

Keywords: central command, cerebral blood flow, hypocapnia, initial orthostatic hypotension, postural tachycardia syndrome
Abstract
Fifty percent of patients with postural tachycardia syndrome (POTS) are hypocapnic during orthostasis related to initial orthostatic hypotension (iOH). We determined whether iOH drives hypocapnia in POTS by low BP or decreased cerebral blood velocity (CBv). We studied three groups; healthy volunteers (n = 32, 18 ± 3 yr) were compared with POTS, grouped by presence [POTS-low end-tidal CO2 (↓ETCO2), n = 26, 19 ± 2 yr] or absence [POTS-normal upright end-tidal carbon dioxide (nlCO2), n = 28, 19 ± 3 yr] of standing hypocapnia defined by end-tidal CO2 (ETCO2) ≤ 30 mmHg at steady-state, measuring middle cerebral artery CBv, heart rate (HR), and beat-to-beat blood pressure (BP). After 30 min supine, subjects stood for 5 min. Quantities were measured prestanding, at minimum CBv, minimum BP, peak HR, CBv recovery, BP recovery, minimum HR, steady-state, and 5 min. Baroreflex gain was estimated by α index. iOH occurred with similar frequency and minimum BP in POTS-↓ETCO2 and POTS-nlCO2. Minimum CBv was reduced significantly (P < 0.05) in POTS-↓ETCO2 (48 ± 3 cm/s) preceding hypocapnia compared with POTS-nlCO2 (61 ± 3 cm/s) or Control (60 ± 2 cm/s). The anticipatory increased BP was significantly larger (P < 0.05) in POTS (8 ± 1 mmHg vs. 2 ± 1) and began ∼8 s prestanding. HR increased in all subjects, CBv increased significantly (P < 0.05) in both POTS-nlCO2 (76 ± 2 to 85 ± 2 cm/s) and Control (75 ± 2 to 80 ± 2 cm/s) consistent with central command. CBv decreased in POTS-↓ETCO2 (76 ± 3 to 64 ± 3 cm/s) correlating with decreased baroreflex gain. Cerebral conductance [meanCBv/mean arterial blood pressure (MAP)] was reduced in POTS-↓ETCO2 throughout. Data support the hypothesis that excessively reduced CBv during iOH may intermittently reduce carotid body blood flow, sensitizing that organ and producing postural hyperventilation in POTS-↓ETCO2. Excessive fall in CBv occurs in part during prestanding central command and is a facet of defective parasympathetic regulation in POTS.
NEW & NOTEWORTHY Dyspnea is frequent in postural tachycardia syndrome (POTS) and is associated with upright hyperpnea and hypocapnia that drives sinus tachycardia. It is initiated by an exaggerated reduction in cerebral conductance and decreased cerebral blood flow (CBF) that precedes the act of standing. This is a form of autonomically mediated “central command.” Cerebral blood flow is further reduced by initial orthostatic hypotension common in POTS. Hypocapnia is maintained during the standing response and might account for persistent postural tachycardia.
INTRODUCTION
The purpose of this study was to determine whether initial orthostatic hypotension (iOH) drives hypocapnia in postural tachycardia syndrome (POTS) by low blood pressure (BP) or decreased cerebral blood flow (CBF). Postural hypocapnia can cause postural tachycardia because hypocapnia produces sinus tachycardia (1–5). Prior observations suggested that reduction of middle cerebral artery (MCA) blood velocity (CBv) preceded hypocapnia (2). Blood flow to the MCA and carotid body originate from the common carotid artery, may change in parallel, and if intermittently reduced, effect intermittent ischemic hypoxia of the carotid body sensitizing and activating the peripheral chemoreflex (3, 4). Hypocapnia ensues, increasing heart rate (HR) and blood pressure (BP) (5). The hypothesis of increased carotid body sensitization received support from our work indicating enhancement of the hypoxic ventilatory response in patients with postural tachycardia syndrome (POTS) (6). Hypocapnia effectively blunted central chemoreflex activity supine and upright (7).
In patients with hypocapnic POTS [end-tidal CO2 (ETCO2) <30 mmHg], reduced CBv is driven by the initial orthostatic hypotension (iOH) (8) that occurs more frequently compared with healthy volunteers during active standing (9). However, these studies did not measure CBv to better understand the physiological production of hypocapnia.
iOH during standing results from the shift of an estimated 500–700 mL from central stores to predominantly venous structures below the diaphragm (10). A brief increase in BP is driven by the mechanical compression of leg and splanchnic venous reserves, BP then falls due to gravitational central hypovolemia producing reflex increases in HR with a local maximum near the BP minimum (nadir) ∼10 s poststand (9). BP is rapidly restored with an initial overshoot in ∼10 s by sympathetic vasoconstriction and splanchnic venoconstriction (11). In contrast, vagal withdrawal occurs within a heartbeat producing rapid reflexively increased HR often prolonged in POTS (12).
iOH is defined as a transient fall of BP on rapid standing exceeding 40 mmHg systolic or 20 mmHg diastolic, although some transient decrease in BP is almost always observed (9). iOH is distinguished from sustained orthostatic hypotension (OH) in which BP progressively falls without recovery after standing (13). iOH findings are reproducible (14). In previous studies, we have shown dynamic and static autoregulation of CBF are less effective in patients with POTS compared with control subjects during orthostatic challenge (15).
We propose that in subjects with hypocapnic POTS, the high frequency of iOH on standing reduces CBv that sensitizes and activates the carotid body by intermittent ischemic hypoxia. We further hypothesize that an excessive fall in CBv may occur in part from a reduction of CBv during central command, i.e., feedforward signals descending from higher centers affecting cardiovascular and respiratory control, in anticipation of standing and is a consequence of impaired autonomic regulation in POTS (16, 17).
METHODS
We recruited healthy control subjects and patients with POTS, a form of chronic orthostatic intolerance (OI) defined as “difficulty tolerating the upright posture because of symptoms that abate once supine” (18). Symptoms of OI include lightheadedness, visual difficulties, impending loss of consciousness, headache, hypotension, tachycardia or bradycardia, pallor, and diaphoresis (19). A diagnosis of POTS requires >3 mo of OI plus persistent excessive upright tachycardia in the absence of sustained hypotension (20). Excessive upright tachycardia is defined in adults by an increase of HR exceeding 30 beats/min (bpm) or to a HR > 120 beats/min during a 10-min orthostatic test (21). An increase >40 beats/min is required for the diagnosis in children less than 19 yr old (22).
Subjects
We prospectively recruited 54 consecutive patients with POTS, ranging in age from 14 to 23 yr over a 2-yr period at the Center for Hypotension at New York Medical College. Approximately half (26/54) gave a history of upright dyspnea. We also recruited 32 healthy nonfainting control subjects, ranging in age from 14 to 22 yr. Patients with POTS were referred for suspected POTS with OI lasting longer than 6 mo. POTS diagnoses were confirmed by a 70° upright tilt test performed at a prior visit. Subjects with POTS were either medication naïve or weaned off all medications for at least 2 wk before being studied.
All subjects provided a medical history and a physical examination with electrocardiography was performed. Cardiovascular disease, systemic disease, and transient infections were always excluded. Patients on long-term bed rest were excluded.
Control subjects were recruited from age-, BMI-, and sex-matched healthy volunteers in whom a 70° upright tilt for 10 min was normal [Control, 18 ± 3 yr, BMI 21 ± 4, n = 32, 3 males; POTS-low end-tidal carbon dioxide (↓ETCO2), 19 ± 2 yr, BMI 20 ± 3, n = 26, 2 males; POTS-normal upright end-tidal carbon dioxide (nlCO2), 19 ± 3 yr, BMI 21 ± 3, n = 28, 2 males; N.S.]. Control subjects reported no illness or orthostatic intolerance and had never fainted. Approximately 50% reported mild dizziness upon rapid standing from the supine position. Exclusion criteria for participation included infectious or systemic illness, competitive athletic training, recent long-term bed rest, hypertension, or pregnancy within the last year. Prior medication, if any, was discontinued for at least 2 wk before participation in this study, except for the use of oral contraceptives. Each female subject was studied during the mid-luteal phase of their menstrual cycle.
All subjects refrained from caffeine, nicotine, or xanthine-containing products for at least 72 h before testing. This study was approved by the Institutional Review Board of New York Medical College. Subjects 18 yr or older signed an informed consent; those younger than 18 yr assented to participate, and their legal guardian signed an informed consent.
Instrumentation.
All subjects were instrumented while lying supine by the research technician. An electrocardiograph measured HR from the interbeat cardiac electrical intervals and beat-to-beat BP were measured using finger photoplethysmography (Finometer; FMS, Amsterdam) placed on the right index finger and corrected for tilt angle, using ModelFlow software (23). Finometer BP readings were automatically calibrated to brachial artery pressure and oscillometric BP measurements. Nasal cannula capnography combined with a pulse oximeter (Smith Medical PM, Waukesha, WI) measured end-tidal CO2 (ETCO2), and oxygen saturation. All subjects were instructed to breathe only through their nostrils; when this did not occur, it was immediately evident on the capnograph signal and subjects were verbally corrected. Respiratory inductive plethysmography (Respitrace, NIMS Scientific, Miami Beach) was only intermittently effective because of rapid standing dislodged transducers. Nevertheless, supine ETCO2 and respiratory intervals, when observed, were perfectly synchronized.
Transcranial Doppler (TCD) (Neurovision; Multigon, Yonkers, NY) measured CBv of the left middle cerebral artery (MCA) using a 2-MHz probe fixed to the subject’s head by a headband. TCD probes are vulnerable to movement from altered MCA insonation focus on standing. Strict successful efforts were made to optimize and stabilize probes at rest and during orthostasis.
Samples of all signals were acquired at 200 samples/s, multiplexed, and A/D converted using USB data acquisition devices DI-706 and DI-720 (DATAQ, Akron Ohio) and analyzed using custom, proprietary, copyrighted software written by one of the authors (J.M.S.).
In previous studies, healthy volunteers had supine ETCO2 values that ranged between 38 and 43 mmHg supine, which decreased with orthostasis but was always ≥33 mmHg. We therefore defined upright hypocapnia during steady-state standing as ETCO2 ≤ 30 mmHg (5). Patients with hypocapnic POTS were designated POTS-↓CO2 and patients with non-hypocapnic POTS were designated POTS-nlCO2. Patients with POTS-↓CO2 uniformly exhibited dyspnea on hyperpnea.
Protocol
All subjects fasted for >3 h before testing. Subjects arrived at our center at 9:00 AM and were prepared for study while supine on a tilt table. Following instrumentation, subjects remained awake and supine for 30 min to acclimate. Baseline HR, BP, ETCO2, and CBv data were collected for 10 min in the supine position to use for HR variability (HRV) and BP variability (BPV) calculations. Subjects stood up over <5 s and remained standing for 5 min while HR, BP, ETCO2, and CBv were continuously monitored. We explained the standing procedure to all study participants who were told that the research technician would approach the tilt table 5–10 s before standing. The act of approach provoked an anticipatory response comprising an increase in HR, and BP in all subjects, an increase in CBv in control subjects and subjects with POTS-nlCO2, but a decrease in CBv in subjects with POTS-↓CO2. Five minutes of standing was chosen because we have not observed vasovagal syncope within this time period, and 5 min sufficed to establish steady-state HR. Some patients with POTS fulfilled criteria for iOH during the standing procedure with brief OI symptoms. After 5 min, subjects returned to supine for recovery during which baseline HR, BP, ETCO2, and CBv were restored.
Event data analysis.
We compared data for control and both POTS groups at baseline, and at events depicted in Fig. 1 which shows BP, HR, and CBv from a representative healthy individual.
Figure 1.

Representative phasic cerebral blood flow velocity (CBFv, A), blood pressure (BP, B), and time averaged heart rate (HR, C), and for a Control subject who had initial orthostatic hypotension (iOH). Marked events are: Baseline; Stand = Standing initiated; Min CBv = the nadir or lowest CBv achieved after standing; Min BP = the nadir or lowest blood pressure achieved after standing; Peak HR = the maximum HR achieved after standing; CBv Recovery = the first local maximum of CBv after standing; BP Recovery = the first local maximum of BP after standing; Min HR (HR Recovery) = the lowest HR after standing; Steady-State HR = HR increases after HR Recovery until it achieves a steady slope.
In time sequence these events are:
Baseline = data collected supine before the anticipatory phase
Stand = standing initiated at end of anticipatory phase
Min CBv = the nadir or lowest mean CBv after standing
Min BP = the nadir or lowest BP achieved after standing
Peak HR = the local maximum HR achieved after standing
CBv Recovery = the first local maximum of mean CBv after standing
BP Recovery = the first local maximum of BP after standing
Min HR (HR Recovery) = the lowest HR after standing
Steady-State HR = HR increases following recovery, at steady-state slope turns downward computed by the running slope differences/t test technique of Zuo et al. (24).
Averaged HR, BP, CBv, ETCO2, and time from standing ± standard deviation were tabulated for each event using a moving average window with a 3 s aperture in Table 1 for POTS-↓CO2, POTS-nlCO2, and control. The start of standing was referenced as time 0. Anticipatory changes in HR, BP, and CBv were measured during the 10 s preceding standing, then averaged, and presented as time 0 (“Standing”). Graphical data in Fig. 2 compare arterial blood pressure, HR, CBv, and ETCO2 at events for representative subjects from each group. Graphical data in Fig. 3 compares mean arterial blood pressure (MAP), mean CBv, and mean cerebral conductance at events for each group averaged over subjects. Cerebral conductance was calculated from the ratio of mean CBv to MAP for each subject at every event.
Table 1.
Time of event, systolic blood pressure, mean cerebral blood velocity, and heart rate
| POTS-↓CO2 < 30 Torr (n = 26) |
POTS-nlCO2 ≥ 30 Torr (n = 28) |
Control (n = 34) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Event | Time | SBP | mCBv | ETCO2 | HR | Time | SBP | mCBv | ETCO2 | HR | Time | SBP | mCBv | ETCO2 | HR |
| Baseline | 116 ± 3 | 76 ± 3 | 36 ± 1*# | 83 ± 4* | 115 ± 4 | 76 ± 2 | 41 ± 1 | 75 ± 3 | 115 ± 4 | 77 ± 2 | 40 ± 2 | 72 ± 2 | |||
| Stand | 0 | 124 ± 3* | 64 ± 3*# | 35 ± 2 | 94 ± 4*# | 0 | 123 ± 3* | 85 ± 2 | 39 ± 2 | 80 ± 3 | 0 | 116 ± 3 | 80 ± 2 | 39 ± 2 | 82 ± 4 |
| CBFv nadir | 9 ± 1 | 83 ± 4 | 48 ± 3*# | 27 ± 2*# | 116 ± 3* | 10 ± 1 | 86 ± 5 | 61 ± 3 | 37 ± 1 | 106 ± 5 | 9 ± 3 | 84 ± 7 | 60 ± 2 | 36 ± 2 | 105 ± 4 |
| BP nadir | 12 ± 1 | 73 ± 2* | 52 ± 3*# | 30 ± 2*# | 117 ± 3* | 12 ± 1 | 73 ± 3* | 64 ± 3 | 37 ± 1 | 112 ± 7 | 10 ± 1 | 82 ± 3 | 61 ± 3 | 37 ± 2 | 106 ± 4 |
| HR peak | 14 ± 2 | 123 ± 7 | 70 ± 5 | 28 ± 1*# | 131 ± 3* | 19 ± 2 | 118 ± 6 | 82 ± 4 | 36 ± 1 | 127 ± 3* | 15 ± 1 | 111 ± 5 | 78 ± 3 | 35 ± 2 | 116 ± 4 |
| CBv recovery | 18 ± 1 | 88 ± 5* | 72 ± 4*# | 27 ± 2*# | 127 ± 4*# | 19 ± 1 | 95 ± 5* | 88 ± 4 | 36 ± 1 | 113 ± 5 | 16 ± 1 | 121 ± 7 | 88 ± 4 | 35 ± 2 | 100 ± 5 |
| BP recovery | 21 ± 2 | 150 ± 6* | 62 ± 5* | 28 ± 2*# | 110 ± 3* | 27 ± 3 | 148 ± 6 | 74 ± 4 | 37 ± 1 | 102 ± 4 | 21 ± 1 | 139 ± 7 | 76 ± 4 | 36 ± 2 | 91 ± 6 |
| HR recovery | 42 ± 8* | 140 ± 6*# | 63 ± 4*# | 28 ± 2*# | 105 ± 4*# | 39 ± 6* | 134 ± 7 | 71 ± 4 | 37 ± 1 | 87 ± 4 | 25 ± 1 | 122 ± 6 | 72 ± 4 | 37 ± 2 | 78 ± 3 |
| Steady state | 85 ± 12* | 135 ± 5*# | 55 ± 4*# | 26 ± 2*# | 115 ± 5* | 101 ± 13* | 120 ± 4 | 68 ± 4 | 37 ± 1 | 105 ± 4* | 55 ± 12 | 122 ± 4 | 72 ± 4 | 37 ± 2 | 90 ± 3 |
| End stand | 300 ± 12 | 136 ± 6*# | 56 ± 4*# | 26 ± 2*# | 116 ± 5* | 300 ± 6 | 121 ± 3 | 67 ± 5 | 35 ± 1 | 107 ± 4* | 300 ± 10 | 120 ± 3 | 71 ± 4 | 37 ± 2 | 89 ± 4 |
All values = means ± SD. Comparisons of study groups on baseline spectral analysis of heart rate and blood pressure variability were conducted using independent t tests for all postural tachycardia syndrome (POTS) vs. controls and for each POTS subgroup vs. controls. Similarly, independent t tests were used for comparing each POTS subgroup. For these univariable tests, there was no adjustment for multiple comparisons. P values are unadjusted for multiple comparisons. BP, blood pressure; CBv, cerebral blood velocity; CBFv, cerebral blood flow velocity; ETCO2, end-tidal CO2; HR, heart rate; mCBv, mean cerebral blood velocity; POTS-↓CO2, postural tachycardia syndrome with low carbon dioxide; POTS-nlCO2, postural tachycardia syndrome with normal upright carbon dioxide; SBP, systolic blood pressure.
P < 0.05 compared with controls; #P < 0.05 compared with baseline.
Figure 2.
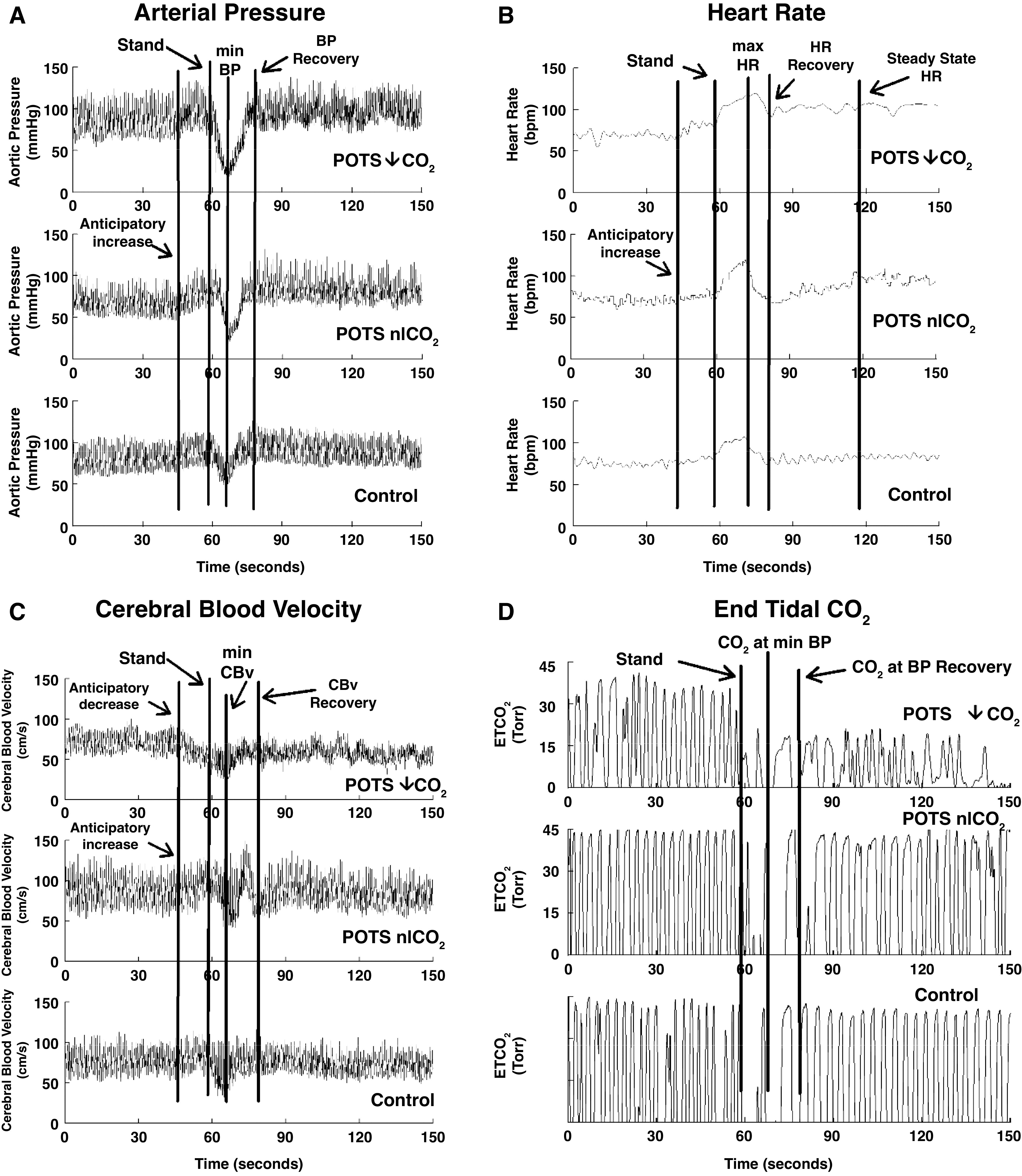
Representative tracings in blood pressure (BP, A), heart rate (HR, B), cerebral blood velocity (CBv, C), and end-tidal CO2 (ETCO2, D) from (top to bottom) postural tachycardia syndrome with low ETCO2 (POTS-↓ETCO2), postural tachycardia syndrome with normal upright ETCO2 (POTS-nlETCO2), and healthy control subjects. Anticipatory increases in BP and HR were present in all. CBv increased in POTS-nlETCO2 but decreased in POTS-↓ETCO2.
Figure 3.

Mean arterial pressure (MAP, A), mean cerebral blood velocity (mCBv, B), and cerebral conductance (C) for postural tachycardia syndrome with low end-tidal carbon dioxide (POTS-↓ETCO2) (in gray), postural tachycardia syndrome with normal upright end-tidal carbon dioxide (POTS-nlETCO2) (in red), and Control (in black). Reduced conductance maintains decreased mCBv in POTS-↓ETCO2. While the middle panel shows no significant differences for a group-by-time interaction, however, main effects for group and time were significant (P < 0.05). The bottom panel shows a significant group-by-time interaction for conductance (P < 0.05). Specifically, the POTS ↓CO2 had significantly a lower mean conductance value at the blood pressure (BP)min time point (adjusted P < 0.01).
HR variability, BP variability, and cardiovagal baroreflex gain.
We previously showed reduced HRV and gain at the operating point of the cardiovagal baroreflex (25). We repeated these calculations in POTS-↓CO2, POTS-nlCO2, and controls using custom software to collect data for at least 10 min (the “baseline period”) based on the spontaneously fluctuating R-R interval and systolic BP signals. For power spectra analysis, we used an autoregressive model based on the methods by Montano et al. (26) and Pagani et al. (27). Signals recorded as discrete time series were converted to an impulse train with equal intervals (28) and we computed the autoregressive power spectra (29). The Yule–Walker equation, modified by the Levinson-Durbin algorithm and succeeded by Anderson’s test, used Akaike’s final prediction error minimization to find the model order. The resultant interval spectrum was transformed to the spectrum of counts via division by the mean signal sequence (28). The defined frequency band total power (0.05–0.40 Hz) was used to calculate individual spectral powers for the low-frequency (LF, 0.05–0.15 Hz), and high-frequency (HF, 0.15–0.40 Hz) power bands. We used the ratio of LF RR interval power to LF BP power [the α index] to describe changes in the cardiovagal baroreflex sensitivity at the operating point (25). HF power corresponds to the respiratory sinus arrhythmia and is vagal dependent but only partly baroreflex dependent (30). LF BP power corresponds to arterial vasomotion and is sometimes used as an index of sympathetic activity (31). LF RR interval power depends on LF BP power transduced by the cardiovagal baroreflex (32). These results are presented in Table 2.
Table 2.
Baseline spectral analysis of heart rate and blood pressure variability
| All Controls | All POTS | Pots ↓ETCO2 | POTS nlETCO2 | |
|---|---|---|---|---|
| Total HRV | 2,838 ± 470 | 1,066 ± 119* | 835 ± 118* | 1,208 ± 125*# |
| LF HRV | 754 ± 143 | 379 ± 65* | 247 ± 58* | 479 ± 96*# |
| HF HRV | 1,444 ± 297 | 389 ± 51* | 339 ± 106* | 428 ± 62* |
| Total BPV | 10.3 ± 2.0 | 11.8 ± 1.3 | 9.4 ± 1.8 | 13.1 ± 1.7 |
| LF BPV | 2.5 ± 0.3 | 3.0 ± 0.3 | 2.9 ± 0.4 | 3.0 ± 0.3 |
| HF BPV | 1.5 ± 0.2 | 1.3 ± 0.1 | 1.5 ± 0.2 | 1.3 ± 0.2 |
| Alpha index | 20.5 ± 2.1 | 15.4 ± 2.0 | 10.9 ± 2.0* | 17.3 ± 2.1# |
Pearson’s correlation was used to assess the linear associations for the spectral analyses. Comparison of study groups on baseline spectral analysis of heart rate and blood pressure variability was conducted using independent t tests for all postural tachycardia syndrome (POTS) vs. controls and for each POTS subgroup vs. controls. Similarly, independent t tests were used for comparing each POTS subgroup. For these univariable tests, there was no adjustment for multiple comparisons. P values are unadjusted for multiple comparisons. HF BPV, high-frequency blood pressure variability; HF HRV, high-frequency heart rate variability; HRV, heart rate variability; LF BPV, low-frequency blood pressure variability; LF HRV, low-frequency heart rate variability; POTS-↓ETCO2, postural tachycardia syndrome with low end-tidal carbon dioxide; POTS-nlETCO2, postural tachycardia syndrome with normal upright end-tidal carbon dioxide.
P < 0.05 compared with all controls; #P < 0.05 ↓ETCO2 compared with POTS nlETCO2.
Statistical methods.
For Tables 1 and 2, comparisons of study groups on baseline spectral analysis of heart rate and blood pressure variability were conducted using independent t tests for all POTS versus controls and for each POTS subgroup versus controls. Similarly, independent t tests were used for comparing each POTS subgroup. For these univariable tests, there was no adjustment for multiple comparisons. For Fig. 3, comparison of POTS groups and controls over the time-ordered events was analyzed as repeated-measures ANOVA (rmANOVA), with time-ordered events considered as a fixed effect. Three dependent variables were considered: MAP, mean CBv, and conductance at each event. Each dependent variable was assessed in a separate model. For these models, we assumed a covariance structure of compound symmetry. To determine whether the pattern of responses over the periods differed between patients with POTS-↓CO2, POTS-nlCO2 and controls, we evaluated a group-by-period interaction term. P values for the interaction term were adjusted using the Greenhouse–Geisser correction. If the interaction term was significant, post hoc comparisons of the study groups used Scheffe’s test to control for multiple comparisons. Significance was established at P < 0.05. To reduce inter-rater variability, all data were collected by the research technician and were analyzed by one investigator who was blinded to subject group membership. Results were calculated using SPSS version 15.
RESULTS
Figure 2 shows representative tracings of (left to right) arterial pressure, HR, CBv, and ETCO2 from among (top to bottom) POTS-↓ETCO2 (n = 26), POTS-nlETCO2 (n = 28), and healthy control (n = 34) subjects. HR also increased in all during anticipatory period. CBv increased in POTS-nlETCO2 and control during the anticipatory period as expected (33, 34) but decreased in POTS-↓ETCO2 despite increasing BP. The overall decrease of CBv from Stand to nadir of CBv was consequently largest in the patient with POTS-↓ETCO2.
Timing of Events Is Prolonged in POTS Compared with Control
Compilation of the data for all subjects is shown in Table 1, and all showed anticipatory increases in BP preceding standing that were smallest in control. Table 1 shows event times, systolic blood pressure (SBP) ± SD, mean CBv ± SD, ETCO2 ± SD, and HR ± SD as dependent variables using the ordinal event scale in POTS-↓ETCO2, POTS-nlETCO2, and healthy control subjects. The time to achieve HR Recovery and Steady-State HR was significantly prolonged for POTS following stand compared with control subjects (P < 0.05).
The BP Nadir Was Lower in All POTS but Afterward Higher in POTS-↓ETCO2. HR Was Increased for All Events for POTS-↓ETCO2
Systolic blood pressure at the Stand was higher (P < 0.05), but at BP Nadir and CBv Recovery was lower in all POTS compared with control (P < 0.05), whereas SBP was thereafter only increased for POTS-↓ETCO2 compared with POTS-nlETCO2 and control (P < 0.05). HR was increased at all events for POTS-↓ETCO2 compared with control (P < 0.05). HR was increased above control for POTS-nlETCO2 at steady state and at the 5-min event (end stand) (P < 0.05).
ETCO2 Was Decreased in POTS-↓ETCO2 at Baseline but Decreased CBv Beginning at Stand
Mean CBv was decreased beginning at Stand for POTS-↓ETCO2 compared with either POTS-nlETCO2 or control (P < 0.05); reduced CBv was initiated preceding the onset of hypocapnia. ETCO2 was decreased in POTS-↓ETCO2 by design but was also significantly reduced at baseline without difference in baseline CBv (P < 0.05).
Mean Arterial Pressure Was Lower than Control in POTS-↓ETCO2 and POTS-nlETCO2 at the BP Nadir but Elevated in POTS-nlETCO2 Thereafter. Mean CBv and Conductance Were Reduced following Stand in POTS-↓ETCO2
Figure 3 demonstrates the dominant role of reduced cerebral blood velocity conductance in maintaining decreased mean CBv (mCBv) in POTS-↓ETCO2. Orthostatic hypertension accompanied upright hypocapnia. As shown in Fig. 3A, no significant differences emerged for main effects of the study group and time or for a group-by-time interaction. Figure 3B shows no significant differences for a group-by-time interaction, however, main effects for group and time were significant (P < 0.05). Of interest, the POTS ↓CO2 group had a lower mean CBv value over all time points compared with the other two study groups (adjusted P < 0.05). Finally, Fig. 3C shows a significant group-by-time interaction for conductance (P < 0.05). Specifically, the POTS ↓CO2 had significantly a lower mean conductance value at the BPmin time point (adjusted P < 0.01).
Baseline Baroreflex Gain as Measured by the α Index Was Reduced for POTS-↓ETCO2 Compared with POTS-nlETCO2 and Control
Measures of HR and BP variability were used to investigate autonomic and baroreflex-related parasympathetic modulation in the frequency domain. Table 2 shows measures of HRV and BPV. The ratio of HR to BP power (the α index of baroreflex gain) and HF-HRV (measures respiratory sinus arrhythmia) reflect different aspects of parasympathetic activity. Total HRV and HF-HRV—are low for all POTS compared with control (P < 0.5) signifying reduced vagal modulation of HR. HF-HRV represents the respiratory sinus arrhythmia driven largely by vagal afferents arising from pulmonary stretch receptors and arterial baroreflex, plus intrinsic rhythmicity of brain respiratory centers (35). The α index, representing baseline cardiovagal baroreflex gain at the operating point of the sigmoidal baroreflex curve, was significantly decreased (P < 0.05) only in POTS-↓ETCO2. A correlational analysis of α and mean CBv resulted in a significant Pearson correlation coefficient of 0.61 (P < 0.001).
DISCUSSION
This study determined whether iOH drives hypocapnia in POTS by low BP or decreased CBv. We had hypothesized that hypocapnic POTS is driven by the high frequency of iOH in POTS on standing producing lower BP. Our data disprove this hypothesis as there is no direct dependence of hypocapnia on the degree of initial hypotension in POTS. However, reduced minimum CBv during early standing was highly related to hypocapnia in POTS, potentially by sensitizing and activating the carotid body via intermittent ischemic hypoxia. We had also hypothesized that an excessive fall in CBv may occur in part from a reduction of CBv during central command in anticipation of standing that is autonomically driven. Our data are consistent with this hypothesis and demonstrate a significant correlation with impaired parasympathetic regulation.
The major findings of this study are that postural hypocapnia in POTS is preceded by a reduction in cerebral conductance and CBv in the middle cerebral artery and that reduced CBv, and conductance begin before the physical act of standing, likely initiated via central command, i.e., descending signals from higher brain centers capable of influencing cardiovascular and respiratory responses, and are maintained throughout the initial standing response. Since iOH occurs with similar frequency in POTS-↓ETCO2 and POTS-nlETCO2, it is unlikely that hypocapnic POTS is driven directly by the Standing reduction in BP. The resulting reduced CBv and cerebral conductance are subsequently sustained by hypocapnia.
Evidence suggests that reduced blood flow in the middle cerebral artery reflects reduced blood flow in the internal carotid artery and also reduced blood flow to the carotid body (36, 37). Reduced blood flow to the carotid body decreases oxygen delivery and results in ischemic or stagnant hypoxia, often seen in heart failure (38). Ischemic hypoxia would occur intermittently upon standing in POTS-↓ETCO2. Intermittent hypoxia is a potent sensitizer of the carotid body chemoreflex (4, 39) and we therefore speculate that this results in hyperventilation and hypocapnia. Hypocapnia also interferes with the central CO2-sensitive chemoreflex (7, 40). Our determination that baseline ETCO2 is reduced in POTS-↓ETCO2 despite no difference in baseline CBv or conductance supports the hypothesis of carotid body sensitization.
Thus, based on our data, we hypothesize that an aberrant response to central command results in the development of hypocapnic POTS. The response of control subjects presumably occurs via a “normal” anticipatory central command response to standing (as a form of exercise), which includes an increase in BP, HR, and CBv (41). A similar response is seen in POTS-nlETCO2. Changes of CBv when BP is increased at the Standing event represents a complex interaction among opposing effects: direct effects of central command should increase CBv as occurs during the initiation of exercise (42), myogenic autoregulation of CBF should constrict cerebral vasculature in response to increased BP to maintain CBF relatively constant (43), and arterial baroreflexes should enhance parasympathetic activity and withdraw sympathetic activity at the level of the cerebral and pial arteries (44). Sympathetic fibers regulate cerebral blood flow only under conditions of adrenergic activation/deactivation (45) and otherwise have controversial effects (46). Muscarinic parasympathetic fibers have also been shown to be relatively ineffectual in producing cerebral artery vasodilation in humans (47). But parasympathetic innervation of cerebral arteries is particularly dense and relies upon cotransmitters in addition to or in preference to acetylcholine (48). Such “noncholinergic parasympathetic nerves” produce diverse vasoactive substances including nitrergic nitric oxide (NO), vasoactive intestinal polypeptide and pituitary adenylate cyclase pathway-activating peptide, with NO functioning as a common vasodilator pathway (49, 50). Thus, increased BP via central command should activate nitrergic nerves producing cerebral vasodilation and enhanced cerebral blood flow—as observed in Control and in POTS-nlCO2, but not in POTS-↓CO2.
Past work suggested that a subset of POTS has a defect in nitrergic parasympathetic activity (51) and blunted cerebral response to NO (52). However, those patients were not sorted by eucapneic or hypocapnic orthostatic responses. In the present study, we have shown that reduced cerebral conductance and postural hypocapnia correlates with decreased α index indicative of parasympathetic baroreflex deficiency in POTS (25). Thus, patients with POTS-↓ETCO2 fail to dilate cerebral vessels, cerebral conductance is reduced, and cerebral blood flow falls.
Therefore, postural hypocapnia does not directly depend on the magnitude of iOH, but rather on the interactions of central command with defective arterial baroreflex-mediated cerebral vasodilation. Anticipatory effects of initial standing on reducing cerebral blood flow are key. We surmise that upon standing in POTS-nlCO2 and control subjects, cerebral blood flow is insufficiently reduced to sensitize carotid body chemoreflexes because nitrergic parasympathetic cerebral innervation is intact.
Limitations
Subjects for this study were recruited as referred to our clinic for evaluation of suspected POTS. There may be recruitment bias that is evident in the age range studied. Therefore, we have no specific insight into how this subgroup of patients with POTS fits into the larger population of patients affected by POTS. Our data are from subjects of a restricted age range and thus conclusions of this study cannot be generalized to different ages.
Transcranial Doppler measures blood velocity through specific large cerebral arteries. The middle cerebral artery was used because it derives its blood flow almost entirely from the carotid artery as does the carotid body. Evidence supports the hypothesis that MCA flow reflects carotid body flow, and that reduced MCA may imply stagnant ischemia of the carotid body. Direct noninvasive measurement of carotid body blood flow is not feasible. We also did not measure transcranial Doppler in both hemispheres because previous work showed that middle cerebral artery CBv was not different between the hemispheres during orthostatic stress (53). Transcranial Doppler only measures cerebral blood velocity rather than flow. However, changes in CBv are thought to accurately reflect changes in cerebral blood flow during orthostasis (54).
Carotid body sensitization was not directly assessed via measurement of the hypoxic ventilatory response. This is a limitation to the explanation of results. At this time, we lacked appropriate instrumentation to perform such testing. Going forward, hypoxic and hyperoxic responses will be tested as well as hypercapnic + hyperoxic responses to better test carotid body sensitization hypothesis and the effects on central chemoreflexes.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
Funding for this project was provided by National Heart Lung and Blood Institute (NHLBI) under Grants RO1 HL134674 and 1R56 HL162752.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.M.S. and M.S.M. conceived and designed research; performed experiments; analyzed data; interpreted results of experiments; prepared figures; drafted manuscript; edited and revised manuscript; approved final version of manuscript.
REFERENCES
- 1. Marshall JM. Peripheral chemoreceptors and cardiovascular regulation. Physiol Rev 74: 543–594, 1994. doi: 10.1152/physrev.1994.74.3.543. [DOI] [PubMed] [Google Scholar]
- 2. Del Pozzi AT, Schwartz CE, Tewari D, Medow MS, Stewart JM. Reduced cerebral blood flow with orthostasis precedes hypocapnic hyperpnea, sympathetic activation, and postural tachycardia syndrome. Hypertension 63: 1302–1308, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stewart JM, Pianosi PT. Postural orthostatic tachycardia syndrome: a respiratory disorder? Curr Res Physiol 4: 1–6, 2021. doi: 10.1016/j.crphys.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prabhakar NR, Kumar GK. Mechanisms of sympathetic activation and blood pressure elevation by intermittent hypoxia. Respir Physiol Neurobiol 174: 156–161, 2010. doi: 10.1016/j.resp.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stewart JM, Pianosi P, Shaban MA, Terilli C, Svistunova M, Visintainer P, Medow MS. Hemodynamic characteristics of postural hyperventilation: POTS with hyperventilation versus panic versus voluntary hyperventilation. J Appl Physiol (1985) 125: 1396–1403, 2018. doi: 10.1152/japplphysiol.00377.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taneja I, Medow MS, Clarke DA, Ocon AJ, Stewart JM. Baroreceptor unloading in postural tachycardia syndrome augments peripheral chemoreceptor sensitivity and decreases central chemoreceptor sensitivity. Am J Physiol Heart Circ Physiol 301: H173–H179, 2011. doi: 10.1152/ajpheart.01211.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guyenet PG, Bayliss DA. Neural control of breathing and CO2 homeostasis. Neuron 87: 946–961, 2015. doi: 10.1016/j.neuron.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stewart JM, Kota A, O'Donnell-Smith MB, Visintainer P, Terilli C, Medow MS. The preponderance of initial orthostatic hypotension in postural tachycardia syndrome. J Appl Physiol (1985) 129: 459–466, 2020. doi: 10.1152/japplphysiol.00540.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wieling W, Krediet CT, van Dijk N, Linzer M, Tschakovsky ME. Initial orthostatic hypotension: review of a forgotten condition. Clin Sci (Lond) 112: 157–165, 2007. doi: 10.1042/CS20060091. [DOI] [PubMed] [Google Scholar]
- 10. Sheriff DD, Nådland IH, Toska K. Role of sympathetic responses on the hemodynamic consequences of rapid changes in posture in humans. J Appl Physiol (1985) 108: 523–532, 2010. doi: 10.1152/japplphysiol.01185.2009. [DOI] [PubMed] [Google Scholar]
- 11. Sagawa K. Baroreflex control of systemic arterial pressure and vascular bed. In: Handbook of Physiology, The Cardiovascular System, edited by Shepherd JT, Abboud FM.. Bethesda, MD: American Physiological Society, 1983, p. 453–496. [Google Scholar]
- 12. Imholz BP, Wieling W, van Montfrans GA, Wesseling KH. Fifteen years experience with finger arterial pressure monitoring: assessment of the technology. Cardiovasc Res 38: 605–616, 1998. doi: 10.1016/s0008-6363(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 13. Goldstein DS, Sharabi Y. Neurogenic orthostatic hypotension: a pathophysiological approach. Circulation 119: 139–146, 2009. doi: 10.1161/CIRCULATIONAHA.108.805887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Borst C, van Brederode JF, Wieling W, van Montfrans GA, Dunning AJ. Mechanisms of initial blood pressure response to postural change. Clin Sci (Lond) 67: 321–327, 1984. doi: 10.1042/cs0670321. [DOI] [PubMed] [Google Scholar]
- 15. Ocon AJ, Medow MS, Taneja I, Clarke D, Stewart JM. Decreased upright cerebral blood flow and cerebral autoregulation in normocapnic postural tachycardia syndrome. Am J Physiol Heart Circ Physiol 297: H664–H673, 2009. doi: 10.1152/ajpheart.00138.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goodwin GM, McCloskey DI, Mitchell JH. Cardiovascular and respiratory responses to changes in central command during isometric exercise at constant muscle tension. J Physiol 226: 173–190, 1972. doi: 10.1113/jphysiol.1972.sp009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Williamson JW, Fadel PJ, Mitchell JH. New insights into central cardiovascular control during exercise in humans: a central command update. Exp Physiol 91: 51–58, 2006. doi: 10.1113/expphysiol.2005.032037. [DOI] [PubMed] [Google Scholar]
- 18. Stewart JM, Boris JR, Chelimsky G, Fischer PR, Fortunato JE, Grubb BP, Heyer GL, Jarjour IT, Medow MS, Numan MT, Pianosi PT, Singer W, Tarbell S, Chelimsky TC; Pediatric Writing Group of the American Autonomic Society. Pediatric disorders of orthostatic intolerance. Pediatrics 141: e20171673, 2018. doi: 10.1542/peds.2017-1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Low PA, Opfer-Gehrking TL, McPhee BR, Fealey RD, Benarroch EE, Willner CL, Suarez GA, Proper CJ, Felten JA, Huck CA. Prospective evaluation of clinical characteristics of orthostatic hypotension. Mayo Clin Proc 70: 617–622, 1995. doi: 10.4065/70.7.617. [DOI] [PubMed] [Google Scholar]
- 20. Sheldon RS, Grubb BP, Olshansky B, Shen W-K, Calkins H, Brignole M, Raj SR, Krahn AD, Morillo CA, Stewart JM, Sutton R, Sandroni P, Friday KJ, Hachul DT, Cohen MI, Lau DH, Mayuga KA, Moak JP, Sandhu RK, Kanjwal K. 2015 Heart rhythm society expert consensus statement on the diagnosis and treatment of postural tachycardia syndrome, inappropriate sinus tachycardia, and vasovagal syncope. Heart Rhythm 12: e41–e63, 2015. doi: 10.1016/j.hrthm.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Plash WB, Diedrich A, Biaggioni I, Garland EM, Paranjape SY, Black BK, Dupont WD, Raj SR. Diagnosing postural tachycardia syndrome: comparison of tilt testing compared with standing haemodynamics. Clin Sci (Lond) 124: 109–114, 2013. doi: 10.1042/CS20120276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singer W, Sletten DM, Opfer-Gehrking TL, Brands CK, Fischer PR, Low PA. Postural tachycardia in children and adolescents: what is abnormal? J Pediatr 160: 222–226, 2012. doi: 10.1016/j.jpeds.2011.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Imholz BP, Settels JJ, van der Meiracker AH, Wesseling KH, Wieling W. Non-invasive continuous finger blood pressure measurement during orthostatic stress compared to intra-arterial pressure. Cardiovasc Res 24: 214–221, 1990. doi: 10.1093/cvr/24.3.214. [DOI] [PubMed] [Google Scholar]
- 24. Zuo B, Li J, Sun C, Zhou X. A new statistical method for detecting trend turning. Theor Appl Climatol 138: 201–213, 2019. doi: 10.1007/s00704-019-02817-9. [DOI] [Google Scholar]
- 25. Stewart JM, Warsy IA, Visintainer P, Terilli C, Medow MS. Supine parasympathetic withdrawal and upright sympathetic activation underly abnormalities of the baroreflex in postural tachycardia syndrome. Hypertension 77: 1234–1244, 2021. doi: 10.1161/HYPERTENSIONAHA.120.16113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Montano N, Ruscone TG, Porta A, Lombardi F, Pagani M, Malliani A. Power spectrum analysis of heart rate variability to assess the changes in sympathovagal balance during graded orthostatic tilt. Circulation 90: 1826–1831, 1994. doi: 10.1161/01.cir.90.4.1826. [DOI] [PubMed] [Google Scholar]
- 27. Pagani M, Montano N, Porta A, Malliani A, Abboud FM, Birkett C, Somers VK. Relationship between spectral components of cardiovascular variabilities and direct measures of muscle sympathetic nerve activity in humans. Circulation 95: 1441–1448, 1997. doi: 10.1161/01.cir.95.6.1441. [DOI] [PubMed] [Google Scholar]
- 28. DeBoer RW, Karemaker JM, Strackee J. Comparing spectra of a series of point events particularly for heart rate variability data. IEEE Trans Biomed Eng 31: 384–387, 1984. doi: 10.1109/TBME.1984.325351. [DOI] [PubMed] [Google Scholar]
- 29. Kay SM, Marple SL Jr.. Spectrum analysis—a modern perspective. Proc IEEE 69: 1380–1419, 1981. doi: 10.1109/PROC.1981.12184. [DOI] [Google Scholar]
- 30. Eckberg DL. Human sinus arrhythmia as an index of vagal cardiac outflow. J Appl Physiol Respir Environ Exerc Physiol 54: 961–966, 1983. doi: 10.1152/jappl.1983.54.4.961. [DOI] [PubMed] [Google Scholar]
- 31. Okamoto LE, Raj SR, Gamboa A, Shibao CA, Arnold AC, Garland EM, Black BK, Farley G, Diedrich A, Biaggioni I. Sympathetic activation is associated with increased IL-6, but not CRP in the absence of obesity: lessons from postural tachycardia syndrome and obesity. Am J Physiol Heart Circ Physiol 309: H2098–H2107, 2015. doi: 10.1152/ajpheart.00409.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goldstein DS, Bentho O, Park MY, Sharabi Y. Low-frequency power of heart rate variability is not a measure of cardiac sympathetic tone but may be a measure of modulation of cardiac autonomic outflows by baroreflexes. Exp Physiol 96: 1255–1261, 2011. doi: 10.1113/expphysiol.2010.056259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsukawa K, Asahara R, Uzumaki M, Hashiguchi Y, Ishii K, Wang J, Smith SA. Central command-related increases in blood velocity of anterior cerebral artery and prefrontal oxygenation at the onset of voluntary tapping. Am J Physiol Heart Circ Physiol 321: H518–H531, 2021. doi: 10.1152/ajpheart.00062.2021. [DOI] [PubMed] [Google Scholar]
- 34. Vianna LC, Araújo CGS, Fisher JP. Influence of central command and muscle afferent activation on anterior cerebral artery blood velocity responses to calf exercise in humans. J Appl Physiol (1985) 107: 1113–1120, 2009. doi: 10.1152/japplphysiol.00480.2009. [DOI] [PubMed] [Google Scholar]
- 35. De Burgh Daly BM. Interactions between respiration and circulation. In: Handbook of Physiology The Respiratory System. Control of Breathing, edited by Fishman AP, Fisher AB, Geiger R. Bethesda, MD: American Physiological Society, 1986, p. 529–594. [Google Scholar]
- 36. Ding Y, Li YL, Schultz HD. Role of blood flow in carotid body chemoreflex function in heart failure 1. J Physiol 589: 245–258, 2011. doi: 10.1113/jphysiol.2010.200584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iturriaga R, Alcayaga J, Zapata P. Contribution of carotid body chemoreceptors and carotid sinus baroreceptors to the ventilatory and circulatory reflexes produced by common carotid occlusion. Acta Physiol Pharmacol Latinoam 38: 27–48, 1988. [PubMed] [Google Scholar]
- 38. Schultz HD, Li YL. Carotid body function in heart failure. Respir Physiol Neurobiol 157: 171–185, 2007. doi: 10.1016/j.resp.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leuenberger UA, Brubaker D, Quraishi SA, Hogeman CS, Imadojemu VA, Gray KS. Effects of intermittent hypoxia on sympathetic activity and blood pressure in humans. Auton Neurosci 121: 87–93, 2005. [Erratum in Auton Neurosci 183: 120, 2014]. doi: 10.1016/j.autneu.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 40. Nattie E, Li A. Central chemoreceptors: locations and functions. Compr Physiol 2: 221–254, 2012. doi: 10.1002/cphy.c100083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sato K, Moriyama M, Sadamoto T. Influence of central command on cerebral blood flow at the onset of exercise in women. Exp Physiol 94: 1139–1146, 2009. doi: 10.1113/expphysiol.2009.048587. [DOI] [PubMed] [Google Scholar]
- 42. Williamson JW. The relevance of central command for the neural cardiovascular control of exercise. Exp Physiol 95: 1043–1048, 2010. doi: 10.1113/expphysiol.2009.051870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Harder DR. Pressure-dependent membrane depolarization in cat middle cerebral artery. Circ Res 55: 197–202, 1984. doi: 10.1161/01.res.55.2.197. [DOI] [PubMed] [Google Scholar]
- 44. van Lieshout JJ, Secher NH. Point: Counterpoint: sympathetic activity does/does not influence cerebral blood flow. Point: sympathetic activity does influence cerebral blood flow. J Appl Physiol (1985) 105: 1364–1366, 2008. doi: 10.1152/japplphysiol.90597.2008. [DOI] [PubMed] [Google Scholar]
- 45. Talman WT, Dragon DN, Ohta H. Baroreflexes influence autoregulation of cerebral blood flow during hypertension. Am J Physiol Heart Circ Physiol 267: H1183–H1189, 1994. doi: 10.1152/ajpheart.1994.267.3.H1183. [DOI] [PubMed] [Google Scholar]
- 46. Strandgaard S, Sigurdsson ST. Point:Counterpoint: sympathetic activity does/does not influence cerebral blood flow. Counterpoint: sympathetic nerve activity does not influence cerebral blood flow. J Appl Physiol (1985) 105: 1366–1367, 2008. doi: 10.1152/japplphysiol.90597.2008a. [DOI] [PubMed] [Google Scholar]
- 47. Hamner JW, Tan CO, Tzeng YC, Taylor JA. Cholinergic control of the cerebral vasculature in humans. J Physiol 590: 6343–6352, 2012. doi: 10.1113/jphysiol.2012.245100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol (1985) 100: 1059–1064, 2006. doi: 10.1152/japplphysiol.00954.2005. [DOI] [PubMed] [Google Scholar]
- 49. Roloff EV, Tomiak‐Baquero AM, Kasparov S, Paton JF. Parasympathetic innervation of vertebrobasilar arteries: is this a potential clinical target? J Physiol 594: 6463–6485, 2016. doi: 10.1113/JP272450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toda N, Ayajiki K, Okamura T. Cerebral blood flow regulation by nitric oxide: recent advances. Pharmacol Rev 61: 62–97, 2009. doi: 10.1124/pr.108.000547. [DOI] [PubMed] [Google Scholar]
- 51. Stewart JM, Medow MS, Minson CT, Taneja I. Cutaneous neuronal nitric oxide is specifically decreased in postural tachycardia syndrome. Am J Physiol Heart Circ Physiol 293: H2161–H2167, 2007. doi: 10.1152/ajpheart.00600.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stewart JM, Medow MS, DelPozzi A, Messer ZR, Terilli C, Schwartz CE. Middle cerebral O2 delivery during the modified Oxford maneuver increases with sodium nitroprusside and decreases during phenylephrine. Am J Physiol Heart Circ Physiol 304: H1576–H1583, 2013. doi: 10.1152/ajpheart.00114.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ocon AJ, Messer ZR, Medow MS, Stewart JM. Increasing orthostatic stress impairs neurocognitive functioning in chronic fatigue syndrome with postural tachycardia syndrome. Clin Sci (Lond) 122: 227–238, 2012. doi: 10.1042/CS20110241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke 31: 1672–1678, 2000. doi: 10.1161/01.str.31.7.1672. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.