

Abstract
The antiviral defense directed by the RNAi pathway employs distinct specificity and effector mechanisms compared with other immune responses. The specificity of antiviral RNAi is programmed by siRNAs processed from virus-derived double-stranded RNA by Dicer endonuclease. Argonaute-containing RNA-induced silencing complex loaded with the viral siRNAs acts as the effector to mediate specific virus clearance by RNAi. Recent studies have provided evidence for the production and antiviral function of virus-derived siRNAs in both undifferentiated and differentiated mammalian cells infected with a range of RNA viruses when the cognate virus-encoded suppressor of RNAi (VSR) is rendered nonfunctional. In this review, we discuss the function, mechanism, and evolutionary origin of the validated mammalian VSRs and cell culture assays for their identification.
Antiviral RNAi response in mammals
Many emerging and re-emerging human infectious diseases, including the current coronavirus disease (COVID-19) pandemic, are caused by viruses with an RNA genome. RNA viruses are best known for their extraordinary abilities to evolve adaptive mutations that promote infection, dissemination, and/or evasion of protein-based host immune mechanisms. Recent studies have revealed a new mammalian defense mechanism against RNA viruses directed by the RNAi pathway [1]. Available data indicate that the mechanism of antiviral RNAi in mammals is most similar to that in fruit flies characterized extensively since 2002 [1,2]: replication of RNA genomes by viral RNA-dependent RNA polymerases (RdRPs) produces double-stranded RNA viral replicative intermediates (vRI-dsRNA; see Glossary), which are processed by Dicer nuclease into siRNAs, thereby triggering Argonaute protein-dependent RNAi to specifically target the invading viral RNAs (Figure 1). Thus, antiviral RNAi is an RNA-based immune mechanism with the specificity programmed by virus-derived siRNAs (vsiRNAs).
Figure 1. Induction and suppression of antiviral RNAi in mammals.
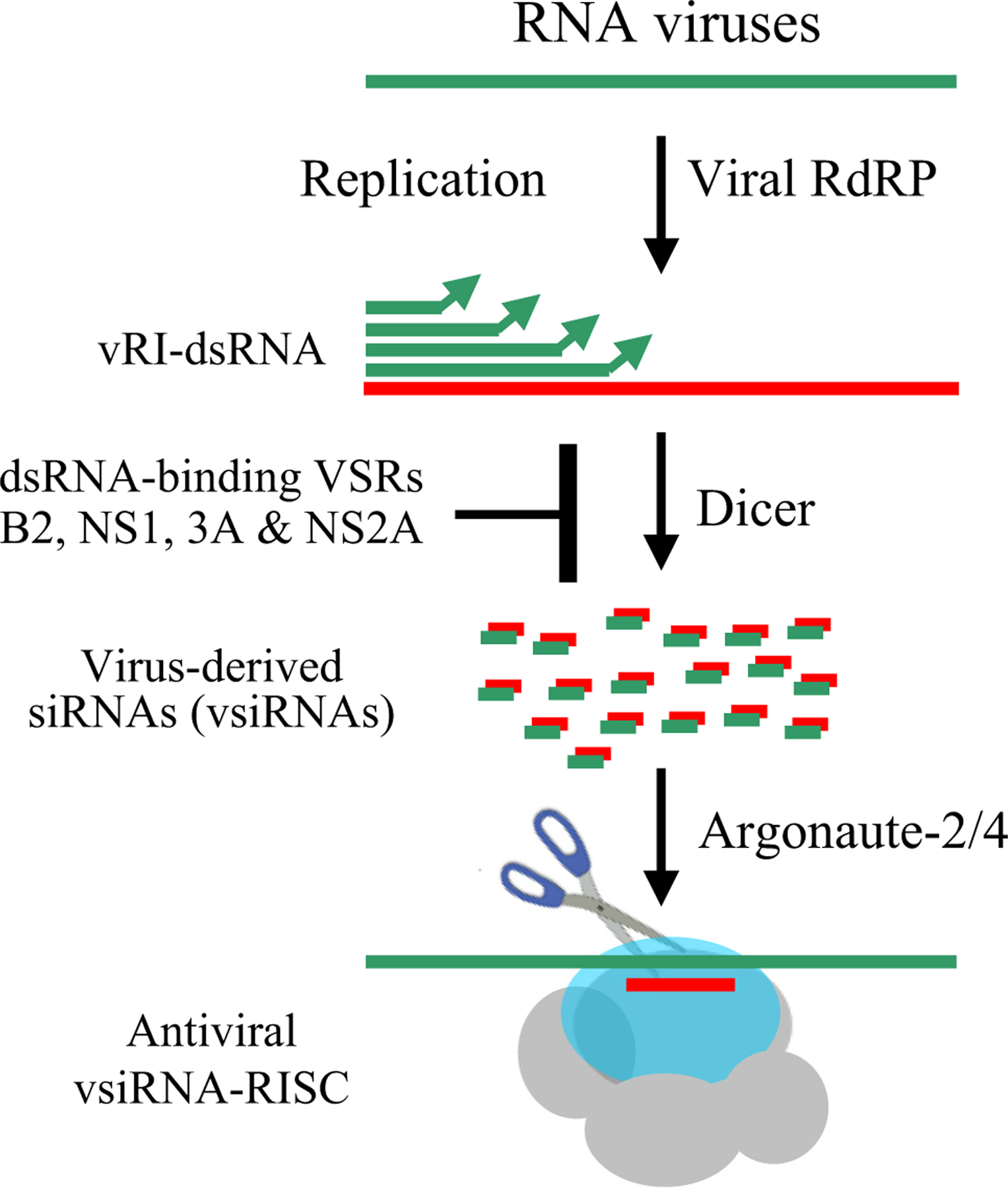
The viral replicative intermediates dsRNA (vRI-dsRNA) synthesized by the RNA-dependent RNA polymerase (RdRP) of RNA viruses are processed by Dicer nuclease into virus-derived siRNAs (vsiRNAs), triggering the assembly of antiviral RISC to target the viral RNAs complementary to the vsiRNAs for RNAi. All of the validated mammalian viral suppressors of RNAi (VSRs) suppress antiviral RNAi by mainly inhibiting the biogenesis of vsiRNAs via dsRNA sequestration.
As reviewed recently [3–10], Dicer-mediated production of abundant vsiRNAs has been documented in differentiated and/or undifferentiated mammalian cells after infection with eight positive- and negative-strand RNA viruses from five families [11–19]. These include Nodamura virus (NoV), encephalomyocarditis virus (EMCV), influenza A virus (IAV), human enterovirus-A71 (EV-A71), Zika virus (ZIKV), dengue virus-2 (DENV2), Sindbis virus, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The vsiRNAs targeting these viruses are all enriched for 22-nucleotide (nt) canonical siRNA duplexes with 2-nt 3′ overhangs [3–10]. Mammals encode one Dicer ribonuclease (RNase) and four Argonaute proteins with a C-terminal RNase H fold, among which only Argonaute-2 (Ago2) retains the RNA slicing activity essential for RNAi [20]. In addition, undifferentiated cells express a novel isoform of Dicer with enhanced ability to process dsRNA into siRNAs [18], which may explain why vsiRNA abundance is reduced in mouse ESCs (mESCs) following differentiation [8,10,12].
Mammalian vsiRNAs are assembled into an RNA-induced silencing complex (RISC) with Argonaute proteins, including Ago2, both in the infected cells and in mice. Similar to cellular miRNAs, the Argonaute-bound vsiRNAs exhibit strong preference for uracil as the 5′-terminal nucleotide [13,17,19,21]. However, these 1U vsiRNAs are more abundant than the three remaining vsiRNA populations combined in total vsiRNAs sequenced without prior Argonaute coimmunoprecipitation from infected adult mice in contrast to those detected in mouse embryonic fibroblasts (MEFs) [21], suggesting in vivo stabilization of the Argonaute-loaded vsiRNAs. Importantly, genetic studies using Dicer and/or Ago2 knockout/knockdown have demonstrated that activation of the vsiRNA response suppresses viral accumulation in several cell culture models for viral infection [11–19,21]. Use of reporter RNAs complementary to vsiRNAs has further shown that production of vsiRNAs activates homology-dependent RNA clearance in infected human cells [14] and that, in response to viral infection, adult mice produce abundant vsiRNAs in Ago2-RISC active to guide specific RNA slicing [21].
The strongest support for an antiviral function of the RNAi pathway in mammals has come from the discovery that despite their small genome sizes, diverse families of RNA viruses encode viral suppressors of RNAi (VSRs) essential for mammalian cell infection. In both undifferentiated and differentiated mammalian cells, the vsiRNA response is potently induced to inhibit viral infection when the cognate VSR is rendered nonexpressing or nonfunctional. Thus, a first step to assess a potential antiviral role of the RNAi pathway against a mammalian virus is to determine whether it encodes a functionally important VSR. This review highlights the key features of assays used in the identification of mammalian VSRs and discusses the function, mechanism, and evolutionary origin of functionally validated VSRs as well as key unanswered questions.
Cell culture assays to identify VSRs
The B2 protein of NoV and the nonstructural protein 1 (NS1) of IAV were first identified as VSRs in an assay established in Drosophila Schneider 2 (S2) cells [22]. In S2 cells, virus genome RNA replication triggers Dicer-2 processing of vRI-dsRNA to initiate the antiviral RNAi pathway requiring not only the slicing-competent fly Ago2 but also the dsRNA-binding protein R2D2 essential for siRNA loading into RISC [23]. A key reagent of the assay is the viral RNA replicon FR1gfp (Figure 2) engineered from the genomic RNA1 of Flock house virus (FHV) in the Nodaviridae. Nodaviral RNA1 encodes the viral RdRP to direct RNA1 self-replication in the absence of RNA2 encoding the capsid protein and the synthesis of a subgenomic RNA (RNA3) to serve as the mRNA for the expression of its B2 protein, a VSR essential for fruit fly infection in vitro and in vivo. FR1gfp was derived from a full-length infectious cDNA clone of FHV RNA1 by replacing most of the B2 coding sequence with that of green fluorescent protein (GFP). As antiviral RNAi induced de novo by FR1gfp RNA replication inhibits GFP expression from the replicon, a putative VSR is identified following visual detection of green fluorescence in S2 cells cotransfected with pFR1gfp and a plasmid expressing a candidate protein (Figure 2).
Figure 2. Cell culture assay for viral suppressor of RNAi (VSR) identification.
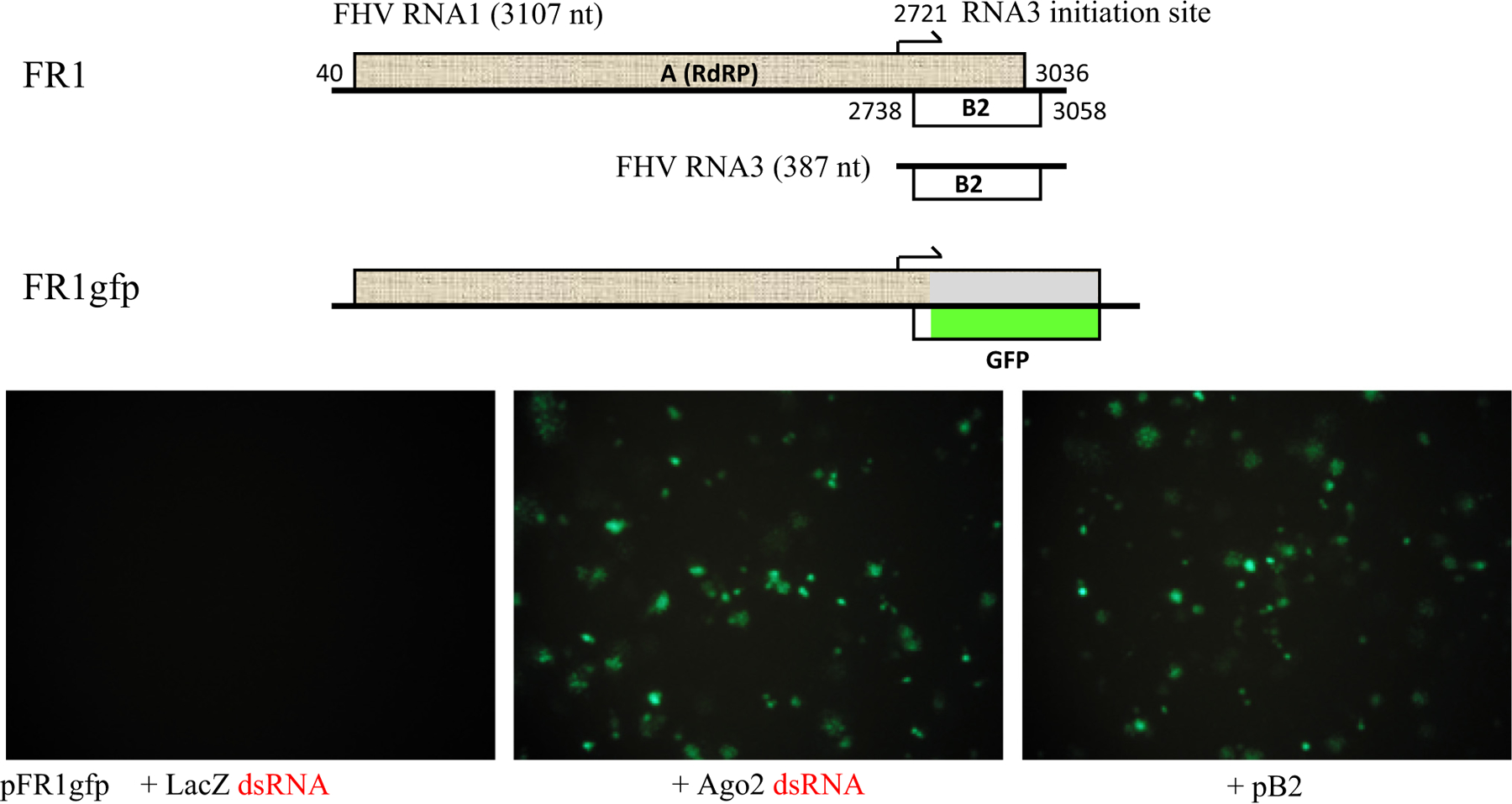
Top: Genomic RNA1 of Flock house virus (FHV and other nodaviruses) encodes two ‘out-of-frame’ overlapped open reading frames (ORFs) for the viral RdRP (protein A) and VSR-B2, translated from RNA1 and its subgenomic RNA (RNA3, synthesized after RNA1 replication), respectively. Nucleotides 2802–3001 of RNA1 as encoded in its infectious cDNA clone pFR1 were replaced with the coding sequence for enhanced green fluorescent protein (eGFP) to give pFR1gfp, allowing inframe fusion of eGFP with a short N-terminal sequence of B2 and translation of the tagged eGFP from the chimeric RNA3. Note the replacement of the C-terminal portion of the viral protein A deleted in FR1gfp by the protein sequence (light gray) translated from the −1 reading frame of eGFP ORF. Bottom: Fruit fly S2 cells displaying eGFP expression are not visible after transfection with pFR1gfp alone (left), but become abundant after RNAi suppression by Ago2 mRNA depletion with Ago2 dsRNA cotransfection (middle) or VSR-B2 expressed from a cotransfected plasmid (right).
Sullivan and Ganem [24] pioneered the use of RNAi artificially induced by small hairpin RNA (shRNA) in human embryonic kidney 293T cells to assay for RNAi suppression. Similar strategies have been used for the discovery of EV-A71 VSR protein 3A [14] and other mammalian VSRs. In these assays, plasmids encoding a reporter gene and a candidate VSR are cotransfected into either mammalian cells with a third plasmid directing shRNA transcription or Drosophila S2 cells with long dsRNA. Analogous to RNAi induction by vRI-dsRNA or synthetic long dsRNA in Drosophila cells, RNAi triggered by shRNA is dependent on Dicer to remove the loop sequence and release the single siRNA duplex for RISC assembly and RNAi. Unlike long dsRNA, shRNA does not trigger translational shutdown in mammalian cells active in the interferon (IFN) response. Thus, a VSR may suppress shRNA-induced RNAi by blocking Dicer processing, RISC assembly, or RISC-mediated repression of the reporter gene expression, which resemble key steps in the antiviral RNAi pathway (Figure 1).
Validated mammalian VSRs
Functional validation of VSRs identified in experimental RNAi suppression assays requires evidence for a specific role of the VSR activity in the context of the cognate viral infection in mammalian cells. Because of the multifunctional nature of viral proteins, the infection-defective phenotype of a VSR-deletion mutant virus may be caused by a defect in a function other than RNAi suppression. Therefore, VSR validation often entails demonstrating specific rescue of infection-defective VSR-deletion mutant viruses by knockout or knockdown of a host RNAi component essential for the biogenesis and/or the antiviral function of the vsiRNAs. To date, mammalian VSRs from four families of RNA viruses, including two mosquito-transmissible viruses, have been functionally validated (Table 1).
Table 1.
Validated mammalian viral suppressors of RNAi
| VSR | Virus | Family | Mechanism | Refs |
|---|---|---|---|---|
| B2 | Nodamura virus | Nodaviridae (+RNA genome) | dsRNA sequestration | [11,12,21,22,24] |
| NS1 | Influenza A virus | Orthomyxoviridae (−RNA genome) | dsRNA sequestration | [13,22,43] |
| 3A | Enterovirus-A71 | Picornaviridae (+RNA genome) | dsRNA sequestration | [14,35] |
| NS2A | Dengue virus 2 | Flaviviridae (+RNA genome) | dsRNA sequestration | [16] |
NoV B2
NoV is a mosquito-transmissible nonenveloped virus lethal to suckling mice and hamsters, as well as insects [25,26]. NoV and FHV are members of the family Nodaviridae that contains two positive-strand genomic RNAs and includes diverse viruses found in insects, fishes, and some vertebrates [26–28]. Much was known about the VSR activity and mechanism of NoV B2 before its functional validation in mammalian cell infection [11,12]. NoV B2 shares sequence, structural, and functional similarities with FHV B2 characterized extensively in fruit fly infection [23] (Figure 3). Self-replicating RNA replicons of NoV were used to first demonstrate viral induction and suppression of antiviral RNAi in mosquito cells and to identify the mosquito Ago2 essential for antiviral defense [22]. Both of the nodaviral B2s are potent suppressors of Ago2-dependent antiviral RNAi in Drosophila and mosquito cells [2,22,29]. In vitro studies have revealed the nodaviral B2 as the first dsRNA-binding protein that suppresses Dicer processing of long dsRNA into siRNA by dsRNA sequestration [24,29,30]. Notably, single residue substitution of an arginine (R) conserved between FHV and NoV B2 proteins with glutamine (Q) is sufficient to abolish both of their activities to bind long dsRNA and to suppress Dicer processing [29,30]. Accordingly, the resulting mutant viruses expressing either no B2 (FHVΔB2/NoVΔB2) or the mutant B2 protein (FHVmB2-R54Q/NoVmB2-R59Q) replicate to high levels only in cells defective in antiviral RNAi. The R-to-Q substitution was chosen because the point mutation introduced into the genome of either NoV or FHV does not alter the sequence of the viral RdRP encoded in the overlapping (−1) reading frame [29,30].
Figure 3. Structural similarities of mammalian double-stranded RNA (dsRNA)-binding viral suppressors of RNAi (VSRs).
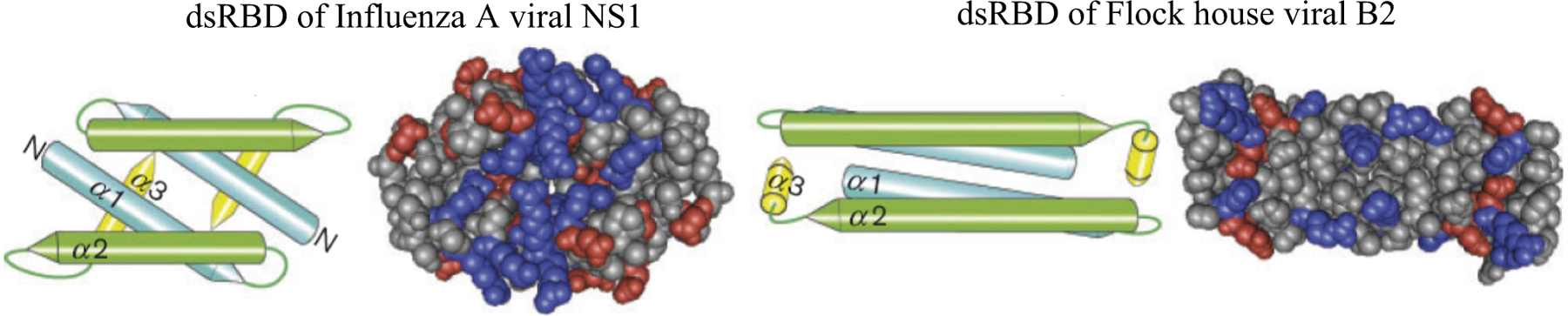
The dsRNA-binding domain (dsRBD) of NS1 (left) and B2 (right) drawn in a cartoon style along with the structures in their space-filling representation, with charged amino acids colored in red for acidic amino acids Asp and Glu and blue for basic amino acids Arg, Lys, and His (right panels). Reprinted, with permission, from [58]. Similar to FHV B2 (right), the 2.5 Å crystal structure of the dsRBD of NoV B2 also shows a four-helix bundle structure capped by two short helices at the C-termini [58].
The first evidence for suppression of mammalian antiviral RNAi by NoV B2 was obtained by comparing virus replication and virus-derived small RNA populations in hamster cells and suckling mice infected with wild-type or B2-deficient NoV [11,31]. For example, lethal NoV infection of suckling mice depends on the VSR activity of B2 to suppress the vsiRNA biogenesis so that the infection with either NoVΔB2 or NoVmB2 is rapidly cleared and accompanied with production of abundant vsiRNAs, especially during in vivo infection [11]. Unlike MEFs, mESCs exhibit deficiencies in innate immunity regulated by IFNs). Nevertheless, NoV infection of both MEFs and mESCs requires B2 suppression of antiviral RNAi because the replicational defects of NoVΔB2 in both cell types are efficiently rescued by RNAi deficiency through genetic depletion of Dicer, Ago2, and/or Ago2’s slicing activity only [12,21].
Genetic inactivation of either the innate immune signaling by types I, II, and III IFNs in the double STAT1 and STAT2 knockout mice or the adaptive immunity by B and T lymphocytes in recombination-activating gene 1 (RAG1)-deficient mice [32] renders adult mice highly susceptible to NoV infection [21]. Strikingly, both mutant strains of adult mice activate antiviral RNAi and clear the infection with either NoVΔB2 or NoVmB2 [21]. Therefore, the VSR activity of B2 is essential for NoV infection in both cell culture and adult mice either active or defective in the IFN-regulated antiviral responses [12,21], demonstrating B2 suppression of antiviral RNAi during in vitro and in vivo infection in both the presence and absence of the IFN signaling. When expressed ectopically, VSR-B2 also efficiently suppresses vsiRNA biogenesis and antiviral RNAi induced by the infection with EV-A71 [14], suggesting that it targets a shared step of antiviral RNAi against distinct RNA viruses. Interestingly, in vivo NoVΔB2 infection triggers systemic spread of vsiRNAs in circulating extracellular vesicles and prevents challenge infection of infant mice by wild-type NoV 2 days after immunization with NoVΔB2 [33]. Thus, VSR-deficient mutant viruses may provide a new generation of live-attenuated virus vaccines to trigger protection prior to the development of adaptive immunity mediated by B and T lymphocytes.
Several lines of evidence indicate that suppression of Dicer processing by VSR-B2 is incomplete. First, VSR-B2 coimmunoprecipitants from BALB/c suckling mice at 3 days postinfection (dpi) with NoV contain canonical, slicing-competent vsiRNAs, which, however, are not enriched for 1U vsiRNAs and are prevented from loading into RISC [19]. Second, total small RNA sequencing without prior VSR-B2 coimmunoprecipitation is sufficient to reveal production of canonical vsiRNAs in C57BL/6 suckling mice at 7 dpi with NoV [19]. Third, the vsiRNAs-RISC purified from NoV-infected, RAG1-deficient adult mice remain active in directing specific vsiRNA-guided RNA slicing in vitro, although less potent than those from NoVΔB2- or NoVmB2-infected mice [21]. Finally, the self-replication of wild-type NoV RNA1 is significantly enhanced in all three lines of RNAi-defective MEFs compared with wild-type MEFs [21], demonstrating that compared with the complete genetic loss-of-function mutation in Dicer or Ago2, antiviral RNAi remains partially active in wild-type MEFs with RNAi suppression by VSR-B2. Moreover, NoV RNA replication potently induces the dsRNA-mediated activation of 2′−5′-oligoadenylate synthetase-RNase L system in Dicer−/− MEFs in both the presence and absence of VSR-B2 [21], which is consistent with the earlier observation that VSR-B2 does not suppress in vivo induction of the known IFN-dependent innate immune responses [11].
Enteroviral 3A
EV-A71 is a nonenveloped positive-strand RNA virus of the genus Enterovirus in the Picornaviridae, which includes many important human and animal pathogens such as poliovirus. The enteroviral genome encodes a large polyprotein that is proteolytically processed into four structural and seven nonstructural proteins. A comprehensive set of experiments show that the nonstructural protein 3A of EV-A71 functions as a VSR to promote infection both in vitro and in vivo [14,34]. VSR-3A exhibits robust VSR activity to suppress both mammalian RNAi induced by shRNA and Drosophila RNAi triggered by either long dsRNA or viral RNA replication. Similar to VSR-B2 [24,29,30], VSR-3A is a dsRNA-binding protein that suppresses Dicer processing by directly sequestering long dsRNA [14]. Consistently, alanine (A) substitution of aspartic acid (D) at position 23 (D23A), conserved among enteroviral 3A proteins, abolishes the activities of VSR-3A in dsRNA binding and dicing suppression. Functional validation of VSR-3A was demonstrated because both Dicer-mediated vsiRNA biogenesis and vsiRNA-dependent RNA degradation are suppressed during the infection of human 293T and rhabdomyosarcoma (RD) cells, primary murine lung fibroblasts (MLFs), or newborn mice with wild-type but not mutant EV-A71 carrying D23A substitution in VSR-3A [14]. Compared with wild-type virus, EV-A71D23A also exhibits replication defects rescued efficiently by RNAi deficiency in 293T cells and MLFs with or without the suppression of the IFN response, demonstrating VSR-3A suppression of antiviral RNAi in both the presence and absence of the IFN signaling [14]. However, it remains unknown whether VSR-3A suppression of antiviral RNAi is incomplete, as suggested by the accumulation of the positive- and negative-strand virus-derived small RNAs with size preference for 21- to 23-nt in suckling mice at 5 days postinfection with wild-type EV-A71 [35].
The D23A substitution also disrupts the dimerization of VSR-3A, suggesting a role for the VSR-3A dimer in dicing suppression via dsRNA sequestration [14]. Notably, synthetic peptides derived from the dimerization domains that disrupt VSR-3A dimerization also potently inhibit its VSR activity to suppress Dicer processing during EV-A71 infection both in vitro and in vivo [9,35]. As a result, therapeutic application of an optimized VSR-3A-targeting peptide protects mice against lethal challenge by not only EV-A71 but also Coxsackievirus-A16 (CV-A16), an enterovirus closely related to EV-A71 [35]. Thus, antiviral RNAi restricts the infection of differentiated cells and mice with not only mutant EV-A71 defective in RNAi suppression but also wild-type EV-A71 so that ongoing RNAi suppression plays an essential role in infection. This finding suggests that antiviral RNAi poses a strong selection pressure to maintain the virus-encoded VSR function in diverse mammalian RNA viruses despite small genome sizes of these viruses. Moreover, these exciting results reveal VSR-3A as a drug target for developing antiviral therapeutics against EV-A71, CV-A16, and related enteroviruses known to cause millions of hand-foot-and-mouth disease cases in children worldwide.
Flaviviral NS2A
DENV2 is a mosquito-transmissible enveloped virus of the genus Flavivirus in the Flaviviridae. Flaviviruses contain a positive-strand RNA genome and encode a single polyprotein processed proteolytically into mature structural and nonstructural proteins. Screening the seven nonstructural proteins of DENV2 for the activity to suppress RNAi in Drosophila S2 cells identified nonstructural 2A (NS2A) as the VSR [16]. NS2As of all four DENV serotypes exhibit similar VSR activity in both fly S2 and mosquito Aag2 cells. Moreover, shRNA-triggered RNAi in human 293T cells is efficiently suppressed by NS2A of DENV2 and three additional flaviviruses including Japanese encephalitis virus (JEV), revealing a new function for the multifunctional flaviviral protein [16,36–39]. Despite sharing no sequence similarity with either nodaviral B2 or enteroviral 3A, NS2A is also a dsRNA-binding VSR that acts to suppress Dicer processing by sequestering long dsRNA [16]. Alanine scanning mutagenesis of the positively charged amino acids conserved among flaviviral NS2A proteins identified the lysine (K) at position 135 as essential for in vitro dsRNA binding and dicing suppression as well as for the suppression of antiviral RNAi triggered by DENV2 infection in mosquito cells [16]. Importantly, the mutant DENV2 carrying K135A substitution in NS2A (DENV2K135A) exhibits a similar defect in the infection of mosquito (Aag2) and mammalian (293T and MLFs) cells that is all efficiently rescued by RNAi deficiency. Dicer deficiency in mosquito and human cells is also sufficient to rescue the mutant JEV expressing a mutant NS2A defective in RNAi suppression [16]. These findings establish a natural function of NS2A to promote flaviviral infection in both mosquito and mammalian cells by suppressing antiviral RNAi [16].
In both human 293T cells and primary MLFs, the vsiRNA production triggered by DENV2 RNA replication is suppressed by wild-type but not the mutant NS2A with K135A substitution [16]. NS2A also suppresses the vsiRNA biogenesis in DENV2-infected mosquito cells; however, dicing suppression by NS2A is incomplete in mosquito cells and appears most effective against the processing of the 5′-terminal vRI-dsRNA [16] as observed previously in the suppression of Dicer processing by VSR-B2 of FHV in fly S2 cells [29]. Interestingly, knockdown of either Dicer-2 or Ago2 in Aag2 mosquito cells markedly enhances the titers of not only DENV2K135A but also wild-type DENV2 (DENV2WT) [16]. Thus, antiviral RNAi remains partially active in DENV2WT-infected mosquito cells despite NS2A expression, further demonstrating incomplete suppression of mosquito antiviral RNAi by NS2A. However, suppression of antiviral RNAi by NS2A in DENV2WT-infected mammalian cells appears complete because Dicer depletion in both human 293 cells and primary MLFs in either the presence or absence of IFN signaling enhances the titers of DENV2K135A but not DENV2WT [16]. Notably, blocking IFN signaling significantly enhances the titers of DENV2WT in both human 293 cells and primary MLFs, regardless of whether Dicer-dependent antiviral RNAi is active [16]. These findings support the conclusion that antiviral RNAi against DENV2 acts in a Dicer-dependent but IFN-I-independent, manner [16]. Moreover, these results also indicate that the VSR activity of NS2A is essential for DENV2 infection in mammalian cells either active or defective in the IFN-regulated antiviral response, revealing an IFN-independent role for the VSR activity for DENV2 infection similar to NoV VSR-B2 [21].
Influenza viral NS1
IAV is an enveloped negative-strand RNA virus in the Orthomyxoviridae and causes annual epidemics and occasional pandemics. NS1 is a multifunctional protein best characterized for its role to antagonize the IFN antiviral response by interacting with dsRNA and host proteins [40]. Although NS1 was identified as a VSR to suppress antiviral RNAi in Drosophila S2 cells [22] and shRNA-induced RNAi in human 293T cells [41], it was not until 2016 that its functional validation in the context of IAV infection was reported [13]. One reason for the delay is that, similar to NoV B2, NS1 potently suppresses Dicer-mediated vsiRNA biogenesis during IAV infection [13]. Thus, an abundant population of vsiRNAs processed from IAV vRI-dsRNA is detectable in human cells after infection with NS1-deficient IAV mutants but not with wild-type IAV. The canonical properties of IAV vsiRNAs made in infected cells become more clearly visible by sequencing the total small RNAs coimmunoprecipitated with AGOs, which served to remove nonspecific small RNAs [13]. It should be pointed out that IAV vRI-dsRNA are not inherently poor substrates of human Dicer, because, in response to the infection with NS1-deficient IAV, human 293T cells in fact produce more vsiRNAs than cellular miRNAs when both are made by human Dicer expressed de novo [19].
The second reason is that IAV is highly sensitive to miRNA regulation of the IFN antiviral response [42], which may explain why the siRNA response to IAV infection exhibits antiviral activity in cells where the host miRNA functionality is not globally perturbed [13,43]. For example, whereas neither IAV nor PR8/delNS1 replicates to higher levels in Dicer−/− 293T cells or Ago2−/− MEFs than their wild-type counterparts, both the wild-type and mutant virus titers increase significantly in Ago4−/− or Ago2D597A MEFs [13,43,44]. Ago4 has redundant functions with Ago1 and Ago3 in binding miRNAs, and, unlike a marked loss of host miRNAs in cells deficient in Dicer or Ago2 protein, the D597A mutation of Ago2 abolishes only its catalytic activity to slice target RNA in RNAi without altering the abundance of most host miRNAs [45–47] characterized [48].
By comparison, Ago4 deficiency promotes significantly higher fold increases in the accumulation of PR8/delNS1 than wild-type IAV-PR8 in MEFs either active or defective in IFN signaling, indicating that NS1 counteracts an antiviral RNAi defense independent of the IFN response [43]. The slicing activity of Ago2 is most likely essential for the same IFN-independent antiviral RNAi because Ago2D597A MEFs exhibit no defects in the IFN antiviral responses and the Ago2D597A mutation enhances accumulation of PR8/delNS1 significantly more than wild-type IAV-PR8 [13]. Together, these findings indicate that NS1 promotes viral infection at least in part by suppressing vsiRNA-mediated antiviral RNAi. Similar to NoV B2, NS1 suppression of mammalian antiviral RNAi is also incomplete because wild-type IAV-PR8 replicates to higher titers in both Ago4−/− and Ago2D597A MEFs than their wild-type counterparts [13,43]. However, it remains unknown whether the dsRNA-binding activity of NS1 is required for the suppression of antiviral RNAi in mammalian cells as has been shown in fly cells [22].
Mammalian VSRs: An example of convergent evolution?
As it is known for plant and insect viruses [49,50], mammalian RNA viruses from diverse families encode VSRs to promote infection by suppressing the siRNA response, providing strong evidence for a broad antiviral activity of the RNAi pathway in mammals. Strikingly, all of the validated mammalian VSRs are dsRNA-binding proteins and suppress antiviral RNAi by inhibiting Dicer-mediated biogenesis of the cognate vsiRNAs, most likely via dsRNA sequestration. Despite limited sequence identity, dsRNA-binding VSRs from the same virus family share strong structural similarity, as illustrated by a conserved set of RNA-binding residues between NoV and FHV B2 proteins, which are less than 30% identical in primary amino acid sequence [51–53]. Furthermore, the homodimerization interface of EV 3A proteins is highly conserved among Enteroviruses [14,35]. However, dsRNA-binding mammalian VSRs from different virus families do not share detectable sequence similarities with each other. Intriguingly, dsRNA binding by VSRs NS1 [54–56], B2 [51–53], and 3A [35,57] all appears to employ two positively charged, antiparallel α-helices as a homodimer [58] (Figure 3). These structures are all distinct from the canonical dsRNA-binding motif found in Dicer, dsRNA-activated protein kinase (PKR), and many cellular dsRNA-binding proteins, which adopt an α1β1β2β3α2 fold [59–61]. Interestingly, PKR and nodaviral B2 exhibit similar dsRNA-binding affinity with dissociation constant values in the nanomolar range [51,62].
The genomic RNA1 of nodaviruses encodes VSR-B2 and the viral RdRP from two ‘out-of-frame’ overlapped open reading frames (ORFs). Phylogenetic and codon usage analyses have shown that similar to VSR-2b of plant cucumoviruses, animal nodaviruses may have acquired VSR-B2 gene more recently by an ‘overprinting’ mechanism [50,63–65], in which the novel ORF B2 is created de novo through spontaneous point mutation inside an existing RdRP progenitor gene, but from the +1 reading frame of the RdRP ORF. Based on these analyses, we propose that VSRs from distinct families of mammalian RNA viruses may have evolved independently as a result of convergent evolution to counteract antiviral RNAi.
Concluding remarks
In this review, we have described mammalian VSRs that inhibit vsiRNA production to promote virus infection of undifferentiated and differentiated mammalian cells in the presence or absence of the IFN signaling. It is clear that our perception of antiviral RNAi as having a limited role in undifferentiated cells has been shifted recently, as more evidence for the induction and suppression of antiviral RNAi in differentiated cells and in vivo has been presented. Nonetheless, numerous outstanding questions remain (see Outstanding questions). One key challenge ahead is to functionally validate mammalian VSRs from additional families of viruses, which is critical to determine whether VSRs are as widespread in mammals as in plants and insects [49,50,66–68]. Many human RNA viruses encode one or more proteins exhibiting various VSR activity in mammalian cells (Table 2). It will be important to investigate whether infection with any of these viruses induces a potent antiviral RNAi response that is suppressed by its cognate VSR.
Outstanding questions.
How widespread are VSRs among families of mammalian RNA viruses? Is the VSR activity a conserved function of homologous proteins within a virus family or order?
Are there additional VSRs that suppress antiviral RNAi to promote infection without interfering with the functionality of host miRNAs and in an IFN-independent manner?
What is the molecular basis for the differential suppression of the Dicer-dependent biogenesis of vsiRNAs, but not host miRNAs, by nodaviral VSR-B2? Is the known interaction of several VSRs with their viral RNA replication complex necessary for efficient suppression of vRI-dsRNA dicing?
Do dsRNA-binding VSRs sequester virus-derived dsRNA from the same or different pool as those detected by host innate immune sensors of the viral nucleic acids?
Are there additional VSRs to promote infection in both the arthropod vectors and mammalian hosts by suppressing antiviral RNAi with the same or distinct mechanisms?
Are there mammalian VSRs that suppress antiviral RNAi by targeting other steps or protein components of the pathway in addition to Dicer processing suppression as dsRNA-binding proteins? Does transsuppression of antiviral RNAi occur between coinfecting viruses? Does mixed infection with viruses encoding VSRs to target the same or different steps of antiviral RNAi enhance infection and virulence in mammals as it is known in plants?
Are there mammalian VSRs exhibiting off-target suppression of host miRNA function to induce disease development in vivo?
Table 2.
Mammalian RNA viral proteins that exhibit viral suppressor of RNAi activity
| VSR | Virus | Family (genome type) | Putative mechanism | Refs |
|---|---|---|---|---|
| 7a | SARS-CoV-1 | Coronaviridae (+RNA) | Unknown | [69] |
| N (nucleocapsid) | SARS-CoV-1/2 | Coronaviridae (+RNA) | dsRNA sequestration | [70,71] |
| NS4B | Dengue virus 1–4 | Flaviviridae (+RNA) | Unknown | [72] |
| Capsid | Zika/yellow fever virus | Flaviviridae (+RNA) | dsRNA sequestration | [73–75] |
| Capsid; NS2 | Hepatitis C virus | Flaviviridae (+RNA) | Dicer binding; dsRNA sequestration | [76,77] |
| Capsid | Semliki Forest virus | Togaviridae (+RNA) | dsRNA sequestration | [78] |
| Capsid | Rubella virus | Matonaviridae (+RNA) | dsRNA sequestration | [79] |
| nsP2; nsP3 | Chikungunya virus | Togaviridae (+RNA) | Unknown | [80] |
| VP30; VP35; VP40 | Ebola virus | Filoviridae. (−RNA) | Dicer binding; dsRNA sequestration; unknown | [11,13,41,81] |
| VP35 | Marburg virus | Filoviridae. (−RNA) | dsRNA sequestration | [13] |
| NSsa | La Crosse virus | Bunyaviridae (−RNA) | Unknown | [82] |
Highlights.
RNA viruses from the Flaviviridae, Nodaviridae, Orthomyxoviridae, and Picornaviridae encode functionally validated viral suppressors of RNAi (VSRs) to promote mammalian cell infection in the presence or absence of the interferon signaling.
Therapeutic targeting of closely related human enteroviral VSRs confers protection in mice against lethal challenges, revealing VSRs as a novel class of antiviral drug targets.
Validated VSRs from different virus families show no sequence similarity, but all suppress Dicer-dependent production of virus-derived siRNAs by sequestering long dsRNA likely as a homodimer with a similar fold of two antiparallel α-helices, suggesting convergent evolution.
Additional families of human RNA viruses, including Ebola virus and SARS-CoV-1 and 2, encode unrelated VSRs awaiting further functional validation.
Acknowledgments
W.X.L. and S.W.D. were supported by National Institutes of Health grant AI141887 (to S.W.D.).
Glossary
- Double-stranded (ds)RNA-binding VSR
VSR that binds to long dsRNA and suppresses Dicer-mediated processing of long dsRNA into siRNAs by dsRNA sequestration
- Double-stranded (ds)RNA viral replicative intermediates (vRI-dsRNA)
RNA replicative intermediates synthesized during the replication of viral RNA genomes by viral RNA-dependent RNA polymerase (RdRP)
- Interferon (IFN)
a group of signaling proteins produced by vertebrates in response to microbes (e.g., viruses and bacteria) to activate specific transcription factors (e.g., STAT proteins), thereby inducing the expression of IFN-stimulated genes (ISGs) such as PKR to establish an antiviral state
- Mammalian VSR
VSR encoded by a virus that infects mammals
- Validated VSR
VSR with its specific activity to suppress RNAi verified as necessary to promote infection with the VSR-encoding virus
- Viral suppressors of RNAi (VSR)
a virus-encoded protein exhibiting the activity to block RNAi induced by long double-stranded RNA (dsRNA), siRNA, or virus replication
- Virus-derived siRNA (vsiRNA)
produced by Dicer-mediated processing of long vRI-dsRNA in an infected cell that guide specific cleavages of its complementary viral RNAs in the antiviral effector complex, RNA-induced silencing complex (RISC)
Footnotes
Declaration of interests
The authors have no interests to declare.
References
- 1.Guo Z et al. (2019) Small RNA-based antimicrobial immunity. Nat. Rev. Immunol 19, 31–44 [DOI] [PubMed] [Google Scholar]
- 2.Li HW et al. (2002) Induction and suppression of RNA silencing by an animal virus. Science 296, 1319–1321 [DOI] [PubMed] [Google Scholar]
- 3.Ding SW et al. (2018) Antiviral RNA interference in mammals. Curr. Opin. Immunol 54, 109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berkhout B (2018) RNAi-mediated antiviral immunity in mammals. Curr. Opin. Virol 32, 9–14 [DOI] [PubMed] [Google Scholar]
- 5.Maillard PV et al. (2019) Slicing and dicing viruses: antiviral RNA interference in mammals. EMBO J. 38, e100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai H and Gu W (2020) Small RNA plays important roles in virus-host interactions. Viruses 12, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi T et al. (2021) Mammalian antiviral systems directed by small RNA. PLoS Pathog. 17, e1010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shahrudin S and Ding SW (2021) Boosting stem cell immunity to viruses. Science 373, 160–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanchez-David RY and Maillard PV (2021) Unlocking the therapeutic potential of antiviral RNAi. Immunity 54, 2180–2182 [DOI] [PubMed] [Google Scholar]
- 10.Jeffrey KL (2021) Upping the ante on mammalian antiviral RNA interference. Cell Host Microbe 29, 1333–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y et al. (2013) RNA interference functions as an antiviral immunity mechanism in mammals. Science 342, 231–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maillard PV et al. (2013) Antiviral RNA interference in mammalian cells. Science 342, 235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y et al. (2016) Induction and suppression of antiviral RNA interference by influenza A virus in mammalian cells. Nat. Microbiol 2, 16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu Y et al. (2017) Human virus-derived small RNAs can confer antiviral immunity in mammals. Immunity 46, 992–1004 [DOI] [PubMed] [Google Scholar]
- 15.Xu YP et al. (2019) Zika virus infection induces RNAi-mediated antiviral immunity in human neural progenitors and brain organoids. Cell Res. 29, 265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu Y et al. (2020) Flavivirus induces and antagonizes antiviral RNA interference in both mammals and mosquitoes. Sci. Adv 6, eaax7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y et al. (2020) The activation of antiviral RNA interference not only exists in neural progenitor cells but also in somatic cells in mammals. Emerg. Microbes Infect 9, 1580–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poirier EZ et al. (2021) An isoform of Dicer protects mammalian stem cells against multiple RNA viruses. Science 373, 231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y et al. (2021) Efficient Dicer processing of virus-derived double-stranded RNAs and its modulation by RIG-I-like receptor LGP2. PLoS Pathog. 17, e1009790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartel DP (2018) Metazoan microRNAs. Cell 173, 20–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han Q et al. (2020) Mechanism and function of antiviral RNA interference in mice. mBio 11, e03278–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li WX et al. (2004) Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc. Natl. Acad. Sci. U. S. A 101, 1350–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding SW (2010) RNA-based antiviral immunity. Nat. Rev. Immunol 10, 632–644 [DOI] [PubMed] [Google Scholar]
- 24.Sullivan CS and Ganem D (2005) A virus-encoded inhibitor that blocks RNA interference in mammalian cells. J. Virol 79, 7371–7379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ball LA et al. (1992) Replication of nodamura virus after transfection of viral RNA into mammalian cells in culture. J. Virol 66, 2326–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scherer WF and Hurlbut HS (1967) Nodamura virus from Japan: a new and unusual arbovirus resistant to diethyl ether and chloroform. Am. J. Epidemiol 86, 271–285 [DOI] [PubMed] [Google Scholar]
- 27.Yang C et al. (2021) Case report and genomic characterization of a novel porcine nodavirus in the United States. Viruses 13, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishikiori M and Ahlquist P (2021) Transmembrane redox regulation of genome replication functions in positive-strand RNA viruses. Curr. Opin. Virol 47, 25–31 [DOI] [PubMed] [Google Scholar]
- 29.Aliyari R et al. (2008) Mechanism of induction and suppression of antiviral immunity directed by virus-derived small RNAs in Drosophila. Cell Host Microbe 4, 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu R et al. (2005) Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature 436, 1040–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sagan SM and Sarnow P (2013) RNAi, antiviral after all. Science 342, 207–208 [DOI] [PubMed] [Google Scholar]
- 32.Mombaerts P et al. (1992) RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y et al. (2022) Mouse circulating extracellular vesicles contain virus-derived siRNAs active in antiviral immunity. EMBO J. 41, e109902 Published online March 28, 2022. 10.15252/embj.2021109902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cullen BR (2017) RNA interference in mammals: The virus strikes back. Immunity 46, 970–972 [DOI] [PubMed] [Google Scholar]
- 35.Fang Y et al. (2021) Inhibition of viral suppressor of RNAi proteins by designer peptides protects from enteroviral infection in vivo. Immunity 54, 2231–2244 e2236 [DOI] [PubMed] [Google Scholar]
- 36.Xie X et al. (2013) Membrane topology and function of dengue virus NS2A protein. J. Virol 87, 4609–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie X et al. (2015) Two distinct sets of NS2A molecules are responsible for dengue virus RNA synthesis and virion assembly. J. Virol 89, 1298–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie X et al. (2019) Dengue NS2A protein orchestrates virus assembly. Cell Host Microbe 26, 606–622 e608 [DOI] [PubMed] [Google Scholar]
- 39.Barnard TR et al. (2021) Molecular determinants of flavivirus virion assembly. Trends Biochem. Sci 46, 378–390 [DOI] [PubMed] [Google Scholar]
- 40.Munoz-Moreno R et al. (2021) Induction and evasion of type-I interferon responses during influenza A virus infection. Cold Spring Harb. Perspect. Med 11, a038414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haasnoot J et al. (2007) The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 3, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witteveldt J et al. (2019) MicroRNA-deficient mouse embryonic stem cells acquire a functional interferon response. eLife 8, e44171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adiliaghdam F et al. (2020) A requirement for Argonaute 4 in mammalian antiviral defense. Cell Rep. 30, 1690–1701 e1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maillard PV et al. (2016) Inactivation of the type I interferon pathway reveals long double-stranded RNA-mediated RNA interference in mammalian cells. EMBO J. 35, 2505–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheloufi S et al. (2010) A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465, 584–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jee D et al. (2018) Dual strategies for Argonaute2-mediated biogenesis of erythroid miRNAs underlie conserved requirements for slicing in mammals. Mol. Cell 69, 265–278 e266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Carroll D et al. (2007) A Slicer-independent role for Argonaute 2 in hematopoiesis and the microRNA pathway. Genes Dev. 21, 1999–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Sastre A et al. (1998) Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252, 324–330 [DOI] [PubMed] [Google Scholar]
- 49.Li WX and Ding SW (2001) Viral suppressors of RNA silencing. Curr. Opin. Biotechnol 12, 150–154 [DOI] [PubMed] [Google Scholar]
- 50.Li F and Ding SW (2006) Virus counterdefense: Diverse strategies for evading the RNA-silencing immunity. Annu. Rev. Microbiol 60, 503–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chao JA et al. (2005) Dual modes of RNA-silencing suppression by Flock House virus protein B2. Nat. Struct. Mol. Biol 12, 952–957 [DOI] [PubMed] [Google Scholar]
- 52.Lingel A et al. (2005) The structure of the flock house virus B2 protein, a viral suppressor of RNA interference, shows a novel mode of double-stranded RNA recognition. EMBO Rep. 6, 1149–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korber S et al. (2009) Structure of the RNA-binding domain of Nodamura virus protein B2, a suppressor of RNA interference. Biochemistry 48, 2307–2309 [DOI] [PubMed] [Google Scholar]
- 54.Liu J et al. (1997) Crystal structure of the unique RNA-binding domain of the influenza virus NS1 protein. Nat. Struct. Biol 4, 896–899 [DOI] [PubMed] [Google Scholar]
- 55.Chien CY et al. (1997) A novel RNA-binding motif in influenza A virus non-structural protein 1. Nat. Struct. Biol 4, 891–895 [DOI] [PubMed] [Google Scholar]
- 56.Chien CY et al. (2004) Biophysical characterization of the complex between double-stranded RNA and the N-terminal domain of the NS1 protein from influenza A virus: Evidence for a novel RNA-binding mode. Biochemistry 43, 1950–1962 [DOI] [PubMed] [Google Scholar]
- 57.Strauss DM et al. (2003) Towards an understanding of the poliovirus replication complex: The solution structure of the soluble domain of the poliovirus 3A protein. J. Mol. Biol 330, 225–234 [DOI] [PubMed] [Google Scholar]
- 58.Marc D (2014) Influenza virus non-structural protein NS1: Interferon antagonism and beyond. J. Gen. Virol 95, 2594–2611 [DOI] [PubMed] [Google Scholar]
- 59.Liu Z et al. (2018) Cryo-EM structure of human Dicer and its complexes with a pre-miRNA substrate. Cell 173, 1191–1203 e1112 [DOI] [PubMed] [Google Scholar]
- 60.Nanduri S et al. (1998) Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 17, 5458–5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fierro-Monti I and Mathews MB (2000) Proteins binding to duplexed RNA: One motif, multiple functions. Trends Biochem. Sci 25, 241–246 [DOI] [PubMed] [Google Scholar]
- 62.Schmedt C et al. (1995) Functional characterization of the RNA-binding domain and motif of the double-stranded RNA-dependent protein kinase DAI (PKR). J. Mol. Biol 249, 29–44 [DOI] [PubMed] [Google Scholar]
- 63.Ding SW et al. (1995) A novel naturally-occurring hybrid gene encoded by a plant RNA virus facilitates long-distance virus movement. EMBO J. 14, 5762–5772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keese PK and Gibbs A (1992) Origins of genes: ‘Big bang’ or continuous creation? Proc. Natl. Acad. Sci. U. S. A 89, 9489–9493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ding SW et al. (1994) New overlapping gene encoded by the cucumber mosaic-virus genome. Virology 198, 593–601 [DOI] [PubMed] [Google Scholar]
- 66.Diaz-Pendon JA and Ding SW (2008) Direct and indirect roles of viral suppressors of RNA silencing in pathogenesis. Annu. Rev. Phytopathol 46, 303–326 [DOI] [PubMed] [Google Scholar]
- 67.Wu Q et al. (2010) Viral suppressors of RNA-based viral immunity: Host targets. Cell Host Microbe 8, 12–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Csorba T et al. (2015) Viral silencing suppressors: Tools forged to fine-tune host-pathogen coexistence. Virology 479–480, 85–103 [DOI] [PubMed] [Google Scholar]
- 69.Karjee S et al. (2010) The 7a accessory protein of severe acute respiratory syndrome coronavirus acts as an RNA silencing suppressor. J. Virol 84, 10395–10401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cui L et al. (2015) The nucleocapsid protein of coronaviruses acts as a viral suppressor of RNA silencing in mammalian cells. J. Virol 89, 9029–9043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mu J et al. (2020) SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci. China Life Sci 63, 1413–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kakumani PK et al. (2013) Role of RNA interference (RNAi) in dengue virus replication and identification of NS4B as an RNAi suppressor. J. Virol 87, 8870–8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Samuel GH et al. (2016) Yellow fever virus capsid protein is a potent suppressor of RNA silencing that binds double-stranded RNA. Proc. Natl. Acad. Sci. U. S. A 113, 13863–13868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zeng J et al. (2021) Functional mapping of AGO-associated zika virus-derived small interfering RNAs in neural stem cells. Front. Cell. Infect. Microbiol 11, 628887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeng J et al. (2020) The zika virus capsid disrupts corticogenesis by suppressing Dicer activity and miRNA biogenesis. Cell Stem Cell 27, 618–632 e619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Y et al. (2006) Hepatitis C virus core protein is a potent inhibitor of RNA silencing-based antiviral response. Gastroenterology 130, 883–892 [DOI] [PubMed] [Google Scholar]
- 77.Zhou H et al. (2020) Hepatitis C virus NS2 protein suppresses RNA interference in cells. Virol. Sin 35, 436–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qian Q et al. (2020) The capsid protein of Semliki Forest virus antagonizes RNA interference in mammalian cells. J. Virol 94. 10.1128/JVI.01233-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu J et al. (2021) The capsid protein of rubella virus antagonizes RNA interference in mammalian cells. Viruses 13. 10.3390/v13020154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mathur K et al. (2016) Analysis of chikungunya virus proteins reveals that non-structural proteins nsP2 and nsP3 exhibit RNA interference (RNAi) suppressor activity. Sci. Rep 6, 38065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fabozzi G et al. (2011) Ebolavirus proteins suppress the effects of small interfering RNA by direct interaction with the mammalian RNA interference pathway. J. Virol 85, 2512–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soldan SS et al. (2005) La Crosse virus nonstructural protein NSs counteracts the effects of short interfering RNA. J. Virol 79, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schnettler E et al. (2010) Diverging affinity of tospovirus RNA silencing suppressor proteins, NSs, for various RNA duplex molecules. J. Virol 84, 11542–11554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garcia-Ruiz H et al. (2018) Tomato spotted wilt virus NSs protein supports infection and systemic movement of a potyvirus and is a symptom determinant. Viruses 10, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]