancer. Naive CD8+ T cells become activated upon recognition of cognate antigen (signal 1) concurrent with engagement of costimulatory receptors (signal 2) and cytokine signaling (signal 3), enabling epigenetic and transcriptional reprogramming to promote cell growth, proliferation, and the acquisition of specialized effector functions1. Activated effector cells exhibit transcriptional and epigenetic heterogeneity, display diverse modes of tissue homing patterns and memory-forming potential, and can be distinguished by expression of markers like IL7R (CD127), KLRG1 and CX3CR12. Memory precursor (MP) cells express higher levels of CD127 and preferentially give rise to the long-lived circulating memory cell pool3. High inflammatory conditions promote the formation of short-lived terminal effector (TE) cells expressing KLRG1 and CX3CR1 that exhibit potent cytotoxic potential but preferentially die following the resolution of infection4–6. Early effector cells (EEC) express low levels of CD127, KLRG1, and CX3CR1, and can give rise to both TE and MP cells6–8. Recent studies have begun to identify distinguishing features of effector cells that preferentially migrate into non-lymphoid tissues to form tissue-resident memory (Trm) cells9,10. Importantly, while the identities of transcription factors (TFs) that promote effector (Id2, Tbx21 (T-bet), Batf), tissue-residency (Runx3, Zfp683, Bhlhe40), and memory cell fates (Eomes, Tcf7, Id3, Myb) are well described1,10–13, the underlying biological processes that regulate the binding and activity of these TFs are largely undefined. Indeed, individual TFs exert overlapping and distinct roles across many cell states (e.g., Tbx21 and Eomes in memory versus exhausted T cells14) highlighting the need to better understand how TF binding site accessibility is regulated to control the behavior of TFs and the differentiation states they specify.
Mammalian SWI/SNF complexes (aka BAF, BRG/BRM-associated factors) are chromatin remodelers that facilitate reorganization of nucleosomes along DNA15. There are three variants: canonical BAF (cBAF), Polybromo-associated BAF (PBAF), and non-canonical BAF (ncBAF), each composed of many shared subunits, but distinguishable by inclusion of unique subunits, including ARID1A/ARID1B (cBAF), PBRM1/ARID2/BRD7 (PBAF), and GLTSCR1/GLTSCR1L/BRD9 (ncBAF)16–18. Enhancer accessibility is particularly sensitive to the loss of ARID1A, a key subunit of the cBAF complex19–21. Normal leukocyte development is accompanied by substantial chromatin remodeling and establishment of novel enhancers; and deletion of either Arid1a or Smarca4 during murine hematopoiesis eliminates the formation of essentially all leukocyte lineages22–24. ARID1A mutations lead to dysregulated gene expression and differentiation patterns and are commonly found in human cancers25,26, including T cell lymphomas26–28. However, it is unknown what role ARID1A plays in differentiating mature T lymphocytes and the generation of CD8+ T cell memory. Given the importance of cBAF in regulating cellular differentiation in diverse contexts, we reasoned that cBAF could play a critical role in regulating the differentiation and functional diversification of activated T lymphocytes, a process that involves extensive chromatin remodeling29,30.
Here we investigated the role of ARID1A-containing cBAF in promoting CD8+ T cell effector differentiation upon acute systemic viral infection. We found that ARID1A was necessary for terminal effector cell differentiation and establishment of long-lived Trm cell populations but was dispensable for circulating memory cell formation. However, Arid1a-deficient circulating memory cells were deficient in production of cytokines, chemokines, and cytotoxic proteins, suggesting that ARID1A was critical for optimizing the functional capabilities of memory cells. Mechanistically, ARID1A was required to induce early expression of TFs that specify TE and Trm fates, as well as to open chromatin at sites where those TFs bind. Arid1a-deficient effector cells displayed reduced global chromatin accessibility and failed to open chromatin regions containing binding sites for ETS, RUNX, bZIP (AP-1), and T-box family transcription factors. Our results demonstrate that ARID1A-containing cBAF complexes act to remodel chromatin that licenses effector differentiation during acute infections and provide insight into how TF recruitment and activity is regulated to instruct the formation of different types of effector and memory CD8+ T cells.
Results
Differentiating antiviral effector CD8+ T cells display highly dynamic accessible chromatin and cBAF occupancy patterns
To better understand the initial waves of CD8+ T cell differentiation during infection, we transferred P14 TCR transgenic cells that recognize the GP33–41 epitope of the LCMV glycoprotein31 into wild-type recipients infected with LCMV-Armstrong, and on days 3 (d3), 5 (d5), and 8 (d8) post-infection (p.i.) we mapped the changes in open chromatin regions (OCRs) and ARID1A binding sites in donor P14 cells using Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq)32 and CUT&RUN33, respectively. In parallel, to assess even earlier changes we performed ATAC-seq on 48-hour in vitro activated P14 cells. We defined 5 OCR clusters corresponding to regions that are unique or conserved across differentiation timespace: (1) Conserved- OCRs conserved at all timepoints (n=21650), (2) Naive- OCRs unique to naive T cells (n=3157), (3) Early Activation- OCRs more selectively found at day 3 p.i. in vivo or 48h post-activation in vitro (n=7883), (4) Activation- OCRs open in all activated cells (n=8781), or (5) Late Activation- OCRs predominantly found at day 5 and day 8 p.i. in vivo effector timepoints (n=1250) (Figures 1A–C). ARID1A binding was highly enriched at OCRs and correlated with the dynamics of open chromatin across the time course of activation (Figures 1A, D; Figure S1A). Cluster annotations revealed that nearly half of Conserved sites align to promoters, while OCRs that were either lost or gained during activation were more frequently found at intergenic and intronic regions (Figure 1C). Consistent with the notion that enhancer rather than promoter accessibility patterns are the predominant determinants of cellular states34, OCRs that opened and closed upon CD8+ T cell activation were predominantly found in enhancers as defined by H3K4me1, H3K27ac35, while promoter-specific H3K4me3 was more strongly enriched at Conserved OCRs than activation-induced OCRs (Figure 1E). In general, regions with increased chromatin accessibility annotated to genes with increased gene expression and vice versa whereas regions with unchanging chromatin accessibility exhibited little changes in gene expression (Figure S1B).
Fig. 1. Differentiating antiviral effector CD8+ T cells exhibit highly dynamic accessible chromatin and cBAF occupancy patterns.
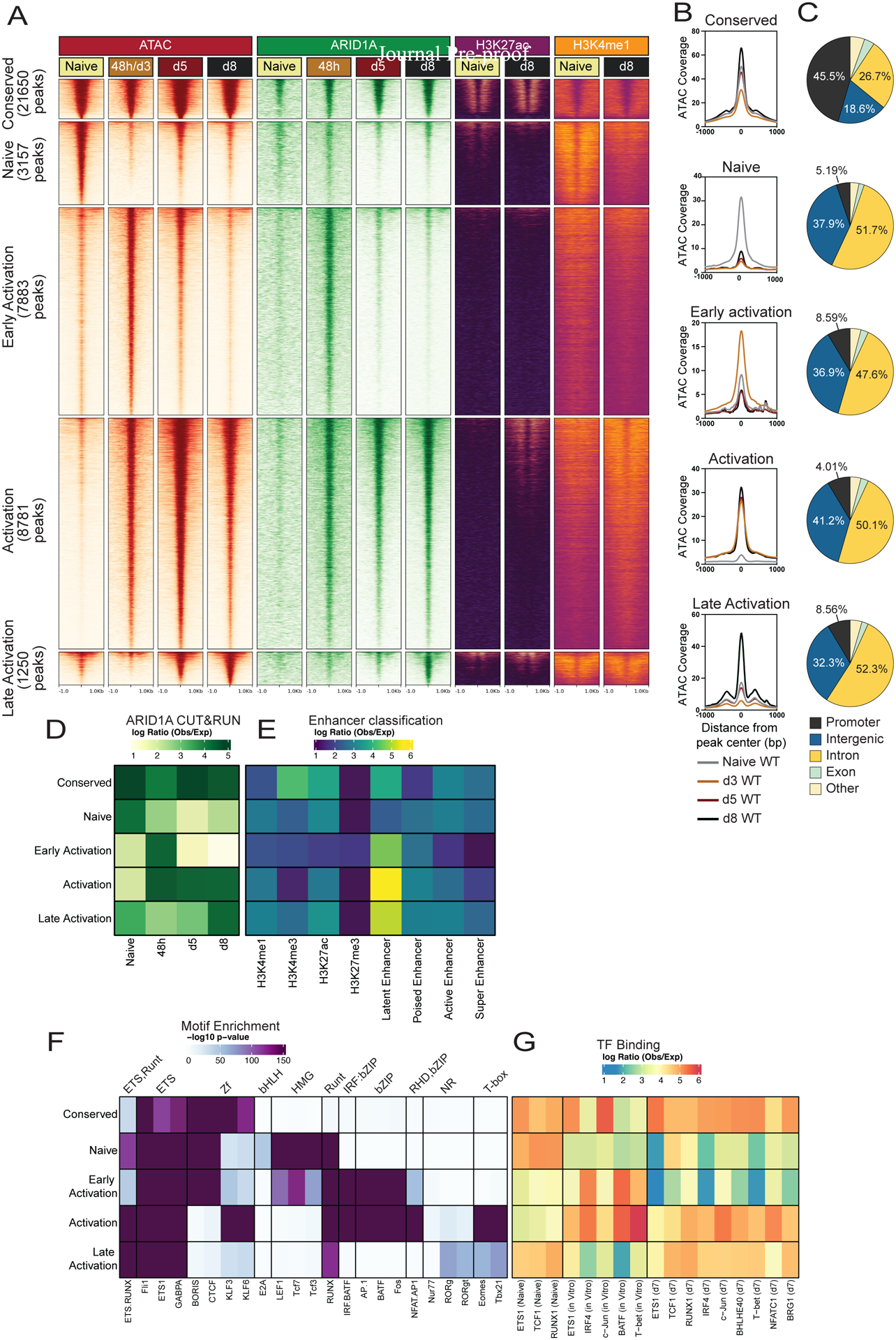
(A) Signal tracks of ATAC-seq, ARID1A C&R, H3K27ac ChIP-Seq and H3K4me1 ChIP-Seq (GSE89036) in naïve, 48h in vitro activated, or d3, d5, or d8 P14 CD8+ T cells from LCMV-Armstrong infection. (B) ATAC-seq signal coverage and (C) annotation of OCRs from clusters in A. (D-G) Using the OCRs from clusters in (A) we correlated those with ARID1A binding (D), histone modifications to define enhancers (E), predicted TF motif enrichment (F) and actual TF binding (observed/expected) of the indicated TFs from public datasets (GSE54191, GSE192390, GSE166718) (G).
TF motif analysis on the five OCR clusters revealed a specific enrichment of HMG motifs (TCF, LEF) in Naive OCRs, while AP-1 (bZIP) motifs were enriched in Early and general Activation OCRs, and the nuclear receptor (NR) and T-box motifs were enriched in the Activation and Late Activation OCRs (Figure 1F; Figure S1C). Next, we overlapped physical TF binding patterns from naive, in vitro activated36,37, and in vivo day 7 CD8+ effector cells38 with our peak clusters (Figure 1G). TF binding of ETS1, TCF1 and RUNX1 in naive cells was primarily restricted to Conserved and Naive OCRs with some binding in Late Activation OCRs, while binding of most TFs including BATF, IRF4, and T-bet strongly overlapped with Early Activation (in vitro activated cells) and Activation (in vitro activated and in vivo d7 cells) induced OCRs. Together, these data outline the temporal kinetics of newly formed enhancers in differentiating virus-specific CD8+ T effector cells and demonstrate that these sites are associated with ARID1A-containing cBAF complexes and distinct sets of TFs at different phases of effector cell differentiation. These findings imply that the cBAF complex plays a central role in shaping the chromatin accessibility landscape and differentiation process in antiviral effector CD8+ T cells.
Arid1a promotes clonal expansion and opening of activation-induced enhancers
To elucidate the function of ARID1A-containing cBAF in activated CD8+ cells while avoiding developmental defects seen with early T cell Arid1a deletions22,24, we crossed Arid1afl/fl mice with Gzmb-Cre+ mice (in which Cre is conditionally active after T cell activation) with Arid1afl/fl mice to generate Gzmb-Cre+ Arid1afl/+ (Arid1acKO) and Gzmb-Cre+ Arid1afl/+ (Arid1acHet) mice39. Gzmb-Cre− Arid1afl/fl or Gzmb-Cre− Arid1afl/+ mice were used as wild-type (WT) littermate controls. We further crossed these mice with P14 TCR transgenic mice (Arid1acKO P14). We then transferred 25,000 wild-type (WT) or Arid1acKO P14 CD8+ T cells into naive congenic recipient mice and infected those mice with LCMV-Armstrong to induce acute viral infection. We confirmed the loss of ARID1A protein in virus-specific CD8+ T cells within 72h of activation in vivo using flow cytometry (Figure S2A).
Clonal expansion of Arid1acKO cells was slightly impaired (~3-fold) early during the expansion phase (day 5 p.i.) and more strongly impaired (~7-fold) at the peak of the effector response (day 8 p.i.) (Figure 2A). P14 CD8+ T cells electroporated with Cas9 Arid1a sgRNA ribonucleoproteins (RNP) similarly expanded less robustly relative to control RNP electroporated cells (Figure S2B). In line with these findings, Arid1acKO cells proliferated slower relative to WT cells as observed using CellTrace violet staining at day 3 p.i. (Figure 2B). Deletion of Arid1a in endogenous effector CD8+ T cells coincided with ~10X higher LCMV titers at day 5 p.i. compared to the Arid1acHet and WT mice (Figure S2C), but then titers were undetectable in all mice by d8 p.i. (data not shown). However, Arid1a deletion had little impact on the numbers of memory CD8+ T cells that formed in the spleen 30–60 days p.i. (Figures 2A and S2B). Thus, there was a strong dependence on ARID1A for the initial effector cell expansion phase, but not for contraction and seeding of the circulating memory pool.
Fig 2. Arid1a promotes clonal expansion and opening of activation-induced enhancers.
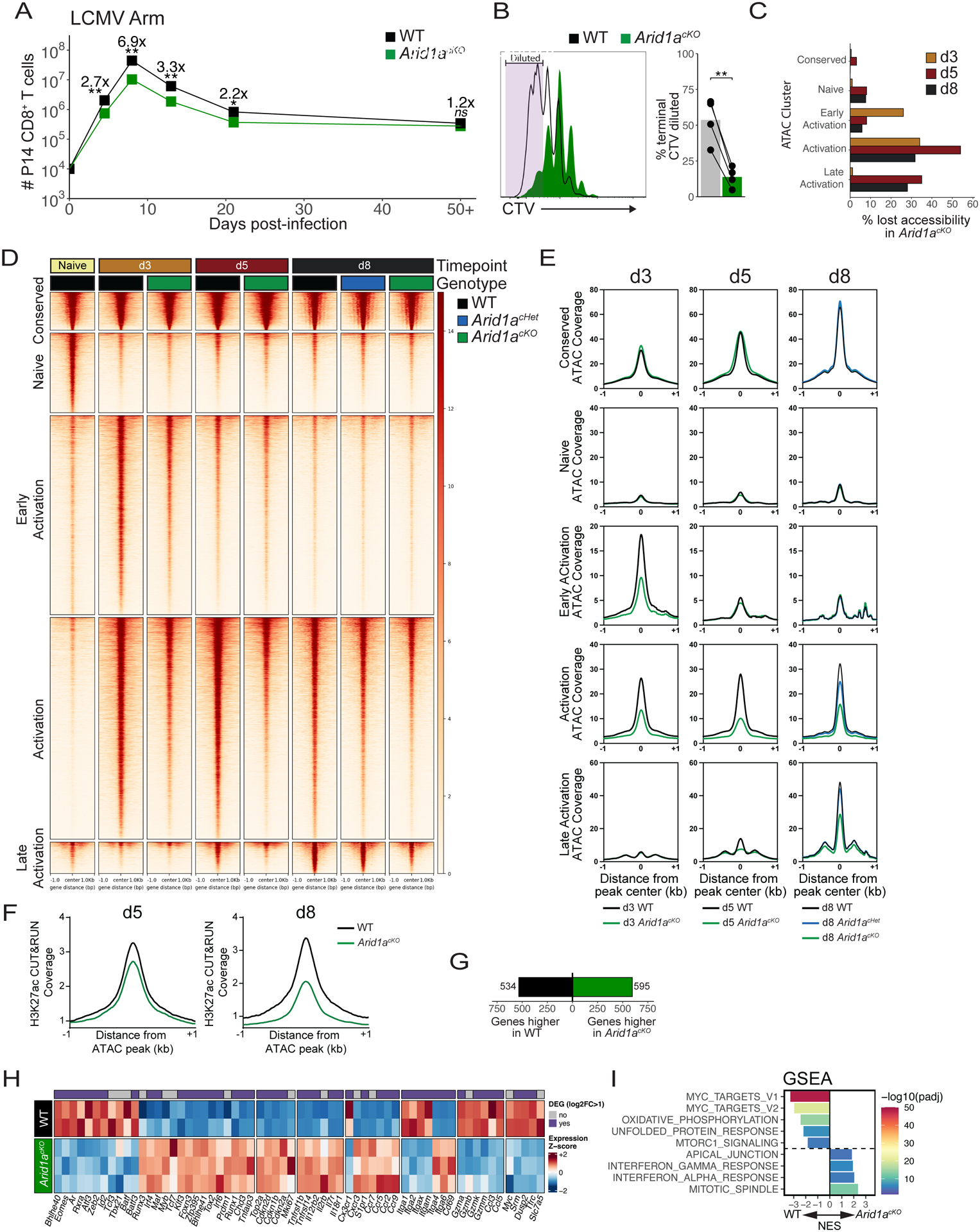
(A) Numbers of WT (black) and Arid1acKO (green) P14 cells after infection with LCMV-Armstrong. n=8 (d4.5), 8–11 (d8), 4 (d13), 4 (d21), 8 (d50+). (B) CellTrace Violet dilution of WT and Arid1acKO P14 cells at 84h p.i. Frequency of cells that divided 7+ times are quantified in the bar graph. (C) Percentage of ATAC-seq peaks lost (2-fold change, FDR<0.05, Benjamini-Hochberg) in Arid1acKO relative to WT cells at d3, d5, and d8 p.i. (D, E) ATAC-seq signal coverage of WT and Arid1acKO P14 cells at d3, d5, and d8 p.i. using the same cluster designations defined in Figure 1A. (F) H3K27ac CUT&RUN signal coverage centered on ATAC-seq peaks in WT vs. Arid1acKO cells. (G-I) WT and Arid1acKO effector cells were isolated at d3 p.i. and compared for differentially expressed genes (DEGs) by RNA-sequencing (>2-fold change, adjusted p-value<0.05, Benjamini-Hochberg). Number of DEGs (G), heatmap of biologically relevant DEGs (H), and gene set enrichment analysis (GSEA) (I) between WT and Arid1acKO effector cells are shown. All heatmap genes shown have an adjusted p-value < 0.05. (I) Top 8 Hallmark gene sets (adjusted p value < 0.01, 10,000 permutations) are shown. ns, not significant; *p<0.05, **p<0.005.
To interrogate how ARID1A-dependent cBAF complexes control chromatin remodeling and enhancer accessibility during CD8+ T cell differentiation we performed ATAC-seq on WT, Arid1acHet, and Arid1acKO P14 cells at d3, d5, and d8 p.i. and assessed chromatin accessibility at the five OCR clusters defined in Figure 1A. As early as day 3 p.i. Arid1acKO cells exhibited defects in chromatin accessibility in ~30–40% (n=2020) of the OCRs in the Early and Activation clusters (Figures 2C–E). By days 5 and 8 p.i., the KO effector cells showed greater loss in ~40–60% (n=4766 (d5), 2789 (d8)) of the Activation and ~30–40% (n=435 (d5), 350 (d8)) of Late Activation OCRs (Figures 2C–E). Several of these OCRs map to activationinduced genes, including effector-associated TFs, Bhlhe40, Tbx21 (T-bet), Zeb2, and Batf (Figure S2D). Sites with reduced accessibility in KO cells were also depleted of H3K27ac deposition indicating that cBAF promotes histone acetyltransferase activity at the enhancers it remodels (Figure 2F). In contrast, accessibility in Conserved OCRs was not affected by Arid1a-deficiency (Figures 2C–E), suggesting that although ARID1A was bound to these promoters and other sites (Figures 1A, D), it was not necessary for maintaining their accessibility. These data demonstrate that cBAF is necessary for the initial opening and/or maintenance of thousands of activation-induced enhancers created during effector CD8+ T cell differentiation.
We next assessed transcriptional changes of d3 early effector WT and KO cells and identified 534 genes significantly lower and 595 genes higher in Arid1acKO cells compared to WT cells (Figure 2G). Interestingly, Arid1acKO cells are markedly deficient in the expression of several effector-associated TFs, including Bhlhe40, Tbx21 (T-bet), Zeb2, and Batf, but expressed higher levels of naive-associated TFs like Maf, Myb, and Tcf7 (Figure 2H). Gene set enrichment analysis (GSEA)40 revealed a strong decrease in Hallmark MYC target genes in Arid1acKO cells (Figure 2I), including Myc, Srm, and Dusp2, as well as other critical regulators of murine T lymphocyte function and cell growth, Tfrc (CD71) and Slc7a5 (CD98) (Figure 2H)41. Additionally, several genes encoding cyclin-dependent kinase inhibitors including Cdkn1b, Cdkn2d, Cdkn2a were more highly expressed in Arid1acKO cells, which together with decreased Myc expression may explain the reduced proliferative rate (Figure 2B). Overall, these data indicate that ARID1A acts early in CD8+ T cell activation to establish OCRs at the enhancers of key T cell differentiation genes, including driver TFs. Failure to open these enhancers and achieve optimal gene expression leads to an overall dampening of early effector CD8+ T cell expansion and differentiation and likely many downstream deficiencies in the activation of subsequent OCRs and late-stage effector differentiation programs.
Arid1a acts in a dose-dependent manner to specify effector subsets
To understand how cBAF regulates the formation of different specialized effector subtypes we first compared WT and Arid1acKO effector cells at 8 days p.i. by flow cytometry and found that the Arid1acKO cells contained very few KLRG1+ and CX3CR1hi cells and were skewed towards a CD127hi MP-like phenotype3 (Figures 3A–B), indicating that terminal effector differentiation was impaired but that cells retained similar potential to give rise to a long-lived circulating memory cell pool. Similar results were obtained in P14 cells in which Arid1a was deleted via Cas9 RNP as well as comparing WT and Arid1acKO endogenous, polyclonal LCMV-specific effector CD8+ T cells (Figures S3A–C). Between 5–20% of KLRG1+ CD127lo TE cells, but not EEC or MP cells, retained ARID1A expression, suggesting the strict requirement for ARID1A for TE differentiation selects for Cre recombinase escapers (Figures S3D–E). Arid1acHet cells displayed an intermediate TE/MP phenotype between that of WT and KO cells (Figures 3A–B), indicating that ARID1A acts in a dose-dependent manner to promote effector cell fates. Arid1acKO effector cells mounted similar cytokine responses as WT cells after GP33–41 peptide stimulation, but produced significantly less IFNγ after stimulation by IL-12 and IL-18 and expressed lower levels of granzyme A (Figures S3F–G). As T-bet is a critical inducer of TE cells6,42, we measured T-bet expression (Figures 3C–D; Figure S3H) and observed ~50% lower T-bet protein in Arid1acKO cells compared to WT cells, consistent with a reduction in Tbx21 transcripts at d3 (Figure 2H). Together, these results indicated that ARID1A-dependent cBAF is needed to permit the differentiation of TE cells, and in the absence of ARID1A, activated T cells fail to upregulate T-bet and preferentially acquired EEC- and MP-like differentiation states.
Fig 3. Arid1a acts in a dose-dependent manner to specify effector subset gene expression patterns.
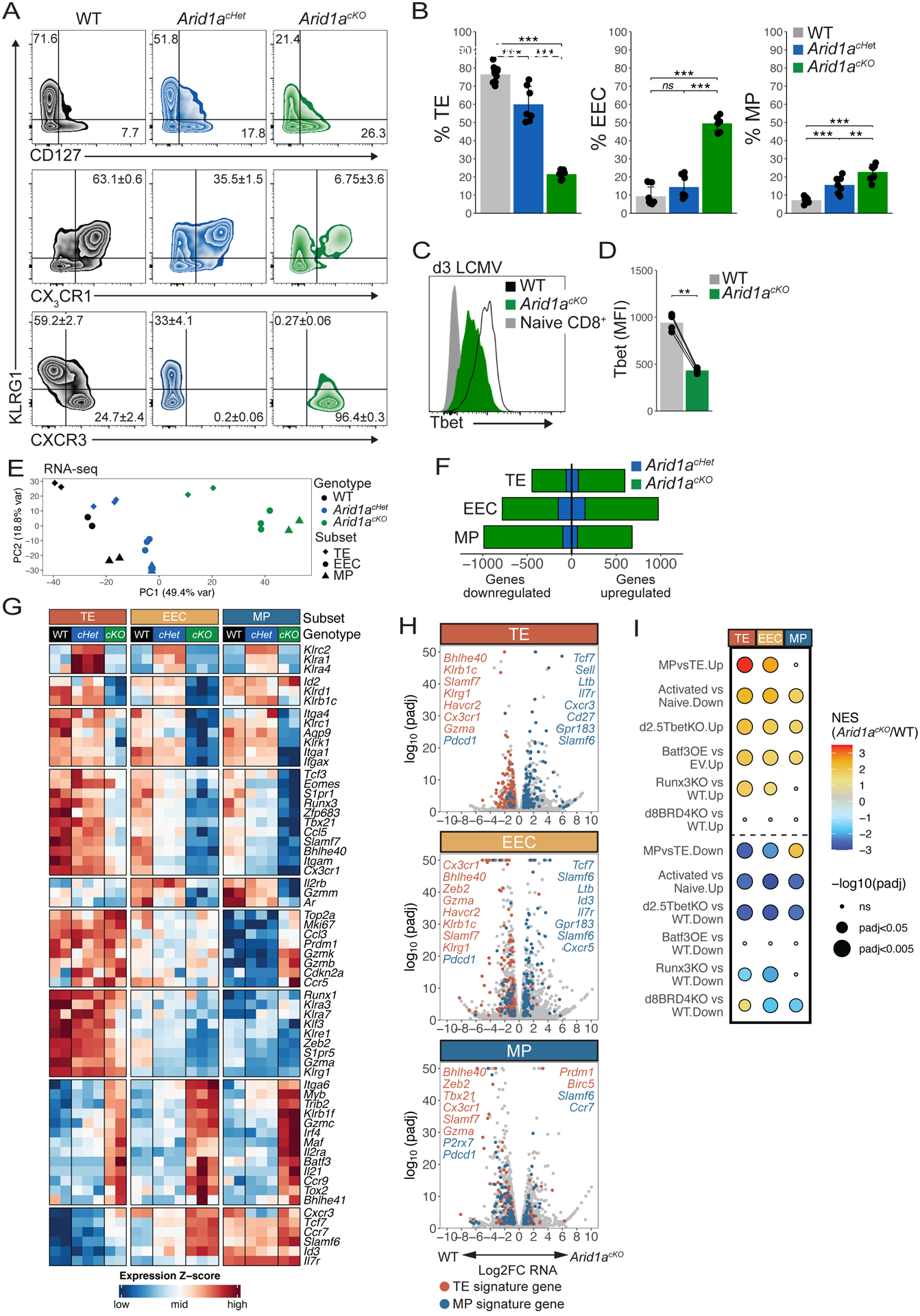
(A,B) Surface marker expression of WT (black, n=11), Arid1acHet (blue, n=7), and Arid1acKO (green, n=7) P14 cells at d8–9 p.i. was analyzed by flow cytometry (A) and mean ± SEM frequencies of TE (KLRG1+CD127−), MP (KLRG1−CD127+), KLRG1+CX3CR1+, KLRG1+CXCR3−, and KLRG1−CXCR3+ cells are shown in bar graphs (B). (C,D) T-bet expression in WT and Arid1acKO P14 cells at d3 p.i. (D) Paired t-test. (E-I) WT, Arid1acKO and Arid1acHet effector cells were isolated at d8 p.i. and sorted based on expression of TE, EEC and MP markers as defined in (A) and differentially expressed genes (DEGs) were identified by RNA-seq. Principal component analysis plot of all the samples (E), number of DEGs (F), heatmap of select DEGs (G), volcano plots of all DEGs including highlighted TE-signature genes (red) and MP-signature genes (blue) (H), and GSEA (I) were used to assess the DEGs in each subset affected by loss of one or two copies of Arid1a. GSEA analysis in (I) directly compared DEGs between WT and Arid1acKO cells and previously published datasets: Activated vs Naive: GSE10739; d2.5TbetKO: PRJNA547650; Batf3OE vs EV: GSE143504; Runx3KO: GSE81888; d8BRD4KO: GSE173515). ns, not significant; *p<0.05, **p<0.005, ***p<0.0005.
We next performed bulk RNA-seq on FACS-purified KLRG1+ CD127lo (TE), KLRG1− CD127lo (EEC), and KLRG1− CD127hi (MP) subsets from WT, Arid1acHet, and Arid1acKO cells at d8 p.i. Principal component analysis (PCA) revealed that the majority of transcriptional variation was explained by the underlying donor cell genotype, and secondarily by subset, indicating that Arid1acHet and Arid1acKO subsets are transcriptionally distinct from their WT cell counterparts (Figures 3E–F). Differential gene expression analysis revealed that 446 TE, 779 EEC, and 986 MP genes were downregulated and 597 TE, 973 EEC, and 679 MP genes were upregulated in Arid1acKO cells relative to WT cells (Figure 3F). DEGs in Arid1acHet cells (downregulated: 61 TE, 151 EEC, 99 MP; upregulated: 75 TE, 150 EEC, 65 MP) were almost entirely a subset of those affected in Arid1acKO cells.
Across all effector subsets, Arid1acKO cells expressed lower levels of several TFs (Tbx21, Zeb2, Eomes, Zfp683, Bhlhe40), trafficking and adhesion molecules (Cx3cr1, S1pr1, Itga1, Itga4) and NK cell-associated genes (Klrk1 (NKG2D), Klrc1 (NKG2A)) relative to the WT cells (Figure 3G). Conversely, Arid1acKO cells expressed higher levels of TFs (Irf4, Myb, Maf, Batf3), chemokine receptors (Ccr9, Ccr7, Ccr5) and the cytokine Il21. In general, Arid1acHet cells had an intermediate gene expression phenotype between WT and Arid1acKO cells, except for a small number of genes (Klrc2, Klra1, Klra4, Lrp6) that exhibited ectopic expression in Arid1acHet cells, but not in Arid1acKO cells (Figure 3G). Additionally, Cxcr3 gene expression and CXCR3 protein was reduced in Arid1acHet cells but increased in the Arid1acKO cells (Figures 3A, G). Analysis of TE- and MP-signature genes within the individual subsets showed that several TE-signature genes such as Tbx21, S1pr5, Cx3cr1, Zeb2, GzmA, and Klrg1 were downregulated and several MP-signature genes like Tcf7, Id3, Cxcr3 were upregulated in Arid1acKO KLRG1+ CD127lo TE cells relative to WT TE cells (Figure 3H). Indeed, GSEA showed that Arid1acKO TE cells were enriched with a more MP-like transcriptional signature relative to WT TE cells (Figure 3I). The failure to activate TE-signature genes and repress MP-signature genes was also observed in the Arid1acKO KLRG1− CD127lo EECs (Figures 3G–H). Conversely, comparison of WT and Arid1acKO KLRG1− CD127hi cells revealed aberrant expression patterns of both MP- and TE-signature genes in Arid1acKO MP cells (Figures 3G–H). Specifically, Arid1acKO MP cells expressed higher levels of MP-signature genes like Ccr7 and Slamf6 and TE-signature genes like Prdm1, Gzmk, GzmB and Ccr5, but lower levels of other MP-signature genes like Gzmm and Aqp9 (Figures 3G–H). In general, the Arid1acKO cells shared gene expression patterns similar to those observed in Tbx21 KO43 and Runx3 KO CD8+ T cells44 (Figure 3I). These data reveal that loss of Arid1a has profound effects on the ability of activated CD8+ T cells to properly engage canonical effector and memory gene expression programs and that Arid1a is haploinsufficient for the expression of many of these genes.
Arid1a-dependent OCRs in d8 effector cells are largely shared across subsets
To better understand how Arid1a regulates gene expression and cell fate specification of CD8+ T cells, we analyzed OCRs in TE, EEC and MP subsets from WT, Arid1acHet, and Arid1acKO cells at d8 p.i. PCA revealed that the principal source of variation in global chromatin accessibility was dictated by donor cell genotype rather than subset (Figure 4A). Critically, comparison of differential OCRs between WT and Arid1acKO subsets showed that nearly all differential OCRs lost accessibility in Arid1acKO cells whereas only a fraction of sites gained accessibility (Figure 4B), indicating that the primary function of cBAF in activated CD8+ T cells is to create and/or maintain OCRs through nucleosome remodeling. Analysis of Arid1acHet cells showed a statistically significant loss (≥2-fold) of chromatin accessibility at ~1000 sites, all of which were also affected in Arid1acKO cells, and essentially no gains (Figures 4B–C). Additionally, nearly every OCR significantly lost in Arid1acKO cells displayed a substantial reduction in accessibility in the Arid1acHet cells (Figure 4C), indicating that the level of accessibility is sensitive to the dosage of cBAF activity, which has clinical relevance to T cell lymphoma where Arid1a haploinsufficiency is commonly observed27. Next, we compared the differential OCRs between TE, EEC and MP subsets to see if loss of Arid1a affected one subset more than another. Arid1a-deficiency impaired the opening of 12,260 OCRs in TE cells, which helps to explain the lack of TE development in the Arid1acKO cells, but surprisingly, even more OCRs (EEC n=16400, MP n=15110) were affected in the EEC- and MP-like subsets (Figures 4B, D). Further, of the OCRs lost in the Arid1acKO d8 effector subsets, the majority were common to all three subsets (n=7802) whereas a minority were subset-specific (TE n=2169, EEC n=2843, MP n=2943) (Figure 4D). Together, these data demonstrate that although Arid1a-deficiency preferentially impaired the formation of KLRG1+ TE cells, there was no biased requirement for cBAF activity in any one subset. Rather, all three subsets required cBAF to create thousands of enhancers both common to and uniquely found in each subset, and loss of cBAF activity resulted in aberrant gene expression patterns and differentiation states in all three subsets.
Fig 4. ARID1A-dependent OCRs in d8 MP, EEC, and TE cells are largely shared across subsets.
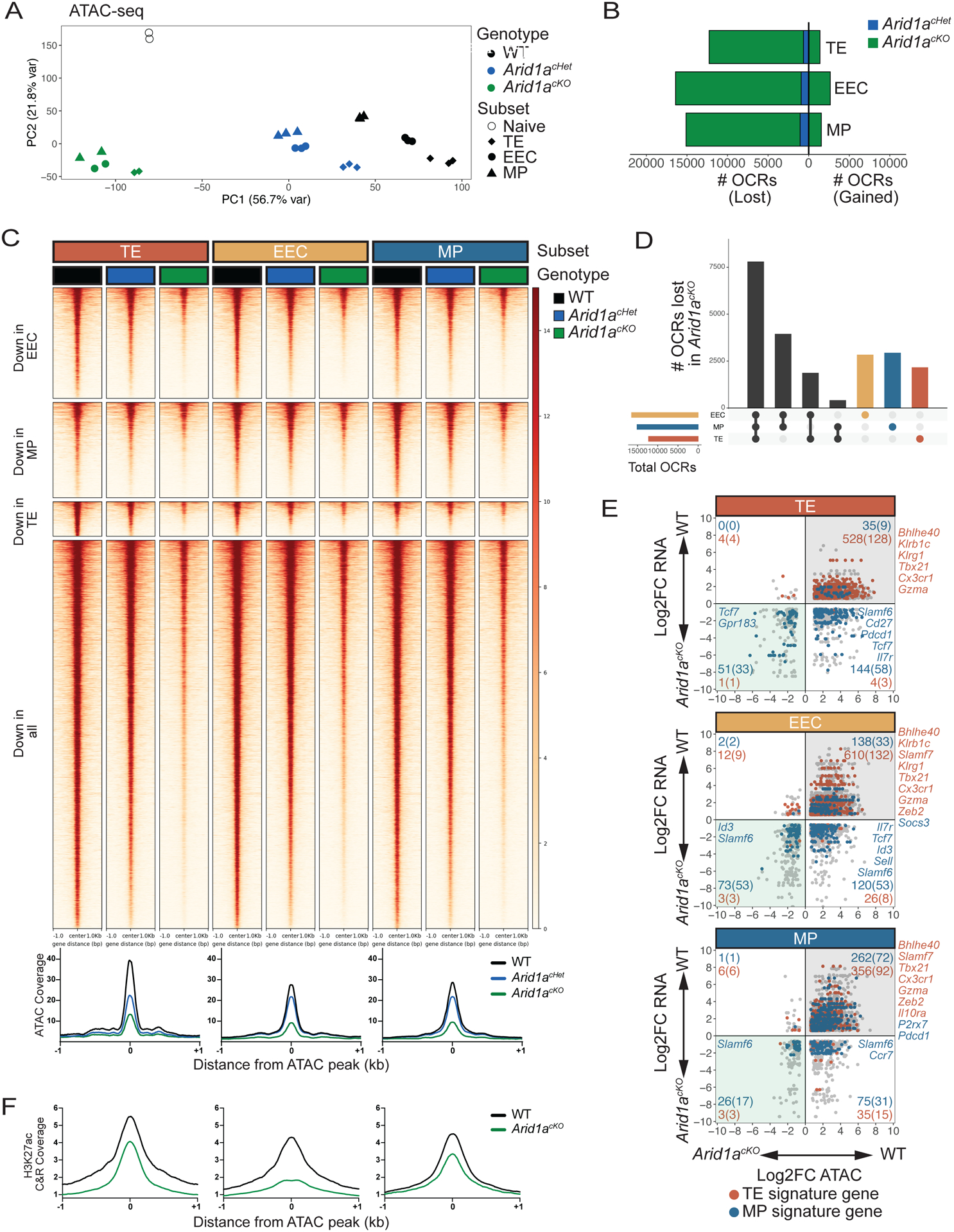
WT, Arid1acKO and Arid1acHet effector cells were isolated at d8 p.i. and sorted based on expression of TE, EEC and MP markers as defined in Figure 3A and differential OCRs were compared by ATAC-sequencing (2-fold change, adjusted p value<0.05, Benjamini-Hochberg). (A) Principal component analysis plot of ATAC-seq from WT, Arid1acHet, and Arid1acKO subsets at d8 post-infection. (B) Number of OCRs lost and gained in Arid1acKO and Arid1acHet subsets relative to WT cells. (C-D) ATAC-seq signal heatmaps of WT, Arid1acHet, and Arid1acKO d8 subsets. OCRs are clustered by whether they are lost in the Arid1acKO relative to WT cells in individual subsets or lost in all three subsets (C) and UpSet plot shows the number of shared and subset-specific OCRs lost in Arid1acKO subsets relative to WT cells (D). (E) Gene expression and paired chromatin accessibility of annotated OCRs in Arid1acKO and WT d8 subsets. TE- and MP-signature genes are highlighted in blue and red, respectively. (F) H3K27ac CUT&RUN signal coverage centered on ARID1A-dependent ATAC-seq peaks in WT vs. Arid1acKO cells TE, EEC, and MP cells.
It is well established that accessibility of cis-regulatory elements strongly correlates with gene expression. Indeed, we found that many DEGs in EEC, TE and MP cells also lost OCR accessibility in the absence of ARID1A (Figure 4E). We focused on the expression of TE- and MP-signature genes within each subset and as predicted there was a strong requirement for Arid1a for both the accessibility and expression of TE- and MP-signature genes in all the subsets (although, by definition, the expression of TE-signature genes in MP cells was considerably lower than in TE cells and vice versa (see Figure 3G)). Most Arid1a-dependent OCRs that were annotated to genes downregulated in Arid1acKO vs. WT cells had reduced H3K27ac, as exemplified by Tbx21, Bhlhe40, Zfp683, and Prdm1 (Figures 4F; Figure S4A). Interestingly, we also observed loss of chromatin accessibility at sites annotated to genes that were upregulated in Arid1acKO EEC, TE, and MP cells (Figure 4E, lower right quadrants), including many MP-signature genes. At such loci (e.g. Sell, Ccr7, Cd9, and Tcf7), H3K27me3 was reduced in Arid1acKO EEC and TE cells suggesting that ARID1A promotes Polycomb-dependent silencing of MP-signature genes in these subsets (Figures S4B). Collectively, these data indicate that ARID1A is required in a dose-dependent manner to generate chromatin accessibility at shared and subset-specific OCRs induced during CD8+ T cell differentiation. Failure to generate these accessible regions in Arid1acKO cells impairs their ability to properly activate ‘proeffector’ genes and regulate ‘pro-memory’ genes, perhaps due to a loss in the binding of TFs and epigenetic modifiers with activating and repressive regulatory properties at ARID1A-dependent accessible sites.
cBAF is required for targeting of T-bet to enhancers in effector CD8+ T cells
To determine which TFs were most dependent on ARID1A activity, we performed TF motif enrichment on the ARID1A-dependent OCRs for each d8 effector subset. This revealed an enrichment of ETS, Runt, bZIP, T-box, and other TF family motifs, suggesting that binding sites for these factors may be less accessible in Arid1a-deficient effector cells, while CTCF motifs were enriched among gained sites (Figure 5A). In support of this, binding of FOSL2, EOMES, BATF, and T-bet37 was selectively and highly enriched at ARID1A-dependent, but not ARID1A-independent OCRs (Figure 5B). To directly examine how certain TFs were affected by ARID1A deficiency we performed CUT&RUN on WT and Arid1acKO d5 effector cells against ETS1, BATF, and T-bet (representing ETS, bZIP, and T-box family members) and mapped their binding at ARID1A-dependent and -independent OCRs (Figure 5C). This showed a significant and strong reduction in ETS1, BATF, and T-bet binding at ARID1A-dependent OCRs, while ETS1 and BATF binding was less affected at ARID1A-independent OCRs. T-bet binding was universally affected genome-wide, likely because of both reduced T-box motif availability and reduced T-bet expression. The TF binding sites most affected by ARID1A-deficiency were predominantly found at intergenic and intronic sites (i.e., enhancers), whereas those bound to ARID1A-independent sites were more frequently annotated to promoters (Figure 5C). These data demonstrate that cBAF-dependent remodeling is predominantly required in activated CD8+ T cells to create enhancers that allow the binding of many key TFs that in turn, govern the gene regulatory networks controlling effector and memory CD8+ T cell differentiation.
Fig 5. cBAF is required for targeting of T-bet to enhancers in effector CD8+ T cells.
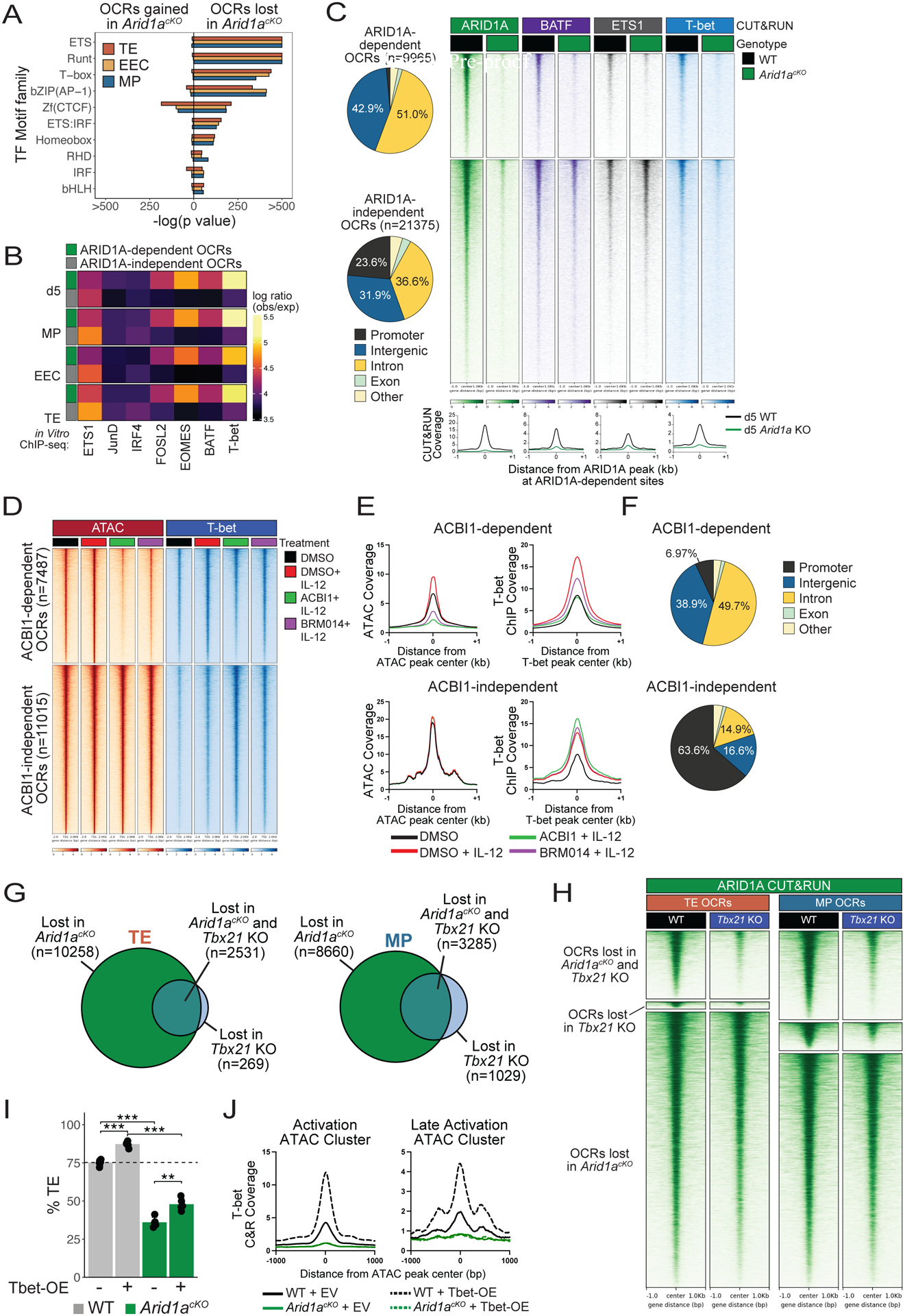
(A-B) As described in Figure 4, OCRs (2-fold change, adjusted p value<0.05, Benjamini-Hochberg) lost in Arid1acKO d5 effector cells or d8 TE, EEC, and MP subsets relative to WT cells were analyzed for enrichment of predicted TF motifs (A) or TF binding signals (observed/expected) of indicated TFs from public ChIP-seq datasets (GSE192390) in ARID1A-dependent (green) and - independent (gray) OCRs (B). (C) Genomic annotations and CUT&RUN signal heatmaps of ARID1A, BATF, ETS1, and T-bet at ARID1A-dependent or -independent OCRs from WT and Arid1acKO effector P14 CD8+ T cells d5 post-infection. (D-F) CD8+ T cells were activated in vitro for 48hrs and then treated with DMSO (black, red), ACBI1 (green) or BRM014 (purple) for 4 hours, and then treated with IL-12 (red, green, purple) for 2 hours, and analyzed for changes in ATAC-seq and T-bet binding by ChIP-seq. Signal coverage heatmaps of ATAC-seq and T-bet ChIP-seq (D) and histograms measuring chromatin accessibility (E) are shown. (F) Genomic annotations of ACBI1-dependent and -independent ATAC-seq OCRs. (G) Overlap of OCRs lost relative to WT (2-fold change, adjusted p value<0.05, Benjamini-Hochberg) in Arid1acKO or Tbx21 KO TE and MP cells. (H) ARID1A CUT&RUN signal heatmap in WT or Tbx21 KO effector cells at d5 p.i. Signal is centered on ARID1A- or Tbet-dependent OCR peaks identified in (G). (I) Retroviral overexpression of T-bet fails to rescue TE formation in Arid1acKO cells at d8 p.i. (J) T-bet CUT&RUN signal coverage histograms at Activation and Late Activation OCRs in WT or Arid1acKO cells transduced with either empty vector (EV) or T-bet overexpression (Tbet-OE) retrovirus at d5 p.i.*p<0.05, **p<0.005, ***p<0.0005.
However, a potential caveat in the experiments above is that the defects in TF binding could stem from several alterations in the ARID1A-deficient cells including reduced expression of certain TFs. Therefore, to bypass some of these confounding issues we used a model of acute disruption of cBAF subunits in in vitro activated T cells with ACBI145, a PROTAC targeting BRG1, BRM, and PBRM1, or BRM01446, an allosteric ATPase inhibitor of BRG1 and BRM. P14 CD8+ T cells were activated in vitro for 48 hours and then treated with either DMSO or ACBI1/BRM014 for 4h prior to 2h treatment with IL-12 to enhance T-bet expression and binding, after which the cells were collected for T-bet ChIP-seq and ATAC-seq. This revealed that BAF complexes were required to maintain chromatin accessibility at ~7500 sites following BAF disruption (Figures 5D–E) and T-bet binding was selectively reduced at these sites. In contrast, T-bet binding was preserved or enhanced at BAF-independent OCRs (Figures 5D–E). Again, cBAF-dependent T-bet binding sites were almost universally annotated to intronic and intergenic elements, while cBAF-independent sites were more highly enriched for promoters (Figure 5F). These data indicate that ATP-dependent cBAF complex remodeling is acutely required to maintain T-bet binding at enhancers in activated CD8+ T cells.
To further assess how T-bet and cBAF may cooperate, we compared the OCRs between WT or Tbx21 KO P14 CD8+ T cells at d8 p.i. and found that Tbx21-deficiency resulted in reduced and gained accessibility at several thousand sites (Figure S5A). Notably, the majority of sites that lost accessibility in Tbx21 KO cells also lost accessibility in Arid1acKO cells (n=2531, 90.4% (TE); n=3285, 76.1% (MP) (Figure 5G), and ARID1A binding was strongly reduced in Tbx21 KO cells at these T-bet/ARID1A co- dependent OCRs (Figure 5H), strongly suggesting that T-bet engages ARID1A to promote accessibility at this subset of activation-induced OCRs. Finally, we tested if the requirement for cBAF to generate TE cells could be overridden by T-bet over-expression. To this end, we over-expressed T-bet in WT or Arid1acKO P14 cells and transferred them into mice infected with LCMV-Armstrong. Intriguingly, T-bet overexpression was unable to restore the ability of Arid1acKO cells to form TE cells (Figure 5I; Figure S5B), indicating that higher levels of T-bet are insufficient to compensate for the loss of cBAF activity due to decreased enhancer accessibility. Furthermore, while T-bet overexpression led to a significant enhancement of T-bet binding in WT cells, the stark inability of T-bet to bind chromatin in Arid1acKO cells was essentially unaltered by T-bet overexpression (Figure 5J), indicating that T-bet is completely reliant on ARID1A to make its binding sites accessible. Collectively, these data demonstrate that cBAF is critical for establishing and maintaining accessibility of thousands of enhancers and binding of several effector-associated TFs, particularly T-bet, in activated CD8+ T cells. Further, while T-bet is necessary and sufficient to induce TE cell differentiation, this activity is entirely reliant on ARID1A.
Arid1a is critical for CD8+ Trm formation
Our results demonstrated that similar numbers of WT and Arid1acKO memory CD8+ T cells formed in the spleen following LCMV-Armstrong infection, yet the genetic and epigenetic analysis of MP-like cells revealed several alterations in the Arid1acKO cells. Therefore, to more broadly understand how Arid1a affected memory T cell development we computationally predicted the TFs most functionally impacted in MP cells by Arid1a-deficiency using the Taiji PageRank algorithm that assesses the global influence of TFs in a given cell state47. This analysis identified ZFP683, BHLHE40, RUNX3, RXRA and SMAD3 in addition to T-bet and EOMES as top-ranked TFs whose activity is predicted to be impaired in Arid1acKO MP cells (Figure 6A; Figure S6A). This was noteworthy because ZFP683, BHLHE40, and RUNX3 are critical to the formation of tissue-resident memory (Trm) cells10,12,13 and indeed, MP cells required ARID1A for promoting expression of both Zfp683 and Bhlhe40, as well as increasing accessibility and H3K27ac deposition at cis-regulatory elements for both genes (Figure 3G; Figure S4A). Furthermore, GSEA showed significant similarity between the DEGs in Arid1acKO and Runx3 KO cells (Figure 3I). These data prompted us to compare the numbers and types of WT and Arid1acKO memory CD8+ T cells that form in lymphoid vs. non-lymphoid organs. In the spleen, we observed similar numbers of circulating WT and Arid1acKO TCM and TEM cells (Figure 6B); however, fewer CD127lo terminal TEM (t-TEM) cells were present in the Arid1acKO cells as expected given that this population descends from TE cells6,48. Circulating Arid1acKO memory cells were also less proficient in cytokine production after restimulation ex vivo (Figure 6C; Figure S6B) and failed to undergo terminal effector differentiation following secondary heterologous infection (Figure 6D), indicating that despite being dispensable for circulating memory cell formation, ARID1A was required for preserving critical memory cell antiviral functions. Arid1acKO memory cells also expressed higher levels of TCF1 (Figure 6E). In contrast to the spleen, in the peripheral tissues such as the liver, small intestine intraepithelial layer (SI-IEL) and salivary gland (SG), there was a profound reduction in the number of Trm cells in the Arid1acKO memory cells relative to the WT controls (Figure 6F; Figure S6C). Further, the memory cells found in the SI-IEL and SG lacking Arid1a failed to upregulate several canonical features of Trm, including CD103, CD49a (Figure 6G; Figure S6D), and Granzyme B (Figure 6H), while expression of CD69 and Granzyme A were comparable with WT Trm expression levels (Figure 6G; Figure S6E). Arid1acKO cells infiltrated into the small intestine at slightly lower rates early during infection (Figure S6F), suggesting that the initial seeding of tissues also contributes to, but does not fully account for the diminished Trm pool. Interestingly, as seen in the circulating memory T cells in the spleen (Figure 6E), Arid1acKO Trm maintained high expression of TCF1 (Figure 6I), a known suppressor of Trm formation49. This is in spite of the fact that TCF1 was similarly repressed in WT and Arid1acKO early effector cells in both lymphoid tissues and in the SIIEL (Figure S6G). However, cBAF activity was seemingly required to sustain repression of TCF1 as effector cells underwent further effector cell diversification (Figure S6H). Taken together, these results demonstrate that despite observing normal numbers of circulating memory CD8+ T cells, Arid1a is required in progenitor cells to open key loci that permit the cells to enter tissues and respond to local cues that induce Trm properties. In summary, we have mapped how cBAF remodels chromatin in virus-specific CD8+ T cells during the first week of infection by creating thousands of enhancers that recruit TFs to cooperatively drive gene expression programs that govern the multitude of effector and memory differentiation states.
Fig 6. ARID1A is critical for Trm formation.
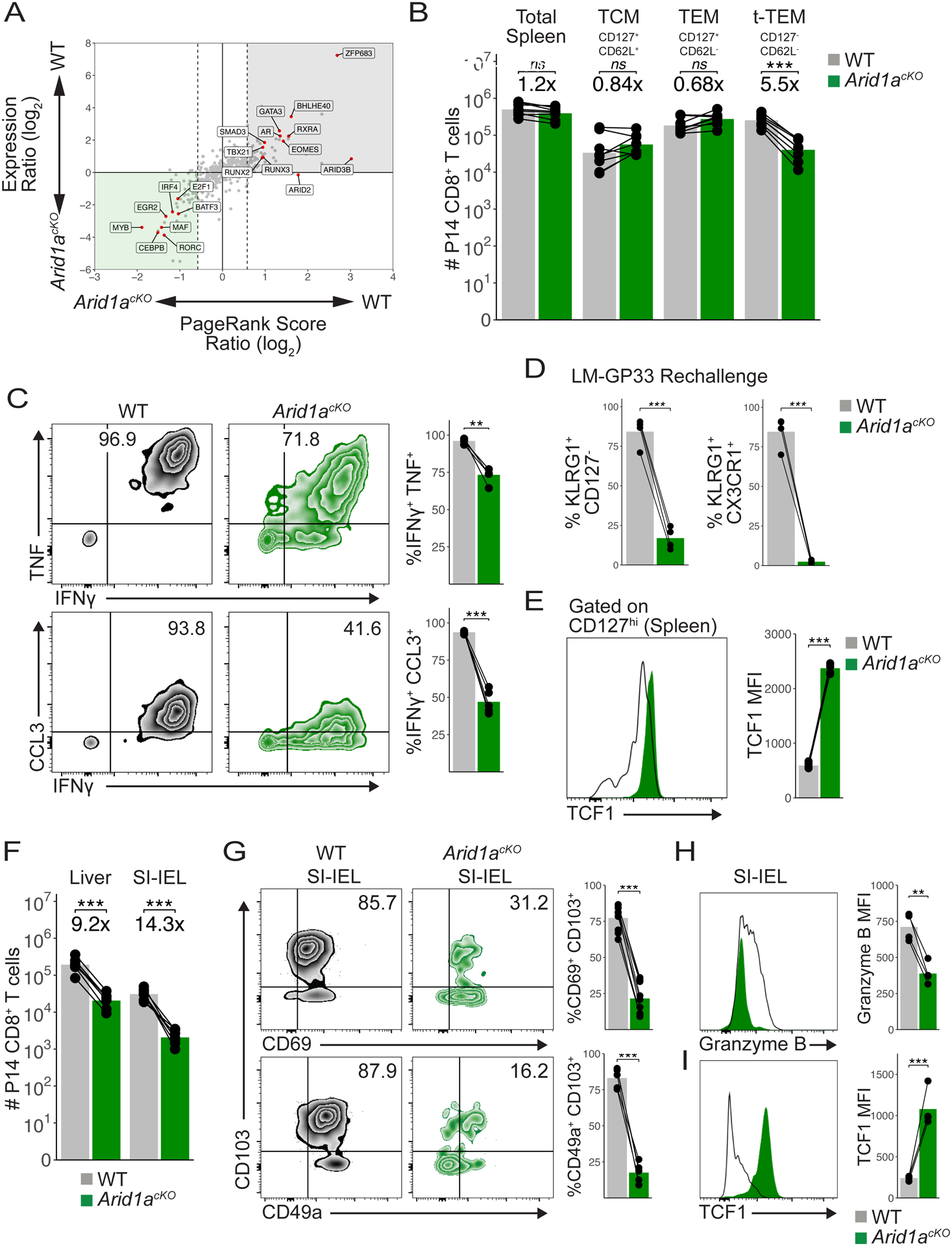
(A) PageRank and mRNA expression analysis of Arid1acKO and WT MP cells from d8 p.i. (B) Absolute numbers of WT (n=8) and Arid1acKO (n=8) P14 splenic memory cells at d30-d60 p.i. (C) Representative cytokine production in WT (n=4) and Arid1acKO (n=4) memory P14 cells in the spleen at d60 p.i. Mean frequency of IFNg+TNF+ (top) and IFNg+CCL3+ (bottom) populations are shown. (D) Frequency of KLRG1+CD127− or KLRG1+CX3CR1+ secondary effector cells 8d p.i. following LM-GP33 infection in mice that previously received CD127+ WT and Arid1acKO LCMV memory P14 cells. (E) TCF1 staining in WT and Arid1acKO CD127hi spleen memory cells at d50 p.i. (F) Absolute numbers of WT and Arid1acKO P14 cells in the liver (n=7) and SI-IEL (n=7) at d50-d60 p.i. (G) Representative flow cytometry plots of WT and Arid1acKO P14 SI-IELs at d60 p.i. Mean frequency of CD69+CD103+ (top) and CD49a+CD103+ (bottom) populations are shown. Granzyme B (H) and TCF1 (I) staining in WT and Arid1acKO P14 SI-IELs at d60 p.i. Paired t-test; *p<0.05, **p<0.005, ***p<0.0005.
Discussion
Effector CD8+ T cell differentiation is a progressive process that occurs over several days to weeks50, with the earliest known cell fate imprinting events occurring as early as the first cell division51. This process is critical to host defense as it generates several types of effector and memory cells that work cooperatively to protect the host from the current infection as well as from future infections. Over the past 15 years the field has discovered many TFs that control the formation of effector and memory CD8+ T cells, but what we don’t understand very well is ‘how these TFs find their binding sites to regulate target gene expression and T cell differentiation?’ The purpose of this study was to characterize how TF binding sites become accessible within activated CD8+ T cells during an immune response by nucleosome remodeling machinery, like the cBAF complex, to govern the spectrum of effector and memory differentiation states produced.
To address this question, we profiled open chromatin profiles over the first week of infection and found that activated CD8+ T cells simultaneously lose accessibility of naive-associated sites while gaining thousands of activation-induced sites in a time-dependent fashion. Loss of cBAF activity strongly prevented the gain of accessibility at activation-induced sites as early as 48–72 hours post activation in vivo, in agreement with a recent report52. However, the loss of naive-associated sites upon T cell activation was not affected by cBAF-deficiency. Most ARID1A-dependent OCRs were annotated as enhancers, and indeed many of these sites required ARID1A to become decorated with H3K27ac and enhance transcriptional activation, indicating that cBAF predominantly shapes the enhancer landscape in activated CD8+ T cells. In contrast, several pro-memory genes like Tcf7, Cd9, Sell, and Ccr7 that become epigenetically silenced via PRC2 (H3K27me3) in late-stage TE cells53,54 had reduced H3K27me3 and were derepressed in the absence of ARID1A. Notably, cBAF directly antagonizes PRC1, resulting in the redistribution of PRC2 upon deletion/disruption of cBAF subunits, impacting H3K27me3 levels and gene expression both positively and negatively55–60. While PRC2 activity is targeted selectively to many pro-memory genes as the effector cells terminally differentiate and lose memory cell potential35,53,54,61, the mechanism of targeting is unclear. Further dissection of the OCRs and TFs that regulate PRC2 targeting in activated CD8+ T cells will illuminate whether cBAF-dependent remodeling is required for the binding of TFs and associated repressors at these sites, particularly for enhancers bound by TE-inducing TFs, such as T-bet, ZEB2, and Blimp-142,62–64, that help to silence such pro-memory loci to enforce terminal differentiation in TE cells.
Our study revealed how cBAF regulates the binding potential of TF families (e.g. ETS, Runt, bZIP, T-box) at enhancers, but not promoters, in activated CD8+ T cells during the first week of infection. Reduced TF binding in Arid1acKO cells can be attributed to a combination of both reduced binding site availability and reduced TF expression. For example, overexpression of T-bet, a factor necessary and sufficient for TE formation, was unable to restore TE differentiation in Arid1a-deficient cells that lacked accessible binding sites. Furthermore, pharmacological cBAF disruption in effector cells reduced both chromatin accessibility and T-bet binding within hours, suggesting that cBAF is also required to maintain late-stage TF activity, consistent with recent reports that loss of chromatin accessibility is observed within minutes of acute BAF degradation65,66. Given the importance of sustained activity by TFs such as FOXO1 for the active maintenance of long-lived memory cells67, future work is thus needed to understand the role of continuous cBAF activity in maintaining the longevity and functional competency of effector and memory T cells.
In the absence of ARID1A, activated CD8+ T cells were defective in developing into TE cells, but not circulating TEM or TCM cells. A recent publication from Guo et al.52 similarly found Arid1a was important for TE cell formation, but in contrast to our findings, they suggested that disruption of cBAF enhanced the formation of memory T cells while we observed no significant difference in memory cells numbers. These differences may be attributed to differences in the timing of Arid1a deletion, or incomplete deletion or generation of Arid1a heterozygous cells in the CRISPR KO setting. Given that they propose a potential translational application of treating activated CD8+ T cells with BAF inhibitors (BD98) to generate more tenacious anti-tumor T cells for adoptive cell therapy, it is worth emphasizing that our study illuminated that ARID1A-deficient memory precursor cells were epigenetically and transcriptionally aberrant and lacked thousands of enhancers. Moreover, our data indicate that ARID1A-deficient effector and memory cells were less proficient at cytokine production and expressed lower levels of Granzyme A, suggesting potential deficiencies in functionality and cytotoxicity. Therefore, ARID1A is evidently dispensable for some aspects of memory cell differentiation (e.g. clonal expansion of IL-7R+ MP-like cells) during LCMV infection, but it is necessary for other key functions and most importantly, for the establishment of tissue residency as elaborated below.
A major finding from our study, which was not explored in Guo et al., is that ARID1A was needed for Trm cell development in non-lymphoid tissues after LCMV infection. Fewer Arid1acKO cells initially infiltrated into the intestines and expressed lower levels of tissue retention molecules, indicating that ARID1A promotes both trafficking into and survival within peripheral tissues. Further, intestinal Arid1acKO cells displayed decreased expression of Trm -signature proteins CD103 and Granzyme B, indicating that these cells were developmentally less mature and likely less protective. Failure of Arid1acKO cells to efficiently form Trm several weeks after infection is likely a consequence of reduced expression of relevant TFs (e.g., Zfp683, Bhlhe40, Runx3) in addition to reduced binding motif accessibility. Runx3 promotes Trm formation in non-lymphoid organs13, but was downregulated in Arid1acKO MP cells, and the Runt (Runx1/2/3) motif is among those most profoundly lost in KO cells. Likewise, Bhlhe40 was identified as a core Trm signature gene13, and was strongly induced in WT effector cells but not in Arid1acKO cells. As was the case with T-bet, overexpression of any individual Trm-associated TF in Arid1acKO cells is unlikely to rescue the Trm formation due to a lack of available binding sites, and because several non-redundant Trm-associated TFs are simultaneously downregulated in these cells. Conversely, Arid1acKO Trm expressed higher levels of TCF1, a factor known to suppress lung Trm formation49. In line with these observations, we observed decreased H3K27me3 deposition at the Tcf7 locus in effector Arid1acKO cells. TGFβ signaling suppresses Tcf7 expression49 and drives Trm formation in many tissues68, and therefore another possible explanation for reduced Trm is that Arid1acKO cells fail to transmit TGFβ signals via SMAD2/3 binding to repress Tcf7, a hypothesis supported by the reduction in SMAD3 activity in MP cells as predicted by PageRank analysis. CD8+ Trm cells provide protection against mucosal infections69 and have promising beneficial prognostic value in human tumors70,71, therefore it will be critical to more carefully examine how BAF inhibitors modulate the differentiation states of TILs in more preclinical models and human T cells as our data demonstrate cBAF is critical for acquisition of Trm differentiation states.
Lastly, this work has high relevance to the understanding of lymphoma etiology because our analysis of Arid1acHet T cells revealed that cBAF operated in a dose-dependent manner to regulate both accessibility and effector cell differentiation states. Most ARID1A mutations in human cancers are heterozygous, and in particular, ARID1A hemizygosity is common to cutaneous T cell lymphoma27. Our data indicate that ARID1A heterozygosity affects accessibility at all sites sensitive to ARID1A loss, resulting in intermediate gene expression changes and cellular phenotypes relative to those of ARID1A-deficient cells. Future work is needed to determine what gene attributes confer sensitivity to ARID1A haploinsufficiency.
In summary, this study characterizes the dynamic regulation of the chromatin landscape in antiviral T cells in vivo through the lens of a chromatin remodeler and ascertains how DNA accessibility is created to orchestrate the transcriptional and epigenetic machinery that governs effector and memory CD8+ T cell differentiation. We anticipate that ARID1A and cBAF carry out similar biological functions in effector CD4+ T cells and other lymphocytes. Indeed, deletion of Arid1a or Smarca4 at various stages of hematopoiesis revealed the critical nature of cBAF in driving development of essentially all hematopoietic lineages22–24 as well as terminal differentiation of CD8+ T cells52,72,73 and regulatory T cells74,75 in tumors. Further, most human pathogenic SNPs map to non-coding regions (i.e. enhancers)76, implying individual enhancers may have a role in modulating gene expression and influencing cell states. This work thus provides data that may clarify the mechanistic basis for certain human immune-related diseases and establishing new opportunities to fine-tune immune cell function via manipulation of cBAF activity in adoptive cell therapies.
Limitations of the study
Our study primarily relied on the use of Granzyme-B Cre to delete Arid1a after T cell activation, and thus we cannot infer what role cBAF may play in early activation (0–48h) events. Secondly, we focused our analysis on memory cells in the spleen and non-lymphoid organs, but it is possible that studying additional lymphoid organs (lymph nodes and bone marrow) may yield further insight into how cBAF more broadly shapes the circulating memory cell pool after infection. Lastly, while we know that cBAF promotes CD8+ T cell expansion, differentiation, and expression of certain cytotoxic molecules (granzyme A) in vivo, we have not experimentally assessed whether cBAF is required for cytotoxic capacity.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Susan M. Kaech (skaech@salk.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All sequencing data from this paper are available in GEO under accession SuperSeries GSE228381.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J were purchased from Jackson Laboratories. P14 mice (Pircher et al., 1987) and Arid1afl/flmice (Chandler et al., 2015) have been previously described. Arid1afl/fl GzmB-Cre mice were generated by crossing P14 mice with Arid1afl/fl and Gzmb-Cre mice. Both female and male mice were used for all studies. Animals were housed in specific-pathogen-free facilities at the Salk Institute. All experimental studies were approved and performed in accordance with guidelines and regulations implemented by the Salk Institute Animal Care and Use Committee.
Infections
Mice were infected with 2×105 PFU LCMV-Armstrong by intraperitoneal injection. For recombinant GP33-expressing Listeria monocytogenes (LM-GP33) infections, mice were injected with 5×104 CFU by retro-orbital injection under anesthesia. LCMV-Armstrong and LM-GP33 stocks were prepared as previously described (Kaech and Ahmed, 2001; Welsh and Seedhom, 2008).
Cell Isolation
Spleens, lymph nodes, and livers were mechanically dissociated with 1mL syringe plungers over a 70um nylon strainer. Livers were resuspended in 8mL 40% isotonic percoll and centrifuged at 800g for 12 minutes at 20C. Spleens and livers were incubated in ammonium chloride potassium (ACK) buffer for 5 minutes. For isolation of small intestinal IEL, Peyer’s patches were first removed by dissection. Intestines were longitudinally cut and then cut into 1cm pieces and washed in PBS. Pieces were incubated in 30mL HBSS with 10% FBS, 10mM HEPES, and 1mM dithioerythritol with vigorous shaking at 37C for 30 minutes. Supernatants were collected, washed, and further isolated using a 40/67% discontinuous percoll density centrifugation for 20 minutes at room temp with no brakes. Salivary glands were minced with scissors and incubated in RPMI with 5% FBS and 0.5mg/mL Collagenase IV, 0.1mg/mL DNAse1, and 2mM CaCl2 for 30 minutes at 37C with gentle shaking, and then strained over 70um nylon strainers.
Flow Cytometry and cell sorting
Cell suspensions were first incubated with eBioscience Fixable Viability Dye eFluor 780 for 5 minutes at room temp. Cells were stained with primary surface antibodies in PBS with 2% FBS, 0.1% NaN3 for 20 minutes on ice. For sorting, staining was performed in PBS with 2% FBS, 2mM EDTA, and 10mM HEPES. DAPI was added to stained cell suspensions at 0.1ug/mL prior to sorting. For experiments involving retroviral transduction, cells were fixed with Biolegend Fixation buffer for 20 minutes at room temp. For intracellular staining, cells were first fixed with eBioscience Foxp3/Transcription Factor Staining buffer fixation/permeabilization buffer for 30 minutes at room temp, and staining was performed in 1X permeabilization buffer for 30 minutes at room temp. Flow cytometry analysis samples were acquired on a BD Symphony A3. Cell sorting was performed on a BD FACSAria Fusion.
In vitro Stimulations and intracellular cytokine staining
Spleen cell suspensions were stimulated in complete RPMI (RPMI with 10% FBS, Pen/strep, L-glutamine, 50uM β-ME) in the presence of 0.1ng/mL GP33–41 peptide or DMSO and brefeldin A for 5 hours at 37C. For cytokine stimulations, cells were cultured for 5 hours with 10ng/mL recombinant mouse IL-12 and IL-18 without brefeldin A, at which point brefeldin A was added for 1 hour prior to staining. Cells were stained for viability and surface markers as described above, and then fixed/permeabilized with cytofix/cytoperm buffer (BD) for 25 minutes at room temp. Intracellular cytokine staining was performed in 1X permeabilization buffer (BD) for 30 minutes at room temp.
Adoptive T cell transfer
Naive WT, Arid1afl/+, or Arid1afl/fl CD8+ P14 cells were isolated by negative selection with biotinylated antibodies against CD4, CD19, B220, MHCII, CD11b, CD11c, CD49b, and Ter119 in MACS buffer, and MojoSort beads were added at 5% v/v for 5 minutes before placing the cell suspension on a magnet and collecting the supernatant. 2.5×104 P14 cells were injected into recipient mice 18–24 hours before infection. For 72h sorting experiments, 5×105 P14 cells were transferred. For proliferation experiments, P14 cells were labeled with 5uM CTV (CellTrace Violet) in warm PBS for 8 minutes at 37C, and 1×106 cells were transferred. For secondary infection experiments, splenocytes and peripheral LNs from 30d LCMV immune P14 chimeric mice were enriched for CD127+ CD8+ cells. Briefly, negative magnetic selection was performed with biotinylated antibodies against CD4, MHCII, Thy1.2, and KLRG1 in MACS buffer, and MojoSort beads were added at 5% v/v for 5 minutes before placing the cell suspension on a magnet and collecting the supernatant. WT and Arid1acKO memory-enriched populations were mixed to form a 1:1 ratio of WT and Arid1acKO P14 memory cells, and then 6×104 memory cells were transferred into naïve recipient mice prior to infection with 1×105 CFU of LM-GP33.
Retrovirus production and transduction
Tbet-MIGR1 was previously described (Joshi et al., 2007). Retrovirus was prepared by transfection of 293T cells with pCL-Eco and T-bet-MIGR1 or EV-MIGRs plasmids using XtremeGene9 transfection reagent. For transductions, P14 cells were stimulated for 18 hours with plate-bound 2ug/mL anti-CD3, 1ug/mL anti-CD28, and 20ng/mL human recombinant IL-2. Supernatant was removed from activated cells, and retroviral supernatants supplemented with polybrene (10ug/mL) were carefully overlayed onto cells, and then cells were centrifuged for 90 minutes at 1500g at 30C. Cells were incubated for 4h at 37C, washed, and adoptively transferred into recipient mice.
Cas9 RNP Electroporation
Cas9/sgRNA mixtures were prepared (0.6uL 62nM recombinant Cas9, 0.3nmol sgRNA, 3.5uL RNAse-free H2O) and incubated for 10 minutes at room temperature. 2–5×106 MACS-purified naive P14 cells were washed in PBS and suspended in 20uL supplemented P3 buffer (Lonza; 16.4uL P3 buffer and 3.6uL Supplement 1). Resuspended cells were immediately mixed with 5uL Cas9/sgRNA mixture and transferred to a Lonza nucleofector strip. Cells were electroporated using the program DN100. 130uL pre-warmed complete RPMI was added to cells prior to incubation for 10 minutes at 37C. Cells were washed extensively with complete RPMI prior to transfer into recipient mice. The sequences of sgRNA used to target Arid1a: UACCCAAAUAUGAAUCAAGG.
ATAC-Seq library preparation and sequencing
ATAC-seq was performed as previously described (Corces et al., 2017). Briefly, 5,000–50,000 viable cells were washed with cold PBS, collected by centrifugation, then lysed in resuspension buffer (RSB) (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2) supplemented with 0.1% NP40, 0.1% Tween-20, and 0.01% digitonin. Samples were incubated on ice for 3 min, then washed out with 1 ml RSB containing 0.1% Tween-20. Nuclei were pelleted by centrifugation at 500g for 10 min at 4°C then resuspended in 50 ul transposition mix (25ul 2x TD buffer, 2.5 ul transposase (100 nM final), 16.5 ul PBS, 0.5 ul 1% digitonin, 0.5 ul 10% Tween-20, 5 ul H2O) and incubated at 37°C for 30 min in a thermomixer with 1000 RPM mixing. DNA was purified using a Qiagen MinElute PCR cleanup kit, then PCR amplified using indexed oligos. The optimal number of amplification cycles for each sample was determined by qPCR. Libraries were size selected using AmpureXP beads and sequenced using an Illumina NextSeq500 for 75bp paired-end reads.
RNA-Seq library preparation and sequencing
Cells were FACS sorted directly into buffer RLT (Qiagen) supplemented with 2-mercaptoethanol and frozen at −80°C. Total RNA was isolated using a Qiagen RNeasy Plus Micro kit according to the manufacturer’s instructions and quantified using an Agilent 4200 TapeStation. Purified RNA was library prepped using a SMART-Seq v4 Ultra Low Input RNA kit. Libraries were quantified and size distribution was determined before 51bp single-end sequencing was performed.
ChIP-seq library preparation and sequencing
Purified polyclonal CD8+ T cells were activated with plate-bound 2ug/mL anti-CD3, 1ug/mL anti-CD28, and 20ng/mL human recombinant IL-2 for 48 hours. Cells were collected and approximately 5×106 cells each were treated with either 1μM ACBI1, 300nM BRM04, or 0.01% DMSO for 4 hours. Recombinant mouse IL-12p70 was added after 4 hours culture for an additional 2 hours. Cells were cross linked in 3 mM disuccinimidyl glutarate for 30 min then in 1% formaldehyde for 10 min at room temperature. After quenching with 125 mM glycine, the cells were washed in 1× PBS, pelleted, flash frozen in liquid nitrogen, and stored at 80 °C. Cell pellets were thawed on ice and incubated in lysis solution (50 mM Hepes KOH pH 8, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, and 0.25% Triton X-100) for 10 min. The isolated nuclei were washed (10 mM TrisHCl pH 8, 1 mM EDTA, 0.5 mM EGTA, and 200 mM NaCl) and sheared in (0.1% SDS, 1 mM EDTA, and 10 mM TrisHCl pH 8) with the Covaris E229 sonicator for 10 min. After centrifugation, chromatin was immunoprecipitated overnight at 4 °C with 1:50 T-bet/TBX21 (Cell Signaling Technology, mAb #97135S). The next day, the antibody-bound DNA was incubated with Protein A+G Dynabeads (Invitrogen) in ChIP buffer (50 mM Hepes KOH pH 7.5, 300 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, and 0.1% SDS), washed, and treated with Proteinase K and RNase A and reverse cross linked. Purified ChIP DNA was used for library generation (NEBNext Ultra II DNA Library Prep Kit for Illumina) according to manufacturer’s instructions.
CUT&RUN library preparation and sequencing
CUT&RUN for murine T cells was performed as previously described (Meers et al., 2019) with modifications (van der Veeken et al., 2020). Briefly, 75–150k cells per sample were washed with PBS, counted, and collected in a V-bottom 96 well plate by centrifugation. Cells were washed once in antibody buffer (buffer 1 (1x permeabilization buffer from eBioscience Foxp3/Transcription Factor Staining Buffer Set diluted in nuclease free water, 1X EDTA-free protease inhibitors, 0.5mM spermidine) containing 2mM EDTA), then incubated with antibodies for 1h on ice. After 2 washes in buffer 1, cells were incubated with pA/G-MNase at 1:4000 dilution in buffer 1 for 1h at 4 degrees. Cells were washed twice in buffer 2 (0.05% (w/v) saponin, 1X EDTA-free protease inhibitors, 0.5mM spermidine in PBS) and resuspended in calcium buffer (buffer 2 containing 2mM CaCl2) to activate the MNase fusion protein. Following a 30 minute incubation on wet ice, 2x stop solution (20mM EDTA, 4mM EGTA in buffer 2) was added and cells were incubated for 12 minutes in a 37 degree incubator to release cleaved chromatin fragments. Supernatants were collected by centrifugation and DNA was extracted using a Qiagen MinElute kit.
CUT&RUN libraries were prepared using a NEBNext Ultra II kit. Histone modification sample libraries were prepared according to the manufacturer’s protocol, and protein factor samples were prepared according to a factor-specific protocol (Liu, 2021). Briefly, protein factor dA-tailing temperature was decreased to 50°C, and the reaction time was increased to 1 hour. After adaptor ligation, 2.1x volume of AMPure XP beads was added to the reaction to ensure high recovery efficiency of short fragments. After 10–12 cycles of PCR amplification, the reaction was cleaned up with 0.8x AMPureXP beads selection to remove PCR products >350bp, followed by a second round of 1.2X AMPureXP bead size-selection to remove PCR products shorter than 150bp. Barcoded libraries were quantified on an Agilent TapeStation and PCR dimers were removed with 1.2X AmpureXP bead size selection. Samples were mixed at equal molar ratios and pooled libraries were loaded to a NextSeq 300 High Output Kit v2 (150 cycles), and sequenced in the NextSeq 300 platform. To enable determination of fragment length, paired-end sequencing was performed (2×42 bp, 6 bp index).
RNA-seq analysis
Single-end 51-bp reads were aligned to the M. musculus mm10 genome using STAR v2.5.3a (Dobin et al., 2013) with default parameters. RNA expression was quantified as raw integer counts using analyzeRepeats.pl in HOMER using the following parameters: -strand both -count exons -condenseGenes -noadj. Differentially expressed genes (DEGs) were identified with getDiffExpression.pl in HOMER using DESeq2 (Love et al., 2014) (cut-offs were set at log2 FC = 0.585 and false discovery rate (FDR) at 0.05 [Benjamini–Hochberg]). GSEA was performed on DEGs against HALLMARK gene sets. For GSEA enrichment plots, fgsea was used with 10,000 permutations = 10,000.
ATAC-Seq analysis
Paired-end 42-bp, or paired-end 75-bp reads were aligned to the M. musculus mm10 genome using STAR (Dobin et al., 2013) with default parameters. ATAC-seq peaks were called using the HOMER findPeaks program using parameters for DNAse-seq (-style dnase). Peaks were called when enriched >4.0-fold over local tag counts. Differentially accessible regions were identified using DESeq2 (Love et al., 2014) by calling getDifferentialPeaksReplicates.pl in HOMER with fold change ≥2.0 or ≤−2.0, FDR < 0.05. Peaks sets were annotated with HOMER, and visualizations were created using deepTools. k-means clustering of ATAC-seq data was performed using DESeq2. Signal heatmaps were generated using deepTools v3.5.1 (Ramírez et al., 2016).
ChIP-Seq analysis
Paired-end 75-bp reads were aligned to the M. musculus mm10 genome using STAR (Dobin et al., 2013) with default parameters. ChIP-seq peaks were called using findPeaks within HOMER using -style factor and an -i input control. Differential ChIP peaks were called using getDiffExpression.pl with fold change ≥ 1.5 or ≤ 1.5, Poisson P value < 0.0001. ChIP-seq peaks were annotated by mapping to the nearest transcription start site (TSS) using the annotatePeaks.pl program. Metaplots were generated using annotatePeaks.pl with parameters -size 2000 and -hist 10. Heatmaps showing overlap of binding sites were generated using mergePeaks in HOMER with flag -matrix, which outputs hypergeometric P values of overlap and the observed/expected ratio of overlap. Peaks sets were annotated with HOMER, and visualizations were created using deepTools.
CUT&RUN analysis
Similar to ChIP-seq analysis, paired-end 75-bp reads were aligned to the M. musculus mm10 genome using STAR (Dobin et al., 2013) with default parameters. Peaks were called with Sparse Enrichment Analysis for CUT&RUN (SEACR) with options -stringent and -norm against an IgG control sample (Meers et al., 2019). Metaplots were generated using annotatePeaks.pl with parameters -size 2000 and -hist 10. Heatmaps showing overlap of binding sites were generated using mergePeaks in HOMER with flag -matrix, which outputs hypergeometric P values of overlap and the observed/expected ratio of overlap. Peaks sets were annotated with HOMER, and visualizations were created using deepTools.
Transcription factor binding motif analysis
Sequences within 100 bp of peak centers were compared to known motifs in the HOMER database using the findMotifsGenome.pl (Heinz et al., 2010) command with default parameters. Random GC content-matched genomic regions were used as background. Enriched motifs are statistically significant motifs in input over background by a P value of less than 0.05 using a cumulative binomial distribution.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using the two-tailed, unpaired, Student’s t-test unless otherwise specified. Each point represented a biological replicate and all data were presented as the mean ± SEM. The P values were represented as follows: ns, not significant, *p<0.05, **p<0.005, ***p<0.0005.
Supplementary Material
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BUV737 CD8a | BD | Cat#612759; RRID: AB_2870090 |
| BV421 CD8a | BioLegend | Cat#100738; RRID: AB_11204079 |
| BV605 CD44 | BD | Cat#563058; RRID: AB_2737979 |
| BUV395 Thy1.1 | BD | Cat#563804; RRID: AB_2632398 |
| BUV496 Thy1.2 | BD | Cat#741046; RRID: AB_2870661 |
| PE CD127 | BioLegend | Cat#135010; RRID: AB_1937251 |
| PE-Cy7 KLRG1 | Invitrogen | Cat#25-5893-82; RRID: AB_1518768 |
| PerCP-Cy5.5 CX3CR1 | BioLegend | Cat#149010; RRID: AB_2564494 |
| BUV395 CXCR3 | BD | Cat#745689; RRID AB_2743174 |
| BV711 CD49a | BD | Cat#564863: RRID: AB_2738987 |
| BUV737 CD69 | BD | Cat#564684; RRID: AB_2738891 |
| BUV395 CD103 | BD | Cat#740238; RRID: AB_2739985 |
| BV480 Va2 | BD | Cat#746615; RRID: AB_2743895 |
| R718 TCRb | BD | Cat#567299 |
| PE Tbet | BioLegend | Cat#644810; RRID: AB_2200542 |
| PE-Cy7 Eomes | Invitrogen | Cat#25-4875-82; RRID: AB_2573454 |
| AF647 Tcf1 | Cell Signaling Technology | Cat#6709S |
| ARID1A | Abcam | Cat#ab182561 |
| BV421 IFNg | BioLegend | Cat#505830; RRID: AB_2563105 |
| PE-Cy7 TNF | BioLegend | Cat#506324; RRID: AB_2256076 |
| APC CCL3 | R&D Systems | Cat#IC450A |
| PE IL-2 | BioLegend | Cat#503808; RRID: AB_315302 |
| PE-Cy7 Granzyme B | Biolegend | Cat#372214; RRID: AB_2728381 |
| PE Granzyme A | ThermoFisher Scientific | Cat#17-5831-82; RRID: AB_2573228 |
| eBioscience™ Fixable Viability Dye eFluor™ 780 | ThermoFisher Scientific | Cat#65-0865-14 |
| CellTrace Violet | ThermoFisher Scientific | Cat#C34557 |
| DAPI | BD | Cat#564907 |
| H3K27ac | Abcam | Cat#4729 |
| H3K27me3 | Cell Signaling Technology | Cat#9733S |
| ARID1A | Cell Signaling Technology | Cat#12354S |
| BATF | Brookwood Biomed | Cat#4003 |
| Tbet | Cell Signaling Technology | Cat#97135S |
| ETS1 | Cell Signaling Technology | Cat#14069 |
| Purified NA/LE Hamster anti-Mouse CD3e | BD | Cat#567114 |
| Purified Hamster anti-Mouse CD28 | BD | Cat#553295; RRID: AB_394764 |
| Bacterial and virus strains | ||
| LCMV Armstrong | Kaech and Ahmed, 2001 | NA |
| Listeria monocytogenes GP33-41 | Ananda Goldrath | NA |
| Chemicals, peptides, and recombinant proteins | ||
| GP33-41 peptide | GenScript | Cat#RP20091 |
| Recombinant Murine IL-12p70 | R&D Systems | Cat#419-ML-010 |
| Recombinant Murine IL-2 | PEPROTECH | Cat# 212-12 |
| Recombinant Murine IL-18 | R&D Systems | Cat#9139-IL-010 |
| ACBI1 | opnMe | NA |
| BRM014 | MedChemExpress | Cat#HY-119374 |
| Brefeldin A | BioLegend | Cat#420601 |
| ACK Lysing Buffer | VWR | Cat#10128-804 |
| eBioscience Foxp3/Transcription factor staining buffer set | ThermoFisher Scientific | Cat#00-5523-00 |
| eBioscience 10X Permeabilization buffer | ThermoFisher Scientific | Cat#00-8333-56 |
| Cytofix/Cytoperm | BD | Cat#554714 |
| RPMI 1640 Medium | GIBCO | Cat#21875034 |
| Fetal bovine serum | Sigma-Aldrich | Cat#F0804 |
| L-Glutamine | GIBCO | Cat#25030081 |
| 2-Mercaptoethanol | GIBCO | Cat#21985023 |
| Penicillin-streptomycin | Invitrogen | Cat#15140148 |
| Collegenase D | Sigma-Aldrich | Cat#11088882001 |
| DnaseI | Sigma-Aldrich | Cat#DN25-5G |
| HBSS | GIBCO | Cat#14025092 |
| Dithioerythritol | EMD Millipore | Cat#233152 |
| 1M HEPES | ThermoFisher Scientific | Cat#15630080 |
| Alt-R S.p. Cas9 Nuclease V3 | IDT | Cat#1081059 |
| pA/G-MNase | Epicypher | Cat#14-1048 |
| Critical commercial assays | ||
| P3 Primary Cell 4D X Kit S | Lonza | V4XP-3032 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE228381 |
| Experimental models: Organisms/strains | ||
| Mouse: granzyme-B-Cre | The Jackson Laboratory | JAX: 003734 |
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: Tg(TcrLCMV)327Sdz (P14) | Pircher et al., 1989 | MGI: 2665105 |
| Mouse: Arid1atm1.1Mag | Chandler et al., 2015 | MGI: 5708705 |
| Mouse: Tbx21 KO | Szabo et al., 2002 | JAX: 004648 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| MIGR1-Tbet | Joshi et al., 2007 | NA |
| Software and algorithms | ||
| STAR | Dobin et al, 2013 | https://github.com/alexdobin/STAR |
| bwa-mem2 | Vasimuddin et al., 2019 | https://github.com/bwa-mem2/bwa-mem2 |
| Homer | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ggplot2 | https://cran.r-proiect.org/web/packages/ggplot2/index.html | |
| deepTools | Ramírez et al., 2016 | https://github.com/deeptools/deepTools/ |
| fgsea | https://github.com/ctlab/fgsea | |
Highlights.
ARID1A-containing cBAF opens enhancers in effector CD8+ T cells during infection
cBAF establishes binding sites of many effector TFs, including T-bet and BATF
cBAF is necessary for late-stage differentiation of TE and Trm cells
Circulating memory cells form without cBAF, but are functionally impaired
Acknowledgements
We thank the Kaech lab for helpful discussion; N. Claffey, M. Liem at the Salk Flow Cytometry core. This work was supported by NIH grants F31HL158235 and F99CA274688 (B.M.), 5T32GM133351 (B.Y.C), 2T32CA009370 (N.S.A.), R37 AI066232 (to S.M.K.) and AI151123 (to D.C.H.), the Pew-Stewart Scholars for Cancer Research (D.C.H.), and the American Cancer Society Research Scholar Award (D.C.H.) as well as fellowships from the Cancer Research Institute (S.M.), Damon Runyon Cancer Research Foundation (T.M.), and the NOMIS Center (D.C.). Additional support came from the Flow Cytometry Core Facility of the Salk Institute with funding from NIH-NCI CCSG: P30 014195 and Shared Instrumentation Grant S10-OD023689 (Aria Fusion cell sorter) and by the NGS Core Facility of the Salk Institute with funding from NIH-NCI CCSG: P30 014195, the Chapman Foundation and the Helmsley Charitable Trust.
Inclusion and Diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
S.M.K. is on the scientific advisory boards and has equity in EvolveImmune Therapeutics, Affini-T Therapeutics, Arvinas, and Pfizer.
References
- 1.Kaech SM, and Cui W (2012). Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol 12, 749–761. 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, and von Andrian UH (2016). The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity 45, 1270–1284. 10.1016/j.immuni.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, and Ahmed R (2003). Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol 4, 1191–1198. 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 4.Cui W, Joshi NS, Jiang A, and Kaech SM (2009). Effects of Signal 3 during CD8 T cell priming: Bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine 27, 2177–2187. 10.1016/j.vaccine.2009.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, Lietzenmayer M, Kroehling L, Takumi A, Kometani K, et al. (2018). KLRG1+ Effector CD8+ T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity 48, 716–729.e8. 10.1016/j.immuni.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Obar JJ, Jellison ER, Sheridan BS, Blair DA, Pham Q-M, Zickovich JM, and Lefrançois L (2011). Pathogen-Induced Inflammatory Environment Controls Effector and Memory CD8+ T Cell Differentiation. J. Immunol 187, 4967–4978. 10.4049/jimmunol.1102335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plumlee CR, Obar JJ, Colpitts SL, Jellison ER, Haining WN, Lefrancois L, and Khanna KM (2015). Early Effector CD8 T Cells Display Plasticity in Populating the Short-Lived Effector and Memory-Precursor Pools Following Bacterial or Viral Infection. Sci. Rep 5, 12264. 10.1038/srep12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Behr FM, Parga-Vidal L, Kragten NAM, van Dam TJP, Wesselink TH, Sheridan BS, Arens R, van Lier RAW, Stark R, and van Gisbergen KPJM (2020). Tissue-resident memory CD8+ T cells shape local and systemic secondary T cell responses. Nat. Immunol 21, 1070–1081. 10.1038/s41590-020-0723-4. [DOI] [PubMed] [Google Scholar]
- 10.Mackay LK, Minnich M, Kragten NAM, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, et al. (2016). Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463. 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 11.Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, Hu J, Hocker JD, Hawk NV, Kapoor V, et al. (2019). The transcription factor c-Myb regulates CD8+ T cell stemness and antitumor immunity. Nat. Immunol 20, 337–349. 10.1038/s41590-018-0311-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, Ye Z, Beckermann KE, Wu Y, Jenkins JW, et al. (2019). The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8+ T Cell Fitness and Functionality. Immunity 51, 491–507.e7. 10.1016/j.immuni.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al. (2017). Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257. 10.1038/nature24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, and Wherry EJ (2012). Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37, 1130–1144. 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hargreaves DC, and Crabtree GR (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21, 396–420. 10.1038/cr.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alpsoy A, and Dykhuizen EC (2018). Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem 293, 3892–3903. 10.1074/jbc.RA117.001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gatchalian J, Malik S, Ho J, Lee D-S, Kelso TWR, Shokhirev MN, Dixon JR, and Hargreaves DC (2018). A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun 9, 5139. 10.1038/s41467-018-07528-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mashtalir N, D’Avino AR, Michel BC, Luo J, Pan J, Otto JE, Zullow HJ, McKenzie ZM, Kubiak RL, St Pierre R, et al. (2018). Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1288.e20. 10.1016/j.cell.2018.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blümli S, Wiechens N, Wu M-Y, Singh V, Gierlinski M, Schweikert G, Gilbert N, Naughton C, Sundaramoorthy R, Varghese J, et al. (2021). Acute depletion of the ARID1A subunit of SWI/SNF complexes reveals distinct pathways for activation and repression of transcription. Cell Rep. 37, 109943. 10.1016/j.celrep.2021.109943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, and Hargreaves DC (2017). Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 6, e30506. 10.7554/eLife.30506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, Park PJ, Shivdasani RA, and Roberts CWM (2017). ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet 49, 296–302. 10.1038/ng.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Astori A, Tingvall-Gustafsson J, Kuruvilla J, Coyaud E, Laurent EMN, Sunnerhagen M, Åhsberg J, Ungerbäck J, Strid T, Sigvardsson M, et al. (2020). ARID1a Associates with Lymphoid-Restricted Transcription Factors and Has an Essential Role in T Cell Development. J. Immunol. Baltim. Md 1950 205, 1419–1432. 10.4049/jimmunol.1900959. [DOI] [PubMed] [Google Scholar]
- 23.Chi TH, Wan M, Lee PP, Akashi K, Metzger D, Chambon P, Wilson CB, and Crabtree GR (2003). Sequential roles of Brg, the ATPase subunit of BAF chromatin remodeling complexes, in thymocyte development. Immunity 19, 169–182. 10.1016/s1074-7613(03)00199-7. [DOI] [PubMed] [Google Scholar]
- 24.Han L, Madan V, Mayakonda A, Dakle P, Woon TW, Shyamsunder P, Nordin HBM, Cao Z, Sundaresan J, Lei I, et al. (2019). Chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice. Leukemia 33, 2291–2305. 10.1038/s41375-019-0438-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kadoch C, and Crabtree GR (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci. Adv 1, e1500447. 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, and Crabtree GR (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet 45, 592–601. 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, Bjornson RD, Maman Y, Wang T, Tordoff J, et al. (2015). Genomic landscape of cutaneous T cell lymphoma. Nat. Genet 47, 1011–1019. 10.1038/ng.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, Xi L, Meng Q, Langridge T, Drummond J, et al. (2015). Genomic profiling of Sézary Syndrome identifies alterations of key T-cell signaling and differentiation genes. Nat. Genet 47, 1426–1434. 10.1038/ng.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scharer CD, Bally APR, Gandham B, and Boss JM (2017). Cutting Edge: Chromatin Accessibility Programs CD8 T Cell Memory. J. Immunol 198, 2238–2243. 10.4049/jimmunol.1602086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott-Browne JP, López-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, and Pereira RM (2016). Dynamic Changes in Chromatin Accessibility Occur in CD8+ T Cells Responding to Viral Infection. Immunity 45, 1327–1340. 10.1016/j.immuni.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pircher H, Michalopoulos EE, Iwamoto A, Ohashi PS, Baenziger J, Hengartner H, Zinkernagel RM, and Mak TW (1987). Molecular analysis of the antigen receptor of virus-specific cytotoxic T cells and identification of a new V alpha family. Eur. J. Immunol 17, 1843–1846. 10.1002/eji.1830171226. [DOI] [PubMed] [Google Scholar]
- 32.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856. 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet 48, 1193–1203. 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott-Browne JP, Pereira RM, Crotty S, Chang JT, Pipkin ME, et al. (2017). Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat. Immunol 18, 573–582. 10.1038/ni.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, et al. (2014). The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol 15, 373–383. 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsao H-W, Kaminski J, Kurachi M, Barnitz RA, DiIorio MA, LaFleur MW, Ise W, Kurosaki T, Wherry EJ, Haining WN, et al. (2022). Batf-mediated epigenetic control of effector CD8+ T cell differentiation. Sci. Immunol 7, eabi4919. 10.1126/sciimmunol.abi4919. [DOI] [PubMed] [Google Scholar]
- 38.Zhong Y, Walker SK, Pritykin Y, Leslie CS, Rudensky AY, and van der Veeken J (2022). Hierarchical regulation of the resting and activated T cell epigenome by major transcription factor families. Nat. Immunol 23, 122–134. 10.1038/s41590-021-01086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacob J, and Baltimore D (1999). Modelling T-cell memory by genetic marking of memory T cells in vivo. Nature 399, 593–597. 10.1038/21208. [DOI] [PubMed] [Google Scholar]
- 40.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marchingo JM, Sinclair LV, Howden AJ, and Cantrell DA (2020). Quantitative analysis of how Myc controls T cell proteomes and metabolic pathways during T cell activation. eLife 9, e53725. 10.7554/eLife.53725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, and Kaech SM (2015). The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J. Exp. Med 212, 2041–2056. 10.1084/jem.20150186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prier JE, Li J, Gearing LJ, Olshansky M, Sng XYX, Hertzog PJ, and Turner SJ (2019). Early T-BET Expression Ensures an Appropriate CD8+ Lineage-Specific Transcriptional Landscape after Influenza A Virus Infection. J. Immunol. Baltim. Md 1950 203, 1044–1054. 10.4049/jimmunol.1801431. [DOI] [PubMed] [Google Scholar]
- 44.Shan Q, Zeng Z, Xing S, Li F, Hartwig SM, Gullicksrud JA, Kurup SP, Van Braeckel-Budimir N, Su Y, Martin MD, et al. (2017). The transcription factor Runx3 guards cytotoxic CD8+ effector T cells against deviation towards follicular helper T cell lineage. Nat. Immunol 18, 931–939. 10.1038/ni.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, Zollman D, Steurer S, Karolyi-Oezguer J, Riedmueller C, et al. (2019). BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol 15, 672–680. 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papillon JPN, Nakajima K, Adair CD, Hempel J, Jouk AO, Karki RG, Mathieu S, Möbitz H, Ntaganda R, Smith T, et al. (2018). Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem 61, 10155–10172. 10.1021/acs.jmedchem.8b01318. [DOI] [PubMed] [Google Scholar]
- 47.Zhang K, Wang M, Zhao Y, and Wang W (2019). Taiji: System-level identification of key transcription factors reveals transcriptional waves in mouse embryonic development. Sci. Adv 5, eaav3262. 10.1126/sciadv.aav3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milner JJ, Nguyen H, Omilusik K, Reina-Campos M, Tsai M, Toma C, Delpoux A, Boland BS, Hedrick SM, Chang JT, et al. (2020). Delineation of a molecularly distinct terminally differentiated memory CD8 T cell population. Proc. Natl. Acad. Sci. U. S. A 117, 25667–25678. 10.1073/pnas.2008571117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu J, Madi A, Mieg A, Hotz-Wagenblatt A, Weisshaar N, Ma S, Mohr K, Schlimbach T, Hering M, Borgers H, et al. (2020). T Cell Factor 1 Suppresses CD103+ Lung Tissue-Resident Memory T Cell Development. Cell Rep. 31, 107484. 10.1016/j.celrep.2020.03.048. [DOI] [PubMed] [Google Scholar]
- 50.Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, Araki K, West EE, Ghoneim HE, Fan Y, et al. (2017). EFFECTOR CD8 T CELLS DEDIFFERENTIATE INTO LONG-LIVED MEMORY CELLS. Nature 552, 404–409. 10.1038/nature25144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang JT, Ciocca ML, Kinjyo I, Palanivel VR, McClurkin CE, DeJong CS, Mooney EC, Kim JS, Steinel NC, Oliaro J, et al. (2011). Asymmetric Proteasome Segregation as a Mechanism for Unequal Partitioning of the Transcription Factor T-bet during T Lymphocyte Division. Immunity 34, 492–504. 10.1016/j.immuni.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo A, Huang H, Zhu Z, Chen MJ, Shi H, Yuan S, Sharma P, Connelly JP, Liedmann S, Dhungana Y, et al. (2022). cBAF complex components and MYC cooperate early in CD8+ T cell fate. Nature. 10.1038/s41586-022-04849-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gray SM, Amezquita RA, Guan T, Kleinstein SH, and Kaech SM (2017). Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8+ T Cell Terminal Differentiation and Loss of Multipotency. Immunity 46, 596–608. 10.1016/j.immuni.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, Yu B, Wehrens EJ, Lopez J, Kim SH, et al. (2017). Early transcriptional and epigenetic regulation of CD8+ T cell differentiation revealed by single-cell RNA-seq. Nat. Immunol 18, 422–432. 10.1038/ni.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Braun SMG, Kirkland JG, Chory EJ, Husmann D, Calarco JP, and Crabtree GR (2017). Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat. Commun 8, 560. 10.1038/s41467-017-00644-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho L, Miller EL, Ronan JL, Ho W, Jothi R, and Crabtree GR (2011). esBAF Facilitates Pluripotency by Conditioning the Genome for LIF/STAT3Signalingand by Regulating Polycomb Function. Nat. Cell Biol 13, 903–913. 10.1038/ncb2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kadoch C, Williams RT, Calarco JP, Miller EL, Weber CM, Braun SMG, Pulice JL, Chory EJ, and Crabtree GR (2017). Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet 49, 213–222. 10.1038/ng.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, Ku WL, Teng M, Cui K, Williams RT, et al. (2017). SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat. Genet 49, 1613–1623. 10.1038/ng.3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stanton BZ, Hodges C, Calarco JP, Braun SMG, Ku WL, Kadoch C, Zhao K, and Crabtree GR (2017). Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet 49, 282–288. 10.1038/ng.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weber CM, Hafner A, Kirkland JG, Braun SMG, Stanton BZ, Boettiger AN, and Crabtree GR (2021). mSWI/SNF promotes Polycomb repression both directly and through genome-wide redistribution. Nat. Struct. Mol. Biol 28, 501–511. 10.1038/s41594-021-00604-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ, Quivy J-P, Almouzni G, and Amigorena S (2018). The epigenetic control of stemness in CD8+ T cell fate commitment. Science 359, 177–186. 10.1126/science.aah6499. [DOI] [PubMed] [Google Scholar]
- 62.Oestreich KJ, and Weinmann AS (2012). T-bet employs diverse regulatory mechanisms to repress transcription. Trends Immunol. 33, 78–83. 10.1016/j.it.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Omilusik KD, Best JA, Yu B, Goossens S, Weidemann A, Nguyen JV, Seuntjens E, Stryjewska A, Zweier C, Roychoudhuri R, et al. (2015). Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med 212, 2027–2039. 10.1084/jem.20150194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, Calame K, and Kaech SM (2009). Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 31, 296–308. 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iurlaro M, Stadler MB, Masoni F, Jagani Z, Galli GG, and Schübeler D (2021). Mammalian SWI/SNF continuously restores local accessibility to chromatin. Nat. Genet 53, 279–287. 10.1038/s41588-020-00768-w. [DOI] [PubMed] [Google Scholar]
- 66.Schick S, Grosche S, Kohl KE, Drpic D, Jaeger MG, Marella NC, Imrichova H, Lin J-MG, Hofstätter G, Schuster M, et al. (2021). Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat. Genet 53, 269–278. 10.1038/s41588-021-00777-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delpoux A, Michelini RH, Verma S, Lai C-Y, Omilusik KD, Utzschneider DT, Redwood AJ, Goldrath AW, Benedict CA, and Hedrick SM (2018). Continuous activity of Foxo1 is required to prevent anergy and maintain the memory state of CD8+ T cells. J. Exp. Med 215, 575–594. 10.1084/jem.20170697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang N, and Bevan MJ (2013). Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696. 10.1016/j.immuni.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schenkel JM, and Masopust D (2014). Tissue-Resident Memory T Cells. Immunity 41, 886–897. 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Amsen D, van Gisbergen KPJM, Hombrink P, and van Lier RAW (2018). Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol 19, 538–546. 10.1038/s41590-018-0114-2. [DOI] [PubMed] [Google Scholar]
- 71.Okła K, Farber DL, and Zou W (2021). Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med 218, e20201605. 10.1084/jem.20201605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Belk JA, Yao W, Ly N, Freitas KA, Chen Y-T, Shi Q, Valencia AM, Shifrut E, Kale N, Yost KE, et al. (2022). Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell, S1535610822002318. 10.1016/j.ccell.2022.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Battistello E, Hixon KA, Comstock DE, Collings CK, Chen X, Rodriguez Hernaez J, Lee S, Cervantes KS, Hinkley MM, Ntatsoulis K, et al. (2023). Stepwise activities of mSWI/SNF family chromatin remodeling complexes direct T cell activation and exhaustion. Mol. Cell 10.1016/j.molcel.2023.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Long L, Wei J, Lim SA, Raynor JL, Shi H, Connelly JP, Wang H, Guy C, Xie B, Chapman NM, et al. (2021). CRISPR screens unveil signal hubs for nutrient licensing of T cell immunity. Nature 600, 308–313. 10.1038/s41586-021-04109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Loo C-S, Gatchalian J, Liang Y, Leblanc M, Xie M, Ho J, Venkatraghavan B, Hargreaves DC, and Zheng Y (2020). A Genome-wide CRISPR Screen Reveals a Role for the Non-canonical Nucleosome-Remodeling BAF Complex in Foxp3 Expression and Regulatory T Cell Function. Immunity 53, 143–157.e8. 10.1016/j.immuni.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Corradin O, and Scacheri PC (2014). Enhancer variants: evaluating functions in common disease. Genome Med. 6, 85. 10.1186/s13073-014-0085-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data from this paper are available in GEO under accession SuperSeries GSE228381.