

Abstract
Proliferating cancer cells secrete a multitude of factors impacting metabolism, interorgan communication, and tumor progression. The distribution of tumor-derived factors to distant organs occurs via the circulation, which provides an extensive reactive surface lined by endothelial cells. Primary tumor-derived proteins impact cancer progression by modulating endothelial cell activation at the (pre-)metastatic niche, which affects tumor cell dissemination as well as the outgrowth of seeded metastatic cells into overt tumors. In addition, new insight indicates that endothelial cell signaling contributes to metabolic symptoms of cancer, including cancer-associated cachexia, opening a new field of vascular metabolism research. This review addresses how tumor-derived factors systemically affect endothelial cell signaling and activation and impact distant organs as well as tumor progression.
Keywords: angiocrine signaling, cachexia, metastasis, systemic signaling, vascular endothelium
Introduction
The notion that endothelial cells (ECs) are merely inactive, at best responsive cells lining the inner surface of blood vessels, has fundamentally changed in recent years. Instead, ECs are increasingly recognized as highly dynamic instructive regulators and modulators of the local microenvironment, creating a unique vascular niche during homeostatic and pathological conditions (1, 2). Recent breakthrough discoveries have further advanced the concepts of angiocrine signaling, EC-derived instructive signals, beyond locally acting paracrine signaling mechanisms toward systemic signaling pathways communicating through the circulation as a regulator of systemic homeostasis (3, 4).
The importance of ECs within the tumor stroma has been highlighted in numerous authoritative reviews in recent years (5, 6). ECs do not only nourish the primary tumor by growing new blood vessels upon angiogenic stimulation but provide a platform for secreted and membrane-bound factors, which interact with cancer cells and contribute to the formation of a growth-promoting tumor-microenvironment. Beyond its local regulatory role, EC-derived angiocrine signaling at distant sites can also affect organ function. Notably, primary tumor-secreted factors, which can act far beyond the tumor boundaries, may systemically affect ECs of distant organs (7). The importance of studying the systemic effects of tumor cells is highlighted by the fact that most cancer-related deaths are due to the systemic secondary impact of the primary tumor such as metastasis, cachexia, and thrombosis (8–10). The effects of modified angiocrine signaling during tumor progression have been primarily studied in the context of the metastatic site; however, the vasculature also impacts processes like coagulation, inflammation, and metabolism, leading to thrombosis and cachexia (FIGURE 1). Thus, studying cancer as a systemic disease has in recent years become an important focus of research to identify and validate novel targets for cancer therapy.
FIGURE 1.

Primary tumor-derived factors systemically affect the distant vasculature to regulate metastasis, thrombosis, and cachexia Primary tumor-derived factors change endothelial cell signaling at the pre-metastatic niche to promote tumor cell dissemination and modulate angiogenesis in metastatic nodules, which affects tumor colonization. Additionally, tumor-derived factors may cause changes in organ function of cancer patients and induce thrombosis in the distant vasculature. During cachexia, cancer cells may regulate endothelial cell signaling to induce adipocyte wasting by activating lipolysis and browning. In skeletal muscle, tumor-derived factors may cause proteolysis and impaired regeneration via abnormal angiocrine signaling. Image was created with BioRender.com, with permission.
In this review, the systemic effects of tumor-derived factors on distant endothelial beds will be described. We propose the concept that ECs should be considered as independent drivers of disease progression impacting the homeostatic signaling of distant organs in response to a systemic trigger, e.g., cancer-derived factors. Angiocrine signaling modulates the host’s macroenvironment by inducing numerous organ-specific microenvironmental changes.
The Effect of Primary Tumor-Secreted Factors on ECs at the Pre-metastatic Site
Metastatic dissemination can be portrayed as a multistep succession of events that eventually leads to the outgrowth of metastatic nodules in distant tissues. As part of this process, cancer cells intravasate into nearby vessels and enter the circulation (11, 12). Subsequently, these circulating tumor cells (CTCs) enter secondary organ sites as disseminated tumor cells that may form metastases. However, these intricate steps critically depend on the local microenvironment, which determines whether tumor cell dissemination can take place. To prepare pre-metastatic sites for tumor cells, primary tumors secrete certain factors, mobilize bone marrow-derived cells (BMCs), and act on the immune system. This enables the formation of a supportive and receptive tissue microenvironment, the so-called pre-metastatic niche (13).
Cellular and molecular dissection of the pre-metastatic niche has revealed that tumor-derived factors can promote metastatic spread in specific, distant organs. Notably, primary tumor-derived vascular endothelial growth factor (VEGF) and placental growth factor (PlGF) contribute to the recruitment of CD11b+ myeloid cells to the lungs before tumor dissemination in Lewis lung carcinoma (LLC) and B16F10 melanoma preclinical mouse models. This results in enhanced recruitment of metastatic cells to the lungs by providing a permissive niche for incoming tumor cells (14). Building on these early landmark findings, the significance of the pre-metastatic niche was studied in more detail in subsequent years and key characteristics of the niche, including immunosuppression, inflammation, angiogenesis, and vascular permeability, have been described (13).
Tumor cells secreting the proinflammatory cytokine IL-1β trigger a granulocyte-colony stimulating factor (G-CSF)-dependent expansion and polarization of neutrophils. Neutrophils thereby acquire the ability to suppress cytotoxic CD8+ T cells, which in turn promotes metastasis formation (15). Moreover, an aggregation of neutrophils was detected in peripheral organs of mammary carcinoma and insulinoma-bearing mice with an accumulation of proinflammatory cytokines such as IL-1β, IL-6, and CXCL1, which contribute to the formation of the pre-metastatic niche (16). The effect of tumor-derived factors on inflammation and immunosuppression has been reviewed in detail elsewhere (13). This review will focus on the effect on ECs and the effector functions of ECs in the target organ.
To promote metastasis, primary tumors may stimulate vascular permeability and angiogenesis in the pre-metastatic niche before the arrival of metastatic tumor cells. This can be mediated via soluble factors or exosomes that are secreted by the primary tumor (13). VEGF, PlGF, transforming growth factor β (TGF-β), tumor necrosis factor α (TNF-α), and G-CSF are some of the most important factors, which have been described to be secreted by the primary tumor and to affect ECs in secondary organs. Primary tumor-derived VEGF specifically upregulates matrix metalloproteinase 9 (MMP9) in pre-metastatic lung ECs via VEGFR-1/Flt-1 tyrosine kinase and thereby promotes lung metastasis by enhancing tumor cell invasion (17). In addition, primary tumor-secreted VEGF may induce EC hyperpermeability in the lungs by accelerating occludin degradation leading to the disruption of tight junctions (18). Subsequently, the primary tumor may induce an increase in total vessel density in the pre-metastatic lung. This effect is mediated by systemically circulating VEGF, which leads to an elevated prostaglandin E2 production in mouse pulmonary microvascular ECs and thereby enhances the adhesion of metastatic 4T1 breast cancer cells (19). In addition to VEGF, PlGF is secreted by the primary tumor and can induce foci of vascular hyperpermeability in the lungs by upregulating E-selectin via FAK in ECs (20). Other factors that were shown to be induced by the primary tumor-expressed proteins VEGF, TGF-β, and TNF-α are the inflammatory chemoattractants S100A8 and S100A9. In the premetastatic lung, they can trigger the accumulation of macrophage antigen 1 (Mac1)-positive myeloid cells (21, 22). Mechanistically, S100A8 and S100A9 induce serum amyloid A 3 (SAA3), which stimulates NF-κB signaling in ECs in a Toll-like receptor 4-dependent manner and thereby facilitates metastasis (23). Further, G-CSF was shown to be overexpressed in several metastatic tumors and to expand Ly6G- and Ly6C-positive granulocytes in the premetastatic niche. These cells produce the Bv8 protein, which is implicated in angiogenesis and stimulates tumor cell migration through activation of the prokineticin receptor 1 (PKR-1) (24).
Despite secreting soluble factors to form a premetastatic niche, primary tumors can also disseminate their cargo in exosomes (25). This was first described in melanoma cells, which release exosomes to prepare sentinel lymph nodes for metastasis (26). Later, it was shown in a pancreatic ductal adenocarcinoma (PDAC) mouse model that tumor-derived exosomes can enhance metastatic burden in the liver. This effect is mediated by macrophage migration inhibitory factor (MIF), which is expressed in exosomes and increases macrophage recruitment from the bone marrow (27). In addition, EC-specific effects of tumor-derived exosomes were reported. In renal cell carcinoma, a subset of tumor-initiating cells (CD105+) releases exosomes, which trigger angiogenesis and promote the formation of a premetastatic niche. These exosomes contain proangiogenic mRNAs and miRNAs that specifically enhance VEGF, MMP2, and VEGFR1 expression in lung ECs (28). These molecules were previously shown to be essential for the formation of a lung premetastatic niche (17, 29). In addition, proteomics analysis of ovarian cancer exosomes revealed activating transcription factor 2 (ATF2) and metastasis-associated protein 1 (MTA1) as exosome-derived proteins that upregulate angiogenesis (30). Furthermore, soluble E-cadherin is released from ovarian cancer in exosomes and induces angiogenesis. Mechanistically, soluble E-cadherin heterodimerizes with VE-cadherin on ECs and thereby activates β-catenin and NF-κB signaling (31). A three-dimensional in vitro system to study the effect of primary tumor-derived factors on the premetastatic niche was introduced in the form of a liver-on-a-chip. By exploiting this system, breast cancer-derived extracellular vesicles were shown to induce endothelial-to-mesenchymal transition (EMT) in liver sinusoidal endothelial cells and destabilize the endothelial barrier, which facilitates metastatic dissemination in the liver (32).
Taken together, several soluble factors or exosomes can, either directly or via the stimulation of immune cells, enhance angiogenesis and vascular permeability at the premetastatic site and thereby prepare distant organs for metastatic colonization (FIGURE 2A).
FIGURE 2.
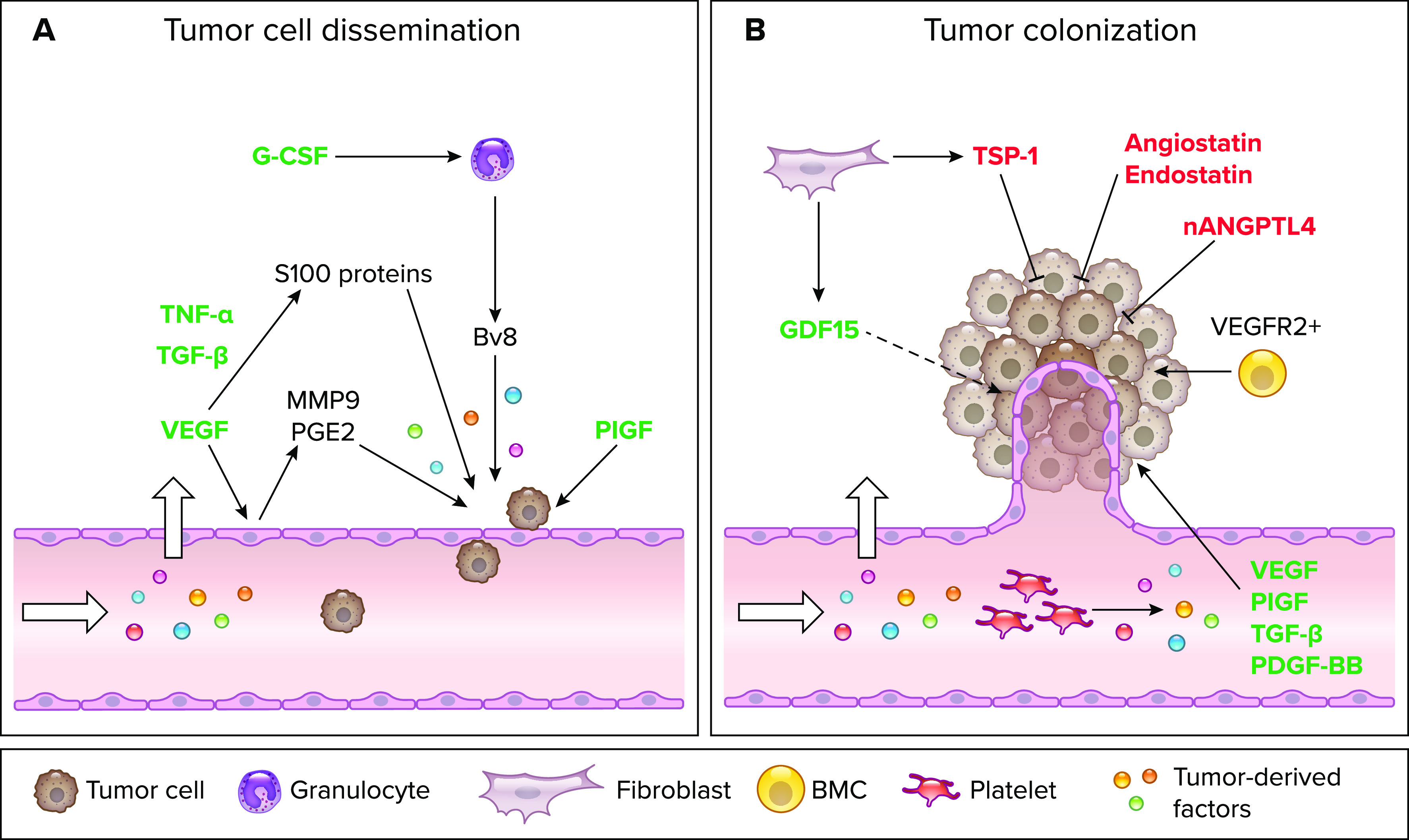
Primary tumor-derived factors affect tumor cell dissemination and colonization at distant sites A: graphical illustration of the effects of primary tumor-derived factors on endothelial cells (ECs) at the premetastatic niche. VEGF, TNF-α, transforming growth factor β (TGF-β), and placental growth factor (PlGF) act directly on ECs at the pre-metastatic niche and thereby induce EC hyperpermeability and enhance tumor cell dissemination. Granulocyte-colony stimulating factor (G-CSF) affects transmigration indirectly via expanding Ly6G+Ly6C+ granulocytes, which activate ECs by secreting Bv8. B: Schematic representation of the effects of primary tumor-derived factors on ECs at the metastatic site. VEGF, PlGF, TGF-β, and PDGF-BB are secreted by tumor-educated platelets and induce angiogenesis thereby promoting tumor colonization. VEGFR2+ bone marrow-derived cells (BMCs) are enhanced and contribute to these processes. Cancer-associated fibroblasts (CAFs) from the primary tumor secrete growth differentiation factor 15 (GDF15) and thrombospondin-1 (TSP-1), which enhance or reduce angiogenesis at the metastatic site, respectively. Molecules such as angiostatin, nANGPTL4, and endostatin are secreted by the primary tumor and directly act on ECs to reduce sprouting and thereby inhibit the outgrowth of metastatic nodules. Tumor-derived factors promoting cancer cell dissemination or colonization are depicted in green and tumor-derived factors inhibiting these processes are highlighted in red. Image was created with BioRender.com, with permission.
The Effect of Primary Tumor-Secreted Factors on Tumor ECs at the Metastatic Site
The final step of the metastatic cascade involves the outgrowth and propagation of disseminated tumor cells into overt tumors at the metastatic site. To form macroscopic metastases, the microenvi-ronment needs to be adapted to favor remodeling of the extracellular matrix, immune cell inhibition, and angiogenesis (7). There have been a number of publications reporting primary tumor-derived angiogenic factors which promote metastatic outgrowth (7, 33, 34). For example, in a luminal breast cancer model, primary tumors can modulate the systemic macroenvironment by loading circulating platelets with a repertoire of proangiogenic cytokines such as VEGF, TGF-β1, PDGF-BB, and PIGF. Subsequently, proangiogenic platelets accumulate at sites of indolent tumors where they aid vessel formation. In addition, in the presence of a primary tumor, the expression of proangiogenic VEGFR2 is enhanced on BMCs from the bone marrow, which promotes the growth of disseminated tumor cells into highly vascularized tumors (33).
A recent report has highlighted for the first time that cancer-associated fibroblasts (CAFs) promote the dissemination of tumor cells. Within this publication, the prostate cancer tumor microenvironment was shown to have a significantly higher expression of growth differentiation factor 15 (GDF15) as compared to the healthy prostate microenvironment. This was attributed to the higher expression of GDF15 in CAFs. Moreover, GDF15-expressing fibroblasts were shown to exert systemic effects on the outgrowth of distant, otherwise indolent, prostate cancer cells (34). Proangiogenic functions of GDF15 have previously been reported in different cell types. For example, in hepatocellular carcinoma, GDF15 acts via AKT, MAPK, and NF-κB signaling to enhance EC proliferation and capillary-like tube formation ability (35). Whether CAF-derived GDF15 promotes the outgrowth of distant micrometastases via a similar mechanism remains to be investigated.
In contrast, there are also a number of publications showing that the primary tumor secretes factors that inhibit the outgrowth of secondary tumor implants and metastasis (36). This phenomenon is referred to as concomitant tumor resistance (CTR) and was first described in 1906 by Ehrlich and Apolant (37). CTR has been experimentally studied in mouse models of melanoma (38, 39), breast cancer (40, 41), lung cancer (42, 43), lymphoma, and fibrosarcoma (44, 45). Different hypotheses have been proposed to explain the phenomenon of CTR. According to these hypotheses, the antimetastatic effects may be due to either the host’s immune system (immunological hypothesis) or systemically acting primary tumor-released antimitogenic and/or antiangiogenic cytokines (angiogenesis-based hypothesis). According to the immunological hypothesis, the primary tumor generates an antitumor immune response that inhibits the outgrowth of a secondary tumor implant (36). This hypothesis has been experimentally demonstrated in mouse models of melanoma and breast cancer. Hence, in a B16F10 melanoma mouse model, the primary tumor was shown to reduce metastasis formation of intravenously injected tumor cells by changing the immune landscape (38) and by reducing circulating platelets (39). In addition, in an EMT6 mammary carcinoma model, the primary tumor was shown to inhibit the metastatic outgrowth of CTCs via activation of CD8+ T cells (40). However, the immunological hypothesis does not provide a compelling explanation for the fact that CTR is also observed in nonimmunogenic tumors (46).
A nonimmunological explanation of CTR relies on the release of antiangiogenic factors by the primary tumor. The angiogenesis-based hypothesis was first proposed by O’Reilly et al. (42). In 1994, they identified the antiangiogenic, primary tumor-secreted factor angio-statin in an LLC mouse model and discovered that the lack of angiostatin after surgical removal of the primary tumor is responsible for the rapid outgrowth of distant metastasis. Mechanistically, angio-statin is supposed to inhibit EC proliferation at the metastatic site, which reduces angiogenesis (42). Subsequent studies identified another antiangiogenic factor, named endostatin, as a primary tumor-secreted factor that suppresses the growth of metastases when administered systemically (47). Endostatin is a proteolytic fragment of type XVIII collagen that induces EC apoptosis by inhibition of cyclin D1. It binds to integrin α5β1 on the surface of ECs, which downregulates the activity of RhoA GTPase leading to the disassembly of the actin cytoskeleton and thereby suppression of angiogenesis (48). Yet another angiogenesis-regulating molecule, thrombospondin-1 (TSP-1), is secreted by a fibrosarcoma primary tumor model in mice and has been proposed to inhibit angiogenesis in the cornea and the growth of metastasis (44). Following these early discoveries, Folkman proposed the contextuality of angiogenic molecules, whereby proangiogenic molecules were believed to be short-lived and locally acting, whereas antiangiogenic molecules were proposed to be long-lived and systemically acting (49, 50). This hypothesis may indeed provide an explanation for the central paradox of CTR: how can a primary tumor-secreted cytokine inhibit angiogenesis at the metastatic site, while having no effect on the primary tumor vasculature itself (36, 51)? According to Hanahan and Folkman (49), the primary tumor-secreted proangiogenic factors stay local, counteract the antiangiogenic molecules and thereby allow an angiogenic switch in the local tumor microenvironment.
Following an intense phase of hype, the field of CTR regulating molecules has made little progress in the last 20 years. This is primarily due to the fact that much of this early work did not stand the test of time and because supposedly CTR-regulating molecules proved translationally disappointing, even though genetic experiments, for example in TSP-1- or collagen XVIII-deficient mice, revealed compelling evidence for angiogenesis-regulating functions of these molecules(52–54).
The concepts of CTR have seen a recent revival with the landmark discovery showing that the primary tumor-derived NH2-terminal fragment of angiopoietin-like 4 (nANGPTL4) inhibits metastasis by reducing vascularity at the metastatic site (55). Mechanistically, nANGPTL4 downregulates Wnt signaling via the syndecan 4 (SDC4) receptor. Conversely, the COOH-terminal fragment cANGLPTL4 stimulates angiogenesis in the primary tumor in an integrin-dependent manner. As ANGPTL4 binds to integrins via the COOH-terminal fragment, the extracellular matrix serves as a reservoir and locally restricts cANGPTL4 to the primary tumor, whereas nANGPTL4 is present systemically. Differential pro- and antiangiogenic effects of the C- and the NH2-terminal fragments, regulated by differential receptor engagement, are in the case of ANGPTL4 regulated by differential expression and presentation of receptors and ligands. Notably, while the proangiogenic COOH-terminal fragment can primarily be detected in primary tumors, the NH2-terminal fragment is predominately detected in the circulation in both tumor-bearing mice and men (55).
Clearly, much more needs to be learned about the mechanisms of CTR, and it is worth reconsidering some of the older literature. For example, by comparing several mouse models of CTR, Franco et al. (45) observed that CTR appears in two peaks. The first peak occurs when the primary tumor is small and is proportional to tumor immunogenicity. The second peak appears in large tumors and does not correlate with tumor immunogenicity. However, it correlates with the activity of serum factors that inhibit the proliferation of tumor cells. This effect is not solely restricted to metastasis. It was shown that a larger primary tumor inhibits the growth of a smaller primary tumor depending on the ratio between tumor sizes: A high ratio leads to inhibition of the smaller tumor and a small ratio leads to stimulation of tumor growth (56).
Importantly, the preclinical findings of CTR are supported by the clinical observation that metastasis occurs often only after the primary tumor has been resected. For instance, pulmonary metastases were shown to be increased in patients with osteosarcoma after primary tumor removal due to an increase in angiogenesis (57). In addition, a rapid metastatic progression is observed in patients with bladder cancer upon radical cystectomy. This was shown to be due to the sudden lack of antiproliferative molecules such as endostatin, angiostatin, and TSP-1, which are highly expressed in human bladder cancer (58). In colorectal cancer patients, metastatic colonization and vascular density in the liver were shown to be increased upon primary tumor removal (59). Overall, these clinical observations validate the preclinical findings that the primary tumor actively limits the growth of metastasis by secreting antiangiogenic cytokines.
As the primary tumor has an effect on metastatic dissemination and colonization, systemic mechanisms must be more critically considered in cancer therapy. Clinical observations suggest that current treatment strategies may enhance systemic changes and eventually promote metastatic colonization. For example, G-CSF is used in cancer patients with myelosuppression induced by chemotherapy. However, as already mentioned, G-CSF induces both a neutrophil-dependent suppression of CD8+ T cells and an expansion of Ly6G+Ly6C+ granulocytes, which enhances tumor cell extravasation and angiogenesis in the pre-metastatic niche, respectively (60). In addition, antiangiogenic therapies can induce the systemic upregulation of off-target molecules such as osteopontin and G-CSF and thereby increase the extravasation potential of tumor cells or support the pre-metastatic niche. Specifically, VEGF receptor tyrosine kinase inhibitors were shown to enhance BMC recruitment to distant organs, inflammatory pathway activation, and altered EC adhesion molecule function, which eventually leads to changes in the endothelial microenvironment and increases extravasation potential for tumor cells (61). Overall, both pro- and antiangiogenic factors have been reported to be secreted by primary tumors to favor or limit metastatic outgrowth, respectively (FIGURE 2B). To optimize strategies to inhibit metastatic progression, it is essential to gain a more detailed molecular and mechanistic understanding of the interplay of the primary tumor with ECs at the meta-static site in different contexts. Toward this end, it is important to compare circulating factors in the serum of healthy versus tumor-bearing patients and to further study their effect on endothelial cells at the metastatic site.
Tumor-Induced Effects on Organs That Do Not Represent the Metastatic Site
While the majority of the scientific literature focuses on the effects of primary tumor-derived factors on metastatic sites, especially the lungs, there is much less known about the effects on the vasculature in organs that do not represent metastatic sites. However, there is solid clinical evidence that patients with cancer have a significantly increased risk of developing thrombosis. This effect is seen in many cancer types such as colorectal, pancreatic, and gastric cancer, a phenomenon commonly referred to as Trousseau syndrome (9). This has been attributed to cancer cells releasing procoagulant and fibrinolytic proteins as well as inflammatory cytokines. In addition, cancer cells are capable of directly adhering to host cells, including ECs, thereby stimulating prothrombotic properties. Eventually, some of these mechanisms, such as fibrin formation and deposition, not only promote thrombotic events but also stimulate tumor progression by creating a more favorable niche (62).
Tissue factor (TF) is the primary cellular initiator of the blood coagulation cascade, which leads to the generation of thrombin with the subsequent formation of a fibrin clot. In many tumor types, TF expression was shown to be induced. In colorectal cancer (CRC), mutations in K-ras and p53 result in constitutive activation of MAPK and phosphatidylinositol 3-kinase (PI3K) signaling, which leads to enhanced TF expression. TF affects ECs via different mechanisms. Results from mouse models of CRC revealed that TF can directly impact tumor angiogenesis through the modulation of VEGF and thrombospondin levels in ECs (63). In addition, alternatively spliced TF activates α6β1 and αVβ3 integrins on ECs and acts as a proangiogenic stimulus by activating focal adhesion kinase PI3K, MAPK, and Akt (64). Thereby, TF triggers angiogenesis in a paracrine fashion by targeting vascular cells. It also induces cytoskeleton remodeling in ECs by stimulating Rac1 and p38 expression leading to enhanced migration of ECs (65). Despite releasing soluble TF, tumor cells can also release exosomes and microparticles containing TF. These microparticles bind to sites of vascular injury and enhance thrombosis (66).
In addition to thrombosis, some cancer patients show limited organ function such as impaired heart and kidney function, which is due to vascular dysfunction. This phenomenon was attributed to the formation of neutrophil extracellular traps (NETs). NETs are extracellular networks consisting of DNA released from neutrophils together with antimicrobial peptides and proteases derived from neutrophil granules and play a role in microbial defense (16). In addition, NETs have been implicated in cancer-associated thrombosis, where tumor cell-secreted G-CSF has been shown to promote NETosis in neutrophils (67). NET accumulation leads to an upregulation of proinflammatory adhesion molecules (ICAM-1, VCAM-1, and E-selectin) as well as proinflammatory cytokines (IL-1β and IL-6) in ECs, which can trigger organ failure due to hypo-perfusion of organs in cancer patients (16). In other pathological conditions, NETs can induce endothelial damage and have cytotoxic effects on ECs. Thereby, NET formation provides a possible explanation for tumor-induced organ failure, which is a substantial cause of morbidity in cancer patients.
Angiocrine Signaling During Cancer-Associated Cachexia
Cancer progression and chronic inflammation lead to massive changes in the body’s metabolism causing an imbalance in energy expenditure. The consequential involuntary body weight loss caused by muscle as well as adipose tissue wasting is defined as cachexia and cannot be completely reversed by dietary supplementation (68). The occurrence of cancer-associated cachexia (CAC) highly depends on the tumor type and correlates with the disease stage (69). Generally, CAC reduces the quality of life and increases morbidity and mortality as well as cancer treatment toxicities (70, 71).
As a multifactorial syndrome, CAC causes changes in many organs and their interorgan and intertissue communication. Muscle wasting is mediated by muscle proteolysis, the degradation of muscle fibers as well as the lack of muscle regeneration due to exhausted muscle stem cells, so-called satellite cells (SCs). The increased expression of the E3 ligases MuFR1 and MAFbx of the ubiquitin-proteasome system is one of the hallmarks of cachectic muscles (72). The metabolic changes in adipose tissue lead to increased lipolysis and adipose tissue browning, which is characterized by an increased expression of the uncoupling protein 1 (UCP1). In adipocytes, UCP1 causes energy wasting due to excessive heat production (73). Thermogenesis is activated in the remaining adult brown adipocytes increasing the energy expenditure. Additionally, the combination of loss of appetite caused by alterations in hypothalamic signaling mechanisms and malabsorption due to changes in the intestinal barrier and the microbiome composition worsens the negative energy balance (74).
Cachexia research has focused on a multitude of organs and tissues at different cachectic stages to comprehend the immense variety of changes caused by cachexia syndrome. Additionally, extensive mouse strain-specific and tumor-specific differences have increased the complexity of CAC research (75). Since ECs are involved in many pathways within tumor progression and the metastatic cascade (as mentioned above), the question arises if ECs influence or are influenced by metabolic conditions like cachexia. Only a few very recent studies have focused on the effect of ECs in cachexia, but in the light of the novelty of this subject, we highlight well-known cachexia markers, which may have an endothelial origin, may change vasculature function, or may lead to altered angiocrine signaling opening a new research direction in the expanding field of CAC.
The Effect of Cytokines on Endothelial Cells During Cancer-Associated Cachexia
Primary tumors secrete various cytokines that have many different effects on the immune system, the surrounding tissue as well as distant sites. Specifically, increased IL-6, IL-8, and TNF-α concentrations are characteristic of CAC (76). Furthermore, tumor-derived exosomes and vesicles have been described to induce skeletal muscle and adipose tissue wasting (77, 78). Here, we will focus on IL-6 due to its pro-inflammatory autocrine and paracrine function in ECs and its distinct roles in CAC.
During the progression of CAC, chronic inflammation mediated by IL-6 and TNF-α has been described to induce white adipose tissue browning (79) and increased lipolysis mediated by STAT3 signaling and the adipose triglyceride lipase (80). However, a systemic study of 136 patients with PDAC concluded that IL-6 only correlates with cancer stage and not with cachexia (81) conceding that IL-6 may mediate cachexia but should not be considered as a biomarker. Cachectic mice have been described to be hypercoagulable partially due to tumor-derived IL-6, which is inducing thrombin expression (82). Likewise, in skeletal muscle, IL-6 plays a pivotal role during CAC regulating mitochondrial dynamics, biogenesis, function, and mitophagy by inducing STAT3 phosphorylation (83). The direct and indirect effects of IL-6-induced STAT3 signaling in CAC have been previously summarized (84).
Since ECs do not express the membrane-bound IL-6 receptor (IL-6R), the soluble IL-6Rα needs to bind to IL-6 to signal through endothelial gp130 activating the expression of STAT3 target genes, notably the production of chemokines [e.g., monocyte chemoattractant protein-1 (MCP-1)] or adhesion molecules (e.g., E-selectin, ICAM-1, and VCAM-1) (85). This alternative signaling via the soluble receptor is described as trans-signaling. Additionally, upon inflammatory stimuli, ECs produce and secrete large amounts of IL-6 increasing its local and systemic concentrations. Tumor-derived IL-6 induces endothelial IL6-STAT3 signaling, which in turn may lead to increased plasma concentrations of cachexia-promoting factors like C-C motif chemokine ligand 2 (CCL2/MCP-1), commonly described in CAC patients (86, 87). The production of IL-6, IL-10, and PAI-1 in ECs is furthermore triggered by IL-6 signaling itself (88).
In 2021, Rupert et al. (89) reported that tumor-derived IL-6 induces a cachectic phenotype in adipose tissue and skeletal muscle via classical and trans-signaling. Primary tumor-secreted IL-6 induced soluble IL-6Rα expression in skeletal muscle, which was transported to the subcutaneous adipose tissue. IL-6Rα promoted IL-6 trans-signaling in white adipose tissue causing increased lipolysis, inflammation, and additional IL-6 production. This activated a feed-forward loop by inducing muscular classical IL-6 signaling stimulating the production of IL-6Rα even more. How endothelial trans-signaling may be involved in this process still needs to be investigated. It is plausible that shed IL-6Rα from the muscle not only activates IL-6 signaling in adipose tissue but also stimulates endothelial IL-6 signaling inducing the expression of STAT3 target genes. Furthermore, tumor-derived TNF-α might lead to an additional increase in plasma IL-6 by its release from ECs. The extent of EC-derived IL-6, endothelial IL-6 signaling and its contribution to tissue wasting and cachexia remains elusive (FIGURE 3A). Generally, it is important to take both, the large interactive surface of blood vessels and the capacity of ECs to amplify tumor-derived signals to modify distant tissues into account.
FIGURE 3.

Endothelial cell signaling pathways during cancer-associated cachexia A: tumor inflammation as well as tumor-derived IL-6 induce endothelial cell (EC) activation and the expression of IL-6 and monocyte chemoattractant protein-1 (MCP-1). Endothelial-derived IL-6 may trigger trans-signaling in adipose tissue causing browning and lipolysis as well as IL-6 release, which in turn activates classical signaling in muscle leading to muscle wasting via mitophagy. Activated skeletal muscle secretes soluble IL-6Rα, which is necessary for trans-signaling in endothelial cells and adipocytes. B: tumor-derived lipocalin-2 (Lcn2) and inflammatory factors may induce Lcn2 signaling in ECs via the 24p3R receptor increasing vascular integrity and decreasing permeability. However, EC activation can also induce Lcn2 expression in ECs, which may cause satellite cell activation and muscle regeneration, insulin resistance, and brown adipose tissue (BAT) activation as well as reduced appetite and food intake. Additionally, Lcn2 induces VEGF release from tumor cells which promotes (tumor) angiogenesis. C: VEGF from cancer cells and myofibers as well as other inflammatory stimuli induces Dll-4 expression and release from ECs. Dll-4-Notch signaling in satellite cells supports their self-renewal and quiescence. In myotubes, Dll-4 signaling causes muscle wasting and disruptions of the sarcomere structure. Image was created with BioRender.com, with permission.
Endothelial Lipocalin 2 Signaling and Secretion
Lipocalin 2 (Lcn2) is a secreted acute-phase protein that is highly expressed upon inflammatory stimulation during bacterial infection (90), heart failure (91), chronic kidney disease (92), and cancer (93). Depending on the pathological condition, Lcn2 has either been described as an important factor of the immune system being induced by cytokines especially via IL-6-Stat3 signaling (90) or as adipokine, promoting insulin resistance (94). The described effects of Lcn2 expression during cancer progression are controversial and highly depend on the tumor type as well as its tissue expression. The differential functions of Lcn2 as an oncogene as well as tumor-suppressor are beyond the scope of this review and have been discussed in some detail elsewhere (95, 96). Since Lcn2 has been described to be involved in several cachexia-affected organs as well as in cancer, its role during CAC becomes of interest.
Mechanistically, Lcn2 forms a complex with MMP9 increasing its stability and thereby promoting extracellular matrix remodeling. Additionally, Lcn2 is involved in iron transport context-dependently modulating the intracellular concentration of iron (95). Generally, its expression can be induced by insulin as well as many other cytokines like TNF-α, IL-1β, and IL-6. Therefore, Lcn2 is considered to be responsive to metabolic stress, nutrients, and cytokines (97). Lcn2 is involved in brown adipose tissue (BAT) activation and regulates its thermogenic activity. It is highly expressed in brown adipocytes after cold exposure and stimulates the expression of the BAT markers proliferator-activated receptor-γ coactivator-1α (PGC-1α) and UCP1 (98). In muscle, Lcn2 is expressed in satellite cells within the first day postinjury playing an important role in muscle regeneration (99). However, its myogenic expression is enhanced in ob/ob mice suffering from sarcopenia as well as in wild-type mice after high-intensity training (100, 101).
During CAC, Lcn2 has been described to be upregulated in neutrophils and was found in high concentrations in the plasma and the cerebrospinal fluid of cancer patients correlating with increased mortality (102). Additionally, Lcn2 is increased in plasma and heart tissue worsening cardiac damage after cachexia-induced cardiomyopathy (103). Due to its ability to cross the blood-brain barrier (BBB), Lcn2 can also inhibit food intake by activating the paraventricular and ventromedial neurons of the hypothalamus (104). Lcn2 binds to the type 4 melanocortin receptor (MC4R) on the POMC neurons in the hypothalamus inhibiting food intake and appetite. The inhibition of Lcn2 and MC4R improved cancer-induced anorexia as well as CAC symptoms (102, 105, 106). A scRNAseq study by Huisman et al. (107) showed that tumor-derived factors change the microenvironment of the medial basal hypothalamus and increase Lcn2 expression in cachectic mice. Additionally, it led in the pancreatic CAC model to induce EC inflammation and high Lcn2 expression in hypothalamic ECs. Recently, Lemecha and colleagues (108) determined an ameliorated cachectic phenotype after the global deletion of Lcn2 in a PDAC mouse model. Tumor-bearing Lcn2 knockout mice showed reduced adipose tissue inflammation, less muscle atrophy, and decreased thermogenesis in brown adipose tissue. The authors concluded that Lcn2 may act as a mediator between adipose tissue and skeletal muscle during PDAC progression.
Generally, the vascular function of Lcn2 has primarily been studied in the brain. Following TNF-α stimulation, Lcn2 reduced vascular permeability by rescuing the expression of ZO-1 and VE-cadherin in brain ECs without having an effect on neutrophil adhesion (109). The modulation of tight and adherent junctions to maintain the BBB was also observed in a transient middle cerebral artery occlusion model in which Lcn2 as well as its receptor 24p3R were expressed in ECs (110), indicating LCN2 signaling in ECs in this model. Additionally, brain ECs induced Lcn2 expression after LPS challenge, which was diminished in IL-6 knockout mice (111). Treatment with Lcn2 increased migration and capillary-like tube formation in rat brain ECs in vitro supporting the notion that Lcn2 promotes neurovascular recovery (112). In a glioma model, Lcn2 expression was induced by tumor-derived exosomes via JAK-STAT3 signaling in bEnd.3 endothelial cells (113). The increase of Lcn2 leads to increased BBB penetration of nanocapsules delivering therapeutic monoclonal antibodies without disrupting their integrity. Apart from its function in neuroinflammation, de-aminated Lcn2 has been described to accumulate in the aorta and arteries of obese mice inducing endothelial dysfunction and hypertension by impairing EC-dependent relaxation and contractions (114, 115). Furthermore, expression of Lcn2 in human breast cancer caused upregulated VEGF expression via hypoxia-inducible factor-1α (116). Lcn2 potentiated VEGF-induced angiogenesis in vivo and may promote tumor progression by inducing breast cancer angiogenesis. The relation between VEGF and Lcn2 was also described in proliferative diabetic retinopathy (117). Endothelial Lcn2 was upregulated in diabetic retinal injury and Lcn2 silencing attenuated diabetic retinopathy progression (118).
In summary, whether the effects of tumor-induced Lcn2 expression on cancer and cachexia progression are based on direct changes in EC function, angiocrine signaling, and vascular integrity or indirect changes in VEGF expression and inflammation remains to be determined (FIGURE 3B). Lcn2 affects vascular function, but it is also expressed by ECs leading to the question of how Lcn2 acts on ECs. Whether EC-derived Lcn2 acts systemically or tissue- and site-specific remains elusive and opens up the question of how much the cellular origin and location of Lcn2 matter during disease progression. Determining the key players of the interplay between different Lcn2-expressing compartments and ECs would lead to new insights into an important pathway of many pathological conditions, especially CAC.
Endothelial Notch Signaling During Cancer-Associated Cachexia
Notch signaling is facilitated by the interaction of Notch ligands (Dll-1, Dll-3, Dll-4, Jag-1, and Jag-2) to one of the four Notch receptors (Notch-1–4). The binding of ligand and receptor activates γ-secretase-induced cleavage and the subsequent release of the intracellular NICD domain activating the transcription of Notch target genes. Notch signaling regulates stem cell function during embryogenesis and adult regeneration. Specifically in ECs, Notch regulates angiogenesis defining tip and stalk cell fates by modulating VEGF sensitivity of ECs (119, 120). During tumor growth, Notch signaling promotes tumor angiogenesis, EMT, and self-renewal of cancer stem cells (121, 122). Endothelial Notch signaling increases metastasis and immune infiltration by promoting transendothelial migration (123) as well as increasing the number of cancer stem cells and circulating tumor cells (124). Additionally, Notch signaling in ECs induces an immunosuppressive microevi-ronment by educating tumor-associated macrophages via CXCL2 (125) making Notch signaling modulation an interesting target for cancer therapy (126).
Notch signaling not only regulates the cancer stem cell pool but also maintains the number of satellite cells (SCs) in the muscle. Notch-3 inhibits SC differentiation and thereby facilitates SC quiescence during homeostasis. However, Notch signaling is also essential during muscle regeneration and the self-renewal of the stem cell pool by regulating the proliferation of SCs after injury (127). Myogenesis is initiated by Notch signaling, which is later replaced by activated Wnt signaling which is necessary for the differentiation in myotubes. The balance of activation and temporal regulation of Notch and Wnt signaling are crucial for effective muscle regeneration (128). The effects of Notch signaling in muscle depend on the cell state leading to controversial results after Notch activation (129, 130). Endothelial Dll-4 is induced by VEGF, which is increased in the plasma in most cancer entities. However, VEGF is also stored in vesicles of skeletal myotubes and can be secreted by exercise, hypoxia, or adenosine release (131). Muscle-derived VEGF regulates the proximity of SCs and ECs, which in turn modulates the quiescent state as well as the self-renewal capacity of SCs via Dll-4-Notch signaling (132). Dysfunctional ECs and impaired angiocrine signaling after metabolic challenge led to less viable and less differentiated human skeletal muscle SCs in vitro (133). Generally, the endothelial-myogenic cross talk, ECs promoting myogenesis and differentiating progenitor cells inducing angiogenesis, is tightly regulated, e.g., by apelin, oncostatin M, and periostin (134).
During CAC, muscle wasting is caused by increased proteolysis as well as defective regeneration mediated by increased Pax7-positive SCs in the skeletal muscle (135). Notch signaling is induced by tumor-derived TNF-α in muscle and tumor tissues during osteosarcoma-induced cachexia. Genetic Notch ablation reduced muscle atrophy and removed the inhibition of muscle differentiation (136). The analysis of sarcoma patient samples revealed decreased muscle regeneration caused by Pax7 and Notch activation in SCs in cachectic versus noncachectic patients (137). Pharmacological inhibition of Notch-Hes1 signaling reduced body weight loss, muscle wasting, and improved survival in a doxo-rubicin-induced cachexia model (138) emphasizing the essential role of Notch signaling in the progression of muscle atrophy. Recently, Fujimaki et al. (139) described that ECs increase Dll-4 release during disuse- and diabetes mellitus-induced muscle atrophy. Upon metabolic and mechanical challenge, the dysfunctional endothelium activated Notch-2 in myofibers leading to reduced muscle mass and disruptions of sarcomere structures. Inhibition of endothelial Dll-4, as well as Notch-2 inactivation in myofibers, prevented muscle wasting induced by metabolic stress (139).
How tumor-derived factors change the EC-muscle cross talk and how exactly Dll-4 expression is induced in ECs during CAC remain to be studied (FIGURE 3C). The maintenance of EC health and the homeostasis of angiocrine signaling during metabolic challenges like CAC may improve the regeneration of muscular function and thereby increase patient survival. The precise mechanisms and direct impact of the endothelium on muscle wasting during cachectic challenges have yet to be elucidated, but the above-mentioned studies implicate a close relation between EC health and muscle regeneration.
Conclusions and Perspectives
The notion that cancer is a systemic disease highlights the importance of designing therapies that target systemic signaling mechanisms to ameliorate systemic cancer-associated changes. ECs affect the local microenvironment by angiocrine signaling and are key players in many pathological conditions, including cancer. However, ECs can amplify received signals systemically and thereby impact the host’s macroenvironment. Changes in one single vascular bed may translate to other sites or even the whole vasculature (4). Fine-tuning and stabilizing the homeostatic signaling of such a vast reactive surface created by the endothelium could help to revert tumor-induced vascular changes and thereby effectively impact metastasis and cachexia. Determining the correlation between vascular health and patient survival as well as disease progression will create new opportunities in cancer research. The transition from vascular dysfunction to a healthy and quiescent vascular bed has the potential to revolutionize treatment strategies. In addition, there is a need to further study the vascular effects of tumor-derived factors in the future to identify biomarkers of disease progression. This will ultimately guide the stratification of therapeutic regimens to predict and improve the survival of cancer patients.
Acknowledgments
Work in the authors’ laboratory was institutionally supported by Heidelberg University and the German Cancer Research Center Heidelberg (DKFZ) as well as extramurally by grants from the Deutsche Forschungsgemeinschaft (Project No. 39404578), the Deutsche Krebshilfe (Project No. 70114532), and the European Research Council (Project No. 787181).
No conflicts of interest, financial or otherwise, are declared by the authors.
S.F.P., D.G., and H.G.A. conceived and designed research; S.F.P. and D.G. prepared figures; S.F.P., D.G., and H.G.A. drafted manuscript; S.F.P., D.G., and H.G.A. edited and revised manuscript; S.F.P., D.G., and H.G.A. approved final version of manuscript.
References
- 1. Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature 529: 316–325, 2016. doi: 10.1038/nature17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pasquier J, Ghiabi P, Chouchane L, Razzouk K, Rafii S, Rafii A. Angiocrine endothelium: from physiology to cancer. J Transl Med 18: 52, 2020. doi: 10.1186/s12967-020-02244-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hasan SS, Fischer A. The endothelium: an active regulator of lipid and glucose homeostasis. Trends Cell Biol 31: 37–49, 2021. doi: 10.1016/j.tcb.2020.10.003. [DOI] [PubMed] [Google Scholar]
- 4. Singhal M, Gengenbacher N, Abdul Pari AA, Kamiyama M, Hai L, Kuhn BJ, Kallenberg DM, Kulkarni SR, Camilli C, Preuss SF, Leuchs B, Mogler C, Espinet E, Besemfelder E, Heide D, Heikenwalder M, Sprick MR, Trumpp A, Krijgsveld J, Schlesner M, Hu J, Moss SE, Greenwood J, Augustin HG. Temporal multi-omics identifies LRG1 as a vascular niche instructor of metastasis. Sci Transl Med 13: eabe6805, 2021. doi: 10.1126/scitranslmed.abe6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, Camps C, Marinez I, Busund LT. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol 6: 209–217, 2011. doi: 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- 6. Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res 316: 1324–1331, 2010. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 7. McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 16: 717–727, 2014. doi: 10.1038/ncb3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dillekås H, Rogers MS, Straume O. Are 90% of deaths from cancer caused by metastases? Cancer Med 8: 5574–5576, 2019. doi: 10.1002/cam4.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost 5: 632–634, 2007. doi: 10.1111/j.1538-7836.2007.02374.x. [DOI] [PubMed] [Google Scholar]
- 10. Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer 2: 862–871, 2002. doi: 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- 11. Gupta GP, Massague J. Cancer metastasis: building a framework. Cell 127: 679–695, 2006. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 12. Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther 5: 28, 2020. doi: 10.1038/s41392-020-0134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell 30: 668–681, 2016. doi: 10.1016/j.ccell.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 14. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafii S, Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438: 820–827, 2005. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJ, Ciampricotti M, Hawinkels L, Jonkers J, de Visser KE. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522: 345–348, 2015. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A, Olsson AK. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res 75: 2653–2662, 2015. doi: 10.1158/0008-5472.CAN-14-3299. [DOI] [PubMed] [Google Scholar]
- 17. Hiratsuka S, Nakamura K, Iwai S, Murakami M, Itoh T, Kijima H, Shipley JM, Senior RM, Shibuya M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2: 289–300, 2002. doi: 10.1016/s1535-6108(02)00153-8. [DOI] [PubMed] [Google Scholar]
- 18. Li R, Qi Y, Jiang M, Zhang T, Wang H, Wang L, Han M. Primary tumor-secreted VEGF induces vascular hyperpermeability in premetastatic lung via the occludin phosphorylation/ubiquitination pathway. Mol Carcinog 58: 2316–2326, 2019. doi: 10.1002/mc.23120. [DOI] [PubMed] [Google Scholar]
- 19. Liu S, Jiang M, Zhao Q, Li S, Peng Y, Zhang P, Han M. Vascular endothelial growth factor plays a critical role in the formation of the pre-metastatic niche via prostaglandin E2. Oncol Rep 32: 2477–2484, 2014. doi: 10.3892/or.2014.3516. [DOI] [PubMed] [Google Scholar]
- 20. Hiratsuka S, Goel S, Kamoun WS, Maru Y, Fukumura D, Duda DG, Jain RK. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc Natl Acad Sci U S A 108: 3725–3730, 2011. doi: 10.1073/pnas.1100446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol 8: 1369–1375, 2006. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 22. Srikrishna G. S100A8 and S100A9: new insights into their roles in malignancy. J Innate Immun 4: 31–40, 2012. doi: 10.1159/000330095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo Y, Ji X, Liu J, Fan D, Zhou Q, Chen C, Wang W, Wang G, Wang H, Yuan W, Ji Z, Sun Z. Effects of exosomes on pre-metastatic niche formation in tumors. Mol Cancer 18: 39, 2019. doi: 10.1186/s12943-019-0995-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, Yu L, Ross J, Korsisaari N, Cao T, Bou-Reslan H, Kallop D, Weimer R, Ludlam MJ, Kaminker JS, Modrusan Z, van Bruggen N, Peale FV, Carano R, Meng YG, Ferrara N. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A 107: 21248–21255, 2010. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sceneay J, Smyth MJ, Moller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev 32: 449–464, 2013. doi: 10.1007/s10555-013-9420-1. [DOI] [PubMed] [Google Scholar]
- 26. Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res 71: 3792–3801, 2011. doi: 10.1158/0008-5472.CAN-10-4455. [DOI] [PubMed] [Google Scholar]
- 27. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, , et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 17: 816–826, 2015. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, Tetta C, Bussolati B, Camussi G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res 71: 5346–5356, 2011. doi: 10.1158/0008-5472.CAN-11-0241. [DOI] [PubMed] [Google Scholar]
- 29. Duda DG, Jain RK. Premetastatic lung “niche”: is vascular endothelial growth factor receptor 1 activation required? Cancer Res 70: 5670–5673, 2010. doi: 10.1158/0008-5472.CAN-10-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yi H, Ye J, Yang XM, Zhang LW, Zhang ZG, Chen YP. High-grade ovarian cancer secreting effective exosomes in tumor angiogenesis. Int J Clin Exp Pathol 8: 5062–5070, 2015. [PMC free article] [PubMed] [Google Scholar]
- 31. Tang MK, Yue PY, Ip PP, Huang RL, Lai HC, Cheung AN, Tse KY, Ngan HY, Wong AS. Soluble E-cadherin promotes tumor angiogenesis and localizes to exosome surface. Nat Commun 9: 2270, 2018. doi: 10.1038/s41467-018-04695-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim J, Lee C, Kim I, Ro J, Kim J, Min Y, Park J, Sunkara V, Park YS, Michael I, Kim YA, Lee HJ, Cho YK. Three-dimensional human liver-chip emulating premetastatic niche formation by breast cancer-derived extracellular vesicles. ACS Nano 14: 14971–14988, 2020. doi: 10.1021/acsnano.0c04778. [DOI] [PubMed] [Google Scholar]
- 33. Kuznetsov HS, Marsh T, Markens BA, Castaño Z, Greene-Colozzi A, Hay SA, Brown VE, Richardson AL, Signoretti S, Battinelli EM, McAllister SS. Identification of luminal breast cancers that establish a tumor-supportive macroenvironment defined by proangiogenic platelets and bone marrow-derived cells. Cancer Discov 2: 1150–1165, 2012. doi: 10.1158/2159-8290.CD-12-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bruzzese F, Hägglöf C, Leone A, Sjöberg E, Roca MS, Kiflemariam S, Sjöblom T, Hammarsten P, Egevad L, Bergh A, Ostman A, Budillon A, Augsten M. Local and systemic protumorigenic effects of cancer-associated fibroblast-derived GDF15. Cancer Res 74: 3408–3417, 2014. doi: 10.1158/0008-5472.CAN-13-2259. [DOI] [PubMed] [Google Scholar]
- 35. Dong G, Zheng QD, Ma M, Wu SF, Zhang R, Yao RR, Dong YY, Ma H, Gao DM, Ye SL, Cui JF, Ren ZG, Chen RX. Angiogenesis enhanced by treatment damage to hepatocellular carcinoma through the release of GDF15. Cancer Med 7: 820–830, 2018. doi: 10.1002/cam4.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ruggiero RA, Bruzzo J, Chiarella P, Bustuoabad OD, Meiss RP, Pasqualini CD. Concomitant tumor resistance: the role of tyrosine isomers in the mechanisms of metastases control. Cancer Res 72: 1043–1050, 2012. doi: 10.1158/0008-5472.CAN-11-2964. [DOI] [PubMed] [Google Scholar]
- 37. Ehrlich P, Apolant H. Experimentelle Carcinomstudien an Mäusen. Jena, Germany: Fischer, 1906. [Google Scholar]
- 38. Kubo H, Mensurado S, Goncalves-Sousa N, Serre K, Silva-Santos B. Primary tumors limit metastasis formation through induction of IL15-mediated cross-talk between patrolling monocytes and NK cells. Cancer Immunol Res 5: 812–820, 2017. doi: 10.1158/2326-6066.CIR-17-0082. [DOI] [PubMed] [Google Scholar]
- 39. Kirstein JM, Hague MN, McGowan PM, Tuck AB, Chambers AF. Primary melanoma tumor inhibits metastasis through alterations in systemic hemostasis. J Mol Med (Berl) 94: 899–910, 2016. doi: 10.1007/s00109-016-1415-2. [DOI] [PubMed] [Google Scholar]
- 40. Piranlioglu R, Lee E, Ouzounova M, Bollag RJ, Vinyard AH, Arbab AS, Marasco D, Guzel M, Cowell JK, Thangaraju M, Chadli A, Hassan KA, Wicha MS, Celis E, Korkaya H. Primary tumor-induced immunity eradicates disseminated tumor cells in syngeneic mouse model. Nat Commun 10: 1430, 2019. doi: 10.1038/s41467-019-09015-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hamilton AM, Parkins KM, Murrell DH, Ronald JA, Foster PJ. Investigating the impact of a primary tumor on metastasis and dormancy using MRI: new insights into the mechanism of concomitant tumor resistance. Tomography 2: 79–84, 2016. doi: 10.18383/j.tom.2016.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79: 315–328, 1994. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 43. Camphausen K, Moses MA, Beecken WD, Khan MK, Folkman J, O'Reilly MS. Radiation therapy to a primary tumor accelerates metastatic growth in mice. Cancer Res 61: 2207–2211, 2001. [PubMed] [Google Scholar]
- 44. Volpert OV, Lawler J, Bouck NP. A human fibrosarcoma inhibits systemic angiogenesis and the growth of experimental metastases via thrombospondin-1. Proc Natl Acad Sci U S A 95: 6343–6348, 1998. doi: 10.1073/pnas.95.11.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Franco M, Bustuoabad OD, di Gianni PD, Goldman A, Pasqualini CD, Ruggiero RA. A serum-mediated mechanism for concomitant resistance shared by immunogenic and non-immunogenic murine tumours. Br J Cancer 74: 178–186, 1996. doi: 10.1038/bjc.1996.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ruggiero RA, Bustuoabad OD, Bonfil RD, Meiss RP, Pasqualini CD. Concomitant immunity in murine tumours of non-detectable immunogenicity. Br J Cancer 51: 37–48, 1985. doi: 10.1038/bjc.1985.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88: 277–285, 1997. doi: 10.1016/S0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 48. Digtyar AV, Pozdnyakova NV, Feldman NB, Lutsenko SV, Severin SE. Endostatin: current concepts about its biological role and mechanisms of action. Biochemistry (Mosc) 72: 235–246, 2007. doi: 10.1134/S0006297907030017. [DOI] [PubMed] [Google Scholar]
- 49. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86: 353–364, 1996. doi: 10.1016/S0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 50. Folkman J. Angiogenesis. Annu Rev Med 57: 1–18, 2006. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 51. Prehn RT. Two competing influences that may explain concomitant tumor resistance. Cancer Res 53: 3266–3269, 1993. [PubMed] [Google Scholar]
- 52. Zak S, Treven J, Nash N, Gutierrez LS. Lack of thrombospondin-1 increases angiogenesis in a model of chronic inflammatory bowel disease. Int J Colorectal Dis 23: 297–304, 2008. doi: 10.1007/s00384-007-0397-5. [DOI] [PubMed] [Google Scholar]
- 53. Wang S, Wu Z, Sorenson CM, Lawler J, Sheibani N. Thrombospondin-1-deficient mice exhibit increased vascular density during retinal vascular development and are less sensitive to hyperoxia-mediated vessel obliteration. Dev Dyn 228: 630–642, 2003. doi: 10.1002/dvdy.10412. [DOI] [PubMed] [Google Scholar]
- 54. Moulton KS, Olsen BR, Sonn S, Fukai N, Zurakowski D, Zeng X. Loss of collagen XVIII enhances neovascularization and vascular permeability in atherosclerosis. Circulation 110: 1330–1336, 2004. doi: 10.1161/01.CIR.0000140720.79015.3C. [DOI] [PubMed] [Google Scholar]
- 55. Hubers C, Abdul Pari AA, Grieshober D, Petkov M, Schmidt A, Messmer T, Heyer CM, Scholch S, Kapel SS, Gengenbacher N, Singhal M, Schieb B, Fricke C, Will R, Remans K, Utikal JS, Reissfelder C, Schlesner M, Hodivala-Dilke KM, Kersten S, Goerdt S, Augustin HG, Felcht M. Primary tumor-derived systemic nANGPTL4 inhibits metastasis. J Exp Med 220: e20202595, 2023. doi: 10.1084/jem.20202595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bruzzo J, Chiarella P, Meiss RP, Ruggiero RA. Biphasic effect of a primary tumor on the growth of secondary tumor implants. J Cancer Res Clin Oncol 136: 1605–1615, 2010. doi: 10.1007/s00432-010-0818-7. [DOI] [PubMed] [Google Scholar]
- 57. Kaya M, Wada T, Nagoya S, Kawaguchi S, Isu K, Yamashita T. Concomitant tumour resistance in patients with osteosarcoma. A clue to a new therapeutic strategy. J Bone Joint Surg Br 86: 143–147, 2004. doi: 10.2106/00004623-200409001-00005. [DOI] [PubMed] [Google Scholar]
- 58. Beecken WD, Engl T, Jonas D, Blaheta RA. Expression of angiogenesis inhibitors in human bladder cancer may explain rapid metastatic progression after radical cystectomy. Int J Mol Med 23: 261–266, 2009. [PubMed] [Google Scholar]
- 59. Peeters CF, de Waal RM, Wobbes T, Westphal JR, Ruers TJ. Outgrowth of human liver metastases after resection of the primary colorectal tumor: a shift in the balance between apoptosis and proliferation. Int J Cancer 119: 1249–1253, 2006. doi: 10.1002/ijc.21928. [DOI] [PubMed] [Google Scholar]
- 60. Okazaki T, Ebihara S, Asada M, Kanda A, Sasaki H, Yamaya M. Granulocyte colony-stimulating factor promotes tumor angiogenesis via increasing circulating endothelial progenitor cells and Gr1+CD11b+ cells in cancer animal models. Int Immunol 18: 1–9, 2006. doi: 10.1093/intimm/dxh334. [DOI] [PubMed] [Google Scholar]
- 61. Ebos JM, Lee CR, Kerbel RS. Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res 15: 5020–5025, 2009. doi: 10.1158/1078-0432.CCR-09-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Falanga A, Russo L, Milesi V, Vignoli A. Mechanisms and risk factors of thrombosis in cancer. Crit Rev Oncol Hematol 118: 79–83, 2017. doi: 10.1016/j.critrevonc.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 63. Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, Coomber BL, Mackman N, Rak JW. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood 105: 1734–1741, 2005. doi: 10.1182/blood-2004-05-2042. [DOI] [PubMed] [Google Scholar]
- 64. Van den Berg YW, van den Hengel LG, Myers HR, Ayachi O, Jordanova E, Ruf W, Spek CA, Reitsma PH, Bogdanov VY, Versteeg HH. Alternatively spliced tissue factor induces angiogenesis through integrin ligation. Proc Natl Acad Sci U S A 106: 19497–19502, 2009. doi: 10.1073/pnas.0905325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ott I, Weigand B, Michl R, Seitz I, Sabbari-Erfani N, Neumann FJ, Schomig A. Tissue factor cytoplasmic domain stimulates migration by activation of the GTPase Rac1 and the mitogen-activated protein kinase p38. Circulation 111: 349–355, 2005. doi: 10.1161/01.CIR.0000153333.52294.42. [DOI] [PubMed] [Google Scholar]
- 66. Geddings JE, Mackman N. Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood 122: 1873–1880, 2013. doi: 10.1182/blood-2013-04-460139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, Scadden DT, Wagner DD. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A 109: 13076–13081, 2012. doi: 10.1073/pnas.1200419109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, Davis M, Muscaritoli M, Ottery F, Radbruch L, Ravasco P, Walsh D, Wilcock A, Kaasa S, Baracos VE. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 12: 489–495, 2011. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 69. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KC. Cancer-associated cachexia. Nat Rev Dis Primers 4: 17105, 2018. doi: 10.1038/nrdp.2017.105. [DOI] [PubMed] [Google Scholar]
- 70. Bachmann J, Ketterer K, Marsch C, Fechtner K, Krakowski-Roosen H, Buchler MW, Friess H, Martignoni ME. Pancreatic cancer related cachexia: influence on metabolism and correlation to weight loss and pulmonary function. BMC Cancer 9: 255, 2009. doi: 10.1186/1471-2407-9-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Marceca GP, Londhe P, Calore F. Management of cancer cachexia: attempting to develop new pharmacological agents for new effective therapeutic options. Front Oncol 10: 298, 2020. doi: 10.3389/fonc.2020.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Johns N, Stephens NA, Fearon KC. Muscle wasting in cancer. Int J Biochem Cell Biol 45: 2215–2229, 2013. doi: 10.1016/j.biocel.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 73. Sun X, Feng X, Wu X, Lu Y, Chen K, Ye Y. Fat wasting is damaging: role of adipose tissue in cancer-associated cachexia. Front Cell Dev Biol 8: 33, 2020. doi: 10.3389/fcell.2020.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Argiles JM, Stemmler B, Lopez-Soriano FJ, Busquets S. Inter-tissue communication in cancer cachexia. Nat Rev Endocrinol 15: 9–20, 2018. doi: 10.1038/s41574-018-0123-0. [DOI] [PubMed] [Google Scholar]
- 75. Geppert J, Walth AA, Exposito RT, Kaltenecker D, Morigny P, Machado J, Becker M, Simoes E, Lima JD, Daniel C, Diaz MB, Herzig S, Seelaender M, Rohm M. Aging aggravates cachexia in tumor-bearing mice. Cancers 14: 90, 2021. doi: 10.3390/cancers14010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Paval DR, Patton R, McDonald J, Skipworth RJ, Gallagher IJ, Laird BJ; Caledonian Cachexia Collaborative. A systematic review examining the relationship between cytokines and cachexia in incurable cancer. J Cachexia Sarcopenia Muscle 13: 824–838, 2022. doi: 10.1002/jcsm.12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fan M, Sun W, Gu X, Lu S, Shen Q, Liu X, Zhang X. The critical role of STAT3 in biogenesis of tumor-derived exosomes with potency of inducing cancer cachexia in vitro and in vivo. Oncogene 41: 1050–1062, 2022. doi: 10.1038/s41388-021-02151-3. [DOI] [PubMed] [Google Scholar]
- 78. Pin F, Beltra M, Garcia-Castillo L, Pardini B, Birolo G, Matullo G, Penna F, Guttridge D, Costelli P. Extracellular vesicles derived from tumour cells as a trigger of energy crisis in the skeletal muscle. J Cachexia Sarcopenia Muscle 13: 481–494, 2022. doi: 10.1002/jcsm.12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Han J, Meng Q, Shen L, Wu G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis 17: 14, 2018. doi: 10.1186/s12944-018-0657-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gandhi AY, Yu J, Gupta A, Guo T, Iyengar P, Infante RE. Cytokine-mediated STAT3 transcription supports ATGL/CGI-58-dependent adipocyte lipolysis in cancer cachexia. Front Oncol 12: 841758, 2022. doi: 10.3389/fonc.2022.841758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ramsey ML, Talbert E, Ahn D, Bekaii-Saab T, Badi N, Bloomston PM, Conwell DL, Cruz-Monserrate Z, Dillhoff M, Farren MR, Hinton A, Krishna SG, Lesinski GB, Mace T, Manilchuk A, Noonan A, Pawlik TM, Rajasekera PV, Schmidt C, Guttridge D, Hart PA. Circulating interleukin-6 is associated with disease progression, but not cachexia in pancreatic cancer. Pancreatology 19: 80–87, 2019. doi: 10.1016/j.pan.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Reddel CJ, Allen JD, Ehteda A, Taylor R, Chen VM, Curnow JL, Kritharides L, Robertson G. Increased thrombin generation in a mouse model of cancer cachexia is partially interleukin-6 dependent. J Thromb Haemost 15: 477–486, 2017. doi: 10.1111/jth.13612. [DOI] [PubMed] [Google Scholar]
- 83. VanderVeen BN, Fix DK, Carson JA. Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: a role for inflammation. Oxid Med Cell Longev 2017: 3292087, 2017. doi: 10.1155/2017/3292087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zimmers TA, Fishel ML, Bonetto A. STAT3 in the systemic inflammation of cancer cachexia. Semin Cell Dev Biol 54: 28–41, 2016. doi: 10.1016/j.semcdb.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Modur V, Li Y, Zimmerman GA, Prescott SM, McIntyre TM. Retrograde inflammatory signaling from neutrophils to endothelial cells by soluble interleukin-6 receptor alpha. J Clin Invest 100: 2752–2756, 1997. doi: 10.1172/JCI119821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dillhoff M, Lewis H, Haverick E, Hart P, Manilchuk A, Pawlik TM, Schmidt C, Talbert E, Guttridge D. Elevated MCP-1 is associated with cancer cachexia in patients with pancreatic ductal adenocarcinoma. HPB 19: S21–S22, 2017. doi: 10.1016/j.hpb.2017.02.200. [DOI] [Google Scholar]
- 87. Talbert EE, Lewis HL, Farren MR, Ramsey ML, Chakedis JM, Rajasekera P, Haverick E, Sarna A, Bloomston M, Pawlik TM, Zimmers TA, Lesinski GB, Hart PA, Dillhoff ME, Schmidt CR, Guttridge DC. Circulating monocyte chemoattractant protein-1 (MCP-1) is associated with cachexia in treatment-naive pancreatic cancer patients. J Cachexia Sarcopenia Muscle 9: 358–368, 2018. doi: 10.1002/jcsm.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, Matsumoto H, Matsuura H, Matsubara T, Shimizu K, Ogura H, Matsuura Y, Kishimoto T. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci U S A 117: 22351–22356, 2020. doi: 10.1073/pnas.2010229117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rupert JE, Narasimhan A, Jengelley DH, Jiang Y, Liu J, Au E, Silverman LM, Sandusky G, Bonetto A, Cao S, Lu X, O'Connell TM, Liu Y, Koniaris LG, Ta Z. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J Exp Med 218: e20190450, 2021. doi: 10.1084/jem.20190450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xu MJ, Feng D, Wu H, Wang H, Chan Y, Kolls J, Borregaard N, Porse B, Berger T, Mak TW, Cowland JB, Kong X, Gao B. Liver is the major source of elevated serum lipocalin-2 levels after bacterial infection or partial hepatectomy: a critical role for IL-6/STAT3. Hepatology 61: 692–702, 2015. doi: 10.1002/hep.27447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yndestad A, Landro L, Ueland T, Dahl CP, Flo TH, Vinge LE, Espevik T, Froland SS, Husberg C, Christensen G, Dickstein K, Kjekshus J, Oie E, Gullestad L, Aukrust P. Increased systemic and myocardial expression of neutrophil gelatinase-associated lipocalin in clinical and experimental heart failure. Eur Heart J 30: 1229–1236, 2009. doi: 10.1093/eurheartj/ehp088. [DOI] [PubMed] [Google Scholar]
- 92. Viau A, El Karoui K, Laouari D, Burtin M, Nguyen C, Mori K, Pillebout E, Berger T, Mak TW, Knebelmann B, Friedlander G, Barasch J, Terzi F. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest 120: 4065–4076, 2010. doi: 10.1172/JCI42004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gomez-Chou SB, Swidnicka-Siergiejko AK, Badi N, Chavez-Tomar M, Lesinski GB, Bekaii-Saab T, Farren MR, Mace TA, Schmidt C, Liu Y, Deng D, Hwang RF, Zhou L, Moore T, Chatterjee D, Wang H, Leng X, Arlinghaus RB, Logsdon CD, Cruz-Monserrate Z. Lipocalin-2 promotes pancreatic ductal adenocarcinoma by regulating inflammation in the tumor microenvironment. Cancer Res 77: 2647–2660, 2017. doi: 10.1158/0008-5472.CAN-16-1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yan QW, Yang Q, Mody N, Graham TE, Hsu CH, Xu Z, Houstis NE, Kahn BB, Rosen ED. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes 56: 2533–2540, 2007. doi: 10.2337/db07-0007. [DOI] [PubMed] [Google Scholar]
- 95. Candido S, Abrams SL, Steelman LS, Lertpiriyapong K, Fitzgerald TL, Martelli AM, Cocco L, Montalto G, Cervello M, Polesel J, Libra M, McCubrey JA. Roles of NGAL and MMP-9 in the tumor microenvironment and sensitivity to targeted therapy. Biochim Biophys Acta 1863: 438–448, 2016. doi: 10.1016/j.bbamcr.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 96. Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta 1826: 129–169, 2012. doi: 10.1016/j.bbcan.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhang Y, Foncea R, Deis JA, Guo H, Bernlohr DA, Chen X. Lipocalin 2 expression and secretion is highly regulated by metabolic stress, cytokines, and nutrients in adipocytes. PLoS One 9: e96997, 2014. doi: 10.1371/journal.pone.0096997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhang Y, Guo H, Deis JA, Mashek MG, Zhao M, Ariyakumar D, Armien AG, Bernlohr DA, Mashek DG, Chen X. Lipocalin 2 regulates brown fat activation via a nonadrenergic activation mechanism. J Biol Chem 289: 22063–22077, 2014. doi: 10.1074/jbc.M114.559104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Rebalka IA, Monaco CM, Varah NE, Berger T, D'Souza DM, Zhou S, Mak TW, Hawke TJ. Loss of the adipokine lipocalin-2 impairs satellite cell activation and skeletal muscle regeneration. Am J Physiol Cell Physiol 315: C714–C721, 2018. doi: 10.1152/ajpcell.00195.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ponzetti M, Aielli F, Ucci A, Cappariello A, Lombardi G, Teti A, Rucci N. Lipocalin 2 increases after high-intensity exercise in humans and influences muscle gene expression and differentiation in mice. J Cell Physiol 237: 551–565, 2022. doi: 10.1002/jcp.30501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Choi EB, Jeong JH, Jang HM, Ahn YJ, Kim KH, An HS, Lee JY, Jeong EA, Lee J, Shin HJ, Kim KE, Roh GS. Skeletal lipocalin-2 is associated with iron-related oxidative stress in ob/ob mice with sarcopenia. Antioxidants (Basel) 10: 758, 2021. doi: 10.3390/antiox10050758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Olson B, Zhu X, Norgard MA, Levasseur PR, Butler JT, Buenafe A, Burfeind KG, Michaelis KA, Pelz KR, Mendez H, Edwards J, Krasnow SM, Grossberg AJ, Marks DL. Lipocalin 2 mediates appetite suppression during pancreatic cancer cachexia. Nat Commun 12: 2057, 2021. doi: 10.1038/s41467-021-22361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Musolino V, Palus S, Latouche C, Gliozzi M, Bosco F, Scarano F, Nucera S, Carresi C, Scicchitano M, von Haehling S, Jaisser F, Hasenfuss G, Anker SD, Mollace V, Springer J. Cardiac expression of neutrophil gelatinase-associated lipocalin in a model of cancer cachexia-induced cardiomyopathy. ESC Heart Fail 6: 89–97, 2019. doi: 10.1002/ehf2.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mosialou I, Shikhel S, Liu JM, Maurizi A, Luo N, He Z, Huang Y, Zong H, Friedman RA, Barasch J, Lanzano P, Deng L, Leibel RL, Rubin M, Nickolas T, Chung W, Zeltser LM, Williams KW, Pessin JE, Kousteni S. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543: 385–390, 2017. doi: 10.1038/nature21697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dallmann R, Weyermann P, Anklin C, Boroff M, Bray-French K, Cardel B, Courdier-Fruh I, Deppe H, Dubach-Powell J, Erb M, Haefeli RH, Hennebohle M, Herzner H, Hufschmid M, Marks DL, Nordhoff S, Papp M, Rummey C, Santos G, Scharer F, Siendt H, Soeberdt M, Sumanovski LT, Terinek M, Mondadori C, Guven N, Feurer A. The orally active melanocortin-4 receptor antagonist BL-6020/979: a promising candidate for the treatment of cancer cachexia. J Cachexia Sarcopenia Muscle 2: 163–174, 2011. doi: 10.1007/s13539-011-0039-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhu X, Callahan MF, Gruber KA, Szumowski M, Marks DL. Melanocortin-4 receptor antagonist TCMCB07 ameliorates cancer- and chronic kidney disease-associated cachexia. J Clin Invest 130: 4921–4934, 2020. doi: 10.1172/JCI138392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Huisman C, Norgard MA, Levasseur PR, Krasnow SM, van der Wijst MG, Olson B, Marks DL. Critical changes in hypothalamic gene networks in response to pancreatic cancer as found by single-cell RNA sequencing. Mol Metab 58: 101441, 2022. doi: 10.1016/j.molmet.2022.101441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lemecha M, Chalise JP, Takamuku Y, Zhang G, Yamakawa T, Larson G, Itakura K. Lcn2 mediates adipocyte-muscle-tumor communication and hypothermia in pancreatic cancer cachexia. Mol Metab 66: 101612, 2022. doi: 10.1016/j.molmet.2022.101612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Du Y, Li W, Lin L, Lo EH, Xing C. Effects of lipocalin-2 on brain endothelial adhesion and permeability. PLoS One 14: e0218965, 2019. doi: 10.1371/journal.pone.0218965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jin M, Kim JH, Jang E, Lee YM, Soo Han H, Woo DK, Park DH, Kook H, Suk K. Lipocalin-2 deficiency attenuates neuroinflammation and brain injury after transient middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 34: 1306–1314, 2014. doi: 10.1038/jcbfm.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hamzic N, Blomqvist A, Nilsberth C. Immune-induced expression of lipocalin-2 in brain endothelial cells: relationship with interleukin-6, cyclooxygenase-2 and the febrile response. J Neuroendocrinol 25: 271–280, 2013. doi: 10.1111/jne.12000. [DOI] [PubMed] [Google Scholar]
- 112. Wu L, Du Y, Lok J, Lo EH, Xing C. Lipocalin-2 enhances angiogenesis in rat brain endothelial cells via reactive oxygen species and iron-dependent mechanisms. J Neurochem 132: 622–628, 2015. doi: 10.1111/jnc.13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Yang C, Wu Y, Wang L, Li S, Zhou J, Tan Y, Song J, Xing H, Yi K, Zhan Q, Zhao J, Wang Q, Yuan X, Kang C. Glioma-derived exosomes hijack the blood-brain barrier to facilitate nanocapsule delivery via LCN2. J Control Release 345: 537–548, 2022. doi: 10.1016/j.jconrel.2022.03.038. [DOI] [PubMed] [Google Scholar]
- 114. Song E, Fan P, Huang B, Deng HB, Cheung BM, Feletou M, Vilaine JP, Villeneuve N, Xu A, Vanhoutte PM, Wang Y. Deamidated lipocalin-2 induces endothelial dysfunction and hypertension in dietary obese mice. J Am Heart Assoc 3: e000837, 2014. doi: 10.1161/JAHA.114.000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liu JT, Song E, Xu A, Berger T, Mak TW, Tse HF, Law IK, Huang B, Liang Y, Vanhoutte PM, Wang Y. Lipocalin-2 deficiency prevents endothelial dysfunction associated with dietary obesity: role of cytochrome P450 2C inhibition. Br J Pharmacol 165: 520–531, 2012. doi: 10.1111/j.1476-5381.2011.01587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yang J, McNeish B, Butterfield C, Moses MA. Lipocalin 2 is a novel regulator of angiogenesis in human breast cancer. FASEB J 27: 45–50, 2013. doi: 10.1096/fj.12-211730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wang H, Lou H, Li Y, Ji F, Chen W, Lu Q, Xu G. Elevated vitreous Lipocalin-2 levels of patients with proliferative diabetic retinopathy. BMC Ophthalmol 20: 260, 2020. doi: 10.1186/s12886-020-01462-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Su X, Zhou P, Qi Y. Down-regulation of LCN2 attenuates retinal vascular dysfunction and caspase-1-mediated pyroptosis in diabetes mellitus. Ann Transl Med 10: 695, 2022. doi: 10.21037/atm-22-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Fernandez-Chacon M, Garcia-Gonzalez I, Muhleder S, Benedito R. Role of Notch in endothelial biology. Angiogenesis 24: 237–250, 2021. doi: 10.1007/s10456-021-09793-7. [DOI] [PubMed] [Google Scholar]
- 120. Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell 16: 196–208, 2009. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 121. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell 34: 536–548, 2018. doi: 10.1016/j.ccell.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 122. Akil A, Gutierrez-Garcia AK, Guenter R, Rose JB, Beck AW, Chen H, Ren B. Notch signaling in vascular endothelial cells, angiogenesis, and tumor progression: an update and prospective. Front Cell Dev Biol 9: 642352, 2021. doi: 10.3389/fcell.2021.642352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, Espinet E, Herpel E, Menuchin A, Chang-Claude J, Hoffmeister M, Gebhardt C, Brenner H, Trumpp A, Siebel CW, Hecker M, Utikal J, Sprinzak D, Fischer A. Endothelial Notch1 activity facilitates metastasis. Cancer Cell 31: 355–367, 2017. doi: 10.1016/j.ccell.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 124. Mendonca L, Trindade A, Carvalho C, Correia J, Badenes M, Gigante J, Duarte A. Metastasis is impaired by endothelial-specific Dll4 loss-of-function through inhibition of epithelial-to-mesenchymal transition and reduction of cancer stem cells and circulating tumor cells. Clin Exp Metastasis 36: 365–380, 2019. doi: 10.1007/s10585-019-09973-2. [DOI] [PubMed] [Google Scholar]
- 125. Alsina-Sanchis E, Mulfarth R, Moll I, Bohn S, Wiedmann L, Jordana-Urriza L, Ziegelbauer T, Zimmer E, Taylor J, De Angelis Rigotti F, Stogbauer A, Giaimo BD, Cerwenka A, Borggrefe T, Fischer A, Rodriguez-Vita J. Endothelial RBPJ is essential for the education of tumor-associated macrophages. Cancer Res 82: 4414–4428, 2022. doi: 10.1158/0008-5472.CAN-22-0076. [DOI] [PubMed] [Google Scholar]
- 126. Moore G, Annett S, McClements L, Robson T. Top Notch targeting strategies in cancer: a detailed overview of recent insights and current perspectives. Cells 9: 1503, 2020. doi: 10.3390/cells9061503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Bjornson CR, Cheung TH, Liu L, Tripathi PV, Steeper KM, Rando TA. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30: 232–242, 2012. doi: 10.1002/stem.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2: 50–59, 2008. doi: 10.1016/j.stem.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 129. Gerrard JC, Hay JP, Adams RN, Williams JC 3rd, Huot JR, Weathers KM, Marino JS, Arthur ST. Current thoughts of Notch‘s role in myoblast regulation and muscle-associated disease. Int J Environ Res Public Health 18: 12558, 2021. doi: 10.3390/ijerph182312558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mourikis P, Tajbakhsh S. Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Dev Biol 14: 2, 2014. doi: 10.1186/1471-213X-14-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hoier B, Hellsten Y. Exercise-induced capillary growth in human skeletal muscle and the dynamics of VEGF. Microcirculation 21: 301–314, 2014. doi: 10.1111/micc.12117. [DOI] [PubMed] [Google Scholar]
- 132. Verma M, Asakura Y, Murakonda BS, Pengo T, Latroche C, Chazaud B, McLoon LK, Asakura A. Muscle satellite cell cross-talk with a vascular niche maintains quiescence via VEGF and Notch signaling. Cell Stem Cell 23: 530–543.e9, 2018. doi: 10.1016/j.stem.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kargl CK, Nie Y, Evans S, Stout J, Shannahan JH, Kuang S, Gavin TP. Factors secreted from high glucose treated endothelial cells impair expansion and differentiation of human skeletal muscle satellite cells. J Physiol 597: 5109–5124, 2019. doi: 10.1113/JP278165. [DOI] [PubMed] [Google Scholar]
- 134. Latroche C, Weiss-Gayet M, Muller L, Gitiaux C, Leblanc P, Liot S, Ben-Larbi S, Abou-Khalil R, Verger N, Bardot P, Magnan M, Chretien F, Mounier R, Germain S, Chazaud B. Coupling between myogenesis and angiogenesis during skeletal muscle regeneration is stimulated by restorative macrophages. Stem Cell Reports 9: 2018–2033, 2017. doi: 10.1016/j.stemcr.2017.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, Wang J, Bloomston M, Muscarella P, Nau P, Shah N, Butchbach ME, Ladner K, Adamo S, Rudnicki MA, Keller C, Coletti D, Montanaro F, Guttridge DC. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 123: 4821–4835, 2013. doi: 10.1172/JCI68523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mu X, Agarwal R, March D, Rothenberg A, Voigt C, Tebbets J, Huard J, Weiss K. Notch signaling mediates skeletal muscle atrophy in cancer cachexia caused by osteosarcoma. Sarcoma 2016: 3758162, 2016. doi: 10.1155/2016/3758162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lu F, Osei-Hwedieh D, Mandell JB, Morales-Restrepo A, Hankins ML, Crasto JA, Ma R, Dinh V, Watters RJ, Weiss KR. Comparison of cachectic and non-cachectic sarcoma patients reveals an important role of Notch signaling in metastasis and myogenesis. Am J Cancer Res 9: 1746–1756, 2019. [PMC free article] [PubMed] [Google Scholar]
- 138. von Grabowiecki Y, Licona C, Palamiuc L, Abreu P, Vidimar V, Coowar D, Mellitzer G, Gaiddon C. Regulation of a Notch3-Hes1 pathway and protective effect by a tocopherol-omega alkanol chain derivative in muscle atrophy. J Pharmacol Exp Ther 352: 23–32, 2015. doi: 10.1124/jpet.114.216879. [DOI] [PubMed] [Google Scholar]
- 139. Fujimaki S, Matsumoto T, Muramatsu M, Nagahisa H, Horii N, Seko D, Masuda S, Wang X, Asakura Y, Takahashi Y, Miyamoto Y, Usuki S, Yasunaga KI, Kamei Y, Nishinakamura R, Minami T, Fukuda T, Asakura A, Ono Y. The endothelial Dll4-muscular Notch2 axis regulates skeletal muscle mass. Nat Metab 4: 180–189, 2022. doi: 10.1038/s42255-022-00533-9. [DOI] [PubMed] [Google Scholar]