

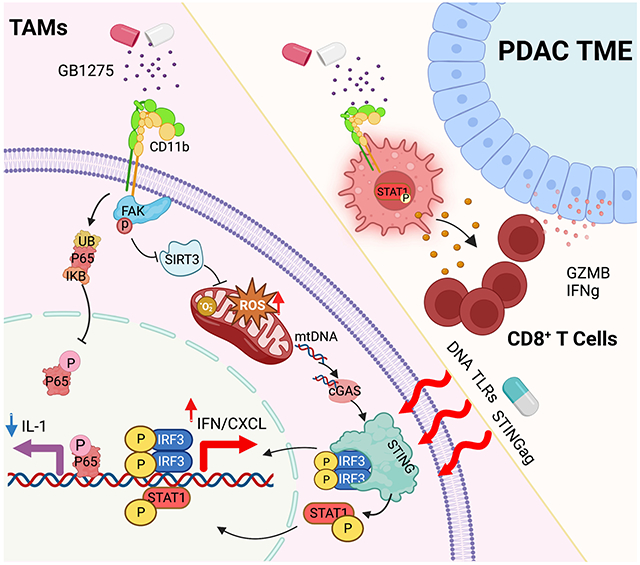
Summary
Chronic activation of inflammatory pathways and suppressed interferon are hallmarks of immunosuppressive tumors. Previous studies have shown that CD11b integrin agonists could enhance anti-tumor immunity through myeloid reprograming, but the underlying mechanisms remain unclear. Herein we find that CD11b-agonists alter tumor-associated macrophage (TAM) phenotypes by repressing NFκB signaling and activating interferon gene expression simultaneously. Repression of NFκB signaling involves degradation of p65 protein and is context independent. In contrast, CD11b agonism induces STING/STAT1 -pathway-mediated interferon gene expression through FAK-mediated mitochondrial dysfunction, with the magnitude of induction dependent on the tumor microenvironment and amplified by cytotoxic therapies. Using tissues from phase I clinical studies, we demonstrate that GB1275 treatment activates STING and STAT1 signaling in TAMs in human tumors. These findings suggest potential mechanism-based therapeutic strategies for CD11b agonists and identify patient populations more likely to benefit.
Keywords: Pancreatic cancer, Tumor-associated macrophages, CD11b, STING, NFκB, immunotherapy
Graphical Abstract
eTOC Blurb
Liu et al., report that CD11b agonists activate STING/STAT/interferon pathways and represses NFκB in in-vitro systems, pancreatic cancer mouse models, and human tumors. Tumor cell death and innate immune agonists synergize with CD11b-agonists, indicating potential mechanism-based therapeutic strategies for translation of CD11b agonist use in cancer patients.
Introduction
The presence of high numbers of tumor-associated macrophages (TAMs) has been associated with poor clinical outcomes in many cancer types 1. In spite of this, TAMs harbor considerable plasticity and can play both pro-tumoral and anti-tumoral roles during therapy 2. In some solid tumors like pancreatic ductal adenocarcinoma (PDAC), dense desmoplastic and fibrotic stroma can drive immunosuppressive and wound healing programs in TAMs 3-5. Thus, reprograming TAMs toward anti-tumor phenotypes is an attractive therapeutic strategy for such cancer types 6.
TAMs are highly plastic to regulate multiple aspects of tumor promotion and restraint 1,7,8. Anti-tumorigenic macrophages are characterized by high expression of tumor necrosis factor (TNFα), IL12a, inducible nitric oxide synthase or MHC molecules, and T cell attracting chemokines, like CXCL9 and CXCL10 9. In contrast, pro-tumorigenic macrophages express high IL-10, the IL-1 decoy receptor, IL-1Ra, arginase-1, and the scavenger receptors CD163, CD204, or CD206 10,11. In human PDAC, TAMs are abundant in the tumor tissues of many patients 12,13. Some of the earliest approaches focused on TAM depletion. In preclinical models, blocking key chemokine receptors (e.g., CCR2 or CXCR1/2) or impairing TAM survival by inhibiting colony stimulating factor-1 receptor (CSF1R) have slowed progression and improved responses to a variety of agents 14-16. However, these approaches tested to date have yet to achieve clinically significant benefits in solid tumors 17. This is likely because these myeloid-ablative strategies are subject to significant compensatory actions by untargeted subsets of myeloid cells that may ultimately limit their therapeutic efficacy in humans 18,19. An alternative strategy is to reprogram TAMs to support anti-tumor immunity.
Previous studies identified CD11b as a candidate target for immunotherapy 20-22. CD11b, composed of the αM (ITGAM) and β2 (CD18) integrins, is widely expressed on multiple myeloid cell subsets. Agonism of CD11b by small molecules modestly reduces the number of tumor-infiltrating immunosuppressive myeloid cells like TAMs, monocytes, and granulocytes; and corresponds with an increase in T cell-mediated tumor control in PDAC and other mouse models 20-22. These data led to early-phase clinical testing in solid tumors (NTC04060342). In this study, we investigated the cellular and molecular mechanism by which CD11b agonists induce antitumor immunity and identified combination therapies to move forward.
Results
Cancer-specific differences in TAM phenotypes.
To understand the unique cancer-specific pathways in TAMs that may impair tumor immunity, we combined publicly available single-cell RNA sequencing (scRNAseq) data sets from 70 tumors from 10 cancer types to identify ~54,000 TAMs. Even after integration, Uniform Manifold Approximation and Projection (UMAP) analysis still showed some segregation of TAMs by tumor types, suggesting the tumor-specific phenotypes and transcriptional features (Figure 1A). We next used gene set enrichment analysis (GSEA) to find cancer-specific phenotypic biases in TAMs. Comparing PDAC TAMs to TAMs in all other cancers, we observed some of the highest levels of the TGFβ, WNT, NFκB, and IL-4/IL-13 pathways and hypoxia and glycolysis gene sets (Figure 1B, S1A-B). These results fit with the highly fibroinflammatory TME that is a hallmark of PDAC. We also observed strong congruence for these pathways across different databases (Figure S1A-B). In contrast, PDAC TAMs had some of the lowest levels of type-1 interferon signaling, antigen presentation, reactive oxygen species (ROS), and oxidative phosphorylation gene sets (Figure 1B, S1A-B). Taken together, these data suggested that in desmoplastic cancers, augmenting interferon and suppressing chronic inflammatory signaling may be key to unlocking tumor immunity.
Figure 1. GB1275 anti-tumor activity is dependent on TAM reprograming.

(A) UMAP visualization of 54,000 TAMs from different cancer types, including PDAC, early gastric cancer (EGC), basal cell carcinoma (BCC), colorectal cancer (CRC), uveal melanoma (UVM), renal cancer (RCA), breast cancer (BRAC), ovarian serous cystadenocarcinoma (OVC), skin cutaneous melanoma (SKCM), and lung adenocarcinoma (LUAD). Clusters are annotated for cancer types.
(B) GSEA identified pathway enrichment in TAMs from each cancer type.
(C) Schematic of in vitro macrophage system.
(D) QPCR mRNA expression analysis of BMDMs treated with TCM for 6 hours. Changes in gene expression are depicted as the fold change from the vehicle.
(E-F) QPCR mRNA expression analysis of BMDMs isolated from wild-type(E) or CD11b-null mice
(F) treated with TCM ± GB1275 for 7 hours. Changes in gene expressions are depicted as the fold change from the vehicle.
(G) WT or CD11b-null mice bearing syngeneic orthotopic KP2 tumors were treated with vehicle or GB1275 for 14 days. Tumor volume and frequencies of tumor-infiltrating TAMs, CD8+ T cells, and Ki67+ CD8+ T cells are depicted (n = 6–8/group).
(H) Mice bearing orthotopic KI tumors treated with lgG+ PBS or αCSF-1 IgG+ Clodronate as depicted. Tumor volume and frequencies of PDAC-infiltrating TAMs, CD8+ T cells, and Ki67+ CD8+ T cells at day 19 are depicted (n = 7–9/group). All graphs depict the mean ± SEM and *denotes a value of p < 0.05 using the two-sided t-test. In vitro data are representative of three independent experiments.
Also see Figure S1.
The anti-tumor activity of CD11b agonists depends on TAMs.
CD11b integrins can regulate both myeloid recruitment into inflamed tissues and cell phenotypes20. In previous studies, CD11b agonists led to reduced tumor-infiltrating myeloid cells and restraint of tumor progression through indirect enhancement of T cell immunity 21. These small molecule agonists were formerly named either LA1 or ADH503 but will be referred to as GB1275 herein, given their current clinical format (Figure S1C and NTC04060342). Using in vitro systems, polarization of bone marrow-derived macrophages (BMDMs) with PDAC tumor cell-conditioned media (TCM) led to reduced Ifnb1 and Cxcl10, but increased Il1 expression (Figure 1C-D). In contrast, adding GB1275 to TCM resulted in upregulation of Ifna1, Ifnb1, and Cxcl9, 10 and 11 but decreased Il1a and Il1b (Figure 1E, Figure S1D, E). These effects were lost in CD11b-null BMDMs (Figure 1F). In vivo, GB1275- treated PDAC tissue also showed increased CXCL10 and 11 and reduced IL-1β (Figure S1F). Taken together, these data suggested CD11b agonism altered the effect of polarization by tumor-derived factors on TAMs. However, it remained unclear if TAM reprograming was critical for efficacy in vivo.
To study this, we first confirmed that GB1275 exhibited anti-tumor activity in vivo through CD11b activation. A syngeneic PDAC cell line (KP2) derived from the genetic p48-Cre;LSL-KrasG12D;Trp53flox/+ mice was orthotopically implanted in wild-type (WT) and CD11b-null mice. As expected, treatment of established tumors with GB1275 repressed PDAC progression, decreased TAM infiltration, and increased CD8+ T cell number and proliferation (Figure 1G, S1G-I). However, these effects were lost in CD11b-null mice (Figure 1G). As CD11b agonists could directly alter macrophage phenotype, we next determined if TAMs were critical for efficacy and anti-tumor immunity. We depleted tissue macrophages using a combination of anti-CSF-1 IgGs and liposomal clodronate (αCSF1/CLD) 23. Mice bearing established syngeneic orthotopic tumors derived from Pdx1-Cre/LSL-KrasG12D/Ink/ArfFlox/Flox mice (KI cells) were treated with αCSF1/CLD, which depleted >80% of TAMs, leading to reduced tumor progression (Figure 1H; S1J). GB1275 reduced KI tumor progression and increased CD8+ CTL infiltration and proliferation, but these effects were lost with TAM depletion (Figure 1H). Together, GB1275 activated CD11b on TAMs to drive anti-tumor immunity and restrain tumor growth.
CD11b-induced activation of TAMs improves T cell effector function.
Prior studies showed that the anti-tumor activity of CD11b agonists was dependent on T cells 21. However, T cells seldom express CD11b. To eliminate potential direct effects on T cells, we assessed CD11b expression and direct impact of GB1275 on T cells. Analysis of p48-Cre;LSL-KrasG12D;Trp53flox/flox(KPC) GEMMs, we found only 2–4% of T cells expressed detectable CD11b, and even positive cells expressed >100-fold less than TAMs (Figure S2A), agreeing with prior human PDAC analysis 21. In vitro, GB1275 did not affect CD8+ T cell proliferation or effector cytokine production (Figure S2B-C). Together, CD11b agonists do not act directly on T cells.
To understand how T cell responses were changed, mass cytometry (CyTOF) was employed on orthotopic KI tumors (Figure 2A-B). UMAP analysis of CyTOF data on PDAC tissues identified several phenotypic subsets of CD8+ and CD4+ T cells (Figure 2C-D, S2D). Across all CD8+ T cell subsets, we observed GB1275 increased granzyme B (GZMB) and Ki67 expressions (Figure 2E). Analyzing changes in CD8+ T cells subsets, we found GB1275 increased proliferative effectors (cluster 5), expressing high levels of CD44, GZMB, T-bet, and Ki67, while CD103+ resident memory (cluster 2) and activated non-proliferative non-effector CD8+ T cells (cluster 3) were reduced (Figure 2F). Notably, across all subsets, we found a trend toward increased PD-1, CTLA-4, and TIM3 expression with treatment (Figure S2E). These data demonstrated that CD11b agonism-induced changes in myeloid cells drove CD8+ CTLs toward proliferative effector phenotypes. To further confirm this, we analyzed orthotopic KP2-OVA tumors (Figure 2G). Consistent with increased effector function, we also observed increased IFNγ positive and TNFα/IFNγ double-positive CD8+ T cells in GB1275-treated PDAC tissues (Figure 2H, S2J). Together, these data suggest that CD11b-agonists indirectly increase proliferative effector CD8+ T cells. By contrast, we observed only limited changes in CD4+ T cells (Figure S2F-I), which agreed with prior data, demonstrating that CD8+, but not CD4+ T cells, are required for CD11b-agonist tumor control 21.
Figure 2. TAMs CD11b activation results in more proliferative effector T cells.

(A) Syngeneic KI orthotopic model and treatment (left). Tumor burden in each group (right) (n = 8/group).
(B) Frequencies of TAMs and CD8+ T cells from A (n = 7/group).
(C) CyTOF UMAP plot of tumor-infiltrating T cells.
(D) Subpopulations of CD8+ T cells and CD4+ T cells.
(E) Median expressions of Ki67 and GZMB in CD8+ T cells.
(F) Percentage of individual subclusters in CD8+ T cells.
(G) Syngeneic KP2-OVA orthotopic model treated with vehicle or GB1275 for 12 days, and frequencies of GZMB+ CD8+ T cells (n = 7/group).
(H) Gate of functional assay of CD8+ T cells from G (left). Percentage of functional CD8+ T cells (right) (n = 7/group). Graphs depict the mean ± SEM and *denotes a value of p < 0.05 using a two-sided t-test for comparisons between two groups.
Also see Figure S2.
CD11b agonism inhibits NFκB signaling directly in TAMs
To understand how CD11b-agonists changed TAM phenotype to support anti-tumor immunity, we performed scRNAseq. We isolated CD45+ cells from syngeneic KP2 PDAC tissue from mice treated with vehicle or GB1275 for 14 days. UMAP analysis distinguished subsets of CD45+ cells (Figure S3A), from which we isolated TAMs/monocytes (Figure 3A). Reclustering of this population identified three subpopulations of TAMs and a monocyte cluster (Figure 3B, S3B). We next identified differentially expressed genes (DEGs) and conducted GSEA on the total TAM/monocyte populations. Consistent with CD11b integrin activation, we observed enrichment of integrin pathways in GB1275-treated TAMs (Figure 3C). Additionally, oxidative phosphorylation and ROS signatures were also increased by GB1275 across all TAM subsets (Figure 3C-D). In contrast, inflammatory response and NFκB signatures were decreased across all subsets of GB1275-treated TAMs (Figure 3C-D). At the gene level, the majority of NFκB/IL-1 signature genes were decreased in TAMs from GB1275-treated mice (Figure 3E, Table S1, S2). Together, these data suggest that CD11b agonism alters TAM phenotypes in vivo.
Figure 3. GB1275 downregulates NFκB/IL-1.

(A) UMAP scRNAseq plots of the whole TAM/Monocytes population (left), Itgam expression in this population (right).
(B) UMAP scRNAseq plots of subclusters from TAM/monocyte population in 14 days-vehicle and GB1275 treated KP2 syngeneic model (left), Right, percentages of individual clusters.
(C) GSEA identified pathway enrichment in the whole TAM/monocyte population (p<0.05).
(D) GSEA identified pathway enrichment in four subclusters (p<0.05).
(E) Volcano plot depicting GB1275- changed differentially expressed genes within the NFκB/IL-1 signaling pathway from the whole TAM/monocyte population.
(F) QPCR mRNA expression analysis of BMDMs treated with GB1275 for 7 hours. Changes in gene expression are depicted as the fold change from the vehicle baseline.
(G) Representative immunoblot for pp65, total p65, pIκB, total IκB, and β-ACTIN (loading control) in BMDMs treated with GB1275 ± TCM for 7 hours.
(H) Representative immunoblot for total p65 and β-ACTIN (loading control) in BMDMs treated with GB1275 for 7 hours after 1 hour-MG132 pretreatment.
(I) BMDMs were treated with GB1275 for 1 or 7 hours after 1 hour-MG132 pretreatment. Immunoblot for p65 from total lysates and polymer-ubiquitin from purified p65 protein.
(J) Representative mpIHC staining for p65, CD11b, F4/80 and CK19 in tumors from 14 days-vehicle and GB1275 treated KPC mice, KP2 (K), and KI (L) orthotopic models. Scale bar, 100μm. Right, percentage of np65+ TAMs (n = 7–8 mice per group).
(M) Kaplan–Meier survival curves for the top 80 downregulated NFκB/ IL-1 signaling-related genes from scRNAseq (E) in TCGA patient dataset for pancreatic adenocarcinoma (PAAD). Graphs show the mean ± SEM; *denotes p < 0.05 using the two-sided t-test for comparisons between two groups or log-rank test for Kaplan–Meier survival curves. In vitro data are representative of three independent experiments.
Consistent with our in vivo observations, in vitro, GB1275 decreased expression of IL-1-associated genes, including Il1a, IL1b, Il18, and Ccl2 in macrophages (Figure 3F), which was dependent on CD11b, but independent of the presence or sources of TCM (Figure S3C-E, S3F). We next assessed downregulation of NFκB by GB1275 in BMDMs under standard culture and with TCM. Under both conditions, GB1275 decreased total and phosphorylated NFκB p65 and IκB protein levels (Figure 3G). Due to its known effect on IL-1 transcription 24,25, we focused on NFκB. First, GB1275 lost its ability to decrease Il1 when NFκB p65 was silenced (Figure S3I-K). Next, we observed no decrease in NFκB p65 mRNA by GB1275 (Figure S3H), suggesting that the regulation was likely through protein stability. To test this, we pretreated macrophages with MG132, a proteasome inhibitor, and found that GB1275 no longer decreased p65 protein (Figure 3H). Moreover, GB1275 increased poly-ubiquitination of p65 (Figure 3I), contributing to proteasomal degradation 26,27. Together, CD11b agonists induce p65 degradation to regulate NFκB/IL-1 signaling.
To verify that CD11b agonists decreased NFκB p65 in vivo, we performed multiplex immunohistochemistry (mpIHC) analysis on PDAC tissues. We serially stained for p65, CD11b, F4/80, and cytokeratin (CK)-19 and found that GB1275 reduced the proportions of TAMs with nuclear p65+ (np65+) in KPC GEMM and orthotopic KP2 and KI tumors (Figure 3J-L). Based on downregulated NFκB/IL-1 signaling by GB1275 in our study, we next analyzed whether this signaling downregulation was related to favorable clinical outcomes in PDAC patients. To accomplish this, we identified a gene-set from the Hallmark NFκB signaling pathway (GeneID=7124) that was downregulated following GB1275 in TAMs in our scRNAseq data (Figure 3E, Table S3). We then segregated PDAC patients from the TCGA with this gene signature and found that low expression was indicative of better survival (Figure 3M). Similarly, lower IL1 gene sets were indicative of better outcomes in PDAC patients (Figure S3O). These data suggest that CD11b agonist suppression of NFκB/IL-1 signaling might lead to favorable clinical outcomes in patients.
CD11b-agonists induce NFκB inactivation in TAMs to inhibit inflammation but not IFN expression.
We next tested if NFκB or IL-1 receptor (IL-1R) was important for interferon induction or T cell enhancement. In vitro, GB1275’s ability to increase Ifn genes in macrophages was independent of p65 expression (Figure S3L). In vivo, we treated PDAC-bearing mice with vehicle or GB1275 ± IL-1R blocking antibodies. As expected, IL-1R blockade slowed tumor progression and decreased immature monocyte and granulocyte infiltration (Figure S3M). However, GB1275’s ability to improve CTL responses and tumor control was not dependent on IL-1R signaling (Figure S3M-N). Thus, CD11b agonist’s downregulation of the NFκB/IL-1 signaling contributed to dampening of inflammation, but not CTL-mediated tumor control.
CD11b agonists activate STING in TAMs.
We next sought to understand how CD11b agonists induce Ifn genes in TAMs. In vitro, the induction of Ifn genes by GB1275 was dependent on TCM polarizing conditions but consistent across multiple cell line-derived TCMs (Figure 1E-F and 4A). To investigate the signaling pathways involved, we performed reversed-phase protein array analyses (RPPA) on BMDMs treated with GB1275 ± TCM. After 4 and 7 hours, GB1275 alone induced 20 and 36 protein changes (Figure S4A, Table S4). But under TCM polarizing conditions, GB1275 induced greater than 60 protein changes at both timepoints, suggesting TCM amplified signaling differences. Importantly, STING protein expression was increased by GB1275 alone, and further increased when under TCM polarizing conditions (Figure 4B, S4B). We chose to focus on STING due to its known role in the induction of Ifn genes.
Figure 4. GB1275 increases IFN/CXCL transcription via STING.

(A) QPCR mRNA expression analysis of BMDMs treated with different TCM ± GB1275 for 7 hours. Changes in gene expression are depicted as the fold change from the vehicle baseline.
(B) Heat map of protein expression in BMDMs treated with vehicle or GB1275 ± TCM for 4 and 7 hours (left). Quantification of STING expression (right).
(C) Heat map of integrin signaling-related protein expression.
(D) QPCR mRNA expression analysis of BMDMs treated with TCM ± GB1275 for 7 hours after 1 hour-FAKi pretreatment. Changes in gene expression are depicted as the fold change from the vehicle baseline.
(E) GSEA identified pathway enrichment in cluster TAM1 in 3B (p<0.05).
(F) Intracellular total ROS in BMDMs stimulated by TCM + GB1275 for different time points (left). Quantification of ROS production (right).
(G) BMDMs were treated by TCM ± GB1275 for 4 hours after 1 hour-FAKi pretreatment. Representative immunoblots for pFAK, SIRT3, and β-ACTIN (loading control).
(H) Intracellular total ROS in BMDMs transfected with Sirt3 siRNA or ctrl siRNA for 24 hours prior to 7-hour TCM ± GB1275 (left). Quantification of ROS production (right).
(I) QPCR analysis of mitochondrial genes released in cytoplasm from BMDMs treated with vehicle or GB1275 ± TCM for indicated time periods. Changes in gene expression are depicted as the fold change from the vehicle baseline.
(J) QPCR mRNA expression analysis of BMDMs treated with TCM ± GB1275 for 7 hours after 1 hour-EtBr pretreatment. Changes in gene expression are depicted as the fold change from the vehicle baseline.
(K) Representative immunoblots for STING, total IRF3, pIRF3, pSTAT1, total STAT1, and β-ACTIN (loading control) in BMDMs treated with TCM ± GB1275 for the indicated time periods (left). Quantification of STING and pIRF3 relative expression (right).
(L) QPCR mRNA expression analysis of BMDMs isolated from wild-type or STING-null mice treated with TCM ± GB1275 for 7 hours. Changes in gene expression are depicted as the fold change from the vehicle baseline.
(M) C57/BL-6 mice were lethally irradiated and adoptively transferred with BM from either wild-type mice or STING-null mice. KP2-syngeneic tumors were established on above mice and treated with vehicle or GB1275 for 14 days. Right, tumor growth curve expressed as percentages of tumor volume changes (n = 7–8/group).
(N) Tumor-infiltrative CD8+ T cell frequencies from the above mice (n = 5–6/group).
(O) The proposed model: GB1275 regulated two separate signaling pathways in macrophages, including p65/IL-1 inhibition and STING/IFN/CXCL axis activation. Graphs show the mean ± SEM; *denotes p < 0.05 using the two-sided t-test between two groups. In vitro data are representative of three independent experiments.
As expected for integrin activation, we observed increased phosphorylated FAK, PyK2, and SRC, and total myosin-IIa in BMDMs after 4-hour GB1275 treatment (Figure 4C, S4C). In keeping with this, the GB1275 ability to induce Ifnb1 and Cxcl10 was lost when FAK was inhibited (Figure 4D). ScRNAseq analysis of TAMs in vivo also suggested that GB1275 activated oxidative phosphorylation and ROS pathways (Figure 3C, 3D, 4E). In agreement with this, we observed increased HSP70 and SOD in BMDMs (Figure 4B) 28,29. These data suggest that CD11b agonists might induce oxidative stress in TAMs. Evaluating this hypothesis in vitro, we found that GB1275 cooperated with TCM to enhance ROS production and this effect relied on FAK signaling (Figure. 4F, S4D). To determine if ROS was critical for IFN gene induction, we pretreated BMDMs with the ROS scavenger, N-acetylcysteine (NAC), and found loss of induction of Ifnb1 and Cxcl10 by GB1275 (Figure S4E). We next hypothesized that Sirtuin-3 (SIRT3) might link integrin-FAK signaling to ROS 30. GB1275 suppressed SIRT3 in BMDMs, which was dependent on FAK (Figure 4G). Furthermore, GB1275 no longer induced ROS production or Ifn genes when SIRT3 was silenced (Figure 4H, S4G). These data suggest that integrin/FAK/SIRT3/ROS signaling is involved in the induction of IFN genes by CD11b-agonists. However, the linkage to STING was unclear.
Previous studies have shown that high ROS could be linked to mitochondrial dysfunction and release of mitochondrial DNA (mtDNA), which could act as a primer for the cGAS-STING pathway 31. Indeed, more mtDNA released into the cytosol following GB1275 treatment, which was amplified by TCM (Figure 4I). When depleting mtDNA by ethidium bromide (EtBr) 32 or DNase1, GB1275 induction of Ifnb1 and Cxcl10 was attenuated (Figure 4J, S4H-I). Finally, analysis of TCM polarized BMDMs showed that GB1275 increased expression of STING and phosphorylation of IRF3 and STAT1 (Figure 4K). When STING expression was lost by siRNA or using STING-null BMDMs, GB1275 was no longer able to increase Ifn gene expression (Figure 4L; S4J-K). Notably, GB1275 downregulated Il1 mRNAs independent of FAK activation, ROS, or STING expression (Figure S4L-N), suggesting two independent pathways under GB1275 regulation (Figure 4O). Together, CD11b agonists led to STING/IFN signaling in “tumor-primed” macrophages.
To determine the importance of STING in GB1275’s function in vivo, we transplanted mice with bone marrow (BM) from control or STING-null mice. After reconstitution, we implanted syngeneic KP2 tumors and found that GB1275’s effects on tumor progression and CD8+ T cells were dependent on STING activity in leukocytes (Figure 4M-N; S4O). Together, these data suggest that CD11b agonists promote tumor immunity through myeloid STING activation.
In BM-transplant studies, GB1275 reduced granulocytes independently of STING expression. However, decreases in TAMs were dependent on BM STING, suggesting that decreased TAM numbers by GB1275 may be linked to turnover following STING activation. Consistent with this possibility, in vitro, prolonged exposure to high doses of GB1275 reduced cell number and phosphorylated AKT and increased cleaved caspase 3, partly dependent on STING expression (Figure S4P-S). This correlated with other studies showing STING/IFN signaling activation led to macrophage turnover33,34.
The expression levels of STING in TAMs correlate with patient outcomes.
Next, we analyzed STING/STAT1 activation by GB1275 in PDAC mouse models. In agreement with in vitro data, KPC tumors, as well as orthotopic KP2 and KI tumors had increased numbers of STING+ and pSTAT1+ total CD11b+ myeloid cells and TAMs (Figure 5A, S5A-C). Simultaneously, we observed a decrease in STING− or pSTAT1− np65+ TAMs by GB1275 (Figure 5A, S5A). These data demonstrate GB1275 activates STING/STAT1 signaling in vivo.
Figure 5. STING activation by GB1275 in mouse models and tumor biopsies.

(A) Representative mpIHC staining for pSTAT1, STING, F4/80, CD11b, and CK19 in tumors from 14-day vehicle and GB1275 treated KPC mice. Scale bar, 100μm. Right, percentages of STING+ CD11b+ cells, STING+ TAMs, pSTAT1+ TAMs, and pSTAT1− p65+ TAMs (n = 7–9 mice per group).
(B) Representative mpIHC staining for STING, CD163, CK19, and CD8α in human PDAC TMAs. Scale bar, 50μm.
(C) Average percentage of STING+ TAMs from the TMAs.
(D) Scatter plot showing Spearman’s correlation between the percentage of STING+ macrophages and CD8+ T cells from B.
(E) Kaplan–Meier survival curves for patients with high STING+ TAM infiltration and low STING+ TAM infiltration from B.
(F) Representative mpIHC staining for STING, pSTAT1, p65, CD163, and pan-keratin (PanK) in 11 paired tumor biopsies from patients. Scale bar, 50μm.
(G) Relative fold changes of STING+, STINGhi macrophages, pSTAT1+, pSTAT1hi macrophages, and nuclear p65+ macrophages in paired pre- and post-groups.
(H) Scatter plots showing Spearman’s correlations between the percentage of STINGhi macrophages and pSTAT1hi macrophages in all 22 tumor biopsies. Scatter plots showing Spearman’s correlation between the percentages of STINGhi macrophages (out of total cells) and CD8+ T cells in all 22 tumor biopsies (right). Graphs show the mean ± SEM; *denotes p < 0.05 using the two-sided t-test between two groups, one sample t-tests, and Wilcoxon tests, or the log-rank test for Kaplan–Meier survival curves.
Next, we studied the importance of STING expression in TAMs in human PDAC. Tissue microarrays (TMAs) from 173 surgical PDAC patients were stained by mpIHC for CD8α, STING, CD163, CD11b, and CK19 (Figure S5D). We defined macrophages as CD11b+, CD163+, and CK19− and found that STING+ TAMs ranged from 1–60% of total TAMs, with an average of 16% of TAMs expressing STING (Figure 5B, C). In PDAC tissues, higher percentages of STING+ TAMs or STING+CD11b+ cells correlated with increased CD8+ T cell infiltration (Figure 5D, S5E). Furthermore, high STING+ TAM infiltration correlated with longer survival (Figure 5E). Together, these data suggest that STING induction in TAMs might be indicative of better T cell responses and improved clinical outcomes.
Biomarker validation in the first-in-human GB1275 clinical trial
To determine if GB1275 could alter STING/STAT1 signaling in human tumors, we evaluated samples from a first-in-human clinical evaluation of the GB1275 [NTC04060342] 35. The Phase 1 portion of the study included a single agent GB1275 dose escalation and safety evaluation either as monotherapy (Regimen A) or in combination with pembrolizumab (Regimen B). Both treatment regimens included patients with advanced treatment refractive solid tumors. In these studies, GB1275 demonstrated good tolerability even at the highest dose level 35. For a subset of patients, tumor tissues were available, and we performed mpIHC for CD11b, CD163, pSTAT1, STING, p65, and pan-cytokeratin (PanK) on 11 paired, pre-treatment and post-treatment biopsy tissues (Figure 5F, Table S5). Compared with pre-treatment tissues, GB1275 elevated the percentages of pSTAT1+ or STING+ TAMs in the majority of patients and across both regimens (Figure 5G; S5F-G). As expected, we observed a correlation between STING and pSTAT1 expression in TAMs; and the percentage of STING+ TAMs correlated with increased CD8+ T cell infiltration (Figure 5H). The p65 expression in TAMs only decreased in 6 out of 11 patients, and the majority was observed in GB1275 single agent (4/6 in Regimen A, 2/5 in Regimen B, Figure S5G). This may not be surprising, as STING/STAT1 activation in vivo may overcome CD11b agonist’s impact on NFκB 36,37. Taken together, CD11b agonists increase STING/STAT1 activation in TAMs in advanced metastatic cancers.
DNA damaging therapies cooperate with CD11b agonism to amplify IFN signaling.
The above data demonstrated that CD11b agonists could activate STING/IFN signaling in TAMs. However, tumor parameters that might define the magnitude of this activation remained unclear. Notably, in vitro, GB1275 maximal induction of STING/IFN signaling in macrophages was dependent on TCM (Figure 4A). Thus, we hypothesized that understanding what factors in TCM augmented STING activation might yield insight into how to further amplify this pathway. To accomplish this aim, we separated TCM into proteins, >3 kilodaltons (kDa), and small molecules, metabolites, <3 kDa (Figure 6A). Next, we assayed whether TCM-derived proteins or small molecules synergized with GB1275. Notably, the TCM fraction containing metabolites and not proteins, enhanced the induction of Ifn genes by GB1275 (Figure 6A). We hypothesized that the small molecules amplifying STING activation might be either whole genomic DNA (gDNA) or mtDNA released by stressed or dying tumor cells in culture. To test this, we analyzed mtDNA and gDNA in TCM from standard culture or when treated with gemcitabine (GEM, Figure S6A). Under untreated conditions, we could readily detect mtDNA but not gDNA in PDAC cell TCM (Figure S6B). However, both mtDNA and gDNA release was observed in GEM treatment PDAC cell TCM (Figure S6B). Moreover, TCM from PDAC cells treated with either GEM (GEM-TCM) or radiation therapy (RT) synergized with GB1275 to induce Ifnb1 and Cxcl10 expression in macrophages (Figure 6B, S6A). Moreover, when depleting DNA in the GEM-TCM with DNase1, GB1275 could no longer increase Ifnb1 or Cxcl10 expression (Figure S6C). These data suggest that even low levels of DNA released from stressed or dying tumor cells augments STING signaling activation by GB1275. Because GEM-treated PDAC cells release more DNA, we determined whether cell-damaging reagents synergized with GB1275 in vivo.
Figure 6. Chemotherapy or radiation therapy combined with GB1275, amplifies STING/IFN signaling.

(A) Concentrated protein (> 3 kDa) and metabolites (< 3 kDa) in TCM were separated by a protein concentrator (left). QPCR mRNA expression analysis of BMDMs treated with different fractions of TCM ± GB1275 for 7 hours. Changes in gene expression are depicted as the fold change from the vehicle baseline (right).
(B) KP2 cells were treated with either gemcitabine (left) or radiation (right). TCM was made from above cells. QPCR mRNA expression analysis of BMDMs treated with the above mentioned TCM ± GB1275 for 7 hours. Changes in gene expression are depicted as the fold changes from the vehicle baseline.
(C) Tumor growth of KP2 syngeneic model treated with vehicle or GB1275 ± chemotherapy (left). Mean percent change in tumor volume on day 12 (n = 9–10/group) (middle). Right, Kaplan–Meier survival analysis (n = 9–10/group).
(D) QPCR mRNA expression analysis of tissue from C (n = 6–8/group). Changes in gene expression are depicted as fold changes from the vehicle baseline.
(E) IFNβ, CXCL10, 11, and IL-1β production in tissues from a syngeneic KP2-OVA orthotopic model treated with vehicle or GB1275 + chemotherapy for 12 days. (n = 7–8 group).
(F) Syngeneic KP2-OVA orthotopic model treated with vehicle, GB1275, chemotherapy, or combination for 12 days. Frequencies of tumor-infiltrating CD8+ T cells, Dex+ CD8+ T cells, and proliferative Dex+ CD8+ T cells (n = 6/group).
(G) Frequencies of Dex+ CD8+ T cells, proliferative CD8+ T cells, and proliferative Dex+ CD8+ T cells in dLN from F.
(H) C57/BL-6 mice were lethally irradiated and adoptively transferred with BM from either wild-type mice or STING-null mice. KP2-OVA orthotopic model was established on above mice and treated with chemotherapy ± GB1275 for 12 days. Frequencies of tumor-infiltrating CD8+ T cells, Dex+ CD8+ T cells, and proliferative Dex+ CD8+ T cells (n = 6/group).
(I) Frequencies of Dex+ CD8+ T cells and proliferative Dex+ CD8+ T cells in dLN from H. The graphs show mean ± SEM; *denotes p < 0.05 using the two-sided t-test between two groups or log-rank test for Kaplan–Meier survival curves. In vitro data are representative of three independent experiments.
Also see Figure S6.
To determine if chemotherapy plus CD11b-agonist could amplify IFN in vivo, we treated PDAC-bearing mice with gemcitabine plus paclitaxel (GEM/PTX, Figure 6C). As expected, GEM/PTX only modestly delayed tumor progression; however, when GEM/PTX was combined with GB1275, we observed reduced tumor burden and improved survival (Figure 6C). In parallel with significant increases in Ifn genes and increased IFNβ, CXCL10, and CXCL11 protein in combination-treated PDAC tissues (Figure. 6D-E). To understand how this combination might impact chemotherapy-induced T cell priming, we analyzed tumor antigen-specific CD8+ T cells in orthotopic KP2-OVA bearing mice. As expected, GB1275 increased the number of total CD8+ T cells as well as OVA-dextramer+ CTLs in PDAC tissues (Figure 6F), which were further improved in combination with chemotherapy (Figure 6F, S6D). Interestingly, only in the combination-treated tumors did we see increased proliferation in OVA-specific CTLs (Figure 6F). Analysis of tumor-draining lymph nodes (dLN) showed that GB1275 enhanced the ability of GEM/PTX to increase/prime tumor-specific CD8+ T cells (Figure 6G; S6E). To determine if priming of T cells by the combination required STING signaling in leukocytes, we transplanted mice with wild-type or STING-null BM. In these mice, in both tumor tissues and dLN, adding GB1275 to GEM/PTX doubled the number and proliferation of tumor-specific CD8+ T cells, which were dependent on STING in BM-derived cells (Figure 6H-I, S6F-G). In contrast, chemotherapy-induced tumor-specific CTLs were not impacted by loss of STING in the BM (Figure 6H-I). Together, GB1275 synergizes with chemotherapy to prime T cell responses.
Innate agonists synergize with CD11b-agonists.
To determine what other strategies could synergize with CD11b agonism, we treated macrophages with GB1275 and sub-saturating doses of either TLR7, TLR9, STING agonists, and compared this to the combination with TCM from GEM-treated PDAC cells. In each condition, GB1275 synergized to dramatically increase Ifn gene expression, with some combinations increasing Ifn genes by over >100-fold (Figure 7A). As expected, TCMs with TLR7-, TLR9-, and STING-agonists, all also increased Il1a, Il1b, and Cd274 expression (Figure 7B), likely through NFκB activation. However, GB1275 given in combination suppressed these inflammatory genes (Figure 7B). Single agent GB1275 also led to extracellular recycling of TLR9 protein, which was required for Cxcl9, 10, and 11 gene inductions in GB1275 plus GEM-TCM combinations (Figure S7A, B), possibly indicating the importance of TLR9 in DNA sensing during chemotherapy combinations. Together, CD11b agonist synergizes with an innate immune agonist to drive IFNs in macrophages.
Figure 7. STING agonist synergizes with GB1275 and remodels TME.
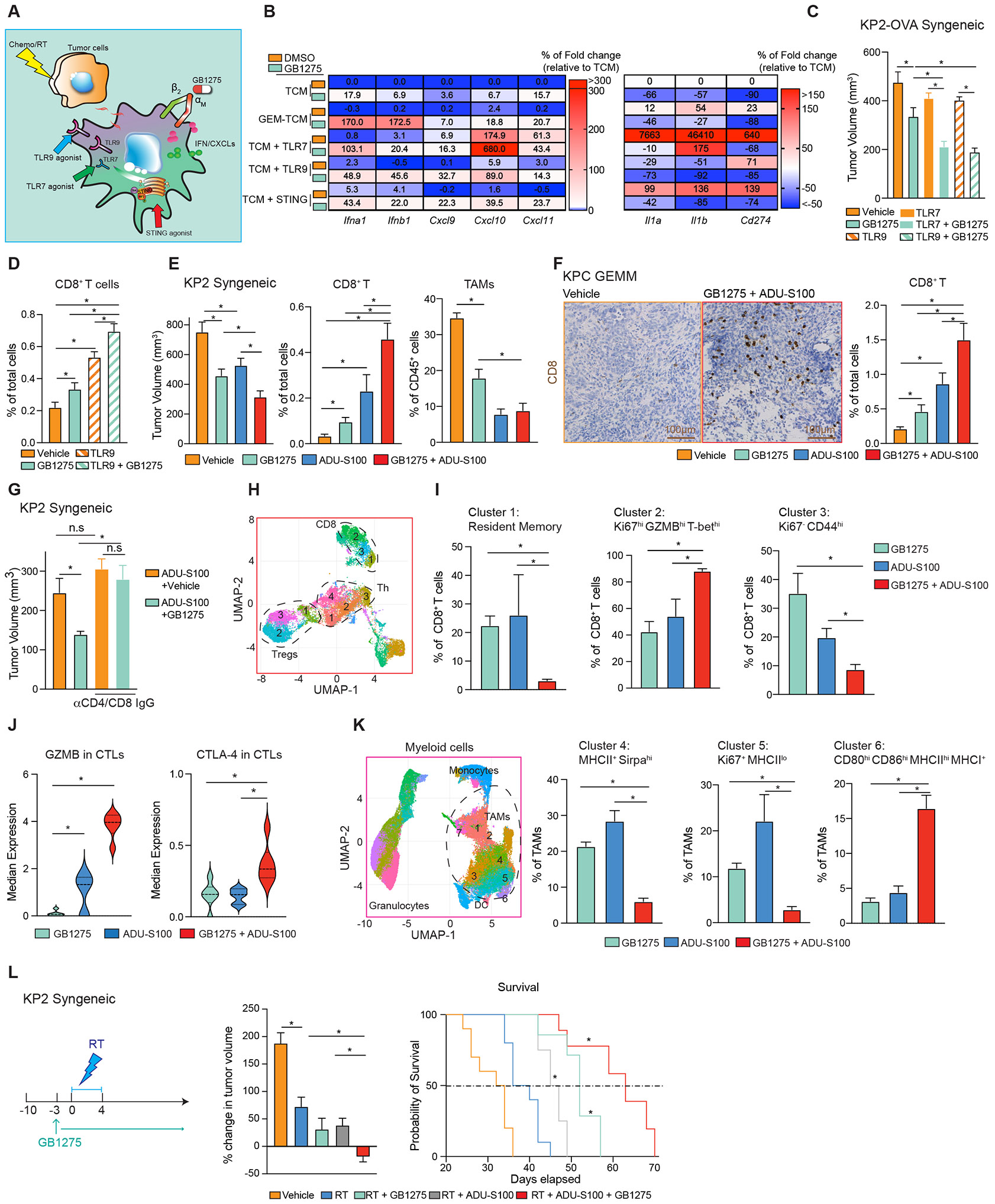
(A) Schematic of several combinations of GB1275.
(B) Heat map of relative mRNA expression from BMDMs after treatment (A). Percentages of changes in gene expression are depicted as the fold change from the vehicle baseline.
(C) Tumor burden from the KP2-OVA syngeneic mouse model treated with vehicle or GB1275 ± TLR7 agonist or TLR9 agonist for 14 days (n = 9–10/group).
(D) Immunohistochemical staining for CD8+ T cells in tumors from 14-day vehicle, GB1275, TLR9 agonist or combination-treated KPC mice. Right, average percentage of CD8+ T.
(E) Tumor burden from the KP2 syngeneic mouse model treated with vehicle or GB1275 ± ADUS100 for 19 days (n = 10/group). Frequencies of tumor-infiltrating CD8+ T cells and macrophages (n = 6/group).
(F) Immunohistochemical staining for CD8+ T cells in tumors from 14-day vehicle, GB1275, ADUS100, or combination-treated KPC mice. Scale bar, 100μm. Right, average percentage of CD8+ T cells (n = 6–7/group).
(G) Tumor burden from KP2 syngeneic mouse model treated with ADU-S100 or combination of GB1275 ± CD4/CD8+ T cell depletion for 12 days (left).
(H) KP2 syngeneic model treated with GB1275, ADU-S100, or combination for 14 days (left). CyTOF UMAP plot of tumor-infiltrating T cells, including CD8+ T cells, Th, and Tregs (n = 6/group).
(I) Percentages of individual subclusters in CD8+ T cells.
(J) Median expressions of GZMB and CTLA-4 in CD8+ T cells.
(K) CyTOF UMAP plot of tumor-infiltrating myeloid cells, including TAMs, granulocytes, monocytes, and dendritic cells from H (n = 6/group) (left). Percentages of individual subclusters in TAMs (right).
(L) Tumor growth of KP2 syngeneic model treated with vehicle or GB1275 ± radiation therapy (6 Gy × 5) ± ADU-S100 (left). Mean percent change in tumor volume on day 12 (n = 10/group) (middle). (right) Kaplan–Meier survival analyses (n =10/group). Graphs show the mean ± SEM; *denotes p < 0.05 using the two-sided t-test between two groups, analysis of variance or log-rank test for Kaplan–Meier survival curves. In vitro data are representative of three independent experiments.
Also see Figure S7.
We next determined if GB1275 improved the efficacy of innate agonists in vivo. KPC GEMMs and syngeneic tumor-bearing mice were treated with vehicle or GB1275 ± TLRs agonists. While both the TLR7 and TLR9 agonists inhibited tumor progression, the combination with GB1275 was superior in syngeneic PDAC models (Figure 7C). In KPC GEMMs, adding GB1275 to the TLR9 agonist decreased tumor burden and increased CD8+ CTL infiltration (Figure 7D, S7C). We next determined the efficiency of GB1275 combined with STING agonist. PDAC-bearing mice were treated with intratumoral injection of the STING agonist, ADU-S100 ± GB1275. Both GB1275 and ADU-S100 suppressed tumor progression, but the combination was superior (Figure 7E). Similarly, both GB1275 and ADU-S100 both increased CD8+ T cell infiltration, but the highest levels were seen in the combination in both syngeneic models and GEMMs (Figure 7E-F). Both STING-agonists and GB1275 reduced TAM numbers (Figure 7E), possibly due to macrophage turnover. Finally, the tumor control of STING agonist alone was not affected by T cell depletion; however, combination was dependent on T cells (Figure 7G). These data suggest that CD11b-agonists and innate immune agonists work synergistically in vivo.
We next studied the immune changes induced by the combination of GB1275 and STING activation. UMAP analysis of T cell CyTOF data identified several populations of CD8+ and CD4+ T cells (Figure 7H, S7D). Compared to single agents, the combination markedly shifted CD8+ T cells toward proliferative effector T-bethi/GZMBhi/Ki67hi phenotype (clusters 2), at the expense of memory (cluster 1) and non-proliferative subsets (cluster 3, Figure 7I; Figure S7E). The combination also significantly increased GZMB expression across CD8+ T cells (Figure 7J). Interestingly, CTLA-4, not PD-1, expression was enhanced in combination (Figure 7J, S7E). Similar to single agent GB1275, the impact on CD4+ T cells was modest. The combination increased activated CD4+ Th cells expressing low PD-1 (cluster 4) and decreased Tregs expressing high PD-1 (cluster 2, Figure S7F-I) Together, in addition to T cell numbers, CD11b-agonists and STING-agonists synergize to enhance effector CD8+ CTLs.
We next analyzed myeloid cells. UMAP analysis identified clusters of TAMs, monocytes, and granulocytes (Figure 7K). In TAMs, the combination of GB1275 and ADU-S100 decreased Sirpαhigh (cluster 4) and Ki67+ MHC-IIlow TAMs (cluster 5) but markedly increased TAMs expressing high levels of CD80, CD86, MHC-II, and MHC-I (cluster 6, Figure 7K, S7J), suggesting a shift from phagocytotic and proliferative TAMs toward antigen presentation. Additionally, PD-L1 expression among TAMs, granulocytes, and conventional dendritic cells (cDCs, Figure. S7K-N) was increased. To verify whether STING signaling activation improved the efficacy of GB1275 in PDAC, we treated PDAC-bearing mice with GB1275 + RT ± STING agonist and observed the combination regressed tumors and improved survival (Figure 7L). These data suggest that GB1275 renders STING agonist more effective at inducing anti-tumor immunity.
Discussion
PDAC is known to be poorly responsive to immunotherapy 38. We assessed a cross-tumor comparison of TAM phenotypes by scRNAseq and found that TAMs in PDAC had lower IFN and antigen presentation signatures. In keeping with this assessment, in PDAC and CRC biopsy tissues, we observed minimal percentages of STING+ or pSTAT1+ TAMs, suggesting weaker IFN signaling in TAMs in these cancers. In contrast, IL-1 and tissue remodeling signatures were higher in PDAC, consistent with the stromal desmoplastic response characteristic of this tumor type. Hence, we postulate inducing IFN signaling in myeloid cells to rescue tumor immunity in some tumor types.
The cGAS-STING signaling pathway has been regarded as important DNA sensing machinery, allowing immune responses to infections, inflammation, and cancer 39. However, activated STING plays a complicated role in cancer 40. On one hand, STING activation induces anti-tumor responses via increased interferon secretion and lymphocyte infiltration, which is promising for cancer immunotherapy 41. STING downregulation could be a factor driving resistance to immune effectors in cancer models 42, and STING signaling activation increases intratumoral T cell number consistent with upregulated IFN signatures 43. Improved productive T cell priming via cDC1s occurs in STING activation conditions 44. These experiments validate that STING /IFN signaling activation has anti-tumorigenic properties. On the other hand, emerging evidence indicated the pro-tumoral role of the cGAS-STING pathway in some cancer models 45,46. STING activation-dependent inflammation could be the major factor driving tumor development 47. STING activation results in TANK-binding kinase-1/NFκB-dependent inflammatory cytokine production 48,49, supporting cancer cell growth and chemoresistance 50. Blood vessel disruption was observed after STING agonist, owing to TNFα secretion 51. The anti-tumoral role of STING activation could be characterized as enhanced IFN production, while the side effects might be from p65-mediated inflammatory cytokine release. In our study, CD11b-agonists regulated two separate signaling pathways, including STING/IFN and p65 inhibition. The latter may decrease side effects of STING activation.
In our PDAC models, blocking p65/IL-1R signaling impacted myeloid cell infiltration and tumor progression, but did not regulate T cell infiltration, possibly suggesting that IFN induction is needed to improve T cell infiltration and/or function. NFκB/IL-1 signature downregulation was related to clinical outcomes of PDAC patients in TCGA; however, the role of NFκB in PDAC is still controversial. NFκB and autophagy signaling activation could reprogram the M2-like phenotype to the M1 phenotype 52,53. TNFα signaling-mediated apoptosis of CD206+ TAMs led to augmented anti-tumor immunity 54. Moreover, non-canonical NFκB signaling activation by cIAP1/2 antagonists stimulated the T cell-TAM axis to inhibit tumor progression. CD40 activation in TAMs infiltrated into tumor tissue, reprogramming tumor stroma 55, and p65 was involved in CD40 activation-induced proinflammatory gene expression 56. On the other hand, NFκB acts as a key link between inflammation and PDAC. Chronic inflammatory cytokine secretions, like IL-1 and IL-6, regulated by p65, led to tumor progression 57. p65 activation-mediated CXCL14 promoted angiogenesis and tumor growth 58, and p65 could regulate HIF1-α and VEGFα to affect epithelial-mesenchymal transition and angiogenesis 59. NFκB inhibition has shown great potential to inhibit PDAC, however, precise regulation of p65 in PDAC is still not well understood.
Previous studies have demonstrated efficacy of GB1275, a CD11b agonist, in preclinical models 20-22,60, and our data identified that GB1275 reprogramed solid tumor immunity through activation of STING-IFN signaling in TAMs. GB1275 entered clinical investigation (NCT04060342), and analyses of STING/pSTAT1 and p65 expression in a limited number of pre- and post-treatment patient tissues are consistent with GB1275 target engagement at the tumor site and suggest the translatability of our mechanistic findings. Specifically, aligned with PDAC model data, GB1275 increased the presence of STING+ and pSTAT1+ TAMs in post-treatment biopsies in the majority of samples tested. In parallel, a reduction in p65+ TAMs in post-treatment biopsies was observed in a subset of treated patients. Pharmacodynamic response heterogeneity may be attributable to differential TAM phenotypes/functional states across the different indications studied, as highlighted by our scRNAseq analysis of TAMs across different cancers. Alternatively, the observed heterogeneity may be highlighting differences in the TME (e.g., the presence of mtDNA), as GB1275-mediated STING activation is context-dependent. Early clinical studies with GB1275 have shown limited efficacy to date 60. Our data on the GB1275 mechanism of action suggests that the anti-tumor response of CD11b agonists could enhance DNA damage-inducing agents, and that future clinical exploration may be warranted.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David G. DeNardo (ddenardo@wustl.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The scRNA sequencing data from PDAC lesions were found at the Gene Expression Omnibus Repository (GEO) accession number GSE220959. All software packages used are publicly available through commercial vendors.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human tumor tissue
TMA studies were conducted on surgically resected PDAC specimens from patients diagnosed at the Department of Pathology at Washington University (St. Louis, MO, USA). To assemble TMAs, clearly defined areas of tumor tissues were demarcated, and two biopsies (1.0 mm in diameter) were taken from each donor block. Four μm paraffin sections were used for multiple immunohistochemistry analyses. All human tissue studies were approved by the Washington University School of Medicine Ethics Committee in accordance with recognized ethical guidelines (IRB# 201704078). Fully automated image acquisition was performed using a Zeiss Axio Scan Z1 Slide Scanner system with a 20× objective (Carl Zeiss, Jena, Germany) to capture whole slide digital images.
Human tumor biopsy sections from the Phase 1 first-in-human clinical trial evaluating GB1275 as monotherapy (Regimen A) and in combination with pembrolizumab (Regimen B) in specified advanced tumors (NCT04060342) were provided by Gossamer Bio 35. Following informed consent, core tissue biopsies were obtained from pre- and post-GB1275 doses from 11 patients. Post-treatment biopsies were obtained prior to week 8 of treatment. For details on tumor type and treatment, see Table S5. Tissues were fixed (10% neutral-buffered formalin, 48 hours) and embedded in paraffin immediately after biopsy. Needle core biopsies were fixed for a minimum of 8 hours.
The genetic mouse PDAC model and other mouse models
KPC (p48-Cre; LSL-KrasG12D/wt; p53Flox/Flox) mice were bred in-house, and C57BL/6 breeders were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). KPC mice were backcrossed to C57BL/6 over six generations and validated as C57BL/6 congenic through single nucleotide polymorphism scanning. The CD11b−/−, STING−/−, and TLR9−/− mice (all on the C57BL/6 background) were purchased from Jackson Laboratories. Mice were maintained in the Washington University Laboratory for Animal Care barrier facility, and all studies involving animals were approved by the Washington University School of Medicine Institutional Animal Studies Committee (protocol #20190856).
Cell lines, pharmacologic compounds and in vivo antibodies
KP2/KP1 cells were derived from a KPC tumor obtained in-house. Kras-INK (KI) cells were derived from Pdx1-Cre/LSL-KrasG12D/Ink/ArfFlox/Flox mice 61. KP2-OVA-GFP cells were generated from KP2 cells transduced with OVA-GFP containing lentivirus and sorted for GFP. Panc-1, Capan-1, CFPAC-1, and HPAC cells were obtained from Dr. Kian H. Lim’s laboratory. All cell lines tested negative for MAP and mycoplasma. GB1275 was provided by Gossamer Bio (San Diego, CA, USA). For animal experiments, GB1275 was administrated at 60 or 120 mg/kg by oral gavage twice a day (BID). GB1275 was dosed at 60 mg/kg in experiments described in Figure 2A-F and 120 mg/kg in all other in vivo experiments. For in vitro experiments, 10μM GB1275 dissolved in dimethyl sulfoxide (DMSO) was used for BMDMs in most experiments. A total of 1 or 5μM of GB1275 was used, as noted in the figure legends. STING agonist (ADU-S100) was purchased from MedChemExpress (Monmouth Junction, NJ, USA) and was administered at 25μg/mouse by intratumoral injection every 4 days. For in vitro experiments, 10nM ADU-S100 (dissolved in DMSO) was treated for BMDMs. Gemcitabine hydrochloride was purchased from Ark Pharm (Arlington Heights, IL, USA) and was administered at 50mg/kg by intravenous injection (i.v) every 5 days. Paclitaxel was purchased from Fresenius Kabi (Bad Homburg, Germany) and was administered at 10mg/kg by i.v. every 5 days. InVivoMAb anti-mouse IL-1R (CD121a) was purchased from BioXCell (Lebanon, NH, USA) (Clone: JAMA-147, BE0256) and was administered at 200μg/mouse, every 3 days by intraperitoneal injection (i.p). For T cell depletion, CD4 or CD8 neutralizing IgG antibodies (αCD4 clone GK1.5; αCD8 clone 2.43, BioXCell) were administered, with the first injection (1 day before GB1275 treatment) containing 400μg and subsequent injections (every 4 days) containing 200μg of each IgG. ODN 1585 -TLR9 ligand and R848 (Resiquimod) (TLR7 agonist) were purchased from InvivoGen (San Diego, CA, USA) and administered at 50μg/mouse by intratumoral injections every 5 days. For in vitro experiments, gemcitabine was treated at 1 or 10μM (10μM was used in Fig. 6B, 1 and 10μM were used in Sup Fig. 6A) dissolved in phosphate-buffered saline for BMDMs. Autophagy inhibitor, 3-MA was purchased from Sigma-Aldrich (St. Louis, MO, USA). The N-acetyl-L-cysteine was purchased from Sigma-Aldrich. VS4718 was used as a FAK1/PyTK2 inhibitor at 1.0μM for Fig. 4G and 0.5μM for the rest experiment. The 2'3'-ethidium bromide solution was purchased from Invitrogen by Thermo Fisher Scientific (Waltham, MA, USA) and was treated as 1.5mg/ mL. MG132 was purchased from MedChemExpress and was treated as 10μM dissolved in DMSO.
Syngeneic model and preclinical animal cohorts
Age-matched 6–8-week-old female C57BL/6 and FVB/NJ mice were used for orthotopic/transplantable mouse models. Syngeneic PDAC tumors were established by surgical implantation, as previously described 61. Approximately, KP2 (200,000), KI (100,000), or KP2-OVA (200,000) cells in 50μL of Cultrex (Trevigen, Gaithersburg, MD, USA) were injected into the pancreas of sex-matched C57BL/6 or FVB/NJ mice as previously described 62. Cohorts of mice were randomized into different treatment groups by gross palpation of tumors in the pancreas. In the transplantable model, KP2 (250,000) or KP2-OVA (250,000) cells in 50μL of Cultrex (Trevigen, Gaithersburg, MD, USA) were injected into each mouse’s back/flank or mammary fat pad (Figure 7L). Cohorts of mice were randomized into different treatment groups by tumor volume from external caliper measurements. Mice were maintained within the Washington University Laboratory for Animal Care barrier facility. All studies involving animals were approved by the Washington University School of Medicine Institutional Animal Studies Committee.
METHOD DETAILS
Tissue harvesting
Mice were euthanized by cardiac perfusion using 15 mL of PBS-heparin under isoflurane anesthesia. When taken for histology, tumor tissues were fixed in 10% neutral-buffered formalin overnight at 4°C. When taken for cellular assays, tumor tissues or respective lymph nodes were manually minced and digested in 15mL of sterile 1× HBSS (Thermo Fisher Scientific) containing 2mg/mL of collagenase A (Roche, Basel, Switzerland) and 1× DNase1 (Sigma-Aldrich) for 30 min at 37°C with constant stirring. Digestion was quenched in 5mL of sterile fetal bovine serum (FBS; Atlanta Biologicals, Flowery Branch, GA, USA), filtered through 40μm nylon mesh, pelleted through centrifugation (2,000rpm for 5min at 4°C), and resuspended in the required media/buffer as single-cell suspensions.
Mass cytometry (CyTOF)
Tumors taken from KI orthotopic models were treated with vehicle or GB1275 for 12 days. Eight tumors from each group were pooled as four samples (two tumors were pooled as one sample). Mouse samples from the KP2 subcutaneous model were treated with GB1275, ADU-S100, or GB1275+ADU-S100 for 14 days. We used six individuals per group. Tumor samples were digested in HBSS supplemented with 2mg/ml_ collagenase A (Roche) and DNase I at 37°C for 30 min with agitation to generate single-cell suspensions. Cell suspensions were counted and stained in 5μM cisplatin per million cells for exactly 3 min on ice and washed with Cy-FACS buffer (PBS, 0.1% BSA, 0.02% NaN3, and 2mM EDTA) twice. Cells were incubated with FcR blocking reagent plus surface-antibody cocktail for 40 min on ice. After incubation, surface marker-stained cells were washed twice with Cy-FACS buffer. Cells were then fixed with 4% paraformaldehyde for 25 min on ice and permeabilized with permeabilization buffer (Invitrogen, Carlsbad, CA, USA) for 40 min containing the intracellular stain cocktail. All antibodies are listed in the Key Resources Table. Cells were then washed twice with PBS and stained with 200μL of DNA intercalator per million cells. Cells were acquired on a CyTOF2 mass cytometer (Fluidigm, South San Francisco, CA, USA) and data were uploaded to Cytobank for further analysis. Events were gated on singlets, live, and CD45+ samples. A maximum of 100,000 events were then visualized using a standard UMAP algorithm. Populations of interest were manually gated and verified based on lineage marker expression.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD44 (IM7) | Leinco | Cat# C382 RRID: AB_2737485 |
| anti-mouse GITR (DTA-1) | BioXcell | Cat# BE0063 RRID: AB_1107688 |
| anti-mouse CD25 (PC61) | Leinco | Cat# C1194 RRID: AB_2737451 |
| anti-mouse CD38 (90) | eBioscience | Cat# 14-0381-82 RRID: AB_467219 |
| anti-mouse CD90 (G7) | Biolegend | Cat# 105202 RRID: AB_313169 |
| anti-mouse Lag-3 (C9B7W) | Leinco | Cat# L306 RRID: AB_2737552 |
| anti-mouse CD27 (LG.7F9) | eBioscience | Cat# 50-124-94 |
| anti-mouse KLRG1 (2F1/KLRG1) | BioXCell | Cat# BE0201 RRID: AB_10949054 |
| anti-mouse CD103 (2E7) | Biolegend | Cat# 121402 RRID: AB_535945 |
| anti-mouse CD4 (GK1.5) | BioXcell | Cat# BE0003-1 RRID: AB_1107636 |
| anti-mouse CD45 (30-F11) | Fluidigm | Cat# 3089005B RRID: AB_2651152 |
| anti-mouse CD62L (MEL-14) | Leinco | Cat# C2118 RRID: AB_2737457 |
| anti-mouse ICOS (C398.4A) | eBioscience | Cat# 14-9949-82 RRID: AB_468637 |
| anti-mouse OX-40 (OX-86) | BioXcell | Cat# BE0031 RRID: AB_1107592 |
| anti-mouse PD-1 (RMP1-30) | eBioscience | Cat# 14-9981-82 RRID: AB_468655 |
| anti-mouse TIGIT (1G9) | BioXcell | Cat# BE0274 RRID: AB_2687797 |
| anti-mouse CD69 (H1.2F3) | eBioscience | Cat# 14-0691-82 RRID: AB_467325 |
| anti-mouse TCRβ (H57-597) | BioXcell | Cat# BE0102 RRID: AB_10950158 |
| anti-mouse CD127 (A7R34) | BioXcell | Cat# BE0065 RRID: AB_1107590 |
| anti-mouse CD39 (Duha59) | Biolegend | Cat# 143802 RRID: AB_11204243 |
| anti-mouse NK1.1 (PK136) | BioXcell | Cat# BE0036 RRID: AB_1107737 |
| anti-mouse CD8α (53-6.7) | Leinco | Cat# C375 RRID: AB_2737478 |
| anti-mouse TCRγδ (GL3) | eBioscience | Cat# 14-5711-82 RRID: AB_467569 |
| anti-mouse Tim3 (RMT3-23) | BioXcell | Cat# BE0115 RRID: AB_10949464 |
| anti-mouse FoxP3 (FJK-16s) | eBioscience | Cat# 14-5773-82 RRID: AB_467576 |
| anti-mouse GATA3 (TWAJ) | eBioscience | Cat# 14-9966-82 RRID: AB_1210519 |
| anti-mouse GranzymeB (GB11) | eBioscience | Cat# MA1-80734 RRID: AB_931084 |
| anti-mouse CTLA-4 (UC10-4B9) | eBioscience | Cat# 50-129-16 |
| anti-mouse Ki67 (8D5) | Novus | Cat# NBP2-22112 |
| anti-mouse TCF1 (812145) | R&D | Cat# MAB8224 |
| anti-mouse ROR-γt (AFKJS-9) | eBioscience | Cat# 14-6988-82 RRID: AB_1834475 |
| anti-mouse Eomes (Dan11mag) | eBioscience | Cat# 50-245-556 |
| anti-mouse T-bet (4B10) | Biolegend | Cat# 644802 RRID: AB_1595503 |
| anti-mouse CD45 (30-F11) | Fluidigm | Cat# 3089005B RRID: AB_2651152 |
| anti-mouse Ki67 (8D5) | Novus | Cat# NBP2-22112 |
| anti-mouse CD11c (N418) | Fluidigm | Cat# 3142003B RRID: AB_2814737 |
| anti-mouse CD68 (FA-11) | Biolegend | Cat# 137001 RRID: AB_2044003 |
| anti-mouse MHC-I (28-14-8) | Fluidigm | Cat# 3144016B RRID: AB_2687831 |
| anti-mouse CD206 (C068C2) | Biolegend | Cat# 141702 RRID: AB_10900233 |
| anti-mouse F4/80 (BM8) | Fluidigm | Cat# 3146008B RRID: AB_2895117 |
| anti-mouse MHC-II (M5/114.15.2) | Biolegend | Cat# 107602 RRID: AB_313317 |
| anti-mouse CD11b (M1/70) | Fluidigm | Cat# 3148003B RRID: AB_2814738 |
| anti-mouse CD172α/SIRPα (P84) | Biolegend | Cat# 144002 RRID: AB_11203711 |
| anti-mouse Ly6C (HK1.4) | Fluidigm | Cat# 3150010B RRID: AB_2895118 |
| anti-mouse Ly6G (1A8) | Fluidigm | Cat# 3151010B |
| anti-mouse CD64 (X54-5/7.1) | Biolegend | Cat# 139301 RRID: AB_10612757 |
| anti-mouse Tim4 (RMT4-54) | Biolegend | Cat# 130002 RRID: AB_1227802 |
| anti-mouse XCR1(Zet) | Biolegend | Cat# 148202 RRID: AB_2563841 |
| anti-mouse CD103 (2E7) | Biolegend | Cat# 121402 RRID: AB_535945 |
| anti-mouse NK1.1 (PK136) | BioXcell | Cat# BE0036 RRID: AB_1107737 |
| anti-mouse Bst2 (120G8) | Novous/imgenx | Cat# DDX0390P-100 RRID: AB_2827525 |
| anti-mouse IRF4 (3E4) | Biolegend | Cat# 646402 RRID: AB_2280462 |
| anti-mouse CD39 (Duha59) | Biolegend | Cat# 143802 RRID: AB_11204243 |
| anti-mouse NK1.1 (PK136) | BioXcell | Cat# BE0036 RRID: AB_1107737 |
| anti-mouse CD83 (Michel-17) | thermofisher scientific | Cat# 14-0831-82 RRID: AB_467357 |
| anti-mouse CD40 (HM40-3) | Fluidigm | Cat# 124601 RRID: AB_1134239 |
| anti-mouse Ox40L (RM134L) | Biolegend | Cat# 108802 RRID: AB_313401 |
| anti-mouse CCR2 (475301) | R&D systems | Cat# MAB55381-100 RRID: AB_2749828 |
| anti-mouse Cx3CR1(SA011F11) | Fluidigm | Cat# 3164023B RRID: AB_2832247 |
| anti-mouse CCR7 (4B12) | Biolegend | Cat# 120101 RRID: AB_389229 |
| anti-mouse PDL2 (TY25) | BioXCell | Cat# BE0112 RRID: AB_10950106 |
| anti-mouse VISTA (MIH63) | Biolegend | Cat# 150202 RRID: AB_2565897 |
| anti-mouse Tim3 (RMT3-23) | BioXcell | Cat# BE0115 RRID: AB_10949464 |
| anti-mouse PD-L1 (10F.9G2) | BioXCell | Cat# BE0101 RRID: AB_10949073 |
| anti-mouse CD80 (16-10A1) | Biolegend | Cat# 104702 RRID: AB_313123 |
| anti-mouse CD135/FLT3 (A2F10) | Thermo Fisher Scientific | Cat# 14-1351-82 RRID: AB_467481 |
| anti-mouse CD86 (GL1) | Fluidigm | Cat# 3172016B RRID: AB_2922923 |
| anti-mouse B220 (RA3-682) | Fluidigm | Cat# 3144011B RRID: AB_2811239 |
| anti-mouse CD45 (30-F11) | eBioscience | Cat# 25-0451-82 RRID: AB_2734986 |
| anti-mouse CD45 (30-F11) | BD Biosciences | Cat# 564225 RRID: AB_2716861 |
| anti-mouse CD45 (30-F11) | BioLegend | Cat# 103184 |
| anti-mouse CD45 (30-F11) | Invitrogen | Cat# 63-045-182 |
| anti-mouse CD3e (145-2C11) | eBioscience | Cat# 17-0031-82 RRID: AB_469315 |
| anti-mouse CD3e (145-2C11) | BD Biosciences | Cat# 563123 RRID: AB_2687954 |
| anti-mouse CD4 (RM4-4) | eBioscience | Cat# 11-0043-82 RRID: AB_464900 |
| anti-mouse CD4 (RM4-4) | BioLegend | Cat# 116022 RRID: AB_2715958 |
| anti-mouse CD8α (53-6.7) | BD Biosciences | Cat# 563786 RRID: AB_2732919 |
| anti-mouse CD8α (53-6.7) | BD Biosciences | Cat# 553035 RRID: AB_398527 |
| anti-mouse Foxp3 (FJK-16s) | eBioscience | Cat# 15-5773-82 RRID: AB_468806 |
| anti-mouse CD19 (eBio1D3) | eBioscience | Cat# 17-0193-82 RRID: AB_1659676 |
| anti-mouse CD11b (M1/70) | eBioscience | Cat# 56-0112-82 RRID: AB_657585 |
| anti-mouse CD11b (M1/70) | Invitrogen | Cat# 56-0112-82 RRID: AB_657585 |
| anti-mouse Ly6C (HK1.4) | eBioscience | Cat# 45-5932-82 RRID: AB_2723343 |
| anti-mouse Ly6C (HK1.4) | BioLegend | Cat# 128006 RRID: AB_1186135 |
| anti-mouse Ly6G (1A8) | BioLegend | Cat# 127608 RRID: AB_1186099 |
| anti-mouse Ly6G (1A8) | BioLegend | Cat# 127614 RRID: AB_2227348 |
| anti-mouse F4/80 (BM8) | eBioscience | Cat# 15-4801-82 RRID: AB_468798 |
| anti-mouse F4/80 (BM8) | Invitrogen | Cat# 25-4801-82 RRID: AB_469653 |
| anti-mouse MHC-II (M5/115.15.2) | eBioscience | Cat# 48-5321-82 RRID: AB_1272204 |
| anti-mouse MHC-II (I-A/I-E) (M5/114.15.2) | Invitrogen | Cat# 58-5321-82 RRID: AB_2811913 |
| anti-mouse Ki67 (SolA15) | eBioscience | Cat# 50-5698-82 RRID: AB_2574235 |
| anti-mouse SIINFEKL Dextramer (N/A) | Immudex | Cat# JD2163 |
| anti-mouse Granzyme B (GB11) | BD Horizon | Cat# 563388 RRID: AB_2738174 |
| anti-mouse IFNγ (XMG1.2) | BioLegend | Cat# 505808 RRID: AB_315402 |
| anti-mouse TNFα (MP6-XT22) | eBioscience | Cat# 11-7321-82 RRID: AB_465418 |
| CD11b (EPR1344) | Abcam | Cat# ab13357 RRID: AB_2650514 |
| F4/80 (D2S9R) | Cell Signaling | Cat# 70076 RRID: AB_2799771 |
| STING (D2P2F) | Cell Signaling | Cat# 13647 RRID: AB_2732796 |
| NFκB p65 (D14E12) | Cell Signaling | Cat# 8242 RRID: AB_10859369 |
| pSTAT1 (D3B7) | Cell Signaling | Cat# 8826 RRID: AB_2773718 |
| CK19 (Troma III) | DSHB | Cat# Uoflowa DSHB TROMA-III C |
| CD8 (D4W2Z) | Cell Signaling | Cat# 98941 RRID: AB_2756376 |
| CD8 (SP16) | Invitrogen | Cat# MA5-14548 RRID: AB_10984334 |
| CD163 (10D6) | Leica Biosystem | Cat# NCL-L-CD163 RRID: AB_2756375 |
| Keratin17/19 (D4G2) | Cell Signaling | Cat# 12434 RRID: AB_2797912 |
| Cytokeratin, Muti (AE1/AE3) | Leica Biosystem | Cat# NCL-L-AE1/AE3-601 RRID: AB_2924990 |
| pAKT (D9E) | Cell Signaling | Cat# 4060 RRID: AB_2315049 |
| pIRF3 (D6O1M) | Cell Signaling | Cat# 29047 RRID: AB_2773013 |
| IRF3 (D83B9) | Cell Signaling | Cat# 4302 |
| STAT1 (D1K9Y) | Cell Signaling | Cat# 14994 RRID: AB_2737027 |
| pFAK | Cell Signaling | Cat# 3283 RRID: AB_2173659 |
| FAK | Cell Signaling | Cat# 3285 RRID: AB_2269034 |
| pp65 (93H1) | Cell Signaling | Cat# 3033 RRID: AB_331284 |
| pIκBα (14D4) | Cell Signaling | Cat# 2859 RRID: AB_561111 |
| IκBα | Cell Signaling | Cat# 9242 RRID: AB_331623 |
| Cleaved Caspase-3 (Asp175) | Cell Signaling | Cat# 9661 RRID: AB_2341188 |
| Ubiquitin (E4I2J) | Cell Signaling | Cat# 43124 RRID: AB_2799235 |
| SIRT3 (D22A3) | Cell Signaling | Cat# 5490 RRID: AB_10828246 |
| β-ACTIN (13E5) | Cell Signaling | Cat# 4970 RRID: AB_2223172 |
| IFNβ | Invitrogen | Cat# PA5-20390 RRID: AB_11155641 |
| CXCL10 (10H11L3) | Invitrogen | Cat# 701225 RRID: AB_2532429 |
| Oligonucleotides | ||
| Primers and siRNA used in this study are list in Table S6 | This paper | N/A |
| Biological Samples | ||
| Human PDAC TMA | Washington University | IRB# 201704078 |
| Tumor biopsies | Gossamer Bio | # NTC04060342 |
| Cultrex Basement membrane extract, Pathclear | Trevigen | Cat# 3432-001-01 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| STING agonist (ML-RR-S2 CDA; ADU-S100) | MedChemExpress | Cat# HY-12885B |
| Gemcitabine hydrochloride | Ark Pharm | Cat# 122111-03-9 |
| Paclitaxel | Fresenius Kabi | #00363323763503 |
| InVivoMAb anti-mouse IL-1R (CD121a) (JAMA-147) | BioXCell | Cat# BE0256 |
| InVivoMAb anti-mouse CD4 (GK1.5) | BioXCell | Cat# BE0003-1 |
| InVivoMAb anti-mouse CD8 (2.43) | BioXCell | Cat# BP0061 |
| ODN 1585 -TLR9 ligand | InvivoGen | Cat# tlrl-1585 |
| R848 (Resiquimod) | InvivoGen | Cat# tlrl-r848-5 |
| Autophagy inhibitor, 3-MA | Sigma-Aldrich | Cat# 5142-23-4 |
| N-acetyl-L-cysteine | Sigma-Aldrich | Cat# 616-91-1 |
| 2'3'- ethidium bromide solution | Invitrogen | Cat# 17898 |
| MG132 | MedChemExpress | Cat# HY-13259 |
| Collagenase A | Sigma/Roche | Cat# 10103586001 |
| DNAse I | Sigma | Cat# 11284932001 |
| Cell Stimulation cocktail (PMA/Iono) | Ebioscience | Cat# 00-4970-93 |
| Brefeldin A | Biolegend | Cat# 420601 |
| Monensin | Biolegend | Cat# 420701 |
| 2′-7′-dichlorodihydrofluorescein diacetate | Sigma-Aldrich | Cat# 287810 |
| Critical Commercial Assays | ||
| E.Z.N.A. Total RNA kit I | Omega | Cat# R6834-02 |
| qScript cDNA Supermix kit | Quantabio | Cat# 95048-500 |
| Taqman Gene Expression Master Mix | Thermo Fisher | Cat# 4370074 |
| BOND Polymer Refine Detection kit Leica #DS9800 | Leica | Cat# DS9800 |
| BOND Intense R Detection kit | Leica | Cat# DS9263 |
| Cytofix kit | BD Bioscience | Cat# 554655 |
| CSF1 neutralizing antibody (Clone 5A1) | BioXCell | Cat# BE0204 |
| PBS Liposomes & Clodronate Liposomes | Liposoma | Cat# CP-005-005 |
| Halt Protease and Phosphatase Inhibitor | Thermo Scientific | Cat# 78442 |
| RIPA Buffer(10x) | Cell Signaling | Cat# UN3082 |
| Mouse Macrophage Nucleofector Kit | Lonza | Cat# VPA-1009 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | Cat# 23225 |
| Recombinant Murine M-CSF | PeproTech | Cat# 315-02 |
| Mouse IFN-beta DuoSet ELISA | R&D Systems | Cat# DY8234-05 |
| Mouse IL-1 beta/IL-1F2 DuoSet ELISA | R&D Systems | Cat# DY401 |
| IP-10/CXCL10 Mouse Matched Antibody Pair | Invitrogen | Cat# BMS6018MST |
| I-TAC/CXCL11 Mouse ELISA Kit | Invitrogen | Cat# EMCXCL11 |
| protein A agarose Beads | Cell Signaling Technology | Cat# 9863 |
| NucleoSpin Tissue Kit | Takara Bio | Cat# 740952.50 |
| SuperSignal™ West Dura Extended Duration Substrate | Thermo Fisher Scientific | Cat# 34076 |
| CellTiter96 Non-Radioactive Cell Proliferation Assay | Promega | Cat# G4002 |
| EasySep Human CD14 Selection Kit | Stemcell Technologies | Cat# 17858 |
| Pierce™ Protein Concentrator PES, 3K MWCO | Thermo Fisher Scientific | Cat# 88525 |
| Experimental Models: Cell Lines | ||
| KP2/KP1; derived from a KPC tumor | This paper; Jiang et al.2016 | N/A |
| KI; derived from Kras-Ink mouse | This paper; Jiang et al.2016 | N/A |
| KP2-OVA-GFP; generated from KP2 cells transduced with OVA-GFP containing lentivirus and sorted for GFP | This paper; VE Lander et at. 2022 | N/A |
| Panc-1 | obtained from Dr. Kian H from ATCC | RRID: CVCL_0480 |
| Capan-1 | obtained from Dr. Kian H from ATCC | RRID: CVCL_0237 |
| CFPAC-1 | obtained from Dr. Kian H from ATCC | RRID: CVCL_1119 |
| HPAC | obtained from Dr. Kian H from ATCC | RRID: CVCL_3517 |
| Experimental Models: Organisms/Strains | ||
| mouse: p48-Cre;KrasLSL-G12D;Trp53fl/fl | Hingorani et al., 2003; Morton et al., 2010 | N/A |
| mouse: C57BL/6J | The Jackson Laboratory | Stock# 000664 |
| mouse: FVB/NCr | Charles River Laboratories | Strain# 559 |
| mouse: B6.129S4-Itgamtm1Myd/J | The Jackson Laboratory | Strain# 003991 |
| mouse: B6(Cg)-Sting1tm1.2Camb/J | The Jackson Laboratory | Strain# 025805 |
| mouse: C57BL/6-TIrgem1.1Ldm/j | The Jackson Laboratory | Strain# 034449 |
| Deposited Data | ||
| scRNASeq data | This paper | GSE220959 |
| Software and Algorithms | ||
| Flowjo v10.7.2 | Flowjo, L.L.C. | Flowjo, L.L.C. RRID:SCR_008520 |
| Prism v9 | Graphpad |
www.graphpad.com RRID:SCR_000306 |
| Docker | Rocker/rstudio:latest | https://hub.docker.com/r/rocker/rstudio |
| cumulusprod/cellranger:4.0.0 | https://hub.docker.com/r/cumulusprod/cellranger/tags | |
| HALO v3.2.1851 | Indica Labs-High Plex Fv4.0.3 | https://indicalab.com/products/high-plex-fl/ |
| Indica Labs-Deconvolution v1.0.4 | ||
| Cytobank | Cytobank, Inc | Wustl.cytobank.org |
| FACSDiva | BD Biosciences | RRID: SCR_001456 |
| Zen | Zeiss | Zeiss.com |
| Fiji v2.0.0 | ImageJ | |
| R v3.6.3 | R Core Team | https://cran.r-project.org/bin/windows/base/old/3.6.3/ |
| Clusterprofiler | https://github.com/YuLab-SMU/clusterProfiler | |
| Seurat v 3.2.0 | https://satijalab.org/seurat/ | |
| Harmony | https://github.com/immunogenomics/harmony | |
| CATALYST | https://github.com/HelenaLC/CATALYST | |
Single-cell RNA sequencing data analysis in humans
Processed count data were downloaded from Gene Expression Omnibus under the following sessions:GSE121636 63 GSE123814 64, GSE139555 65, GSE145370 66, GSE154826 67, GSE155698 68, and GSE176078 69. Ovarian data were downloaded from a code repository 70. Expression data were imported into R, v4.1.0 (R Foundation for Statistical Computing, Vienna, Austria) using the Seurat (v4.1.0) R package 71. Cells were filtered for PTPRC expression to ensure immune cells. Additional quality control filtering was based on the percentage of mitochondrial genes < 10% of counts and removal of cells with a feature number greater than 2.5 of the standard deviation of all cells. Myeloid cells were isolated using the scGate (v1.0.0) R package 72 using the “MoMacDC” model. After isolation, manual removal of monocytes utilized feature counts and canonical markers. Dimensional reduction to produce a UMAP (RunUMAP) utilized the standard Seurat workflow, with the addition of data harmonization (RunHarmony) using the harmony (v0.1.0) R package 73 using sequencing run and cancer type as the grouping variables. Both the UMAP calculation and neighbor identification used 15 harmonized dimensions. Single-cell gene enrichment was performed using the UCell implementation 74 in the escape R package 63 across the Hallmark and C2 libraries in the Molecular Signaling Database 75. Statistical testing across all cancer types was performed using the Kruskal-Wallis test, with individual comparisons calculated with the pairwise Wilcoxon test. Adjusted p-values for significance testing were based on the total number of pairwise Wilcoxon results using the Bonferroni correction for multiple hypothesis testing.
Single-cell RNA sequencing data analysis in mice
Single-cell analysis was performed as previously described 76,77. Briefly, PDAC tissues were taken from vehicle-treated, GB1275-treated KP2 subcutaneous pancreatic tumors, and at 14 days post-treatment. Immune cells (CD45+) were sorted by an Aria-II cell sorter (BD Biosciences, San Jose, CA, USA). Each sample was generated from a pool of three mice per treatment group, and two total libraries were sequenced.
Sorted cells from each sample were encapsulated into droplets and libraries were prepared using Chromium Single Cell 3’v3 Reagent kits according to the manufacturer’s protocol (10× Genomics, Pleasanton, CA, USA). The generated libraries were sequenced by a NovaSeq 6000 sequencing system (Illumina, San Diego, CA, USA) to an average of 50,000 mean reads per cell. Cellranger mkfastq pipeline (10× Genomics) was used to demultiplex illumine base call files to FASTQ files. Files from samples were demultiplexed with > 97% valid barcodes, and > 94% q30 reads. Afterwards, fastq files from each sample were processed with Cellranger counts and aligned to the mm10 reference (Version 3.1.0, 10× Genomics) to generate the feature barcode matrix.
The filtered feature with barcode matrices were loaded into Seurat as objects. For each Seurat object, genes that were expressed in less than three cells and cells that expressed less than 1,000 or more than 6,000 genes, were excluded. Cells with greater than 10% mitochondrial RNA content were also excluded, resulting in 10,933 cells in the vehicle and 12,086 in the GB1275-treated group. SCTransform with default parameters was used on each individual sample to normalize and scale the expression matrix against the sequence depths and percentages of mitochondrial genes. Cell cycle scores and the corresponding cell cycle phase for each cell were calculated and assigned after SCTransform based on the expression signatures for S and G2/M genes (CellCycleScoring). The differences between the S phase score and G2/M score were regressed out by SCTransform on individual samples. Variable features were calculated for each sample independently and ranked, based on the number of samples they were independently identified (SelectIntegrationFeatures). The top 3,000 shared variable features were used for multi-set canonical correlation analysis to reduce dimensions and identify projection vectors that defined shared biological states among samples and maximized overall correlations across datasets. Mutual nearest neighbors (pairs of cells, with one from each dataset) were calculated and identified as “anchors” (FindIntegrationAnchors). Multiple datasets were then integrated based on these calculated “anchors” and guided order trees with default parameters (IntegrateData). Multiple datasets were then integrated based on using Harmony Integration (RunHarmony). Principle component analysis (PCA) was performed on the 3,000 previously calculated genes (function RunPCA). A UMAP dimensional reduction was performed on the scaled matrix using the first 30 PCA components to obtain a two-dimensional representation of cell states. Then, these defined 30 dimensionalities were used to refine the edge weights between any two cells based on Jaccard similarity (FindNeighbors) and were used to cluster cells through FindClusters functions, which implemented shared nearest neighbor modularity optimization. To characterize clusters, the FindAllMarkers function with log-fold threshold = 0.25 and minimum 0.25-fold difference and MAST test results were used to identify signatures alone with each cluster. Then, the TAMs were selected, and the top 3,000 variable features were recalculated to recluster. DEGs between the two groups were calculated for each dataset with min.pct of 0.1 and logfc. threshold of 0.01 and MAST test (FindMarkers). Then, the differentially expressed gene (DEG) lists from each dataset were filtered with a value of p < 0.05 and ranked based on fold change. These ranked gene sets were fed into GSEA to test for Gene Ontology terms, Kyoto Encyclopeida of Genes and Genomes pathways, Reactome Database, and the Molecular Signatures Database gene sets with false discover rate (FDR) < 0.05 in ClusterProfiler 78.
Mouse tissue isolation and flow cytometry
Following tissue digestion, single-cell suspensions were resuspended in flow cytometry buffer (PBS containing 1% BSA), FcR blocked with rat α-mouse CD16/CD32 antibodies (Ebioscience, Santa Clara, CA, USA) for 10 min and pelleted by centrifugation. Where applicable, CD8+ T cells specific for antigen OVA were labeled by incubating cell suspension with H2Kb::SIINFEKL-specific MHC-I dextramer (1:5; Immudex protocol) for 10min at room temperature prior to extracellular staining. Single cells were consequently labeled with 100μL of fluorophore-conjugated α-mouse extracellular antibodies at recommended dilutions for 25 min on ice. Intracellular staining for intracellular markers was conducted subsequently using the EBioscience Transcription Factor Staining buffer set, according to manufacturer’s instructions. All antibodies are listed in Key Resources Table. Data were acquired on an X-20 (BD Biosciences) or Cytek Aurora (Fremont, CA, USA) and analyzed using FlowJo software (v10).
For ex-vivo T cell functional (cytokine release) assays, following tissue digestion, primary tumor cell suspensions containing 1 million cells were incubated in 96-well plates with 1μM Brefeldin A (Biolegend, San Diego, CA, USA) and 2μM Monensin Solution (Biolegend) and 1× Stimulation Cocktail (eBioscience) for 6 hours at 37°C and 5% CO2. After incubation, cells were labeled with fluorophore-conjugated anti-mouse antibodies as described above. For in vitro T cell functional assays, following spleen smashing and single-cell suspension procedures, cells were incubated and treated as ex-vivo T cell function assays in the presence of GB1275 at different doses for indicated periods. For in vitro T cell proliferation assay, T cells from the spleen were labeled by CFSE (5μM, Invitrogen by Thermo Fisher Scientific), and treated with GB1275 at different doses for 2 days. After incubation, cells were labeled with fluorophore-conjugated anti-mouse antibodies as above. Data were acquired on Cytek Aurora and analyzed using FlowJo software (v10).
Immunohistochemistry and mpIHC
Tissues were fixed in 10% neutral formalin for 24 hours and embedded in paraffin after graded-ethanol dehydration and sectioned into 6-μm sections using a microtome. Automated staining of tissues was carried out on the BOND RXm (Leica Biosystems, Wetzlar, Germany) following dewaxing and appropriate antigen retrieval. Immunostaining was chromogenically visualized using the Bond Polymer Refine Detection (DS9800, Leica Biosystems). Slides were dehydrated through graded ethanol, followed by xylene, then mounted using xylene-based Cytoseal (Thermo Fisher). Primary antibodies are listed in (Key Resources Table).
For IHC, whole-tissue scans at 10× or 20× magnification were obtained on a Zeiss Axio Scan Z1 brightfield/fluorescence Slide Scanner. Whole-tissue scans were analyzed with HALO software (Indica Labs, Albuquerque NM, USA) using Area quantification, Cytonuclear, or HighPlex modules.
For mpIHC of mouse PDACs, embedded tissues were sectioned into 6- um sections and loaded into BOND RXm (Leica Biosystems) for a series of staining, including using antibodies to pSTAT1, STING, p65, CD11b, F4/80, and CK19. Human TMA slides were stained with antibodies to CD8, STING, CD163, CD11b, and CK19. Tumor biopsy slides were stained with antibodies to CD8, STING, pSTAT1, CD163, CD11b, p65, and PanK. Based on antibody host species, default manufacturer protocols were used (IntenseR and Polymer Refine), containing antigen retrieval with citrate buffer, goat serum and peroxide block, primary antibody incubation, post-primary incubation, and chromogenically visualized with an AEC substrate (Abcam, Cambridge, UK). Between every two cycles of staining, the slides were manually stained for hematoxylin, then scanned by Axio Scan.Z1 (Zeiss, Jena, Germany). The slides were then destained by a gradient of ethanol plus a 2% hydrochloride wash and blocked with extra avidin/biotin (Vector Laboratories, Burlingame, CA, USA) and a Fab fragment block (Jackson Laboratory, Bar Harbor, ME, USA). Prior to starting another staining cycle. Citrate-based antigen retrieval was performed before each staining cycle.
For mpIHC, images of the same specimen, but using different stains, were cropped into multiple segments by Zen software (Zeiss). Each segment was then deconvoluted (Deconvolution, Version 1.0.4; Indica Labs) for individual stains and fused using HALO software with the default manufacturer’s settings. Markers of interest were pseudo-colored and quantified using High Plex FL software in HALO software.
Macrophage depletion
To deplete tissue-resident macrophages, 6–8-week-old FVB/NJ mice were treated with three doses of CSF1 neutralizing antibody (clone 5A1; BioXCell, Lebanon, NH, USA) (1mg, 0.5mg, and 0.5mg on days 3, 10, and 17; Figure 1H) and three doses of clodronate-containing liposomes (Liposoma, Groningen, The Netherlands; 200μL/each on days 4, 11, and 18). Control mice were treated with the same doses/volume of IgG (clone HRPN, BioXCell) and control liposomes (or PBS as indicated, Liposoma). On day 0, mice were implanted orthotopically with 100,000 KI cells and then treated with GB1275 as previously described.
Small Animal Radiation Therapy (RT)
Ten days post-tumor implantation, cohorts of mice were randomized into different treatment groups using gross tumor volumes. Mice were given daily fractionated doses of RT for 5 days (6 Gy × 5) using the Small Animal Radiation Research Platform (SARRP200; Xstrahl Life Sciences). Mice were placed on the irradiation platform one at a time and fitted with a nose cone for isoflurane anesthesia. Cone beam computed tomography (CT) imaging was performed for each individual mouse to pinpoint tumors, and the images were imported into Muriplan and used to select an isocenter. The tumor was then irradiated to 6 Gy using anterior-posterior-opposed beams using the 10 mm × 10 mm collimator at a dose rate of 3.9 Gy/min. Mice were monitored over 2 weeks for signs of radiation sickness or weight loss. DietGel recovery gel was provided for a 14-day window immediately following radiation therapy in survival studies.
For in vitro radiation experiments, an RS2000 160 kV X-ray irradiator using a 0.3mm copper filter (Rad Source Technologies) was used. PDAC cells were irradiated to 8Gy.
Bone marrow transplantation
Six- to eight-week-old C57BL/6 mice were exposed to irradiation dosed at 800Gy (400Gy × 2). Animals were subsequently injected with 5 × 106 bone marrow cells from either WT or STING-null mice. A syngeneic PDAC model was established with these mice after 4 weeks of recovery.
Isolation of bone marrow-derived macrophages
BM-MACs were isolated following the protocol described previously 79. Marrow cells were isolated by flushing femurs and tibias from C57BL/6J mice and cultured in DMEM/F12 medium (Lonza, Basel, Switzerland) containing 10% FBS, penicillin/streptomycin (Gibco) and 20ng/mL macrophage colony–stimulating factor (M-CSF, PeproTech, Cranberry, NJ, USA). After 5 days in culture, adherent macrophages were harvested and seeded on 10 μg/mL fibronectin-coated (Sigma-Aldrich) plates for different experiments. On day 6, macrophages were pretreated with DMEM/F12 medium (Lonza) containing 1% FBS and DMSO or GB1275 (1, 5, or, 10μM dissolved in DMSO for different experiments) for 1 hour followed by removal of the old media. Cells were incubated in DMEM/F12 medium (Lonza) containing 1% FBS or fresh tumor-conditioned media (TCM) (Note: should not be frozen or kept at 4°C for long time periods), containing GB1275 or DMSO at indicated doses for 1, 4, or 6 hours. In the whole process, 20ng/mL macrophage colony–stimulating factor was always added to the different media. After indicated periods of stimulation, we removed the media, and rinsed the cells with PBS prior to RNA isolation or other experiments.
RNA isolation and real-time PCR
Total RNA was extracted from tissue or cells, using an E.Z.N.A. Total RNA Kit (Omega Bio-tek, Norcross, GA). Complementary DNAs (cDNAs) were synthesized using qScript cDNA SuperMix (QuantaBio, Beverley, MA, USA). Quantitative real-time PCR Taqman primer-probe sets (Applied Biosystems, Waltham, MA, USA) were used (Table S6), and the relative gene expression was determined on an ABI 7900HT quantitative PCR machine (ABI Biosystems) using Taqman Gene Expression Master Mix (Applied Biosystems). The comparative threshold cycle method was used to calculate fold changes in gene expression, which were normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), TATA-box binding protein (TBP), and/or hypoxanthine phosphoribosyl transferase (HPRT) as reference genes.
Reverse-Phase Protein Array (RPPA)
Cell extracts were lysed using (RIPA) lysis buffer [25mM Tris-HCl pH 7.5, 150mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS] supplemented with protease and phosphatase inhibitors (Roche, Basel, Switzerland). Samples were then submitted to the MD Anderson Cancer Center for the RPPA assay (RPPA CORE 11192019_169).
Cytokine assay
Tumor tissues from the KP2-OVA-GFP orthotopic PDAC model were lysed by Procartaplex lysis buffer (Thermo Fisher Scientific, EPX-99999-000) (50μL lysis buffer per 10mg tissue) supplemented with protease and phosphatase inhibitors (Roche). Protein concentration from each sample was adjusted to 5mg/mL. Supernatants were harvested after different stimulations of BMDMs for 12 hours. IFNβ, CXCL10, CXCL11, and IL-1β from PDAC tissue and IL-1β from supernatants were measured by enzyme-linked immunosorbent assay (ELISA). For tissue, the amount of IFNβ (DY8234-05, R&D Systems, Minneapolis, MN, USA), IL-1β (DY-401, R&D Systems), CXCL10 (BMS6018MST, Invitrogen, Thermo Fisher Scientific), and CXCL11 (EMCXCL11, Invitrogen, Thermo Fisher Scientific) was shown as pg per mg protein in tissue. For supernatants, the amount of IL-1β was normalized by protein levels in its cell lysate.
Western immunoblot and immunoprecipitation (IP)
Cell lysates were harvested using radioimmunoprecipitation assay (RIPA) lysis buffer [25 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% SDS] supplemented with protease and phosphatase inhibitors (Roche). Protein from supernatant was isolated by methanol and chloroform, and then dissolved by 1% SDS sample buffer. Protein samples were resolved in Tris-glycine sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS/PAGE) gels and transferred to polyvinylidene difluoride membranes (Invitrogen). After blocking in 1× TBST buffer with 5% w/v bovine serum albumin (BSA), membranes were probed with primary antibodies against pp65, tp65, pIκB, tIκB, STING, plRF3, tIRF3, pSTAT1 tSTAT1, SIRT3, IFNβ, CXCL10, and β-actin overnight at 4°C (Key Resources Table). Membranes were washed three times in 1× TBST and probed with HRP-conjugated secondary antibody for 1 hour at RT. Membranes were developed with Pierce ECL Western Blotting Substrates (Pierce Chemical, Dallas, TX, USA) and detected using a ChemiDoc XRS+ system (Bio-Rad, Hercules, CA, USA). For immunoprecipitation, cell lysates were incubated with p65 antibody overnight at 4°C followed by incubation in protein A agarose (Cell Signaling Technology, Danvers, MA, USA) for 3 hours. The beads were washed five times with lysis buffer and then used for western immunoblotting.
Isolate released mtDNA
The mtDNA was isolated following a ptreviously described protocol 80. Briefly, after different treatments, the cells were lysed by 1% NP40 for 15 min on ice. The lysates were centrifuged at 13,000 rpm for 15 min at 4°C. Supernatants were transferred to the isolated mtDNA. A NucleoSpin Tissue Kit (Takara Bio USA, San Jose, CA, USA) was adopted to purify mtDNA from the cytosolic fraction according to the manufacturer’s instructions. After extracting DNA from the cytosolic fraction, real-time PCR was employed to measure cytosolic mitochondrial DNA. The comparative threshold cycle method was used to calculate fold changes in gene expression, which were normalized to the expression of the 18S rRNA reference gene.
Measuring ROS production
Briefly, after different treatments, cells were harvested and incubated with 10μM 2′-7′-dichlorodihydrofluorescein diacetate (Sigma-Aldrich) in serum-free medium for 20min at 37°C in the dark. After incubation, samples were washed twice with cold PBS. Data were acquired on an X-20 (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Cell proliferation assay
Cell proliferation assays were performed by using the CellTiter96 Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Briefly, 20,000 BMDMs/well were seeded into 96-well plates and incubated overnight. After different treatments, dye solution was added to live cultures for 4 hours at 37°C. Absorbance was measured at 570 nm on a Multiskan GO plate reader (Thermo Fisher Scientific).
In vitro siRNA treatment
Small interfering RNAs (siRNAs) targeting mouse Rela, Sting1, Sirt3 and Tlr9 were purchased from Integrated DNA Technologies (Coralville, IA, USA). Sequences as listed in the Table S6. The siRNA transfections for primary BMDMs were performed using a Mouse Macrophage Nucleofector™ Kit (Lonza, Basel, Switzerland) and Nucleofector™ 2b Device (Lonza) with prewritten program Y-001 for BMDMs following the manufacturer’s instructions. RNA and protein from transfected primary cells were harvested 24 hours after the transfections.
Isolation of human CD14+ monocyte-derived macrophages
Leukoreduction chambers from normal donors were obtained from the BJH Pheresis Center (Washington University). Human peripheral blood mononuclear cells (PBMCs) were isolated using Dr. Fehniger’s laboratory protocol (Washington University). Briefly, chamber eluate was mixed well with PBS containing 1 unit/mL heparin, centrifuged with brakes-off, and the “buffy layer” was isolated. The tube was centrifuged at 1,800rpm for 10min and pellets were incubated in 1× RBC lysis buffer. After subsequent centrifugation at 1,300rpm for 4min, PBMC pellets were resuspended in RPMI media. Human CD14+ monocytes were sorted from PBMCs using an EasySep Human CD14 Selection Kit (Stemcell Technologies, Vancouver, Canada) according to the manufacturer’s instructions. CD14+ monocyte isolation purity of > 90% was confirmed by flow cytometry. CD14+ monocytes were cultured in RPMI medium containing 10% FBS, penicillin/streptomycin (Gibco, Gaithersburg, MD, USA) and 50 ng/mL macrophage colony–stimulating factor (M-CSF; PeproTech, Waltham, MA, USA) on 10 μg/mL fibronectin-coated plates (Sigma-Aldrich). After 7 days in culture, adherent macrophages were used for different experiments.
Separation of TCM
A total of 10 mL TCM was filled into the top tube of protein concentrators (3,000 molecular weight cut-off; Thermo Fisher Scientific) and centrifuged at 4,600 × g for 1 hour at 4°C until 80%–90% of the solution from the top tube was in the bottom tube. Concentrated proteins (> 3,000) were in the top tube, and the metabolites (< 3,000) were in the bottom tube. The different fractions of the TCM were diluted to their original volume with media, then used to treat BMDMs.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
All statistical analyses were performed using Prism software (GraphPad, San Diego, CA, USA), with input from a Biostatistics Core expert at Washington University. All data are representative of at least two independent experiments, unless specifically noted. Sample size was precalculated to satisfy power requirements (with > 85% confidence) in most experiments and is specified in the figure legends wherever applicable. To accomplish randomization for orthotopic or syngeneic tumor experiments, animals were sorted by a blinded investigator, with tumor sizes in ascending order and then the groups were assigned in descending order. Each group was checked post hoc to verify that there was no statistical difference in average starting tumor size. Data are shown as the mean ± SEM, unless otherwise noted. Statistical tests such as the unpaired parametric Student’s t-test, analysis of variance (Bonferroni multiple comparisons), one sample t-test, and Wilcoxon test were used based on the normality of data. For survival analyses, the log-rank (Mantel-Cox) test was used. A value of p < 0.05 was considered statistically significant for all studies; n.s denotes not significant.
Supplementary Material
Differentially expressed gene list in TAM/Mo GB1275 vs Vehicle, Related to Figure 3. Differential gene expression analysis results in whole TAMs/monocytes population after GB1275 treatment, comparing with vehicle.
Protein changes after GB1275 treatment by RPPA, Related to Figure 4. Relative protein expression in BMDMs treated with vehicle or GB1275 ± TCM for 4 and 7 hours.
Oligonucleotides, related to STAR Methods.
Highlights.
CD11b agonists activates STING/STAT1 and represses of NFκB in TAMs.
STING/pSTAT1 activation in TAMs enhances anti-tumor T cell immunity.
GB1275 increases pSTAT1 and STING in TAMs in patient tumor tissues.
Tumor cell death and innate immune agonists synergize with CD11b-agonists.
Acknowledgments
DGD was supported by NCI R01CA273190 R01CA177670, R01CA203890, P30CA09184215, P50CA196510, WUSM CCSG grant and the BJC Cancer Frontier Fund. JKS was supported by S10OD020136-01. The author would like to thank Gossamer Bio for providing reagents and clinical samples.
Inclusion and Diversity statement
We support inclusive, diverse and equitable conduct of research.
Footnotes
Declaration of interests
DGD is on the scientific advisory board for Adhaere, 149.Bio and Gossamer Bio. R.E.O, A.V.G, J.M. and L.C. are employees from Gossamer Bio.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DeNardo DG, and Ruffell B (2019). Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 19, 369–382. 10.1038/s41577-019-0127-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biswas SK, and Mantovani A (2010). Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11, 889–896. 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 3.Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, and Friess H (2012). The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol 9, 454–467. 10.1038/nrgastro.2012.115. [DOI] [PubMed] [Google Scholar]
- 4.Ruffell B, and Coussens LM (2015). Macrophages and therapeutic resistance in cancer. Cancer Cell 27, 462–472. 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmid MC, and Varner JA (2012). Myeloid cells in tumor inflammation. Vasc Cell 4, 14. 10.1186/2045-824X-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu H, Jiao F, Han T, and Wang LW (2015). Functional significance of macrophages in pancreatic cancer biology. Tumour Biol 36, 9119–9126. 10.1007/s13277-015-4127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biswas SK, and Mantovani A (2010). Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature Immunology 11, 889–896. 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 8.Lecoultre M, Dutoit V, and Walker PR (2020). Phagocytic function of tumor-associated macrophages as a key determinant of tumor progression control: a review. J Immunother Cancer 8. ARTN e001408 10.1136/jitc-2020-001408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez FO, Gordon S, Locati M, and Mantovani A (2006). Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177, 7303–7311. 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 10.Gordon S, and Taylor PR (2005). Monocyte and macrophage heterogeneity. Nat Rev Immunol 5, 953–964. 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 11.Gordon S, and Martinez FO (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604. 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 12.Kurahara H, Shinchi H, Mataki Y, Maemura K, Noma H, Kubo F, Sakoda M, Ueno S, Natsugoe S, and Takao S (2011). Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res 167, e211–219. 10.1016/j.jss.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 13.Yoshikawa K, Mitsunaga S, Kinoshita T, Konishi M, Takahashi S, Gotohda N, Kato Y, Aizawa M, and Ochiai A (2012). Impact of tumor-associated macrophages on invasive ductal carcinoma of the pancreas head. Cancer Sci 103, 2012–2020. 10.1111/j.1349-7006.2012.02411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Y, Hawkins WG, and DeNardo DG (2015). Regramming myeloid responses to improve cancer immunotherapy. Oncoimmunology 4, e974399. 10.4161/2162402X.2014.974399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE, et al. (2018). Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 67, 1112–1123. 10.1136/gutjnl-2017-313738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. (2013). Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73, 1128–1141. 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE, et al. (2018). Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 67, 1112-+. 10.1136/gutjnl-2017-313738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, Greenberg PD, and Hingorani SR (2014). Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 63, 1769–1781. 10.1136/gutjnl-2013-306271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, Jacobs RC, Ye J, Patel AA, Gillanders WE, et al. (2017). Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 10.1136/gutjnl-2017-313738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmid MC, Khan SQ, Kaneda MM, Pathria P, Shepard R, Louis TL, Anand S, Woo G, Leem C, Faridi MH, et al. (2018). Integrin CD11b activation drives anti-tumor innate immunity. Nat Commun 9, 5379. 10.1038/s41467-018-07387-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, Breden MA, Li X, Krisnawan VE, Khan SQ, et al. (2019). Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Transl Med 11. 10.1126/scitranslmed.aau9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao QY, Lu L, Du GW, Wang ZH, Li XT, and Ju F (2020). 15-hydroxy-6 alpha,12-epoxy-7 beta,10 alpha H,11 beta H-spiroax-4-ene-12-one sensitizes rectal tumor cells to anti-PD1 treatment through agonism of CD11b. Immunopharm Immunot 42, 358–365. 10.1080/08923973.2020.1778722. [DOI] [PubMed] [Google Scholar]
- 23.Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, et al. (2017). Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 47, 323–338 e326. 10.1016/j.immuni.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bloom MJ, Saksena SD, Swain GP, Behar MS, Yankeelov TE, and Sorace AG (2019). The effects of IKK-beta inhibition on early NF-kappa-B activation and transcription of downstream genes. Cell Signal 55, 17–25. 10.1016/j.cellsig.2018.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davoodi H, Hashemi SR, and Seow HF (2012). Increased NFk-B activity in HCT116 colorectal cancer cell line harboring TLR4 Asp299Gly variant. Iran J Allergy Asthma Immunol 11, 121–132. 011.02/ijaai.121132. [PubMed] [Google Scholar]
- 26.Hu X, Zhang H, Zhuang L, Jin G, Yang Q, Li M, Sun W, and Chen F (2020). Ubiquitin-Fold Modifier-1 Participates in the Diabetic Inflammatory Response by Regulating NF-kappaB p65 Nuclear Translocation and the Ubiquitination and Degradation of IkappaBalpha. Drug Des Devel Ther 14, 795–810. 10.2147/DDDT.S238695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pereira SG, and Oakley F (2008). Nuclear factor-kappaB1: regulation and function. Int J Biochem Cell Biol 40, 1425–1430. 10.1016/j.biocel.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Moriyama-Gonda N, Igawa M, Shiina H, Urakami S, Shigeno K, and Terashima M (2002). Modulation of heat-induced cell death in PC-3 prostate cancer cells by the antioxidant inhibitor diethyldithiocarbamate. BJU Int 90, 317–325. 10.1046/j.1464-410x.2002.02810.x. [DOI] [PubMed] [Google Scholar]
- 29.Doganlar O, and Doganlar ZB (2015). Responses of Antioxidant Enzymes and Heat Shock Proteins in Drosophila to Treatment with a Pesticide Mixture. Arch Biol Sci 67, 869–876. 10.2298/Abs141031046d. [DOI] [Google Scholar]
- 30.Dai L, Xie Y, Zhang W, Zhong X, Wang M, Jiang H, He Z, Liu X, Zeng H, and Wang H (2021). Weighted Gene Co-Expression Network Analysis Identifies ANGPTL4 as a Key Regulator in Diabetic Cardiomyopathy via FAK/SIRT3/ROS Pathway in Cardiomyocyte. Front Endocrinol (Lausanne) 12, 705154. 10.3389/fendo.2021.705154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riley JS, Quarato G, Cloix C, Lopez J, O'Prey J, Pearson M, Chapman J, Sesaki H, Carlin LM, Passos JF, et al. (2018). Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J 37. 10.15252/embj.201899238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu M, Shi Y, Wei X, Yang Y, Zhou Y, Hao X, Zhang N, and Niu R (2007). Depletion of mitochondrial DNA by ethidium bromide treatment inhibits the proliferation and tumorigenesis of T47D human breast cancer cells. Toxicol Lett 170, 83–93. 10.1016/j.toxlet.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 33.Klaas L, Vier J, Gentle IE, Hacker G, and Kirschnek S (2021). Diversity of cell death signaling pathways in macrophages upon infection with modified vaccinia virus Ankara (MVA). Cell Death Dis 12. ARTN 1011 10.1038/s41419-021-04286-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murthy AMV, Robinson N, and Kumar S (2020). Crosstalk between cGAS-STING signaling and cell death. Cell Death Differ 27, 2989–3003. 10.1038/s41418-020-00624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park H, Bendell JC, Messersmith WA, Rasco DW, Bono JSD, Strickler JH, Zhou L, Carter LL, Bruey J-M, Li J, et al. (2021). Preliminary clinical and biologic results of GB1275, a first-in-class oral CD11b modulator, alone and with pembrolizumab, in advanced solid tumors (KEYNOTE A36). Journal of Clinical Oncology 39, 2505–2505. 10.1200/JCO.2021.39.15_suppl.2505. [DOI] [Google Scholar]
- 36.Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D'Silva DB, Moghaddas F, Tailler M, Lawlor KE, Zhan Y, et al. (2020). TBK1 and IKKepsilon Act Redundantly to Mediate STING-Induced NF-kappaB Responses in Myeloid Cells. Cell Rep 31, 107492. 10.1016/j.celrep.2020.03.056. [DOI] [PubMed] [Google Scholar]
- 37.Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jonsson KL, Jakobsen MR, Nevels MM, Bowie AG, and Unterholzner L (2018). Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-kappaB Signaling after Nuclear DNA Damage. Mol Cell 71, 745–760 e745. 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balachandran VP, Beatty GL, and Dougan SK (2019). Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 156, 2056–2072. 10.1053/j.gastro.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burdette DL, and Vance RE (2013). STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 14, 19–26. 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 40.Kwon J, and Bakhoum SF (2020). The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 10, 26–39. 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang M, Chen P, Wang L, Li W, Chen B, Liu Y, Wang H, Zhao S, Ye L, He Y, and Zhou C (2020). cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol 13, 81. 10.1186/s13045-020-00916-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan YS, Sansanaphongpricha K, Xie Y, Donnelly CR, Luo X, Heath BR, Zhao X, Bellile E, Hu H, Chen H, et al. (2018). Mitigating SOX2-potentiated Immune Escape of Head and Neck Squamous Cell Carcinoma with a STING-inducing Nanosatellite Vaccine. Clin Cancer Res 24, 4242–4255. 10.1158/1078-0432.CCR-17-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. (2018). PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep 25, 2972–2980 e2975. 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuertes MB, Woo SR, Burnett B, Fu YX, and Gajewski TF (2013). Type I interferon response and innate immune sensing of cancer. Trends Immunol 34, 67–73. 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, Jacob L, Patwa R, Shah H, Xu K, et al. (2016). Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498. 10.1038/nature18268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, Munn D, and Mellor AL (2016). STING Promotes the Growth of Tumors Characterized by Low Antigenicity via IDO Activation. Cancer Res 76, 2076–2081. 10.1158/0008-5472.CAN-15-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahn J, Xia T, Konno H, Konno K, Ruiz P, and Barber GN (2014). Inflammation-driven carcinogenesis is mediated through STING. Nat Commun 5, 5166. 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tanaka Y, and Chen ZJ (2012). STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5, ra20. 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abe T, and Barber GN (2014). Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol 88, 5328–5341. 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khoo LT, and Chen LY (2018). Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep 19. 10.15252/embr.201846935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. (2015). Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11, 1018–1030. 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan HY, Wang N, Man K, Tsao SW, Che CM, and Feng Y (2015). Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis 6. ARTN e1942 10.1038/cddis.2015.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu M, O'Connor RS, Trefely S, Graham K, Snyder NW, and Beatty GL (2019). Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47-mediated 'don’t-eat-me' signal. Nat Immunol 20, 265–275. 10.1038/s41590-018-0292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jaynes JM, Sable R, Ronzetti M, Bautista W, Knotts Z, Abisoye-Ogunniyan A, Li D, Calvo R, Dashnyam M, Singh A, et al. (2020). Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci Transl Med 12. 10.1126/scitranslmed.aax6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun WJ, Huhn RD, Song WR, Li DG, Sharp LL, et al. (2011). CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 331, 1612–1616. 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng T, Wang M, Chen L, Guo Y, Chen Z, and Wu J (2018). Increased expression of CD40/TRAF1 and activation of nuclear factor-kappakappaB-dependent proinflammatory gene expression in collagen-induced arthritis. Scand J Rheumatol 47, 455–460. 10.1080/03009742.2018.1432684. [DOI] [PubMed] [Google Scholar]
- 57.Dougan M, Li D, Neuberg D, Mihm M, Googe P, Wong KK, and Dranoff G (2011). A dual role for the immune response in a mouse model of inflammation-associated lung cancer. J Clin Invest 121, 2436–2446. 10.1172/JCI44796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wente MN, Mayer C, Gaida MM, Michalski CW, Giese T, Bergmann F, Giese NA, Buchler MW, and Friess H (2008). CXCL14 expression and potential function in pancreatic cancer. Cancer Lett 259, 209–217. 10.1016/j.canlet.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 59.Pramanik KC, Makena MR, Bhowmick K, and Pandey MK (2018). Advancement of NF-kappaB Signaling Pathway: A Novel Target in Pancreatic Cancer. Int J Mol Sci 19. 10.3390/ijms19123890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, Kumar V, Armstrong AR, Carmona-Rivera C, Dorschner JM, et al. (2017). CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J Clin Invest 127, 1271–1283. 10.1172/JCI88442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. (2016). Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22, 851–860. 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim MP, Evans DB, Wang H, Abbruzzese JL, Fleming JB, and Gallick GE (2009). Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 4, 1670–1680. 10.1038/nprot.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borcherding N, Vishwakarma A, Voigt AP, Bellizzi A, Kaplan J, Nepple K, Salem AK, Jenkins RW, Zakharia Y, and Zhang W (2021). Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun Biol 4, 122. 10.1038/s42003-020-01625-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259. 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, Chiang EY, Iftikhar H, O'Gorman WE, Au-Yeung A, et al. (2020). Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 579, 274–278. 10.1038/s41586-020-2056-8. [DOI] [PubMed] [Google Scholar]
- 66.Zheng Y, Chen Z, Han Y, Han L, Zou X, Zhou B, Hu R, Hao J, Bai S, Xiao H, et al. (2020). Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment. Nat Commun 11, 6268. 10.1038/s41467-020-20019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, Chang C, Walker L, Lansky A, Le Berichel J, et al. (2021). Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 39, 1594–1609 e1512. 10.1016/j.ccell.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Steele NG, Carpenter ES, Kemp SB, Sirihorachai V, The S, Delrosario L, Lazarus J, Amir ED, Gunchick V, Espinoza C, et al. (2020). Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat Cancer 1, 1097–1112. 10.1038/s43018-020-00121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, Thennavan A, Wang C, Torpy JR, Bartonicek N, et al. (2021). A single-cell and spatially resolved atlas of human breast cancers. Nat Genet 53, 1334–1347. 10.1038/s41588-021-00911-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laumont CM, Wouters MCA, Smazynski J, Gierc NS, Chavez EA, Chong LC, Thornton S, Milne K, Webb JR, Steidl C, and Nelson BH (2021). Single-cell Profiles and Prognostic Impact of Tumor-Infiltrating Lymphocytes Coexpressing CD39, CD103, and PD-1 in Ovarian Cancer. Clin Cancer Res 27, 4089–4100. 10.1158/1078-0432.CCR-20-4394. [DOI] [PubMed] [Google Scholar]
- 71.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andreatta M, Berenstein AJ, and Carmona SJ (2022). scGate: marker-based purification of cell types from heterogeneous single-cell RNA-seq datasets. Bioinformatics. 10.1093/bioinformatics/btac141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andreatta M, and Carmona SJ (2021). UCell: Robust and scalable single-cell gene signature scoring. Comput Struct Biotechnol J 19, 3796–3798. 10.1016/j.csbj.2021.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lander VE, Belle JI, Kingston NL, Herndon JM, Hogg GD, Liu X, Kang LI, Knolhoff BL, Bogner SJ, Baer JM, et al. (2022). Stromal Reprogramming by FAK Inhibition Overcomes Radiation Resistance to Allow for Immune Priming and Response to Checkpoint Blockade. Cancer Discov 12, 2774–2799. 10.1158/2159-8290.CD-22-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zuo C, Baer JM, Knolhoff BL, Belle JI, Liu X, Alarcon De La Lastra A, Fu C, Hogg GD, Kingston NL, Breden MA, et al. (2023). Stromal and therapy-induced macrophage proliferation promotes PDAC progression and susceptibility to innate immunotherapy. J Exp Med 220. 10.1084/jem.20212062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, and DeNardo DG (2014). CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 74, 5057–5069. 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bronner DN, and O'Riordan MX (2016). Measurement of Mitochondrial DNA Release in Response to ER Stress. Bio Protoc 6. 10.21769/BioProtoc.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differentially expressed gene list in TAM/Mo GB1275 vs Vehicle, Related to Figure 3. Differential gene expression analysis results in whole TAMs/monocytes population after GB1275 treatment, comparing with vehicle.
Protein changes after GB1275 treatment by RPPA, Related to Figure 4. Relative protein expression in BMDMs treated with vehicle or GB1275 ± TCM for 4 and 7 hours.
Oligonucleotides, related to STAR Methods.
Data Availability Statement
The scRNA sequencing data from PDAC lesions were found at the Gene Expression Omnibus Repository (GEO) accession number GSE220959. All software packages used are publicly available through commercial vendors.