


Keywords: distal tubule K+ transport, flow sensitivity, hyperkalemia, hypokalemia, kaliuresis
Abstract
The Ca2+-permeable transient receptor potential vanilloid type 4 (TRPV4) channel serves as the sensor of tubular flow, thus being well suited to govern mechanosensitive K+ transport in the distal renal tubule. Here, we directly tested whether the TRPV4 function is significant in affecting K+ balance. We used balance metabolic cage experiments and systemic measurements with different K+ feeding regimens [high (5% K+), regular (0.9% K+), and low (<0.01% K+)] in newly created transgenic mice with selective TRPV4 deletion in the renal tubule (TRPV4fl/fl-Pax8Cre) and their littermate controls (TRPV4fl/fl). Deletion was verified by the absence of TRPV4 protein expression and lack of TRPV4-dependent Ca2+ influx. There were no differences in plasma electrolytes, urinary volume, and K+ levels at baseline. In contrast, plasma K+ levels were significantly elevated in TRPV4fl/fl-Pax8Cre mice on high K+ intake. K+-loaded knockout mice exhibited lower urinary K+ levels than TRPV4fl/fl mice, which was accompanied by higher aldosterone levels by day 7. Moreover, TRPV4fl/fl-Pax8Cre mice had more efficient renal K+ conservation and higher plasma K+ levels in the state of dietary K+ deficiency. H+-K+-ATPase levels were significantly increased in TRPV4fl/fl-Pax8Cre mice on a regular diet and especially on a low-K+ diet, pointing to augmented K+ reabsorption in the collecting duct. Consistently, we found a significantly faster intracellular pH recovery after intracellular acidification, as an index of H+-K+-ATPase activity, in split-opened collecting ducts from TRPV4fl/fl-Pax8Cre mice. In summary, our results demonstrate an indispensable prokaliuretic role of TRPV4 in the renal tubule in controlling K+ balance and urinary K+ excretion during variations in dietary K+ intake.
NEW & NOTEWORTHY The mechanoactivated transient receptor potential vanilloid type 4 (TRPV4) channel is expressed in distal tubule segments, where it controls flow-dependent K+ transport. Global TRPV4 deficiency causes impaired adaptation to variations in dietary K+ intake. Here, we demonstrate that renal tubule-specific TRPV4 deletion is sufficient to recapitulate the phenotype by causing antikaliuresis and higher plasma K+ levels in both states of K+ load and deficiency.
INTRODUCTION
K+ is the major intracellular cation with only a minor fraction (∼1–2%) of it being present in the extracellular compartments. Such uneven distribution necessitates a tight control of plasma K+ levels following food intake since the ratio of intracellular to extracellular K+ concentration is the major determinant of the plasma membrane potential and, thus, the electrical activity of both excitable cells (neurons, cardiomyocytes, skeletal muscle cells, and smooth muscle cells) and nonexcitable cells (epithelial and endothelial cells) (1, 2). Indeed, even small deviations from normal plasma K+ concentration (typically 4.5 mM) are associated with an increased risk of cardiovascular morbidity and mortality in various cohorts of patients (3).
Kidneys play a critical role in maintaining systemic K+ balance by excreting ∼90% of dietary K+ load (1, 2, 4). Likewise, up to 99% of filtered K+ is reabsorbed by the renal tubule during fasting periods/low dietary K+ intake to maintain total body K+ content (4). The aldosterone-sensitive distal nephron, and primarily the collecting duct, is the chief site for controlling K+ transport to match dietary K+ intake with urinary K+ excretion (2). Under unstressed conditions, the most abundant principal cells use electrogenic Na+ reabsorption via the epithelial Na+ channel (ENaC) to secrete K+ into the lumen via renal outer medullary K+ (ROMK) channels (5). Upon dietary K+ load, K+ secretion can be drastically augmented by the flow-dependent activation of large-conductance K+ (BK) channels in both principal and intercalated cells, so-called flow-induced K+ secretion (FIKS), with disruption of this mechanism leading to hyperkalemia (6–9). On the other side of the spectrum, reabsorption via H+-K+-ATPase (α1- and α2-isoforms) on the apical membrane in intercalated cells and, to a lesser extent, principal cells function in the condition of K+ deficiency (10, 11). Consistently, mice with H+-K+-ATPase deletion exhibit augmented urinary K+ excretion leading to a distorted systemic K+ balance (12).
The transient receptor potential vanilloid type 4 (TRPV4) channel is a mechanosensitive Ca2+-permeable channel that is activated by increased fluid flow and hypotonicity in a variety of cells endogenously expressing the channel (13–16). TRPV4 is broadly expressed in different tissues and organs, including the central and peripheral nervous system, eye, endothelium, and different epithelia such as the skin, alveoli, trachea, bile ducts, pancreas, and sweat glands with the kidney having the highest levels of the channel (17–24). Here, TRPV4 functional activity was found mainly in the proximal tubule and aldosterone-sensitive distal nephron (25, 26). The accumulated evidence unequivocally suggests that TRPV4 mediates shear stress-induced Ca2+ influx, thus being a flow and pressure sensor in these segments of the renal tubule (25, 27–30).
Taking into consideration the flow-dependent nature of K+ secretion in the collecting duct (9, 31) and increased ultrafiltrate delivery to the distal nephron during both dietary K+ load and deficiency (32, 33), it is possible that mechanosensitive TRPV4 is perfectly positioned to govern K+ transport at this site. Accordingly, we have previously demonstrated that mice with global TRPV4 deletion develop hyperkalemia when fed a high-K+ diet exhibiting lower urinary K+ levels and reduced BK channel activity in split-opened collecting ducts (28). We also observed that renal K+ conservation is facilitated under these conditions, with augmented H+-K+-ATPase activity in knockout mice during systemic K+ depletion (34).
In the present study, we investigated mice with targeted deletion of TRPV4 specifically in the renal tubule to determine whether TRPV4 expression in the kidney controls urinary K+ excretion during varying K+ intake and to test whether tubule-specific expression of the channel is required for the modulation of systemic K+ balance. Previous studies have shown that conditional ablation of TRPV4 eliminates mechanical and temperature sensitivity in a glial cell type that plays a central role in retinal K+ homeostasis and secretion (35). Here, we found that deletion of TRPV4 in the renal tubule is sufficient to distort adaptation to both high and low K+ intake by reducing urinary K+ levels and subsequently leading to significantly higher plasma K+ levels under these conditions.
METHODS
Reagents
All chemicals and materials were from Sigma (St. Louis, MO), VWR (Radnor, PA), Fisher (Waltham, MA), and Tocris (Ellisville, MO) unless noted otherwise and were at least of reagent grade.
Research Animals
Animal use and welfare adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals following protocols reviewed and approved by the Animal Care and Use Committee of the University of Texas Health Science Center (Houston, TX).
Transgenic mice with loxP sites surrounding exon 6 encoding the first transmembrane domain of TRPV4 were generated from embryonic stem cell clones (KOMP Project CSD79331) using standard Cre-LoxP technology. Mice were crossed with ROSA-Flp (Stock No. 012930, The Jackson Laboratory) to excise the selection cassette. The genotyping primers for ROSA-Flp were as follows: 5′- CGAAGAGTTTGTCCTCAACCG-3′ and 5′- CTGGCTTCTGAGGACCG-3′ (for detection of the ROSA Flp allele at 195 bp), and the genotyping primers for ROSA wild-type (WT) were as follows: 5′- AATCTGTGGGAAGTCTTGTCC-3′ and 5′- CTGGCTTCTGAGGACCG-3′ (for detection of the ROSA WT allele at 193 bp). To generate renal tubule-specific TRPV4 knockout, mice homozygous for the TRPV4 loxP allele (TRPV4fl/fl) were further crossed with Pax8tm1.1(cre)Mbu mice (Stock No. 028196, The Jackson Laboratory). The genotyping primers were as follows: 5′- TAAGAGAGAGTACACTGGAATGTCCTAGC-3′ and 5′- TATACGAAGTTATACGTGTCGACAACG-3′ (for detection of the TRPV4 floxed and WT alleles at 666 and 527 bp, respectively) and 5′- GAGATGGCGCAACGCAATTAATG-3′ and 5′- ACACAAGACAACACACAACAACCCC-3′ (for detection of the control allele at 291 bp). The genotyping primers for Pax8-cre were as follows: 5′- AGCTGGCCCAAATGTTGCTGG-3′ and 5′- CCCTCCTAGTTGATTCAGCCC-3′ to detect a product at 673 bp. Cycling parameters for PCR were as follows: first segment, 94°C, 5 min (1 cycle); second segment, 94°C, 15 s; third segment, 65– 55 (↓1°C/cycle)°C, 30 s; fourth segment, 72°C, 40 s (40 cycles); and fifth segment: 72°C, 5 min (1 cycle). The representative PCR analysis is shown in Fig. 1A.
Figure 1.
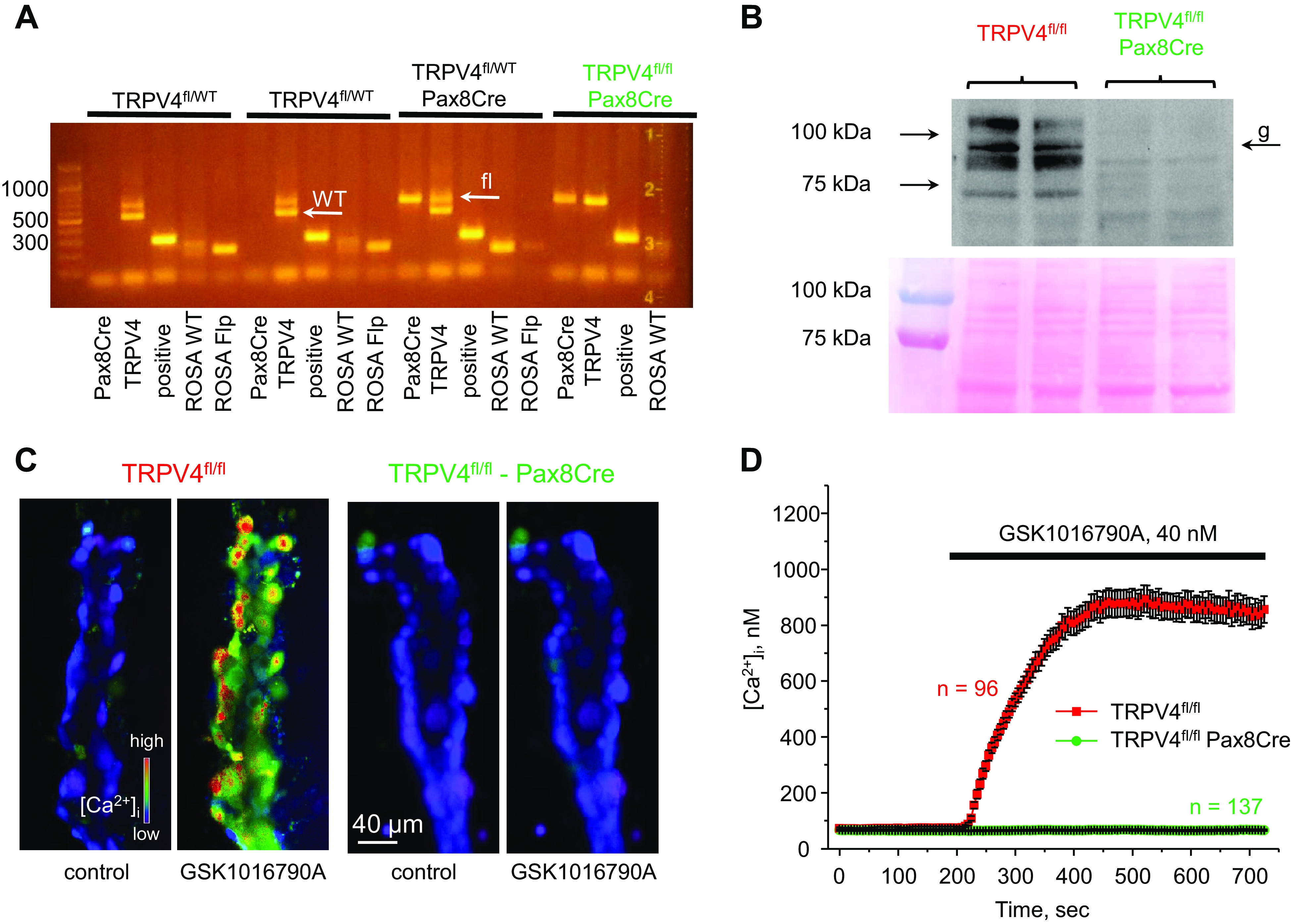
Verification of successful transient receptor potential vanilloid type 4 (TRPV4) deletion in the renal tubule. A: representative PCR genotype analysis of transgenic littermates probed with primers for Pax8Cre, TRPV4, and ROSA Flp, as described in methods. The desired genotype (TRPV4fl/fl-Pax8Cre, highlighted in green) contained positive reaction for Pax8, the presence of an upper loxP (fl) allele, and the absence of a lower wild-type (WT) allele for TRPV4, as indicated by white arrows. B, top: representative Western blot probed with anti-TRPV4 antibodies from whole kidney lysates of TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice (1 female and 1 male) showing the complete absence of a TRPV4-reporting signal. TRPV4 appeared as a duplet of bands around 95 kDa with the higher band representing a heavy mannitol sugar glycosylation form (shown as “g”). Each line represents an individual mouse. B, bottom: Ponceau red staining of the same nitrocellulose membrane demonstrating equal protein loading. C: representative pseudocolor images of intracellular Ca2+ concentration ([Ca2+]i) changes (blue: low and red: high) in isolated split-opened collecting ducts loaded with the Ca2+-sensitive dye Fura2 at baseline (left) and following 5-min application of 40 nM of the TRPV4 agonist GSK1016790A (right) from TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice. D: averaged time courses of [Ca2+]i changes upon application of 40 nM GSK1016790A (shown by the horizontal bar) in individual collecting duct cells within split-opened area from TRPV4fl/fl (red) and TRPV4fl/fl-Pax8Cre (green) mice. Each time point is shown as means ± SE. Numbers of individual cells are shown. Data were obtained from four different collecting ducts isolated from three different mice (2 females and 1 male) for each group.
Systemic Measurements
Three-month-old littermate TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice (male and female in equal proportions when possible) were acclimated for 3 days in metabolic cages (3600M021, Techniplast, West Chester, PA) with free access to water and regular rodent chow (0.9% K+, 0.3% Na+, and 0.5% Cl−, TD.7012 Envigo, Madison, WI). Following acclimation, 24-h urine samples were collected and assessed. Mice were fed a K+-deficient diet (<0.01% K+, 0.3% Na+, and 0.45% Cl−, TD.88239, Envigo) or a high-K+ diet (5% K+, 0.3% Na+, and 5% Cl−, TD.150699, Envigo) for 2 consecutive days to fit a Monday-to-Friday schedule. There were comparable ∼10% decreases in total body weight on the high-K+ diet (Supplemental Fig. S1A) and no changes in body weight on the K+-deficient diet (Supplemental Fig. S1B) in TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice, suggesting a similar food intake for both strains. To assess long-term effects, conventionally housed mice were fed with the respective diet for 4 days and placed into metabolic cages for 2 days, and the actual measurements were performed on day 7. Urinary K+ concentration was measured using a Jenway PFP7 Flame photometer (Bibby Scientific, Burlington, NJ), and the reported values were normalized to animal body weight. There were no apparent differences in urinary K+ excretion upon manipulations with dietary K+ intake between male and female in TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice, as provided in Supplemental Fig. S2, A and B, respectively. To measure plasma K+, ∼20 µL blood was collected in conscious animals by bleeding into heparinized glass capillaries (Cat. No. 22–362-566, Fisher). Sealed capillaries were centrifuged at 1,300 g for 5 min to separate plasma. As shown in Supplemental Fig. S3, there was no hemolysis in all collected samples from TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice. To monitor acid-base status, blood samples (∼500 μL) were taken by terminal cardiac puncture in anesthetized animals with samples being immediately processed with an OPTI CCA-TS 2 blood gas and electrolyte analyzer (OPTI Medical Systems, Roswell, GA) supplied with E-Lyte CCA cassettes.
Western Blot Analysis
Immediately after dissection, kidneys were placed on ice, decapsulated, and homogenized in three volumes of ice-cold lysis buffer containing 50 mM Tris·Cl, 5 mM EDTA, and 1% Triton X-100 (pH 7.5) supplemented with Complete Mini protease and PhosSTOP phosphatase inhibitor cocktails (Roche Diagnostics, Indianapolis, IN). Homogenates were centrifuged at 1,000 g for 15 min at 4°C, and the sediment was discarded. Protein concentration was determined with a Bradford assay using BSA as the standard. Samples (40 μg/lane) were separated on 9% polyacrylamide gels at 150 V for 90 min and transferred to a nitrocellulose membrane for 1.5 h at 100 V. Nitrocellulose membranes were incubated with either primary anti-TRPV4 antibodies (rabbit polyclonal, 1:1,000, Cat. No. ACC-034, Alomone Labs, Jerusalem, Israel), anti-αENaC antibodies (rabbit polyclonal, 1:1,000, Cat. No. SPC-403, Stress Marq Biosciences), anti-αBK or KCa1.1 antibodies (rabbit polyclonal, 1:500, Cat. No. APC-021, Alomone Labs), or anti-H+-K+-ATPase β-subunit antibodies (rabbit polyclonal, 1:500, Cat. No. ab176992, Abcam) overnight at 4°C. Upon washout (3 times for 10 min in Tris-buffered saline-Tween), the membrane was incubated with peroxidase-conjugated goat anti-rabbit (1:10,000, Cat. No. NC9448271, Jackson ImmunoResearch) secondary antibodies for 1 h at room temperature. Ponceau red staining was used to verify equal protein load in different samples.
Isolation and Split Opening of Cortical Collecting Ducts
The procedure for the isolation of cortical collecting ducts from mouse kidneys suitable for fluorescent intracellular Ca2+ concentration ([Ca2+]i) and intracellular pH (pHi) measurements closely followed the protocols previously published by our group (34, 36). Kidneys were cut into thin slices (<1 mm) with slices placed into ice-cold bath solution containing (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.35). Cortical collecting ducts were visually identified by their morphological features (pale color and coarse surface) and by their postexperimental staining with anti-AQP2. Collecting ducts were mechanically isolated from kidney slices by microdissection using watchmaker forceps under a stereomicroscope. Isolated collecting ducts were attached to 5 × 5 mm cover glasses coated with poly-l-lysine. A coverglass containing a collecting duct was placed in a perfusion chamber mounted on an inverted Nikon Eclipse Ti-S microscope and perfused with bath solution at room temperature. Cortical collecting ducts were further split opened with two sharpened micropipettes, controlled with different micromanipulators, to reliably monitor [Ca2+]i and pHi signals from individual cells within a monolayer. Collecting ducts were used within 2 h of isolation.
Intracellular Ca2+ and pH Measurements
Split-opened cortical collecting ducts were loaded with either Fura-2 by incubation with 2 μM Fura-2/AM or 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF) by incubation with 15 μM BCECF-AM in the bath solution for 40 min at room temperature followed by a washout with the bath solution for an additional 10 min. Collecting ducts were placed in an open-top imaging study chamber (RC-26GLP, Warner Instruments, Hamden, CT) with a bottom coverslip viewing window, and the chamber was attached to the microscope stage of a Nikon Ti-S Wide-Field Fluorescence Imaging System (Nikon Instruments, Melville, NY) integrated with a Lambda XL light source (Sutter Instrument, Novato, CA) and QIClick 1.4 megapixel monochrome charge-coupled device camera (QImaging, Surrey, BC, Canada) via NIS Elements 4.3 Imaging Software (Nikon Instruments). Cells were imaged with a ×40 Nikon Super Fluor objective, and regions of interest were drawn for individual cells.
For [Ca2+]i measurements, the Fura-2 fluorescence intensity ratio was determined by excitation at 340 and 380 nm and calculating the ratio of the emission intensities at 511 nm in the usual manner every 5 s. Changes in the ratio were converted into changes in intracellular Ca2+, as we have previously described in great detail (37). Similarly, the BCECF fluorescence intensity ratio was determined by excitation at 495 and 440 nm and calculating the ratio of the emission intensities at 520 nm every 5 s. Changes in the ratio were converted into changes in pHi by performing a calibration in high-K+ solutions (145 mM KCl) with predefined pH (6.0, 7.0, and 8.0, adjusted by HCl and KOH, respectively) in the presence of 15 µM nigericin, as was originally described (38). Minor BCECF and Fura-2 bleaching during the timeline of experiments was corrected. Experiments were performed under permanent perfusion of a solution containing (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES at a rate of 1.5 mL/min. On average, six individual collecting ducts (30–50 cells in each) from three mice were used for each experimental set. For intracellular acidification, a solution containing (in mM) 40 NH4Cl, 110 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES was applied to the recording chamber for 2 min. The rate of recovery after acidification was calculated as a linear slope of the initial pHi recovery rate (usually 30 s) from the lowest pHi values for each cell after removal of the acidification pulse, as we have previously described (34, 36, 39). Acid-secreting (A-type) and base-secreting (B-type) intercalated cells were distinguished by their opposite changes in pHi levels (alkalization and acidification) in response to application of the basolateral ClC-K2 Cl− channel blocker 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB; 100 µM), as we have previously described (36).
Immunofluorescent Microscopy (Kidney Sections)
Freshly isolated kidneys were decapsulated and placed into cryotubes with embedding medium (Andwin Scientific Tissue-Tek CRYO-OCT Compound 4583, Cat. No. 14–373-65, Torrance, CA). The kidneys were flash-frozen in liquid nitrogen for 3–5 min. Transverse cut 6-µm thick sections were made on the CM 1850 cryostat (Leica, Buffalo Grove, IL). Sections were allowed to warm to room temperature, washed three times in PBS, and fixed with 10% neutral-buffered formalin for 15 min. After extensive washout, kidney sections were permeabilized with 0.1% Triton X-100 (Cat. No. 56H0850, Sigma Aldrich, St. Louis, MO) for 10 min and treated with 10% normal goat serum for 1 h at room temperature. Sections were incubated overnight at 4°C with rabbit anti-mouse anti-AQP2-ATTO Fluor-550 (1:200, Cat. No. AQP2-002-AO, Alomone Labs) and rabbit anti-mouse anti-TRPV4 (1:500, Cat. No. ACC-034, Alomone Labs) antibodies. After being washed with PBS for 20 min at room temperature, samples were incubated with goat anti-rabbit Alexa 488 F(ab′) secondary antibodies (1:1,000, Cat. No. A1182668, Invitrogen, Eugene, OR). After being washed with PBS for 20 min at room temperature, nuclei were stained with DAPI (0.5 µg/mL) for 10 min at room temperature. Samples were mounted with Fluoromount mounting media (Thermo Fisher Scientific, Pittsburg, PA). Labeled kidney sections were imaged with a Nikon A1R confocal microscope, as we have previously described (36). In brief, samples were excited with 405-, 488-, and/or 561-nm laser diodes, and emission was captured with a 16-bit Cool SNAP HQ2 camera (Photometrics) interfaced to a PC running NIS Elements software.
Immunofluorescent Microscopy (Split-Opened Collecting Ducts)
Following pHi measurements, split-opened collecting ducts were fixed with 10% neutral-buffered formalin (Cat. No. PFNBF240, AzerScientific, Morgantown, PA) for 15 min at room temperature. After fixation, samples were permeabilized by the addition of 1% SDS (Cat. No. L4390, Sigma Aldrich) in PBS for 10 min and washed in PBS two times for 10 min. Nonspecific staining was blocked with 10% normal goat serum (Cat. No. NBP223475, Novus Biologicals, Centennial, CO) in PBS for 1 h at room temperature. Samples were incubated overnight at 4°C in the dark with rabbit anti-mouse anti-AQP2 antibodies (1:4,000, Cat. No. AQP2-002, Alomone Labs), washed with PBS, and incubated with goat anti-rabbit Alexa 594 F(ab′) secondary antibodies (1:1,000, Cat. No. 111–587-003, Jackson ImmunoResearch) for 60 min at room temperature. After being washed with PBS (2 times for 5 min), samples were stained with DAPI (300 nM concentration, Cat. No. 5.08741.0001, MilliporeSigma, Calbiochem, San Diego, CA) to visualize nuclei. Samples were mounted with Fluoromount mounting media (Thermo Fisher Scientific). Samples were imaged with a Nikon A1R confocal microscope using 405- and 561-nm laser diodes, and emission was captured with a 16-bit Cool SNAP HQ2 camera (Photometrics) interfaced to a PC running NIS Elements software. Maximal intensity projections were created from serial confocal images with 0.75 µm increments on the vertical axis.
Data Analysis
All summarized data are reported as means ± SE. Statistical comparisons were made using one-way ANOVA with a post hoc Tukey’s test or one-way repeated-measures ANOVA with a post hoc Bonferroni test. P values of <0.05 were considered significant.
RESULTS
Creation of Mice with Renal Tubule-Specific Deletion of the TRPV4 Channel
We first deleted the TRPV4 channel specifically in the renal tubule to examine potential pathophysiological ramifications on systemic K+ balance. Transgenic TRPV4fl/fl mice with loxP sites around exon 6 encoding the first transmembrane domain were crossed with mice bearing Pax8 Cre recombinase, a commonly accepted specific promoter of epithelial cells of the renal tubule (40). Figure 1A shows representative PCR analyses exemplifying the successful creation of the desired TRPV4fl/fl-Pax8Cre genotype. Western blot assessment demonstrated the complete absence of the TRPV4-reporting signal in whole kidney homogenates of TRPV4fl/fl-Pax8Cre mice (Fig. 1B). Using immunofluorescent microscopy in kidney sections of control TRPV4fl/fl mice, we found that TRPV4 was most apparent in the AQP2-positive collecting duct and connecting tubule segments with a weaker signal being also detected in AQP2-negative, likely proximal, tubule regions (Supplemental Fig. S4), which is consistent with previously reported sites of channel expression (25, 26). In contrast, no specific staining was detected in kidney sections from TRPV4fl/fl-Pax8Cre mice. We next verified the lack of TRPV4 activity in freshly isolated split-opened collecting ducts. Application of a potent and selective TRPV4 agonist, GSK1016790A (40 nM), drastically increased [Ca2+]i in all cells of TRPV4fl/fl mice but failed to affect [Ca2+]i in cells from TRPV4fl/fl-Pax8Cre mice (Fig. 1C). The respective time courses of [Ca2+]i changes in response to GSK1016790A application are shown in Fig. 1D. Overall, our results shown in Fig. 1 and Supplemental Fig. S4 provide compelling evidence of the successful ablation of functional TRPV4 expression in the renal tubule using the Pax8cre promoter.
TRPV4fl/fl-Pax8Cre Mice Exhibit Impaired Adaptation to Dietary K+ Intake
Mice with deleted TRPV4 in the renal tubule (TRPV4fl/fl-Pax8Cre) did not exhibit apparent deficiencies during development, having comparable body weight with respective TRPV4fl/fl littermates: 25.8 ± 0.8 g (n = 13 animals) versus 24.9 ± 0.5 g (n = 14 animals) for 3-mo-old male mice, respectively. Moreover, there were no significant differences in acid-base balance and major plasma electrolytes at baseline (Table 1). We have previously shown that global TRPV4 deletion can lead to notable changes in plasma K+ levels during variations in dietary K+ intake (28). Here, we tested whether selective deletion of TRPV4 in the renal tubule is sufficient to affect plasma K+ levels. As shown in Fig. 2, TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice had comparable plasma K+ levels when fed a regular-K+ diet (0.9% K+): 4.11 ± 0.06 and 3.97 ± 0.05 mM/L, respectively. In contrast, 1-wk treatment with a high-K+ diet (5% K+) had only a minor effect on plasma K+ levels in TRPV4fl/fl mice (4.16 ± 0.08 mM/L) but significantly increased plasma K+ levels in TRPV4fl/fl-Pax8Cre mice to 4.85 ± 0.08 mM/L (Fig. 2). Furthermore, 1-wk treatment with a K+-deficient diet (<0.01% K+) decreased plasma K+ levels in TRPV4fl/fl mice (2.75 ± 0.09 mM/L), whereas only a minor nonsignificant reduction was seen in TRPV4fl/fl-Pax8Cre mice (3.78 ± 0.09 mM/L). The observed effects were similarly observed in both male and female mice. As provided in Supplemental Fig. S3, the samples used for analysis showed a lack of hemolysis. We did not detect notable differences in plasma Na+ and Cl− as well as acid-base status, namely, pH and , between TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice on both high- and low-K+ diets (Table 1). Overall, these results demonstrate that deletion of TRPV4 in the renal tubule leads to increased plasma K+ levels with both high- and low-K+ diets.
Table 1.
Comparison of plasma electrolytes in TRPV4fl/fl TRPV4fl/fl-Pax8Cre mice kept on low-, regular-, and high-K+ diets for 7 days
| <0.01% K+ Diet, TD.88238 |
0.9% K+ Diet, TD.7012 |
5% K+ Diet, TD.150699 |
||||
|---|---|---|---|---|---|---|
| TRPV4fl/fl | TRPV4fl/fl-Pax8Cre | TRPV4fl/fl | TRPV4fl/fl-Pax8Cre | TRPV4fl/fl | TRPV4fl/fl-Pax8Cre | |
| Number of animals tested | 7 (4 females and 3 males) | 5 (3 females and 2 males) | 6 (3 females and 3 males) | 7 (4 females and 3 males) | 7 (3 females and 4 males) | 8 (4 females and 4 males) |
| Plasma Na+, mM | 146.0 ± 1.3 | 143.6 ± 0.6 | 144.9 ± 0.7 | 145.3 ± 0.7 | 145.7 ± 1.4 | 146.3 ± 0.6 |
| Plasma Cl−, mM | 112.0 ± 0.8 | 110.7 ± 1.0 | 110.3 ± 0.9 | 110.7 ± 0.5 | 111.3 ± 0.7 | 112.4 ± 0.6 |
| Plasma , mM | 23.2 ± 0.7 | 24.2 ± 1.0 | 24.3 ± 0.8 | 25.5 ± 0.8 | 24.4 ± 0.9 | 24.3 ± 0.9 |
| Plasma pH | 7.29 ± 0.02 | 7.32 ± 0.02 | 7.33 ± 0.02 | 7.34 ± 0.02 | 7.34 ± 0.03 | 7.29 ± 0.03 |
TRPV4, transient receptor potential vanilloid type 4.
Figure 2.

Mice with renal tubule-selective transient receptor potential vanilloid type 4 (TRPV4) deletion exhibit increased plasma K+ levels on low and high K+ intake. Shown is a summary graph of plasma K+ levels in TRPV4fl/fl (red) and TRPV4fl/fl-Pax8Cre (green) mice kept on low (<0.01% K+), regular (0.9% K+), and high (5% K+) K+ intake for 7 days. Males and females are indicated as triangles and circles, respectively (6 female mice and 6 male mice in each group). *Significant increase (P < 0.05, one-way ANOVA with a post hoc Tukey’s test) vs. respective values of TRPV4fl/fl mice, as indicated by the horizontal bars. Supplemental Fig. S3 provides verification of the lack of hemolysis in the tested samples.
TRPV4 Deletion in the Renal Tubule Causes K+ Retention by the Kidney
We next performed metabolic cage experiments to compare the patterns of urinary excretion of K+ in TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice in response to increases and decreases in K+ intake. There were no significant differences in 24-h urinary K+ levels (Fig. 3A) and urinary volume (Fig. 3B) when both strains were fed a regular-K+ diet (0.9% K+). High-K+ diet induced comparable increases in urinary volume in both strains. Importantly, urinary K+ levels were significantly higher in TRPV4fl/fl mice than those in TRPV4fl/fl-Pax8Cre mice during the first and second days of high-K+ diet (5% K+; Fig. 3A). However, no significant differences in urinary K+ levels were found on day 7, suggesting that a new steady state had been achieved. Although urinary aldosterone levels were similar in control and the first 2 days of high-K+ diet, TRPV4fl/fl-Pax8Cre mice had a significantly higher aldosterone-to-creatinine ratio than TRPV4fl/fl mice on day 7 (Fig. 4), suggesting the development of a compensatory response to hyperkalemia in knockout mice (Fig. 2).
Figure 3.

Mice with transient receptor potential vanilloid type 4 (TRPV4) deletion in the renal tubule demonstrate transient-deficient kaliuresis to dietary K+ load. Summary graphs showing the comparison of 24-h urinary K+ levels (A) and urinary volume (B) in TRPV4fl/fl mice (red, 2 females and 6 males) and TRPV4fl/fl-Pax8Cre mice (green, 4 females and 6 males) maintained on a regular-K+ diet (0.9% K+) and in response to a high-K+ diet (5% K+) for 1, 2, and 7 days. Each dot represents a measurement from an individual animal. Males and females are indicated as triangles and circles, respectively. *Significant changes (P < 0.05, one-way ANOVA with a post hoc Tukey’s test) vs. the respective values of TRPV4fl/fl mice, as indicated by the horizontal bars.
Figure 4.

Chronic dietary K+ load increases urinary aldosterone levels in mice with transient receptor potential vanilloid type 4 (TRPV4) deletion in the renal tubule. Shown is a summary graph of comparison of 24-h urinary aldosterone in TRPV4fl/fl mice (red, 2 females and 3 males) and TRPV4fl/fl-Pax8Cre mice (green, 2 females and 3 males) maintained on a regular-K+ diet (0.9% K+) and in response to a high-K+ diet (5% K+) for 1, 2, and 7 days. Each dot represents a measurement from an individual animal. Males and females are indicated as triangles and circles, respectively. *Significant increase (P < 0.05, one-way ANOVA with a post hoc Tukey’s test) vs. the respective values of TRPV4fl/fl mice, as indicated by the horizontal bars.
Aldosterone is known to increase ENaC activity, thus augmenting a driving force for K+ secretion via ROMK channels as well as stimulating flow-induced K+ secretion via BK channels (2, 9, 41). To determine the underlying mechanism of renal K+ retention in TRPV4fl/fl-Pax8Cre mice, we next monitored expression of αENaC expression with Western blot analysis in control and knockout mice fed a high-K+ diet. The recent detailed characterization of commercially available αENaC antibodies convincingly demonstrated that the aldosterone-induced full-length αENaC band (around 100 kDa) is located just above an intense nonspecific band (42). Indeed, the Western blot from whole kidney homogenates probed against αENaC exhibited the same pattern with the upper band reporting the full-length form (Fig. 5A). As shown in Fig. 5B, TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice had similar αENaC expression, which argues against defective ROMK-dependent K+ secretion in knockout mice. In contrast to constitutively active ROMK channels, BK-dependent K+ secretion requires elevations of [Ca2+]i (41). We have previously shown that single-channel BK activity is markedly reduced in mice with globally deleted TRPV4 (28). Thus, we next tested whether tubule-specific TRPV4 knockout also affects BK channel levels. As shown on the actual Western blot from whole kidney homogenates from TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice kept on a high-K+ diet (Fig. 5C) and the respective summary graph, there was a minor but significant (P = 0.007) 10% increase in the pore-forming α-subunit of BK in knockout mice. Overall, our results suggest that the inability to stimulate Ca2+-activated BK channels due to TRPV4 deletion but not changes in ENaC-dependent Na+-driving force, alterations in flow (comparable urinary output), or diminished mineralocorticoid input is responsible for reduced kaliuresis in TRPV4fl/fl-Pax8Cre mice during K+ load.
Figure 5.

Transient receptor potential vanilloid type 4 (TRPV4) deletion does not change epithelial Na+ channel (ENaC) and large-conductance K+ channel (BK) levels. A, top: actual Western blot probed with anti-αENaC antibodies from whole kidney lysates of TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice maintained on a high-K+ diet (5% K+) for 7 days. The full-length αENaC band around 100 kDa as well as a bright nonspecific band are indicated by arrows. Each line represents an individual animal (2 females and 2 males). A, bottom: Ponceau red staining of the same nitrocellulose membrane demonstrating equal protein loading. B: summary graph comparing full-length αENaC expression in TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice from the conditions shown in A. Intensity values were normalized to the total signal of the respective lines in Ponceau red staining. Males and females are indicated as triangles and circles, respectively. C, top: actual Western blot probed with anti-αBK antibodies from whole kidney lysates of TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice maintained on a high-K+ diet (5% K+) for 7 days. Each line represents an individual animal (2 females and 2 males). C, bottom: Ponceau red staining of the same nitrocellulose membrane demonstrating equal protein loading. D: summary graph comparing αBK expression in TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice from the conditions shown in A. Intensity values were normalized to the total signal of the respective lines in Ponceau red staining. *Significant increase (P < 0.05, one-way ANOVA with a post hoc Tukey’s test).
In the next set of experiments, we examined renal K+ conservation in TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice in response to a K+-deficient diet (<0.01% K+). As shown in Fig. 6A, TRPV4fl/fl-Pax8Cre mice had significantly lower urinary K+ levels during the first and second day of K+ restriction than TRPV4fl/fl littermates. Interestingly, mild but significant reductions in urinary K+ levels were also present on day 7, suggesting augmented K+ reabsorption in the collecting duct in knockout mice compared with control mice. Low-K+ diet induced a progressive rise in urinary output, which become significant at day 7 (Fig. 6B). At the same time, there were no changes in urinary volume between TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice during the whole testing period. Apically expressed H+-K+-ATPase has been recognized to play a central role in K+ reabsorption in the collecting duct in the condition of K+ deficiency (11, 43). Since both gastric and nongastric isoforms of H+-K+-ATPase are present in collecting duct cells (10, 11), we next compared expression of β subunit of the pump in whole kidney homogenates of TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice kept on regular- and K+-deficient diets for 7 days. As shown in the representative Western blot (Fig. 7A) and summary graph (Fig. 7B), β H+-K+-ATPase levels were markedly higher in TRPV4fl/fl-Pax8Cre mice on the regular-K+ diet and particularly on the K+-deficient diet.
Figure 6.

Transient receptor potential vanilloid type 4 (TRPV4)fl/fl-Pax8Cre mice demonstrate sustained augmented renal K+ conservation in response to systemic K+ deficiency. Summary graphs showing a comparison of 24-h urinary K+ levels (A) and urinary volume (B) in TRPV4fl/fl mice (red, 4 females and 6 males) and TRPV4fl/fl-Pax8Cre mice (green, 2 females and 7 males) maintained on a regular-K+ diet (0.9% K+) and in response to a low-K+ diet (<0.01% K+) for 1, 2, and 7 days. Each dot represents a measurement from an individual animal. Males and females are indicated as triangles and circles, respectively. *Significant changes (P < 0.05, one-way ANOVA with a post hoc Tukey’s test) vs. the respective values of TRPV4fl/fl mice, as indicated by the horizontal bar.
Figure 7.

H+-K+-ATPase levels are increased in transient receptor potential vanilloid type 4 (TRPV4)fl/fl-Pax8Cre mice on both regular and low K+ intake. A, top: representative Western blot probed with anti-H+-K+-ATPase β-subunit antibodies from whole kidney lysates of TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice maintained on a regular-K+ diet (0.9% K+) and a low-K+ diet (<0.01% K+) for 7 days. Each line represents an individual animal. A, bottom: Ponceau red staining of the same nitrocellulose membrane demonstrating equal protein loading. B: summary graph comparing H+-K+-ATPase β-subunit expression in TRPV4fl/fl mice (3 females and 3 males) and TRPV4fl/fl-Pax8Cre mice (3 females and 3 males) from the conditions shown in A. Intensity values were normalized to the total signal of the respective lines in Ponceau red staining. Males and females are indicated as triangles and circles, respectively. *Significant changes (P < 0.05, one-way ANOVA with a post hoc Tukey’s test) vs. the respective values of TRPV4fl/fl mice, as indicated by the horizontal bars.
TRPV4 Deletion Causes Increased H+ Extrusion as an Index of H+-K+-ATPase Activity in the Collecting Duct in Response to Systemic K+ Deficiency
In the next set of experiments, we examined the functional consequences of increased β H+-K+-ATPase expression in TRPV4fl/fl-Pax8Cre mice. Since H+-K+-ATPase-dependent K+ reabsorption is coupled to proton secretion, we next compared the rates of pHi recovery after ammonium pulse in freshly isolated split-opened collecting ducts loaded with the pH-sensitive dye BCECF from TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice (Fig. 8A). Previous studies have reported H+-K+-ATPase expression in both A-type and B-type secreting intercalated cells and, to a lower level, principal cells (11, 45). To dissect cell type-specific changes in H+-K+-ATPase, we applied an inhibitor of basolateral Cl− conductance, NPPB (100 µM), following the intracellular acidification-recovery protocol (Fig. 8A). We have recently demonstrated that this maneuver induces opposite actions on pHi, namely, increases and decreases, in A-type and B-type intercalated cells, respectively, while having no effect in principal cells (36). Principal cells were additionally identified by postexperimental labeling for AQP2 (Fig. 8A) (36). As shown in the averaged time courses of pHi changes, the recovery after acidification was faster in all cell types in collecting ducts of TRPV4fl/fl-Pax8Cre mice compared with cells from TRPV4fl/fl mice kept on a K+-deficient diet (Fig. 8B). The accelerated kinetics in TRPV4fl/fl-Pax8Cre cells is indicative of increased functional expression of H+-K+-ATPase. Somewhat unexpectedly, the recovery rates after acidification were comparable in all cell types of TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice kept on a regular-K+ diet (0.9% K+; Fig. 8C) despite considerable higher β H+-K+-ATPase levels in the latter (Fig. 7). Of note, dietary K+ deficiency increased the recovery rate and presumably H+-K+-ATPase activity in A-type intercalated cells of TRPV4fl/fl mice alone, with no significant differences detected in B-type and principal cells (Fig. 8C). In contrast, the recovery rates after acidification were significantly accelerated in all cell types in TRPV4fl/fl-Pax8Cre mice upon comparison of regular- and low-K+ diets. Taken together, our data demonstrate augmented H+ excretion in collecting duct cells, which is indicative of higher H+-K+-ATPase activity in TRPV4fl/fl-Pax8Cre mice than in TRPV4fl/fl mice in the state of K+ deficiency. These findings provide a mechanistic explanation for decreased urinary K+ excretion (Fig. 6) and higher plasma K+ levels (Fig. 2) in knockout mice.
Figure 8.
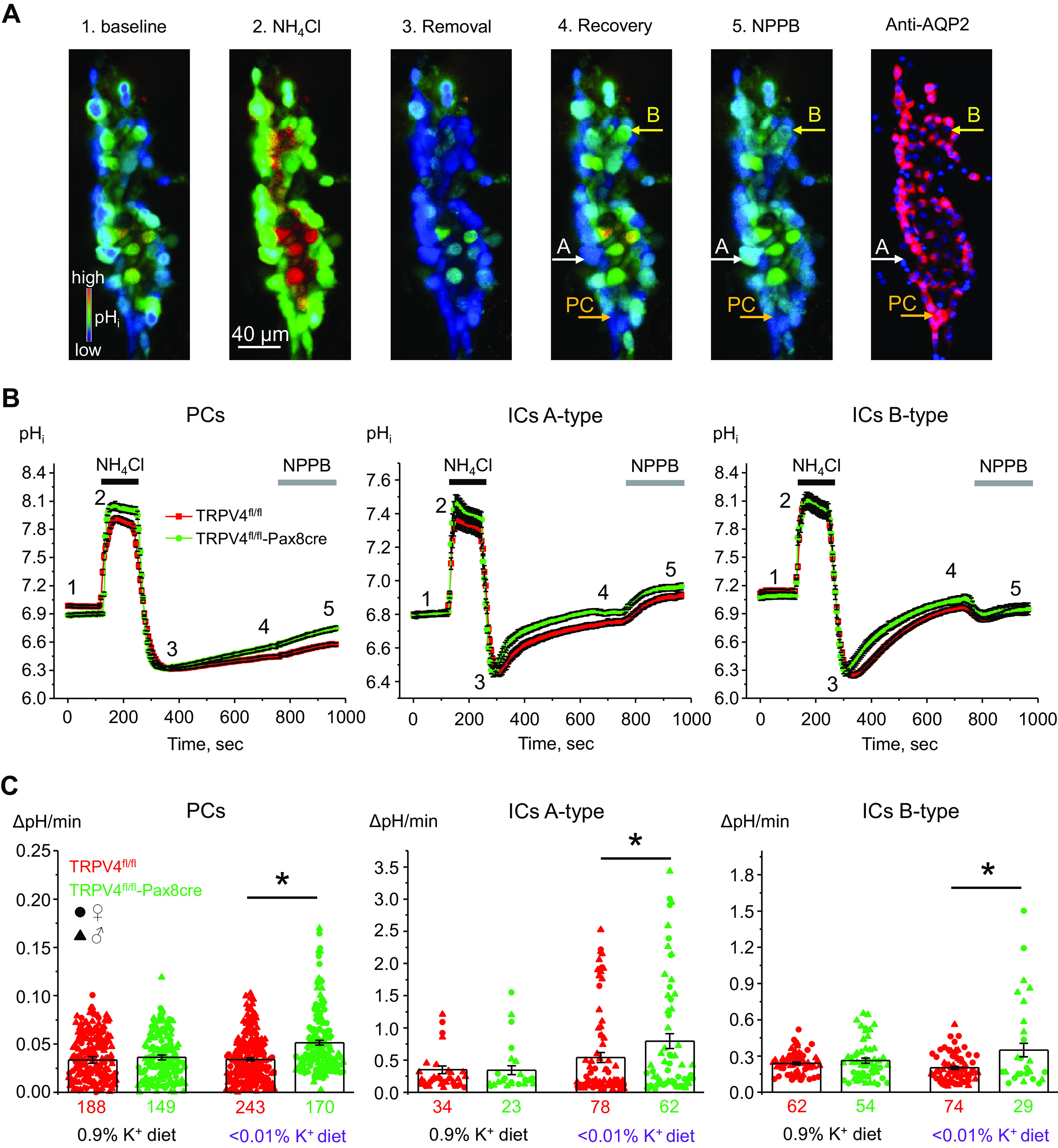
Mice with transient receptor potential vanilloid type 4 (TRPV4) deletion in the renal tubule have increased H+-K+-ATPase-mediated intracellular pH (pHi) recovery after intracellular acidification in freshly isolated split-opened collecting ducts. A: representative pseudocolor images (blue: acidic and red: alkali) of pHi in a split-opened cortical collecting duct loaded with the pH-sensitive dye 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein at baseline (44), upon application of 40 mM NH4Cl (25), immediately after NH4Cl removal (40), upon recovery to baseline pHi values (6), and following application of the ClC-K2 blocker 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB) to distinguish A-type and B-type intercalated cells (ICs) (1). A confocal micrograph of the same split-opened collecting duct probed with anti-aquaporin-2 (AQP2; pseudocolor red) to identify principal cells (PCs) is shown on the right. Examples of A-type (responding with alkalization to NPPB) and B-type (responding with acidification to NPPB) ICs and PCs (no change in pHi to NPPB) are depicted with white, yellow, and orange arrows, respectively. Nuclear DAPI staining is shown as pseudocolor blue. B: summary graphs comparing the time course of pHi changes in PCs (left), A-type ICs (middle), and B-type ICs (right) upon application of 40 mM NH4Cl (shown by black horizontal line) and NPPB (shown by shaded horizontal bars) in cortical collecting ducts from TRPV4fl/fl mice (red dots) and TRPV4fl/fl-Pax8Cre mice (green dots) fed a low-K+ diet for 7 days. The time points shown in A are marked as 1–5. C: summary graphs of the recovery after acidification in individual PCs (right), A-type ICs (middle), and B-type ICs (right) within split-opened areas of collecting ducts from TRPV4fl/fl mice (red dots) and TRPV4fl/fl-Pax8Cre mice (green dots) fed a regular-K+ diet (0.9% K+) and low-K+ diet (<0.01% K+) for 7 days. The rate was calculated for each individual cell (shown as dots) as a linear slope of the initial pHi recovery from the lowest pHi value after 40 mM NH4Cl removal. Numbers of cells in each group are shown. In average, six different collecting ducts from three different mice of each strain were used for each condition (1 female and 2 males or 2 females and 1 male). Cells from male and female are indicated as triangles and circles, respectively. *Significant changes (P < 0.05, one-way ANOVA with a post hoc Tukey’ test) vs. the respective values of TRPV4fl/fl mice, as indicated by the horizontal bars.
DISCUSSION
In the present report, we provide the first direct evidence that functional expression of TRPV4 in the renal tubule is a physiologically relevant component of systemic K+ homeostasis. Taking advantage of newly created TRPV4fl/fl-Pax8Cre mice with selective deletion of the channel in the kidney, we demonstrate that this manipulation leads to augmented renal K+ retention manifested as lower urinary K+ excretion during both high and low K+ intake (Fig. 9). At the whole body level, the antikaliuretic effects of TRPV4 deletion manifest as significantly higher plasma K+ levels compared with values seen in control littermates having no Pax8Cre expression.
Figure 9.

Principle scheme summarizing effects on K+ balance in transient receptor potential vanilloid type 4 (TRPV4)fl/fl and TRPV4fl/fl-Pax8Cre (specific deletion in the renal tubule) mice in response to low and high dietary K+ intake. Increases and decreases vs. the unstressed condition are shown as upward and downward arrows with their numbers reflecting the respective changes in magnitude. ∼No significant changes were detected.
TRPV4 is a well-recognized Ca2+-permeable mechanosensitive channel, which can be activated by direct physical distortion of the plasma membrane (27, 46) as well as by mechanical stimulation of a plasma membrane-associated signaling cascade(s), most commonly phospholipase A2 (47, 48). TRPV4 was originally cloned as a putative osmosensor (18), which, in combination with its abundant expression in the kidney, was hypothesized to play a role in urinary concentrating ability. Here, TRPV4 expression was primarily observed in the proximal tubule and collecting duct segments (25, 26). However, global TRPV4 deletion did not have any effect on urinary volume and osmolarity at baseline and after water deprivation (19, 26). Consistently, we showed that specific TRPV4 deletion does not change urinary volume during variations in dietary K+ intake (Figs. 3B and 6B). Furthermore, we have previously demonstrated that either genetic ablation or pharmacological inhibition abolish [Ca2+]i elevations to increased flow but not in response to hypotonicity in freshly isolated split-opened collecting ducts (25, 28, 49).
The flow-dependent nature of TRPV4 stimulation suggests its functional coupling with Ca2+-activated BK channels to mediate FIKS in collecting duct during dietary K+ load (6, 9, 41). Indeed, a high-K+ diet increases TRPV4 and BK levels in the collecting duct largely in an aldosterone-dependent manner (2, 28). Thus, coadministration of a mineralocorticoid receptor blocker, spironolactone, precluded elevations in renal TRPV4 expression by high-K+ diet. Mice with global TRPV4 deletion had decreased single-channel BK activity and developed hyperkalemia when fed a high-K+ diet (28). In the present report, we demonstrated that selective deletion of TRPV4 in the renal tubule is sufficient to recapitulate this phenotype. Thus, we showed significantly reduced urinary K+ levels in TRPV4fl/fl-Pax8Cre mice during the initial adaptation period to 5% K+ diet (Fig. 3A) with restoration of urinary K+ levels at day 7 at the expense of significantly higher plasma K+ (Fig. 2) and increased aldosterone levels (Fig. 4). At the same time, there were no significant differences in urinary flow rate (Fig. 3B) and αENaC expression (Fig. 5, A and B) as an index of ROMK-dependent K+ secretion in knockout animals and their littermate controls. The comparable αENaC levels in TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice despite higher aldosterone in the latter most likely indicate saturation of the response. Consistently, there were no differences in urinary volume and Na+ excretion in WT and TRPV4−/− mice subjected to dietary salt restriction (26). Altogether, this demonstrates an indispensable role of renal tubule TRPV4 in promoting FIKS, which cannot be compensated by any other mechanisms outside the kidney. It should be noted, though, that Pax8 would also delete TRPV4 in the proximal tubule. However, it is unlikely that it can contribute to the observed antikaliuretic effects since inhibition of proximal tubule transport is prokaliuretic (50). In this segment, TRPV4 is expressed mainly on the basolateral membrane (26, 27). It has been previously shown that pressure-induced TRPV4 activation stimulates albumin uptake likely via megalin-dependent endocytosis (26, 27). Furthermore, there is no experimental evidence indicating any significant role of TRPV4 in mediating electrolyte transport in the proximal tubule.
The collecting duct plays a critical role in adaptation to day-to-day variations in water and electrolyte intake (5, 51, 52). It has a high plasticity in part due to the presence of different cell types: principal and intercalated cells having distinct physiological roles and transport profiles (5, 51). The less abundant intercalated cells are generally recognized for governing pH homeostasis due their ability to secrete acids (A-type) or bases (B-type) with their ratio in the collecting duct being dictated by systemic acid-base insults (36, 51). In addition, intercalated cells are responsible for K+ reabsorption in the state of K+ deficiency (11). Apically localized H+-K+-ATPase underlies K+ uptake at the expense of H+ extrusion to the tubule lumen to minimize urinary K+ loss. H+-K+-ATPase is similarly present in both A-type and B-type intercalated cells, and its expression is stimulated by low K+ intake (10, 45). Importantly, dietary K+ restriction also increases urinary flow (Fig. 6) indicative of TRPV4 activation. In this case, stimulation of TRPV4 could occur in a with-no-lysine kinase (WNK)-dependent manner, which is known to be activated by K+ deficiency (53). Indeed, we have recently reported that WNK1 is critical to increasing TRPV4 expression and activity in collecting duct cells (30). Our metabolic cage balance experiments revealed significantly lower urinary K+ levels in TRPV4fl/fl-Pax8Cre mice than in TRPV4fl/fl mice subjected to a low-K+ diet, with these differences persisting at least to day 7 (Fig. 6A). Thus, it is reasonable to assume that the long-lasting K+ retention is responsible for minimal of any decreases in plasma K+ levels in knockout animals, whereas controls developed prominent hypokalemia due to a less efficient K+ conservation (Fig. 2). Moreover, we found significantly higher H+-K+-ATPase levels in TRPV4fl/fl-Pax8Cre mice fed the regular-K+ diet and particularly the K+-deficient diet (Fig. 7). We have also provided functional evidence of augmented H+ extrusion in intercalated (both A-type and B-type) and principal cells in freshly isolated split-opened collecting ducts from TRPV4-deficient mice. Using the protocol of pHi recovery after acidification as an index of H+-K+-ATPase activity, we observed significantly accelerated rates in TRPV4fl/fl-Pax8Cre mice on a low-K+ diet compared with the values seen in genetic controls (Fig. 8, B and C). Of note, dietary K+ restriction increased recovery only in A-type intercalated cells of TRPV4fl/fl mice, whereas the differences also became significant in B-type intercalated cells and principal cells of TRPV4fl/fl-Pax8Cre mice. It should be noted that H+ exporting systems other than H+-K+-ATPase are present in collecting duct cells, mainly Na+/H+ exchanger 1 and V-ATPase (51, 54), thus also contributing to the measured pHi recovery. However, none of these transporters are expected to be upregulated by dietary K+ deficiency, pointing to H+-K+-ATPase as the underlying cause for the accelerated H+ extrusion in collecting duct cells of TRPV4fl/fl-Pax8Cre mice. Indeed, we have previously shown accelerated H+-K+-ATPase-dependent pHi extrusion in mice with global TRPV4 deletion when Na+/H+ exchanger 1 and V-ATPase were blocked with EIPA and bafilomycin, respectively (34). Interestingly, we did not detect significant differences in pHi recovery in TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice fed a regular diet (Fig. 8C), despite higher H+-K+-ATPase levels in knockout mice under this condition (Fig. 7). Since a low-K+ diet is thought to stimulate both expression and trafficking of H+-K+-ATPase to the apical plasma membrane (11), it is possible that intracellular localization of the pump is predominantly cytosolic in mice fed a regular-K+ diet. This is consistent with the lack of differences in urinary K+ excretion between TRPV4fl/fl and TRPV4fl/fl-Pax8Cre mice under this condition (Figs. 3A and 6A). The molecular mechanism whereby TRPV4 deletion increases H+-K+-ATPase functional activity remains to be ascertained in future investigations. Previous studies have demonstrated a critical role of elevations in cAMP levels in augmenting H+-K+-ATPase activity and expression in the collecting duct via CREB and Epac 1/Rap-1/Raf-B/ERK pathways but not via protein kinase A-[Ca2+]i signaling (55, 56). We documented that TRPV4 is a critical determinant of intracellular Ca2+ homeostasis in rodent and human collecting duct cells with its deletion or inhibition leading to a reduction in basal [Ca2+]i levels (29, 57). Interestingly, a reduction of [Ca2+]i is associated with cAMP-dependent activation of the B-Raf-B/ERK pathway in these cells (44, 58, 59). Thus, it is plausible to propose that the observed augmentation of H+-K+-ATPase expression and activity in collecting ducts from TRPV4fl/fl-Pax8Cre mice could be due to an increased cAMP-dependent signaling event.
Overall, we show that the augmented renal K+ retention in TRPV4fl/fl-Pax8Cre mice recapitulates the phenotype observed upon global TRPV4 deletion, which we have previously reported (34), thus demonstrating the supremacy of TRPV4 expression in renal tubules for K+ balance not only upon K+ load but also during systemic K+ deficiency. Disturbances in plasma K+ levels are common comorbific and potentially life-threatening complications in patients with kidney diseases (1). Thus, the incidence of hyperkalemia is greatly increased in patients with chronic kidney disease having a glomerular filtration rate of <60 mL/min/1.73 m2 (60, 61), in part due to decreased distal Na+ delivery and flow to stimulate FIKS. Hypokalemia is often observed in patients with hyperaldosteronism or undergoing an antihypertensive regimen containing thiazide diuretics (62), which increase flow in K+-secreting collecting duct segments. Moreover, both hyperkalemia and hypokalemia can occur in patients after dialysis due to malnutrition, K+ content in the dialysate, etc. (for a review, see Ref. 1). The present study demonstrates the potency of TRPV4 function in the renal tubule to affect renal K+ excretion and to control K+ levels (Fig. 2). Specifically, we show physiologically meaningful prokaliuretic actions of TRPV4 in promoting K+ secretion and inhibiting K+ reabsorption in the collecting duct during high- and low K+-diets, respectively. Taking into consideration that TRPV4 expression is highest in the renal tissue, it might be promising to pharmacologically stimulate TRPV4, particularly when glomerular filtration rate is low, to mimic FIKS activation and decrease plasma K+ levels. Likewise, concomitant administration of thiazide diuretics with TRPV4 blockers would mitigate urinary K+ losses to prevent the development of hyperkalemia. We are looking forward that such strategies targeting TRPV4 function could be implemented into the clinic to better control K+ homeostasis and plasma K+ levels in the future.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.22246414.v1.
GRANTS
This research was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK117865 and DK119170; American Heart Association Grants EIA35260097 (to O.P.) and AHA-23POST1020372 (to K.P.); National Eye Institute Grants EY031817, EY027920, and P30EY014800 (to D.K.), and an unrestricted Grant from Research to Prevent Blindness to the Department of Ophthalmology at the University of Utah.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.K. and O.P. conceived and designed research; A.S., K.P., O.Z., V.N.T., M.K., and O.P. performed experiments; A.S., K.P., O.Z., V.N.T., M.K., and O.P. analyzed data; A.S., K.P., O.Z., V.N.T., M.L., D.K., and O.P. interpreted results of experiments; A.S., K.P., O.Z., V.N.T., and O.P. prepared figures; O.P. drafted manuscript; A.S., K.P., O.Z., V.N.T., M.K., M.L., D.K., and O.P. edited and revised manuscript; A.S., K.P., O.Z., V.N.T., M.K., M.L., D.K., and O.P. approved final version of manuscript.
REFERENCES
- 1. Clase CM, Carrero JJ, Ellison DH, Grams ME, Hemmelgarn BR, Jardine MJ, Kovesdy CP, Kline GA, Lindner G, Obrador GT, Palmer BF, Cheung M, Wheeler DC, Winkelmayer WC, Pecoits-Filho R; Conference Participants. Potassium homeostasis and management of dyskalemia in kidney diseases: conclusions from a kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 97: 42–61, 2020. doi: 10.1016/j.kint.2019.09.018. [DOI] [PubMed] [Google Scholar]
- 2. Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 10: 1050–1060, 2015. doi: 10.2215/CJN.08580813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hayes J, Kalantar-Zadeh K, Lu JL, Turban S, Anderson JE, Kovesdy CP. Association of hypo- and hyperkalemia with disease progression and mortality in males with chronic kidney disease: the role of race. Nephron Clin Pract 120: c8–16, 2012. doi: 10.1159/000329511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol 71: 381–401, 2009. doi: 10.1146/annurev.physiol.010908.163241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. Clin J Am Soc Nephrol 10: 135–146, 2015. doi: 10.2215/CJN.05760513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carrisoza-Gaytan R, Ray EC, Flores D, Marciszyn AL, Wu P, Liu L, Subramanya AR, Wang W, Sheng S, Nkashama LJ, Chen J, Jackson EK, Mutchler SM, Heja S, Kohan DE, Satlin LM, Kleyman TR. Intercalated cell BKα subunit is required for flow-induced K+ secretion. JCI Insight 5:e130553, 2020. doi: 10.1172/jci.insight.130553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grimm PR, Irsik DL, Settles DC, Holtzclaw JD, Sansom SC. Hypertension of Kcnmb1−/− is linked to deficient K secretion and aldosteronism. Proc Natl Acad Sci USA 106: 11800–11805, 2009. doi: 10.1073/pnas.0904635106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holtzclaw JD, Grimm PR, Sansom SC. Role of BK channels in hypertension and potassium secretion. Curr Opin Nephrol Hypertens 20: 512–517, 2011. doi: 10.1097/MNH.0b013e3283488889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Woda CB, Bragin A, Kleyman TR, Satlin LM. Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol 280: F786–F793, 2001. doi: 10.1152/ajprenal.2001.280.5.F786. [DOI] [PubMed] [Google Scholar]
- 10. Greenlee MM, Lynch IJ, Gumz ML, Cain BD, Wingo CS. The renal H,K-ATPases. Curr Opin Nephrol Hypertens 19: 478–482, 2010. doi: 10.1097/MNH.0b013e32833ce65f. [DOI] [PubMed] [Google Scholar]
- 11. Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS. The renal H+-K+-ATPases: physiology, regulation, and structure. Am J Physiol Renal Physiol 298: F12–F21, 2010. doi: 10.1152/ajprenal.90723.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Greenlee MM, Lynch IJ, Gumz ML, Cain BD, Wingo CS. Mineralocorticoids stimulate the activity and expression of renal H+,K+-ATPases. J Am Soc Nephrol 22: 49–58, 2011. doi: 10.1681/ASN.2010030311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Everaerts W, Nilius B, Owsianik G. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol 103: 2–17, 2010. doi: 10.1016/j.pbiomolbio.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 14. Mamenko M, Zaika O, Boukelmoune N, O'Neil RG, Pochynyuk O. Deciphering physiological role of the mechanosensitive TRPV4 channel in the distal nephron. Am J Physiol Renal Physiol 308: F275–F286, 2015. doi: 10.1152/ajprenal.00485.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol 286: C195–C205, 2004. doi: 10.1152/ajpcell.00365.2003. [DOI] [PubMed] [Google Scholar]
- 16. Pochynyuk O, Zaika O, O'Neil RG, Mamenko M. Novel insights into TRPV4 function in the kidney. Pflugers Arch 465: 177–186, 2013. doi: 10.1007/s00424-012-1190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lapajne L, Rudzitis CN, Cullimore B, Ryskamp D, Lakk M, Redmon SN, Yarishkin O, Krizaj D. TRPV4: cell type-specific activation, regulation and function in the vertebrate eye. Curr Top Membr 89: 189–219, 2022. doi: 10.1016/bs.ctm.2022.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liedtke W, Choe Y, Martí-Renom MA, Bell AM, Denis CS, Sali A, Hudspeth AJ, Friedman JM, Heller S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103: 525–535, 2000. doi: 10.1016/s0092-8674(00)00143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc Natl Acad Sci USA 100: 13698–13703, 2003. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, Lieben L, Torrekens S, Moermans K, Vanden Bosch A, Bouillon R, Nilius B, Carmeliet G. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab 8: 257–265, 2008. doi: 10.1016/j.cmet.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 21. Muramatsu S, Wakabayashi M, Ohno T, Amano K, Ooishi R, Sugahara T, Shiojiri S, Tashiro K, Suzuki Y, Nishimura R, Kuhara S, Sugano S, Yoneda T, Matsuda A. Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation. J Biol Chem 282: 32158–32167, 2007. doi: 10.1074/jbc.M706158200. [DOI] [PubMed] [Google Scholar]
- 22. Rosenbaum T, Benítez-Angeles M, Sánchez-Hernández R, Morales-Lázaro SL, Hiriart M, Morales-Buenrostro LE, Torres-Quiroz F. TRPV4: a physio and pathophysiologically significant ion channel. Int J Mol Sci 21: 3837, 2020. doi: 10.3390/ijms21113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Toft-Bertelsen TL, MacAulay N. TRPing to the point of clarity: understanding the function of the complex TRPV4 ion channel. Cells 10: 165, 2021. doi: 10.3390/cells10010165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 76: 387–417, 2007. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berrout J, Jin M, Mamenko M, Zaika O, Pochynyuk O, O'Neil RG. Function of TRPV4 as a mechanical transducer in flow-sensitive segments of the renal collecting duct system. J Biol Chem 287: 8782–8791, 2012. doi: 10.1074/jbc.M111.308411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Janas S, Seghers F, Schakman O, Alsady M, Deen P, Vriens J, Tissir F, Nilius B, Loffing J, Gailly P, Devuyst O. TRPV4 is associated with central rather than nephrogenic osmoregulation. Pflugers Arch 468: 1595–1607, 2016. doi: 10.1007/s00424-016-1850-5. [DOI] [PubMed] [Google Scholar]
- 27. Gualdani R, Seghers F, Yerna X, Schakman O, Tajeddine N, Achouri Y, Tissir F, Devuyst O, Gailly P. Mechanical activation of TRPV4 channels controls albumin reabsorption by proximal tubule cells. Sci Signal 13: eabc6967, 2020. doi: 10.1126/scisignal.abc6967. [DOI] [PubMed] [Google Scholar]
- 28. Mamenko MV, Boukelmoune N, Tomilin VN, Zaika OL, Jensen VB, O'Neil RG, Pochynyuk OM. The renal TRPV4 channel is essential for adaptation to increased dietary potassium. Kidney Int 91: 1398–1409, 2017. doi: 10.1016/j.kint.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tomilin V, Reif GA, Zaika O, Wallace DP, Pochynyuk O. Deficient transient receptor potential vanilloid type 4 function contributes to compromised [Ca2+]i homeostasis in human autosomal-dominant polycystic kidney disease cells. FASEB J 32: 4612–4623, 2018. doi: 10.1096/fj.201701535RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tomilin VN, Pyrshev K, Khayyat NH, Zaika O, Pochynyuk O. With-no-lysine kinase 1 (WNK1) augments TRPV4 function in the aldosterone-sensitive distal nephron. Cells 10: 1482, 2021. doi: 10.3390/cells10061482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu W, Wei Y, Sun P, Wang WH, Kleyman TR, Satlin LM. Mechanoregulation of BK channel activity in the mammalian cortical collecting duct: role of protein kinases A and C. Am J Physiol Renal Physiol 297: F904–F915, 2009. doi: 10.1152/ajprenal.90685.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rieg T, Vallon V, Sausbier M, Sausbier U, Kaissling B, Ruth P, Osswald H. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int 72: 566–573, 2007. doi: 10.1038/sj.ki.5002369. [DOI] [PubMed] [Google Scholar]
- 33. Wen D, Cornelius RJ, Sansom SC. Interacting influence of diuretics and diet on BK channel-regulated K homeostasis. Curr Opin Pharmacol 15: 28–32, 2014. doi: 10.1016/j.coph.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tomilin V, Mamenko M, Zaika O, Wingo CS, Pochynyuk O. TRPV4 deletion protects against hypokalemia during systemic K+ deficiency. Am J Physiol Renal Physiol 316: F948–F956, 2019. doi: 10.1152/ajprenal.00043.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matsumoto H, Sugio S, Seghers F, Krizaj D, Akiyama H, Ishizaki Y, Gailly P, Shibasaki K. Retinal detachment-induced Müller glial cell swelling activates TRPV4 ion channels and triggers photoreceptor death at body temperature. J Neurosci 38: 8745–8758, 2018. doi: 10.1523/JNEUROSCI.0897-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pyrshev K, Khayyat NH, Stavniichuk A, Tomilin VN, Zaika O, Ramkumar N, Pochynyuk O. ClC-K2 Cl− channel allows identification of A- and B-type of intercalated cells in split-opened collecting ducts. FASEB J 36: e22275, 2022. doi: 10.1096/fj.202200160R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mamenko M, Zaika O, O'Neil RG, Pochynyuk O. Ca2+ imaging as a tool to assess TRP channel function in murine distal nephrons. Methods Mol Biol 998: 371–384, 2013. doi: 10.1007/978-1-62703-351-0_29. [DOI] [PubMed] [Google Scholar]
- 38. Thomas JA, Buchsbaum RN, Zimniak A, Racker E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18: 2210–2218, 1979. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- 39. Zaidman NA, Tomilin VN, Hassanzadeh Khayyat N, Damarla M, Tidmore J, Capen DE, Brown D, Pochynyuk OM, Pluznick JL. Adhesion-GPCR Gpr116 (ADGRF5) expression inhibits renal acid secretion. Proc Natl Acad Sci USA 117: 26470–26481, 2020. doi: 10.1073/pnas.2007620117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bouchard M, Souabni A, Busslinger M. Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis 38: 105–109, 2004. doi: 10.1002/gene.20008. [DOI] [PubMed] [Google Scholar]
- 41. Liu W, Morimoto T, Woda C, Kleyman TR, Satlin LM. Ca2+ dependence of flow-stimulated K secretion in the mammalian cortical collecting duct. Am J Physiol Renal Physiol 293: F227–F235, 2007. doi: 10.1152/ajprenal.00057.2007. [DOI] [PubMed] [Google Scholar]
- 42. Mutchler SM, Shi S, Whelan SCM, Kleyman TR. Validation of commercially available antibodies directed against subunits of the epithelial Na+ channel. Physiol Rep 11: e15554, 2023. doi: 10.14814/phy2.15554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Verlander JW, Moudy RM, Campbell WG, Cain BD, Wingo CS. Immunohistochemical localization of H-K-ATPase α2c-subunit in rabbit kidney. Am J Physiol Renal Physiol 281: F357–F365, 2001. doi: 10.1152/ajprenal.2001.281.2.F357. [DOI] [PubMed] [Google Scholar]
- 44. Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM Jr, Grantham JJ. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int 66: 964–973, 2004. doi: 10.1111/j.1523-1755.2004.00843.x. [DOI] [PubMed] [Google Scholar]
- 45. Lynch IJ, Greenlee MM, Gumz ML, Rudin A, Xia SL, Wingo CS. Heterogeneity of H-K-ATPase-mediated acid secretion along the mouse collecting duct. Am J Physiol Renal Physiol 298: F408–F415, 2010. doi: 10.1152/ajprenal.00333.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Servin-Vences MR, Moroni M, Lewin GR, Poole K. Direct measurement of TRPV4 and PIEZO1 activity reveals multiple mechanotransduction pathways in chondrocytes. eLife 6: e21074, 2017. doi: 10.7554/eLife.21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Toft-Bertelsen TL, Yarishkin O, Redmon S, Phuong TTT, Križaj D, MacAulay N. Volume sensing in the transient receptor potential vanilloid 4 ion channel is cell type-specific and mediated by an N-terminal volume-sensing domain. J Biol Chem 294: 18421–18434, 2019. doi: 10.1074/jbc.RA119.011187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA 101: 396–401, 2004. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tomilin VN, Mamenko M, Zaika O, Ren G, Marrelli SP, Birnbaumer L, Pochynyuk O. TRPC3 determines osmosensitive [Ca2+]i signaling in the collecting duct and contributes to urinary concentration. PLoS One 14: e0226381, 2019. doi: 10.1371/journal.pone.0226381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tomilin VN, Pyrshev K, Stavniichuk A, Hassanzadeh Khayyat N, Ren G, Zaika O, Khedr S, Staruschenko A, Mei FC, Cheng X, Pochynyuk O. Epac1−/− and Epac2−/− mice exhibit deficient epithelial Na+ channel regulation and impaired urinary Na+ conservation. JCI Insight 7: e145653, 2022. doi: 10.1172/jci.insight.145653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roy A, Al-Bataineh MM, Pastor-Soler NM. Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 10: 305–324, 2015. doi: 10.2215/CJN.08880914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Staruschenko A. Regulation of transport in the connecting tubule and cortical collecting duct. Compr Physiol 2: 1541–1584, 2012. doi: 10.1002/cphy.c110052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wall SM, Verlander JW, Romero CA. The renal physiology of pendrin-positive intercalated cells. Physiol Rev 100: 1119–1147, 2020. doi: 10.1152/physrev.00011.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Laroche-Joubert N, Marsy S, Luriau S, Imbert-Teboul M, Doucet A. Mechanism of activation of ERK and H-K-ATPase by isoproterenol in rat cortical collecting duct. Am J Physiol Renal Physiol 284: F948–F954, 2003. doi: 10.1152/ajprenal.00394.2002. [DOI] [PubMed] [Google Scholar]
- 56. Xu X, Zhang W, Kone BC. CREB trans-activates the murine H+-K+-ATPase α2-subunit gene. Am J Physiol Cell Physiol 287: C903–C911, 2004. doi: 10.1152/ajpcell.00065.2004. [DOI] [PubMed] [Google Scholar]
- 57. Zaika O, Mamenko M, Berrout J, Boukelmoune N, O'Neil RG, Pochynyuk O. TRPV4 dysfunction promotes renal cystogenesis in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 24: 604–616, 2013. doi: 10.1681/ASN.2012050442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int 63: 1983–1994, 2003. doi: 10.1046/j.1523-1755.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 59. Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 279: 40419–40430, 2004. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 60. Drawz PE, Babineau DC, Rahman M. Metabolic complications in elderly adults with chronic kidney disease. J Am Geriatr Soc 60: 310–315, 2012. doi: 10.1111/j.1532-5415.2011.03818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Foley RN, Wang C, Ishani A, Ibrahim HN, Collins AJ. Creatinine-based glomerular filtration rates and microalbuminuria for detecting metabolic abnormalities in US adults: the National Health and Nutrition Examination Survey 2003–2004. Am J Nephrol 28: 431–437, 2008. doi: 10.1159/000112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rodenburg EM, Visser LE, Hoorn EJ, Ruiter R, Lous JJ, Hofman A, Uitterlinden AG, Stricker BH. Thiazides and the risk of hypokalemia in the general population. J Hypertens 32: 2092–2097, 2014. doi: 10.1097/HJH.0000000000000299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.22246414.v1.
Data Availability Statement
Data will be made available upon reasonable request.