

Abstract
Objective: In Friedreich’s ataxia (FRDA), the most affected tissues are not accessible to sampling and available transcriptomic findings originate from blood-derived cells and animal models. Herein, we aimed at dissecting for the first time the pathophysiology of FRDA by means of RNA-sequencing in an affected tissue sampled in vivo. Methods: Skeletal muscle biopsies were collected from seven FRDA patients before and after treatment with recombinant human Erythropoietin (rhuEPO) within a clinical trial. Total RNA extraction, 3′-mRNA library preparation and sequencing were performed according to standard procedures. We tested for differential gene expression with DESeq2 and performed gene set enrichment analysis with respect to control subjects. Results: FRDA transcriptomes showed 1873 genes differentially expressed from controls. Two main signatures emerged: (1) a global downregulation of the mitochondrial transcriptome as well as of ribosome/translational machinery and (2) an upregulation of genes related to transcription and chromatin regulation, especially of repressor terms. Downregulation of the mitochondrial transcriptome was more profound than previously shown in other cellular systems. Furthermore, we observed in FRDA patients a marked upregulation of leptin, the master regulator of energy homeostasis. RhuEPO treatment further enhanced leptin expression. Interpretation: Our findings reflect a double hit in the pathophysiology of FRDA: a transcriptional/translational issue and a profound mitochondrial failure downstream. Leptin upregulation in the skeletal muscle in FRDA may represent a compensatory mechanism of mitochondrial dysfunction, which is amenable to pharmacological boosting. Skeletal muscle transcriptomics is a valuable biomarker to monitor therapeutic interventions in FRDA.
Introduction
Friedreich’s ataxia (FRDA, OMIM #229300) is a rare multisystemic disorder affecting up 1:25 000 people in the Caucasians (1). In the majority of cases, the disease is triggered by biallelic GAA-repeat expansions within the first intron of the frataxin (FXN) gene (2).
Pathologically expanded FXN alleles are associated with enhanced heterochromatinization (3), hypermethylation (4) and transcription repression, resulting in reduced levels of an otherwise functional protein (5). An inverse correlation exists between GAA repeat length and FXN levels (2). FXN is a mitochondrial protein involved in the biosynthesis of iron–sulphur clusters (ISC), the prosthetic groups of several respiratory chain enzymes and DNA-repair proteins (6–9). FXN deficiency results in defective ISC synthesis, impaired oxidative phosphorylation and mitochondrial failure with iron accumulation (10).
Clinically, FRDA presents with a progressive afferent ataxia due to primary affection of the dorsal root ganglia, posterior column of the spinal cord, peripheral sensory nerves and later of the cerebellum (11). Hypertrophic cardiomyopathy and skeletal deformities are classical comorbidities (11). Diabetes mellitus, as well as hearing and visual loss, may occur in later stages (11). As classical mitochondriopathies, FRDA features also subclinical myopathy, as evident in functional and histological studies (12–15). Myopathy contributes to the weakness and marked fatigue, which overtly outlasts the burden expected from the ataxic syndrome.
Key aspects of FRDA pathophysiology remain elusive. First, it is unknown how the deficiency of a ubiquitous protein leads to a selective dysfunction of few organs. The mechanisms underlying the neurological progression are likewise unclear. Histopathological studies revealed a hypoplasia of the affected neural structures, without major neurodegenerative aspects (16), a finding which does not explain the delayed onset of ataxia after birth and the steady progression thereafter.
Transcriptomic studies allow to define the molecular signature of a biological system at a given time point, thus offering a unique insight into the pathophysiology of a disease. Since the key affected cell types in FRDA are inaccessible to sampling in vivo, previous studies have been performed in blood cells and fibroblasts (17,18), i.e. tissues without a disease phenotype. Transcriptomic approaches in murine models of FRDA (19–21) bear shortcomings, since they do not fully recapitulate the human phenotype. Lai et al. tried to overcome these limitations using induced pluripotent stem cells (iPSC)-derived sensory neurons but failed to replicate a clear disease signature in this model, probably due to the early maturation stage (21).
Herein, we report the first gene expression profiling generated by RNA-sequencing in skeletal muscle biopsies of genetically confirmed FRDA patients. Muscle biopsies were obtained before and after treatment with recombinant human erythropoietin (rhuEPO) in the frame of a clinical trial (14,22). Comparing patients’ transcriptomes with matched controls, we delineated for the first time the transcriptomic signature of FRDA in an affected tissue, in vivo. In the present study, findings from differential expression (DE) analysis are compatible with a double-hit pathophysiology, namely with a transcriptional/translational downregulation and a mitochondrial derangement. Moreover, DE dynamics after rhuEPO highlighted the potential of skeletal muscle transcriptomics as valuable biomarker in early-stage trials in FRDA.
Results
Six male and one female patients with a mean age of 40 years at the time of examination (range 26–59) were included (Fig. 1A). All patients carried two expanded FNX alleles, with an average length of 584 ± 337 repeats in the shorter GAA expansions (GAA1). Mean disease duration was 18 ± 8 years and mean age at disease onset was 22 ± 8 years. The mean SARA (Scale for assessment and rating of ataxia) score at the time of the biopsy was 23 ± 8 points, representing a moderate to advance ataxia stage.
Figure 1.
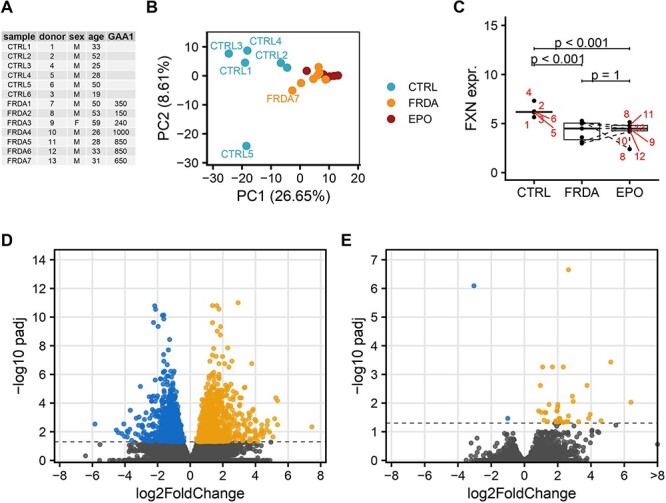
Transcriptomic analysis of skeletal muscle biopsies of FRDA patients before and after rhu-EPO treatment as compared to controls. (A) FRDA patients and controls are listed along their Patient_ID and demographic data. (B) PCA of the 500 most variable genes shows a separation of gene expression profiles according to disease status (blue controls, orange FRDA before rhuEPO treatment, red FRDA after rhuEPO treatment) along principal component 1 (PC1). (C) Boxplots of normalized expression values of FXN in control, FRDA and post-treatment FRDA samples. Patient ID is shown in red. Adjusted P-values from DESeq2 DE analysis. (D, E): Volcano plot of differentially expressed genes in FRDA samples compared to control samples (D) and of rhuEPO-treated FRDA patients compared to matched pre-treatment samples (E). Genes with an adjusted P-value lower than 0.05 were considered significant, genes upregulated in FRDA are shown in orange and downregulated genes are shown in blue.
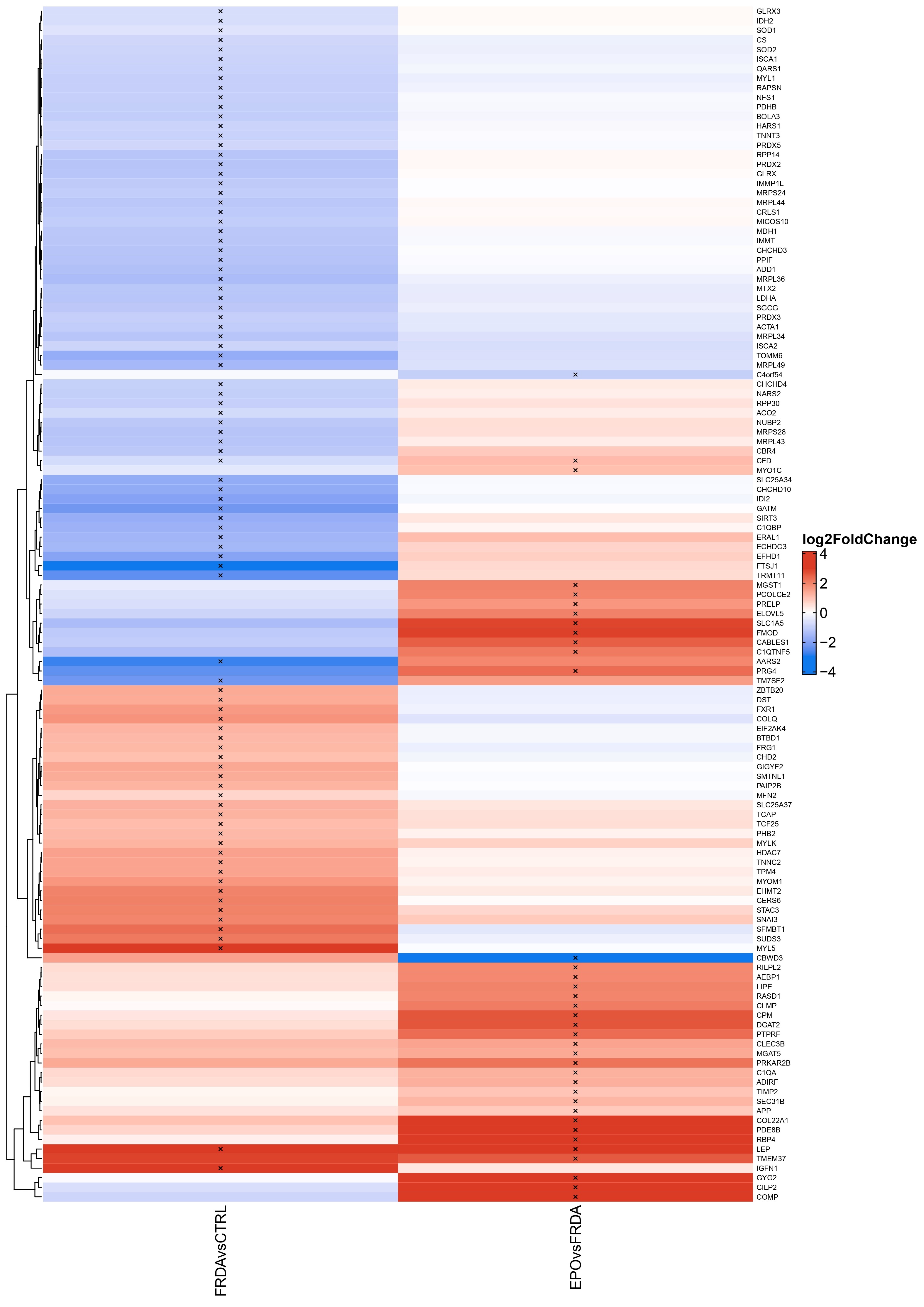
For each biopsy mRNA preparation, ∼6.2-million sequencing reads were generated. Approximately, 86% of them were mapped to the human genome/to exons (56%). Exploratory data analysis by means of principal component analysis (PCA) showed a clear separation of FRDA and control samples based on the expression of the 500 most variable genes (Fig. 1B). FXN expression was >3.5-fold decreased in FRDA samples comparing to controls (log2-fold change = −1.94, P < 0.001, Fig. 1C). FXN expression inversely correlated with the length of the shorter GAA repeat expansion (Spearman rank correlation = −0.61, P = 0.14, Fig. 2) in FRDA samples. Overall, 1873 genes were differentially expressed between FRDA and controls (with a false discovery rate < 0.05) with 1144 genes showing a >2-fold change in expression (Fig. 1D). Across the DE genes set, 918 down-regulated and 955 were up-regulated in FRDA as compared to controls. The DE genes addressed in more details in the manuscript are reported also in Table 1 and in the heatmap in Supplementary Material, Figure S1. The complete list is provided in Supplementary Material, Table S1. Gene set enrichment analysis assigned the DE genes in 257 overrepresented pathways (123 down- and 134 up-regulated), as shown in Fig. 3 and Supplementary Material, Table S2.
Figure 2.
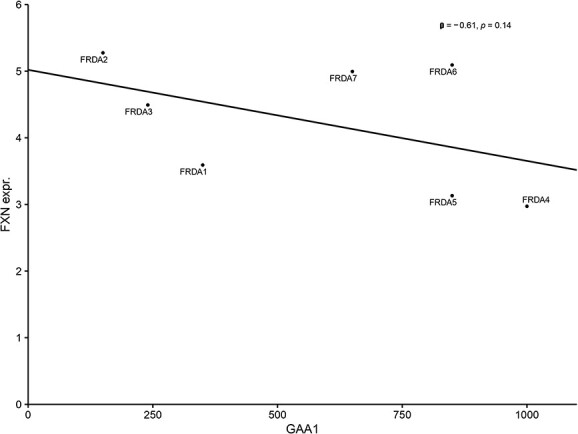
Correlation between the GAA1 repeats and FXN expression in FRDA patients.
Table 1.
DE genes cited in the results are listed according to their functional domain, along with log2FC and adjusted P values
| DOMAIN | Definition | log2FC | Adjusted P-value |
|---|---|---|---|
| ISC assembly factors/scaffold | |||
| BOLA3 | bolA family member 3 | −1.06 | <0.01 |
| NFS1 | NFS1 cysteine desulfurase | −1.02 | <0.01 |
| NUBP2 | nucleotide binding protein 2 | −1.15 | <0.01 |
| GLRX3 | glutaredoxin 3 | −0.69 | 0.02 |
| ISCA1 | iron–sulfur cluster assembly 1 | −0.90 | <0.001 |
| ISCA2* | iron–sulfur cluster assembly 2 | −0.89 | 0.01 |
| Krebs cycle | |||
| CS | citrate synthase | −0.85 | <0.0001 |
| IDH2 | isocitrate dehydrogenase (NADP(+)) 2 | −0.69 | <0.01 |
| ACO2 | Aconitase 2 | −0.76 | <0.001 |
| PDHB* | pyruvate dehydrogenase E1 subunit beta | −1.03 | <0.001 |
| MDH1 | malate dehydrogenase 1 | −1.18 | <0.001 |
| Mitochondrial transport proteins | |||
| IMMP1L | inner mitochondrial membrane peptidase subunit 1 | −1.11 | <0.01 |
| MTX2 | metaxin 2 | −1.20 | <0.0001 |
| SLC25A34 | solute carrier family 25 member 34 | −1.80 | 0.02 |
| TOMM6 | translocase of outer mitochondrial membrane 6 | −1.78 | 0.01 |
| Mitochondrial structural/regulatory proteins | |||
| CHCHD10 | coiled-coil-helix-coiled-coil-helix domain containing 10 | −1.83 | <0.0001 |
| CHCHD3 † | coiled-coil-helix-coiled-coil-helix domain containing 3 | −1.27 | <0.01 |
| CHCHD4 | coiled-coil-helix-coiled-coil-helix domain containing 4 | −0.99 | 0.04 |
| EFHD1 | EF-hand domain family member D1 | −1.97 | <0.0001 |
| IMMT | inner membrane mitochondrial protein | −1.19 | <0.0001 |
| MICOS10 | mitochondrial contact site and cristae organizing system subunit 10 | −1.06 | <0.0001 |
| PPIF | peptidylprolyl isomerase F | −1.30 | <0.01 |
| SIRT3 | sirtuin 3 | −1.74 | <0.0001 |
| Mitochondrial ribosomes | |||
| C1QBP † | complement C1q binding protein | −1.66 | <0.0001 |
| ERAL1 | era like 12S mitochondrial rRNA chaperone 1 | −1.59 | 0.02 |
| MRPL34 | mitochondrial ribosomal protein L34 | −1.21 | <0.0001 |
| MRPL36 | mitochondrial ribosomal protein L36 | −1.41 | <0.0001 |
| MRPL43* | mitochondrial ribosomal protein L43 | −1.22 | <0.0001 |
| MRPL44* | mitochondrial ribosomal protein L44 | −1.18 | <0.01 |
| MRPL49* † | mitochondrial ribosomal protein L49 | −1.52 | <0.01 |
| MRPS24 | mitochondrial ribosomal protein S24 | −1.06 | <0.0001 |
| MRPS28 | mitochondrial ribosomal protein S28 | −1.25 | 0.002 |
| Upregulated mitochondrial DE genes | |||
| MFN2* | mitofusin 2 | 0.66 | 0.03 |
| PHB2 | prohibitin 2 | 1.15 | <0.01 |
| SLC25A37 | solute carrier family 25 member 37 | 1.25 | <0.0001 |
| tRNA synthesis and maturation | |||
| AARS2 | alanyl-tRNA synthetase 2, mitochondrial | −2.81 | <0.01 |
| HARS1 | histidyl-tRNA synthetase 1 | −0.90 | <0.01 |
| NARS2* | asparaginyl-tRNA synthetase 2, mitochondrial | −1.04 | <0.01 |
| QARS1* | glutaminyl-tRNA synthetase 1 | −0.94 | <0.001 |
| FTSJ1 | FtsJ RNA 2′-O-methyltransferase 1 | −3.31 | <0.01 |
| RPP14 | ribonuclease P/MRP subunit p14 | −1.25 | <0.0012 |
| RPP30 | ribonuclease P/MRP subunit p30 | −1.02 | 0.04 |
| TRMT11 | tRNA methyltransferase activator subunit 11–2 | −2.48 | <0.01 |
| Antioxidant defence | |||
| GLRX | glutaredoxin | −1.23 | 0.02 |
| PRDX2* | periredoxin 2 | −1.26 | <0.001 |
| PRDX3 | periredoxin 3 | −1.02 | <0.01 |
| PRDX5* † | periredoxin 5 | −0.85 | <0.01 |
| SOD1 | superoxide dismutase 1 | −0.57 | 0.04 |
| SOD2 | superoxide dismutase 2 | −0.87 | <0.01 |
| Transcription & translation repression | |||
| EHMT2 | euchromatic histone lysine methyltransferase 2 | 1.99 | 0.02 |
| EIF2AK4 | eukaryotic translation initiation factor 2 alpha kinase 2 | 1.21 | <0.0001 |
| GIGYF2 | GRB10 interacting GYF protein 2 | 1.40 | <0.0001 |
| HDAC7 | histone deacetylase 7 | 1.50 | <0.0001 |
| PAIP2B | poly(A) binding protein interacting protein 2B | 1.19 | 0.01 |
| SFMBT1* | scm like with four mbt domains 1 | 2.30 | <0.0001 |
| SUDS3 | SDS3 homolog, SIN3A corepressor complex component | 2.15 | <0.001 |
| TCF25 | transcription factor 25 | 1.03 | <0.01 |
| Lipid metabolism | |||
| CBR4 | carbonyl reductase 4 | −1.15 | <0.0001 |
| CERS6 | ceramide synthase 6 | 2.04 | <0.0001 |
| CRLS1* | cardiolipin synthase 1 | −1.12 | <0.01 |
| ECHDC3 | enoyl-CoA hydratase domain containing 3 | −1.53 | <0.01 |
| IDI2 | isopentenyl-diphosphate delta isomerase 2 | −2.00 | <0.0001 |
| LEP † | leptin | 4.15 | <0.01 |
| TM7SF2 | transmembrane 7 superfamily member 2 | −2.27 | 0.02 |
| Skeletal muscle specific | |||
| ACTA1 | actin alpha 1, skeletal muscle | −1.07 | <0.0001 |
| ADD1 | adducin 1 | −1.34 | <0.0001 |
| BTBD1 | BTB domain containing 1 | 1.15 | 0.02 |
| CHD2 | chromodomain helicase DNA binding protein 2 | 1.02 | <0.0001 |
| COLQ | collagen like tail subunit of asymmetric acetylcholinesterase | 1.72 | 0.02 |
| DST | dystonin | 1.36 | <0.0001 |
| FRG1 | FSHD region gene 1 | 1.17 | <0.0001 |
| FXR1 | FMR1 autosomal homolog 1 | 1.63 | <0.0001 |
| GATM | glycine amidinotransferase | −2.26 | <0.0001 |
| IGFN1 | immunoglobulin like and fibronectin type III domain containing 1 | 3.86 | <0.001 |
| LDHA | lactate dehydrogenase A | −1.23 | 0.03 |
| MYL1 | myosin light chain 1 | −1.01 | <0.01 |
| MYL5 | myosin light chain 5 | 3.04 | <0.0001 |
| MYLK | myosin light chain kinase | 1.20 | 0.02 |
| MYOM1 | myomesin 1 | 1.68 | <0.0001 |
| RAPSN | receptor associated protein of the synapse | −1.01 | 0.01 |
| SGCG | sarcoglycan gamma | −1.14 | <0.001 |
| SMTNL1 | smoothelin like 1 | 1.34 | <0.01 |
| SNAI3 | snail family transcriptional repressor 3 | 1.94 | <0.001 |
| STAC3 | SH3 and cysteine rich domain 3 | 2.00 | <0.0001 |
| TCAP | titin-cap | 1.23 | <0.01 |
| TNNC2 | troponin C2, fast skeletal type | 1.47 | <0.001 |
| TNNT3 | troponin T3, fast skeletal type | −0.93 | 0.02 |
| TPM4 | tropomyosin 4 | 1.47 | <0.01 |
| ZBTB20 | zinc finger and BTB domain containing 20 | 1.37 | <0.0001 |
Asterisks (*) and crosses (†) indicate the genes that were found to be differentially expressed (with the same net effect towards down- and up-regulation) also in FRDA fibroblasts and blood cells, respectively.
Figure 3.
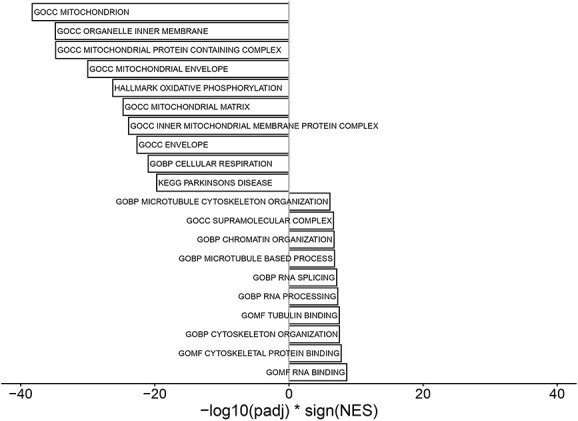
GSEA. Barplot of the 10 most significantly up- and downregulated gene sets in FRDA versus control samples.
Downregulation of mitochondrial transcriptome and translational machinery
Pathways related to mitochondrial composition and function dominated the set of downregulated DE genes (GO:0005739, Mitochondrion, P < 0.0001, see also Fig. 3 and Supplementary Material, Table S2). Mitochondrial DE genes were associated with manifold aspects of mitochondrion structure and functioning, including oxidative phosphorylation, electron transport chain, ISC assembly/binding, mitochondrial structure, Krebs cycle, mitochondrial gene expression and translation. The most downregulated gene set (GOCC_INNER_MITOCHONDRIAL_PROTEIN_COMPLEX) includes the respiratory chain complexes from I to V. Downregulated DE genes included Krebs cycle enzymes (CS, IDH2, ACO2, MDH1, PDHB), mitochondrial transport protein (IMMP1L, MTX2, SLC25A34, TOMM6), structural/regulatory genes (CHCHD10, CHCHD3, CHCHD4, EFHD1, IMMT, MICOS10, PPIF, SIRT3). Apart from FXN, further key iron–sulphur clusters assembly factors/scaffold (BOLA3, NFS1, NUBP2, GLRX3, ISCA1, ISCA2) were also significantly less represented in FRDA transcriptomes. Several cytosolic ISC-binding proteins involved in DNA replication and repair, such as helicases, polymerases, endonuclease, did not display a differential expression between FRDA and controls (RTEL1, BRIP1, PRIM2, POLD1, REV3L, NTHL1, MUTYH, DNA2, ELP3, ABCE1).
Mitochondrial ribosome constituents (MRPL34, MRPL36, MRPL43, MRPL44, MRPL49, MRPS24, MRPS28) and regulatory proteins (C1QBP, ERAL1) represented a further dominating cluster in the DE set. Cytoplasmic ribosomal components were also globally downregulated although to a lesser extent than their mitochondrial counterparts. Similarly, genes related to tRNA maturation (FTSJ1, RPP14, RPP30, TRMT11), cytosolic and mitochondrial aminoacyl-tRNA synthetases genes (AARS2, NARS2, QARS1, HARS1) also clustered under down-regulated DE genes.
Both mitochondrial (SOD2, PRDX3, PRDX5) and cytoplasmic key antioxidant defence components (SOD1, GLRX, PRDX2) listed in the downregulated DE genes.
We did not find differences concerning the expression of master regulators of the mitochondrial biogenesis (PPARGC1A/PGC-1α, NFE2L2/NRF2, TFAM, UCP2) between FRDA and controls.
Only few mitochondrial genes showed an upregulation in FRDA transcriptomes. These included Mitoferrin-1 (SLC25A37), an iron importer at the internal mitochondrial membrane required for ISC synthesis, as well as PHB2/prohibitin-2 and MFN2/mitofusin-2, two proteins involved in the regulation of mitochondrial architecture and fission-to-fusion balance.
Upregulation of transcription and chromatin modification-related terms
Gene set enrichment analysis delineated chromatin/RNA binding and microtubule organization items as most significantly upregulated clusters in FRDA transcriptomes. Multiple overlapping pathways related to mRNA processing, splicing/spliceosome regulation were upregulated in FRDA. Concerning chromatin modification, overrepresented items included several histone binding proteins and enzymes related to histone de-/methylation as well as de-/acetylation. Herein, emerged a cluster of regulatory terms with a net repressor effect on transcription (EHMT2, HDAC7, TCF25, SFMBT1, SUDS3). Interestingly, also translation repressors (EIF2AK4, GIGYF2, PAIP2B) listed to the upregulated DE genes.
Differential expression of lipid metabolism genes in FRDA
DE analysis highlighted an enrichment in genes related to lipid metabolism. Downregulated genes were involved in the biosynthesis of fatty acid (CBR4, ECHDC3), cholesterol (IDI2, TM7SF2) and cardiolipin (CRLS1). Upregulated terms included enzymes involved in the synthesis of phospholipids and ceramides, including CERS6, whose products play a role in inflammatory response and mitochondrial fragmentation (23). Notably, leptin (LEP) was markedly upregulated in FRDA compared to controls (log2FC = 4.34, P = 0.027). We did not find differences in the expression of the PGC-1α/PPARγ pathway-related lipogenesis genes SCD, ACSS2, ACLY, AACS and ELOVL3.
Changes in skeletal-muscle-specific transcriptome in FRDA
Several skeletal-muscle-specific genes (24) appeared to be differentially expressed in FRDA transcriptomes compared to controls. The majority of muscle-specific DE genes were upregulated and included regulatory genes controlling myogenesis and myoblastic differentiation (ZBTB20, BTBD1, CHD2, FRG1, FXR1, IGFN1, SNAI3), key structural proteins (COLQ, DST, MYL5, MYBPC2, MYLK, MYOM1, TCAP, TTN) and regulators of contraction (SMTNL1, STAC3, TNNC2, TPM4). Downregulated DE genes included structural proteins (ACTA1, ADD1, MYL1, SGCG, TNNT3, RAPSN) and key enzymes in muscle metabolism (GATM, LDHA).
Effect of rhuEPO treatment on skeletal muscle transcriptome in FRDA
With respect to rhuEPO treatment, 37 DE genes emerged between baseline and post-treatment, of which 35 were upregulated (Figs 1E, 4 and Supplementary Material, Table S1).
Figure 4.
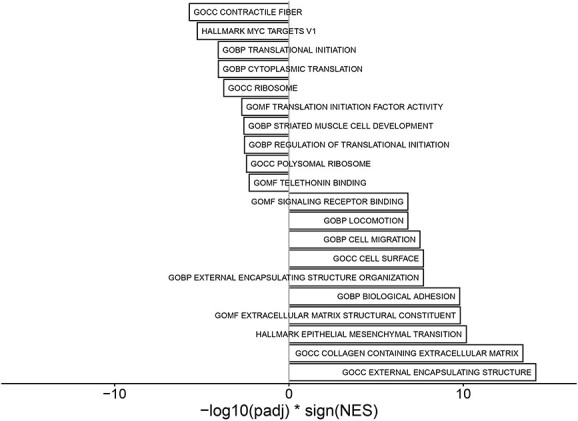
GSEA. Barplot of the 10 most significantly up- and downregulated gene sets in rhuEPO-treated versus untreated FRDA samples.
Notably, RhuEPO treatment led to a further strong upregulation of leptin compared to baseline (log2FC = 3.25, P = 0.04, Fig. 5). Extracellular-matrix-related elements were also markedly upregulated (COMP, log2FC = 6.40, P < 0.01; CILP2, log2FC = 4.60, P = 0.04; COL22A1, log2FC = 3.77, P < 0.01, FMOD log2FC = 2.93, P < 0.01). Further upregulated DE genes included GYG2 (log2FC = 5.19, P < 0.001), coding for glycogenin-2 an enzyme involved in the synthesis of glycogen in muscle, as well as ELOVL5 (log2FC = 2.01, P = 0.01) and DGAT2 (log2FC = 2.64, P = 0.03), enzymes involved in the elongation of fatty acids and hydrolysis from triglycerides, respectively. FXN mRNA level did not display consistent changes after rhuEPO treatment (Fig. 1B).
Figure 5.
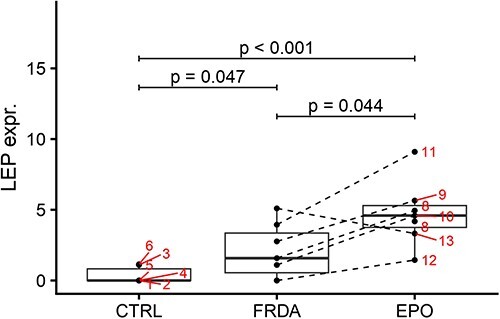
Boxplots of log-transformed normalized expression values of LEP in control, FRDA and post-treatment FRDA samples. Patient ID is shown. Adjusted P-values from DESeq2 DE analysis.
Discussion
Clinical and basic research in FRDA has advanced at a remarkable pace and has recently led to a first positive signal concerning therapeutics development (25). Cumulating evidence gained mostly from disease models and easily accessible tissues (e.g. fibroblasts) provided the basis for these advancements. Nonetheless, the key link between FXN deficiency and the resulting peculiar phenotype remains elusive and the definition of a therapeutic approach with a major impact on the disorder is still an unmet need. In this light, we aimed at dissecting the biology of an affected tissue in the setting of full-blown disease in vivo. We fulfilled this aim by applying RNA-sequencing in skeletal muscle samples obtained within a clinical trial investigating the effects of rhuEPO in genetically confirmed FRDA patients with overt disease (14,22).
Skeletal muscle transcriptomics showed profound and widespread DE changes in FRDA compared to controls. Overall FRDA samples showed (1) a net shift towards downregulation of the global mitochondrial transcriptomes as well as the mitochondrial and cytosolic translational machinery and (2) an upregulation of genes involved in transcription and chromatin regulation, especially of repressors.
The recognition of FRDA as a mitochondrial disorder dates back to early studies revealing a deficiency of mitochondrial ISC-proteins in affected tissues (7). The mitochondrial signature of FRDA has been corroborated by multiple lines of evidence in disease models and in vivo (7,10,12). From a pathophysiological point of view, mitochondrial damage is believed to be a downstream effect consequent to FXN deficiency and ISC synthesis failure. Independent observations postulated progressive mitochondrial damage as main trigger of the clinical progression (26,27). Notably, a recent study in iPSC-derived sensory neurons (21) failed to reproduce a mitochondrial phenotype in this model, most likely due to the early maturation stage of the cells. Previous transcriptomic studies in patients’ derived tissues have been carried out in peripheral mononuclear blood cells (PBMCs) and fibroblasts (17,18,28). Also in these cells, FXN deficiency was associated with a definite transcriptional signature, oriented towards down-regulation. PBMCs and above all fibroblasts from FRDA patients displayed also a downregulation of several mitochondrial components, including mitochondrial ribosomal components, respiratory chain enzymes, membrane carriers and mitochondrial aminoacyl-tRNA synthetases genes (17,18). In comparison, the present findings in skeletal muscle display a far more extensive alteration of the global mitochondria-related transcriptome, as highlighted in the gene set enrichment analysis (see Fig. 3 and Supplementary Material, Table S2). Herein, terms involving manifold aspects of mitochondrial function dominated the list of downregulated pathways. Our data emphasize the association of an advanced disease with a profound mitochondrial failure, as opposed to a presymptomatic state. They furthermore underpin the severe mitochondrial signature in clinically affected tissues as compared to cellular systems without a disease phenotype. The very recent approval of a NRF2 pathway activator as first drug for the treatment of FRDA underscores the central role of the mitochondrial failure in the pathophysiology of the disease (29).
Our skeletal muscle samples did not display the definite transcriptional downregulation in DNA replication and repair enzymes found in blood derived cells and fibroblasts (17,18,28). DE genes from this category in skeletal muscle were both downregulated (ERCC1, ERCC2) and upregulated (NBN, PMS1, RAD50, TOP1). Changes in lipid metabolism genes were highlighted also in previous studies, though related DE genes in PBMCs and fibroblasts only marginally overlap with the present findings in skeletal muscle. Comparing with previous observation in mice (20), the present analysis in skeletal muscle biopsies did not show a clear downregulation of genes related to aerobic, slow-twitch fibers. Instead, comparing with skeletal muscle fiber signatures from reference sequencing data (30), FRDA patients displayed a downregulation of TNNT3, a troponin typical of anaerobic, fast-twitch, type II fibers.
Transcriptomics data reproduce only the first step of the biological processes that ultimately lead to the definition of a cell- and time-specific protein set. Gene expression profiling data cannot anticipate the ultimate molecular consequences at a cellular level and must be interpreted in the light of available evidence. According to the most obvious assumption, the extensive downregulation of nuclear-coded mitochondrial mRNA in skeletal muscle in FRDA may simply reflect a marked reduction in the mitochondria pool in full-blown, advanced disease and thus a reduced need for mitochondrial proteins. Assuming that ISC deficiency and mitochondrial failure are a downstream effect, we wondered though if a compensatory feedback should take place at a transcriptional level, for example with an upregulation of ISC machinery proteins. DE changes in skeletal muscle specific genes seems to recapitulate such a feedback response to an ongoing damage, with upregulation of myodifferentiation terms and structural proteins. It remains speculative if a global downregulation shift in FRDA samples may play a role in the observed severe repression of mitochondrial transcriptome, an effect that may be more evident in the skeletal muscle, due to the enrichment in mitochondria. We discuss this hypothesis considering that upregulated DE genes in FRDA skeletal muscle clustered in the RNA processing and chromatin regulation, with repressor terms especially emerging. Notably, expanded FNX alleles have been clearly associated with hypermethylation, enhanced heterochromatinization and transcription repression but, to the best of our knowledge, it has not been investigated if these changes spread outside the FXN locus. Our findings argue against a downregulation of the mitochondrial biogenesis at a transcriptional level, as described in murine models (20).
Interestingly, skeletal muscle transcriptomics highlighted an upregulation of LEP, coding for the hormone leptin, as a possible compensatory mechanism for the mitochondrial failure in FRDA. Leptin is a master regulator of energy homeostasis and cumulative research highlighted its protective effect in the setting of mitochondrial dysfunction (31–33). Especially, leptin appears to improve the mitochondrial fusion-to-fission balance, for instance hindering the downregulation of Mitofusin-2 (31). This latter mitochondrial gene was also upregulated in our FRDA samples compared to controls. Notably, LEP is upregulated also in FRDA-derived lymphoblastoid cell lines and in the skeletal muscle frataxin-deficient mice (17). A recent study also reported an elevated leptin plasma levels in the KIKO mouse model of FRDA (34), which has been interpreted as a sign of a dysmetabolic phenotype. To this concern, circulating leptin has a different function comparing to the locally produced leptin, which exerts tissue-protective effects. Rhu-EPO treatment resulted in an additional strong upregulation of leptin in FRDA (in sum ~250-fold compared to controls). Besides protective properties, leptin synthetized in the skeletal muscle appears to have paracrine effects resulting in the stimulation of local angiogenesis (35). Indeed leptin, such as EPO, can be induced by tissue hypoxia (36). Moreover, EPO itself can stimulate production of leptin and other cytokines in human adipose tissue, supporting regenerative processes and tissue reparation (37). Concordant with such effects, rhuEPO led both to the upregulation of leptin as well as of terms related to the extracellular matrix remodeling. An increased muscle capillary densities has also been detected in FRDA patients treated with rhuEPO (14). Overall, rhuEPO treatment resulted mostly in an upregulation of gene expression, although far more restricted than in its primary targets, the hematopoietic tissue (38). Clinical trials with rhuEPO did not result in a clinically meaningful improvement in ataxia parameters (39), and currently EPO-derivatives are no more in the treatment pipeline of FRDA. Nonetheless, transcriptomics data before and after rhuEPO treatment are valuable for the design of future studies. Indeed, our findings point out the leptin pathway as a druggable target in FRDA, as already shown in other models of mitochondrial dysfunction (31,33). Moreover, our data underline the great potential of skeletal muscle biopsy transcriptomics as real-time biomarker in FRDA, which is sensitive to pharmacological intervention.
As already addressed, RNA-sequencing data offer insight into a very early stage of the pathophysiological cascade and concomitant evaluation of proteomic and metabolomic data are required to delineate the final picture of a disease model at a cellular level. For instance, post-transcriptional mechanisms are implied in the impairment in the NRF2-related pathway, which is central to the pathophysiology of FRDA (40). However, comparing to the currently available metabolomics approach, RNA-sequencing enables to capture the global transcriptional state of a cell and not only the state of a predefined subset of genes. Our data, generated by means of such a comprehensive approach, hint at a double-hit pathology in FRDA, with a profound, mitochondrial failure, progressing over time and a transcriptional/translational repression upstream. Successful therapeutic approaches might need to target both aspects to produce a major effect on the disease burden.
Materials and Methods
Study population and ethical issues
Seven genetically confirmed patients were recruited within a registered clinical trial investigating the effect of rhuEPO in FRDA conducted at the Center for rare movement disorders of the Medical University of Innsbruck. Study procedures as well as the detailed processing of the biopsies have been already published elsewhere (14). FRDA patients received 3000 international units of rhuEPO subcutaneously three times per week during a study period of 8 weeks. This pilot study was designed to evaluate the effect of rhuEPO on biomarkers (histological, biochemical and assays of mitochondrial enzymes, transcriptomics). Within the trial, biopsies from the gastrocnemius muscle were obtained before and after the treatment with rhuEPO. They underwent open biopsy under local anesthesia of the right gastrocnemius muscle before treatment and of the left gastrocnemius muscle after treatment.
Skeletal muscle biopsies with unremarkable findings from six matched controls were available for comparison of transcriptomics data. The local institutional review board approved the study (Approval number, UN 3152_LEK). Patients and controls gave written informed consent to the study and for publication.
RNA extraction and analysis
Total RNA was extracted using Trizol (ThermoFisher #15596026) with the PureLink RNA Micro Scale Kit (ThermoFisher #12183016) according to the manufacturer’s protocol. RNA integrity was analyzed on an Agilent Tapestation with the High Sensitivity RNA Screen Tapes (Agilent #5067-5579). RNA with RIN > 7 was submitted to the Innsbruck Medical University NGS core facility for Quantseq 3′mRNA-Seq (Lexogen, Vienna, Austria) library preparation, following the manufacturer’s instructions. The resulting libraries were multiplexed and sequenced with an Ion Proton Sequencer (Ion torrent/Fisher Scientific, Austria) using ion Hi-Q chemistry.
FASTQC 0.11.9 was used to check the sequencing quality of raw data. Sequencing reads were trimmed of low-quality bases and poly-A tails with CUTADAPT 4.0 (41) using a quality threshold of 20 (−q), a minimum overlap of 10 (−O), a maximum error rate of 0.15 (−e), a minimum read length of 10 (−m) and the adapter sequence A{100} (−a). The nf-core (42) rnaseq preprocessing pipeline version 3.6 (10.5281/zenodo.6327553) was used with the trimming step disabled for the alignment of reads with STAR 2.7.9a (43) to the GRCh38 human reference genome and for the quantification of aligned reads with SALMON 1.5.2 (44). NGSCheckMate 1.0.0 (45) was used to confirm the proper pairing of matched samples derived from the same patients. Raw counts were imported into R 4.1.3 with tximport 1.22.0 (46) and DESeq2 1.34.0 (47) was used with IHW 1.22.0 (48) to identify differentially expressed genes. Changes in gene expression are reported as log2 fold changes (log2FC). Gene set enrichment analysis was performed on ranked DESeq2 output with clusterProfiler 4.2.2 (49) using HALLMARK, GO, KEGG or EPO-related gene sets retrieved with msigdbr 7.4.1 from the Molecular Signatures Database 7.4 (50). Summed z-scaled gene expression values of mitochondria-localized proteins as annotated in MitoCarta 3.0 were used as surrogates of mitochondrial function and tested for differences between conditions using a Wilcoxon rank sum test. All code to reproduce the analysis is available on GitHub (https://github.com/icbi-lab/indelicato_2023). RNA sequencing data were deposited in GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE226646).
Supplementary Material
{kind=link}
Acknowledgements
Elisabetta Indelicato, Sylvia Boesch, Wolfgang Nachbauer, Andreas Eigentler and Matthias Amprosi are members of the European Reference Network for Rare Neurological Diseases—Project ID No 739510. Elisabetta Indelicato is supported by a grant of the Friedreich’s Ataxia Research Alliance (FARA).
Conflict of Interest statement. The authors have no conflict of interests to report.
Contributor Information
Elisabetta Indelicato, Center for Rare Movement Disorders Innsbruck, Medical University of Innsbruck, Innsbruck 6020, Austria; Department of Neurology, Medical University of Innsbruck, Innsbruck 6020, Austria.
Alexander Kirchmair, Biocenter, Institute of Bioinformatics, Medical University of Innsbruck, Innsbruck 6020, Austria.
Matthias Amprosi, Center for Rare Movement Disorders Innsbruck, Medical University of Innsbruck, Innsbruck 6020, Austria; Department of Neurology, Medical University of Innsbruck, Innsbruck 6020, Austria.
Stephan Steixner, Institute of Hygiene and Medical Microbiology, Medical University of Innsbruck, Innsbruck 6020, Austria.
Wolfgang Nachbauer, Center for Rare Movement Disorders Innsbruck, Medical University of Innsbruck, Innsbruck 6020, Austria; Department of Neurology, Medical University of Innsbruck, Innsbruck 6020, Austria.
Andreas Eigentler, Center for Rare Movement Disorders Innsbruck, Medical University of Innsbruck, Innsbruck 6020, Austria; Department of Neurology, Medical University of Innsbruck, Innsbruck 6020, Austria.
Nico Wahl, Institute for Neuroscience, Medical University of Innsbruck, Innsbruck 6020, Austria.
Galina Apostolova, Institute for Neuroscience, Medical University of Innsbruck, Innsbruck 6020, Austria.
Anne Krogsdam, Biocenter, Institute of Bioinformatics, Medical University of Innsbruck, Innsbruck 6020, Austria.
Rainer Schneider, Institute of Biochemistry, Leopold Franzens University, Innsbruck 6020, Austria; Center of Molecular Biosciences Innsbruck (CMBI), Innsbruck 6020, Austria.
Julia Wanschitz, Department of Neurology, Medical University of Innsbruck, Innsbruck 6020, Austria.
Zlatko Trajanoski, Biocenter, Institute of Bioinformatics, Medical University of Innsbruck, Innsbruck 6020, Austria.
Sylvia Boesch, Center for Rare Movement Disorders Innsbruck, Medical University of Innsbruck, Innsbruck 6020, Austria; Department of Neurology, Medical University of Innsbruck, Innsbruck 6020, Austria.
References
- 1. Vankan, P. (2013) Prevalence gradients of Friedreich's ataxia and R1b haplotype in Europe co-localize, suggesting a common Palaeolithic origin in the Franco-Cantabrian ice age refuge. J. Neurochem., 126, 11–20. [DOI] [PubMed] [Google Scholar]
- 2. Campuzano, V., Montermini, L., Molto, M.D., Pianese, L., Cossee, M., Cavalcanti, F., Monros, E., Rodius, F., Duclos, F., Monticelli, A.et al. (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science, 271, 1423–1427. [DOI] [PubMed] [Google Scholar]
- 3. Saveliev, A., Everett, C., Sharpe, T., Webster, Z. and Festenstein, R. (2003) DNA triplet repeats mediate heterochromatin-protein-1-sensitive variegated gene silencing. Nature, 422, 909–913. [DOI] [PubMed] [Google Scholar]
- 4. Rodden, L.N., Chutake, Y.K., Gilliam, K., Lam, C., Soragni, E., Hauser, L., Gilliam, M., Wiley, G., Anderson, M.P., Gottesfeld, J.M., Lynch, D.R. and Bidichandani, S.I. (2021) Methylated and unmethylated epialleles support variegated epigenetic silencing in Friedreich ataxia. Hum. Mol. Genet., 29, 3818–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gottesfeld, J.M. (2019) Molecular mechanisms and therapeutics for the GAA.TTC expansion disease Friedreich ataxia. Neurotherapeutics, 16, 1032–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen, O.S., Hemenway, S. and Kaplan, J. (2002) Inhibition of Fe-S cluster biosynthesis decreases mitochondrial iron export: evidence that Yfh1p affects Fe-S cluster synthesis. Proc. Natl. Acad. Sci. U. S. A., 99, 12321–12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rotig, A., deLonlay, P., Chretien, D., Foury, F., Koenig, M., Sidi, D., Munnich, A. and Rustin, P. (1997) Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet., 17, 215–217. [DOI] [PubMed] [Google Scholar]
- 8. Shan, Y., Napoli, E. and Cortopassi, G. (2007) Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum. Mol. Genet., 16, 929–941. [DOI] [PubMed] [Google Scholar]
- 9. Thierbach, R., Drewes, G., Fusser, M., Voigt, A., Kuhlow, D., Blume, U., Schulz, T.J., Reiche, C., Glatt, H., Epe, B., Steinberg, P. and Ristow, M. (2010) The Friedreich's ataxia protein frataxin modulates DNA base excision repair in prokaryotes and mammals. Biochem. J., 432, 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gonzalez-Cabo, P. and Palau, F. (2013) Mitochondrial pathophysiology in Friedreich's ataxia. J. Neurochem., 126, 53–64. [DOI] [PubMed] [Google Scholar]
- 11. Parkinson, M.H., Boesch, S., Nachbauer, W., Mariotti, C. and Giunti, P. (2013) Clinical features of Friedreich's ataxia: classical and atypical phenotypes. J. Neurochem., 126, 103–117. [DOI] [PubMed] [Google Scholar]
- 12. Lodi, R., Cooper, J.M., Bradley, J.L., Manners, D., Styles, P., Taylor, D.J. and Schapira, A.H. (1999) Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proc. Natl. Acad. Sci. U. S. A., 96, 11492–11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lodi, R., Hart, P.E., Rajagopalan, B., Taylor, D.J., Crilley, J.G., Bradley, J.L., Blamire, A.M., Manners, D., Styles, P., Schapira, A.H. and Cooper, J.M. (2001) Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich's ataxia. Ann. Neurol., 49, 590–596. [PubMed] [Google Scholar]
- 14. Nachbauer, W., Boesch, S., Reindl, M., Eigentler, A., Hufler, K., Poewe, W., Loscher, W. and Wanschitz, J. (2012) Skeletal muscle involvement in Friedreich ataxia and potential effects of recombinant human erythropoietin administration on muscle regeneration and neovascularization. J. Neuropathol. Exp. Neurol., 71, 708–715. [DOI] [PubMed] [Google Scholar]
- 15. Nachbauer, W., Boesch, S., Schneider, R., Eigentler, A., Wanschitz, J., Poewe, W. and Schocke, M. (2013) Bioenergetics of the calf muscle in Friedreich ataxia patients measured by 31P-MRS before and after treatment with recombinant human erythropoietin. PLoS One, 8, e69229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koeppen, A.H., Becker, A.B., Qian, J. and Feustel, P.J. (2017) Friedreich ataxia: hypoplasia of spinal cord and dorsal root ganglia. J. Neuropathol. Exp. Neurol., 76, 101–108. [DOI] [PubMed] [Google Scholar]
- 17. Coppola, G., Burnett, R., Perlman, S., Versano, R., Gao, F., Plasterer, H., Rai, M., Sacca, F., Filla, A., Lynch, D.R.et al. (2011) A gene expression phenotype in lymphocytes from Friedreich ataxia patients. Ann. Neurol., 70, 790–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Napierala, J.S., Li, Y., Lu, Y., Lin, K., Hauser, L.A., Lynch, D.R. and Napierala, M. (2017) Comprehensive analysis of gene expression patterns in Friedreich's ataxia fibroblasts by RNA sequencing reveals altered levels of protein synthesis factors and solute carriers. Dis. Model. Mech., 10, 1353–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coppola, G., Choi, S.H., Santos, M.M., Miranda, C.J., Tentler, D., Wexler, E.M., Pandolfo, M. and Geschwind, D.H. (2006) Gene expression profiling in frataxin deficient mice: microarray evidence for significant expression changes without detectable neurodegeneration. Neurobiol. Dis., 22, 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coppola, G., Marmolino, D., Lu, D., Wang, Q., Cnop, M., Rai, M., Acquaviva, F., Cocozza, S., Pandolfo, M. and Geschwind, D.H. (2009) Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich's ataxia. Hum. Mol. Genet., 18, 2452–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lai, J.I., Nachun, D., Petrosyan, L., Throesch, B., Campau, E., Gao, F., Baldwin, K.K., Coppola, G., Gottesfeld, J.M. and Soragni, E. (2019) Transcriptional profiling of isogenic Friedreich ataxia neurons and effect of an HDAC inhibitor on disease signatures. J. Biol. Chem., 294, 1846–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nachbauer, W., Wanschitz, J., Steinkellner, H., Eigentler, A., Sturm, B., Hufler, K., Scheiber-Mojdehkar, B., Poewe, W., Reindl, M. and Boesch, S. (2011) Correlation of frataxin content in blood and skeletal muscle endorses frataxin as a biomarker in Friedreich ataxia. Mov. Disord., 26, 1935–1938. [DOI] [PubMed] [Google Scholar]
- 23. Hammerschmidt, P., Ostkotte, D., Nolte, H., Gerl, M.J., Jais, A., Brunner, H.L., Sprenger, H.G., Awazawa, M., Nicholls, H.T., Turpin-Nolan, S.M.et al. (2019) CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation in obesity. Cell, 177, 1536–1552.e23. [DOI] [PubMed] [Google Scholar]
- 24. Uhlen, M., Fagerberg, L., Hallstrom, B.M., Lindskog, C., Oksvold, P., Mardinoglu, A., Sivertsson, A., Kampf, C., Sjostedt, E., Asplund, A.et al. (2015) Proteomics. Tissue-based map of the human proteome. Science, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 25. Lynch, D.R., Chin, M.P., Delatycki, M.B., Subramony, S.H., Corti, M., Hoyle, J.C., Boesch, S., Nachbauer, W., Mariotti, C., Mathews, K.D.et al. (2021) Safety and efficacy of Omaveloxolone in Friedreich ataxia (MOXIe study). Ann. Neurol., 89, 212–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh, I., Faruq, M., Padma, M.V., Goyal, V., Behari, M., Grover, A., Mukerji, M. and Srivastava, A.K. (2015) Investigation of mitochondrial DNA variations among Indian Friedreich's ataxia (FRDA) patients. Mitochondrion, 25, 1–5. [DOI] [PubMed] [Google Scholar]
- 27. Karthikeyan, G., Santos, J.H., Graziewicz, M.A., Copeland, W.C., Isaya, G., Van Houten, B. and Resnick, M.A. (2003) Reduction in frataxin causes progressive accumulation of mitochondrial damage. Hum. Mol. Genet., 12, 3331–3342. [DOI] [PubMed] [Google Scholar]
- 28. Haugen, A.C., Di Prospero, N.A., Parker, J.S., Fannin, R.D., Chou, J., Meyer, J.N., Halweg, C., Collins, J.B., Durr, A., Fischbeck, K.et al. (2010) Altered gene expression and DNA damage in peripheral blood cells from Friedreich's ataxia patients: cellular model of pathology. PLoS Genet., 6, e1000812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(2023) Reata pharmaceuticals announces FDA approval of SKYCLARYS™ (Omaveloxolone), the first and only drug indicated for patients with Friedreich’s ataxia. in press. https://www.reatapharma.com/investors/news/news-details/2023/Reata-Pharmaceuticals-Announces-FDA-Approval-of-SKYCLARYS-Omaveloxolone-the-First-and-Only-Drug-Indicated-for-Patients-with-Friedreichs-Ataxia/default.aspx.
- 30. Oskolkov, N., Santel, M., Parikh, H.M., Ekstrom, O., Camp, G.J., Miyamoto-Mikami, E., Strom, K., Mir, B.A., Kryvokhyzha, D., Lehtovirta, M.et al. (2022) High-throughput muscle fiber typing from RNA sequencing data. Skelet. Muscle, 12, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng, Y., Buchan, M., Vitanova, K., Aitken, L., Gunn-Moore, F.J., Ramsay, R.R. and Doherty, G. (2020) Neuroprotective actions of leptin facilitated through balancing mitochondrial morphology and improving mitochondrial function. J. Neurochem., 155, 191–206. [DOI] [PubMed] [Google Scholar]
- 32. Finocchietto, P., Perez, H., Blanco, G., Miksztowicz, V., Marotte, C., Morales, C., Peralta, J., Berg, G., Poderoso, C., Poderoso, J.J. and Carreras, M.C. (2022) Inhibition of mitochondrial fission by Drp-1 blockade by short-term leptin and Mdivi-1 treatment improves white adipose tissue abnormalities in obesity and diabetes. Pharmacol. Res., 178, 106028. [DOI] [PubMed] [Google Scholar]
- 33. Supale, S., Thorel, F., Merkwirth, C., Gjinovci, A., Herrera, P.L., Scorrano, L., Meda, P., Langer, T. and Maechler, P. (2013) Loss of prohibitin induces mitochondrial damages altering beta-cell function and survival and is responsible for gradual diabetes development. Diabetes, 62, 3488–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turchi, R., Tortolici, F., Guidobaldi, G., Iacovelli, F., Falconi, M., Rufini, S., Faraonio, R., Casagrande, V., Federici, M., De Angelis, L.et al. (2020) Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis., 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nwadozi, E., Ng, A., Stromberg, A., Liu, H.Y., Olsson, K., Gustafsson, T. and Haas, T.L. (2019) Leptin is a physiological regulator of skeletal muscle angiogenesis and is locally produced by PDGFRalpha and PDGFRbeta expressing perivascular cells. Angiogenesis, 22, 103–115. [DOI] [PubMed] [Google Scholar]
- 36. Ambrosini, G., Nath, A.K., Sierra-Honigmann, M.R. and Flores-Riveros, J. (2002) Transcriptional activation of the human leptin gene in response to hypoxia. Involvement of hypoxia-inducible factor 1. J. Biol. Chem., 277, 34601–34609. [DOI] [PubMed] [Google Scholar]
- 37. Sabbatini, M., Bosetti, M., Borrone, A., Moalem, L., Taveggia, A., Verna, G. and Cannas, M. (2016) Erythropoietin stimulation of human adipose tissue for therapeutic refilling releases protective cytokines. J. Tissue Eng., 7, 2041731416671278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang, G., Kitaoka, T., Crawford, A., Mao, Q., Hesketh, A., Guppy, F.M., Ash, G.I., Liu, J., Gerstein, M.B. and Pitsiladis, Y.P. (2021) Cross-platform transcriptomic profiling of the response to recombinant human erythropoietin. Sci. Rep., 11, 21705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mariotti, C., Nachbauer, W., Panzeri, M., Poewe, W., Taroni, F. and Boesch, S. (2013) Erythropoietin in Friedreich ataxia. J. Neurochem., 126, 80–87. [DOI] [PubMed] [Google Scholar]
- 40. Lynch, D.R. and Farmer, G. (2021) Mitochondrial and metabolic dysfunction in Friedreich ataxia: updaete on pathophysiological relevance and clinical interventions. Neuronal Signal, 5, NS20200093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin, M. (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. Journal, 17, 10–12. [Google Scholar]
- 42. Ewels, P.A., Peltzer, A., Fillinger, S., Patel, H., Alneberg, J., Wilm, A., Garcia, M.U., Di Tommaso, P. and Nahnsen, S. (2020) The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol., 38, 276–278. [DOI] [PubMed] [Google Scholar]
- 43. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Patro, R., Duggal, G., Love, M.I., Irizarry, R.A. and Kingsford, C. (2017) Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods, 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee, S., Lee, S., Ouellette, S., Park, W.Y., Lee, E.A. and Park, P.J. (2017) NGSCheckMate: software for validating sample identity in next-generation sequencing studies within and across data types. Nucleic Acids Res., 45, e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Soneson, C., Love, M.I. and Robinson, M.D. (2015) Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res, 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ignatiadis, N., Klaus, B., Zaugg, J.B. and Huber, W. (2016) Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods, 13, 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., Feng, T., Zhou, L., Tang, W., Zhan, L.et al. (2021) clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Camb), 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liberzon, A., Subramanian, A., Pinchback, R., Thorvaldsdottir, H., Tamayo, P. and Mesirov, J.P. (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics, 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.