


Abstract
A selected ribozyme ligase, L1, has been engineered to respond to small organic effectors. Residues important for ribozyme catalysis were mapped to a compact core structure. Aptamers that bound adenosine and theophylline were appended to the core structure, and the resultant aptazymes proved to be responsive to their cognate effectors. Rational sequence substitutions in the joining region between the aptamer and the ribozyme yielded aptazymes whose activities were enhanced from 800–1600-fold in the presence of 1 mM ATP or theophylline, respectively. However, when an anti-flavin aptamer was appended to the core ribozyme structure flavin-responsivity was minimal. The joining region between the aptamer and the ribozyme core was randomized and a series of negative and positive selection steps yielded aptazymes that were activated by up to 260-fold in the presence of 100 µM FMN. The selected joining regions proved to be ‘communication modules’ that could be used to join other aptamers to the ribozyme core to form aptazymes. These results show that ribozyme ligases can be readily engineered to function as allosteric enzymes, and reveal that many of the techniques and principles previously demonstrated during the development of hammerhead aptazymes may be generalizable.
INTRODUCTION
Breaker and his co-workers have previously demonstrated that catalysis by the hammerhead ribozyme can be made dependent on molecular effectors by the simple expedient of appending aptamers to non-essential stem regions. Since the structure of the hammerhead ribozyme had been determined, these authors could understand the mechanism of effector modulation in terms of ligand-induced conformational changes. While the allosteric transitions primarily involved simple changes in secondary structure, it was unclear if such strategies would also immediately prove successful if applied to other ribozymes, especially ribozymes where little or no advance structural information is available.
As a starting point for the generation of aptazyme ligases we chose the L1 ligase, a ribozyme originally identified by in vitro selection (1). The chief advantage of the L1 ligase is its small size, 130 nt, since we suspected that ligand-induced conformational changes would have a proportionately larger influence on a small, relatively unstable ligase as opposed to a larger, more structurally robust ligase (such as the Group I intron or the Bartel Class I ligase; 2). While other small ligases could also have been utilized, such as the hairpin ribozyme or the Bartel Class II ligase, the activity of the L1 ligase was already known to be modulated by an oligonucleotide effector (1). Thus, it seemed likely that the activity of the L1 ligase might also be modulated by other molecular effectors.
MATERIALS AND METHODS
Sequences and primers
The sequence of the L1 ligase is GGA CUU CGG UCC AGU GCU CGU GCA CUA GGC CGU UCG ACC AUG UGG GUC CGC UGC CAG CGG CAA UCU GGC AUG CUA UGC GGA ACC UUC ACA UCU UAG ACA GGA GGU UAG GUG CCU CGU GAU GUC CAG UCG C. The substrate used for ligation assays was S28A [(dA)22-ugcacu; RNA in lowercase] unless otherwise stated. The standard primers used to amplify the L1 ligase were 42.90w (TTCTAATACGACTCACTATAGGACTTCGGTCCAGTG-CTCGTG; T7 promoter underlined) and 18.90a (GCGACTGGACATCACGAG).
Synthesis of pools and mutants
The D90 doped pool was prepared using solid phase DNA synthesis methodologies as previously described (1). The D90 pool was based upon the sequence of the L1 ribozyme but with the internal 90 residues synthesized with a 37% degeneracy and the primer binding sites synthesized without mutation (TTCGGTCCAGTGCTCGTG-cac tag gcc gtt cga cca tgt ggg tcc gct gcc agc ggc aat ctg gca tgc tat gcg gaa cct tca cat ctt aga cag gag gtt agg tgc-CTCGTGATGTCCAGTCGC, degenerate residues in lowercase; non-degenerate residues in uppercase).
Templates for flavin (3) and chloramphenicol (4) aptazymes were each synthesized on two 0.2 µmol columns [FMNn8: GGA CTT CGG TCC AGT GCT CGT GCA CTA GGC CGT TCG ACC (N3–4) AGG ATA TGC TTC GGC AGA AGG (N3–4) CTT AGA CAG GAG GTT AGG TGC CTC GTG ATG TCC AGT CGC; CAMn8: GGA CTT CGG TCC AGT GCT CGT GCA CTA GGC CGT TCG ACC N(3–4) ACA GTG AAA AAA GAC GTG TGA ATG TCA CAC TGA AAA AA N(3–4) CTT AGA CAG GAG GTT AGG TGC CTC GTG ATG TCC AGT CGC; aptamer domains are underlined, N = A, G, C or T). The 3′ half of the pool, including the first randomized section, was synthesized with three random positions on column 1 and four random positions on column 2. The synthesis was then paused, the columns removed from the synthesizer, opened, and the resins mixed and again split into two columns. The synthesis was then resumed and the 5′ side of the random connecting stem synthesized with three random positions on column 1 and four random positions on column 2. When the synthesis was complete, the resins were mixed to create the final pool templates.
Templates for Stem C deletion variants were made on a DNA synthesizer and PCR amplified using the standard L1 PCR primers (42.90w and 18.90a). Stem B deletion variants were generated by PCR amplification of an L1 DNA template using the standard 3′ PCR primer and a nested 5′ primer (TTC TAA TAC GAC TCA CTA TAG GAC CTC GGC GAA AGC CGT TCG ACC A) to introduce the deletion. Composite deletions were created with a combination of nested 5′ primers and Stem C templates.
In vitro selection of ribozymes
The general selection procedure for ligase ribozymes has been previously described (1). Very briefly, pool RNA was denatured in water followed by the addition of ligation buffer (30 mM Tris, pH 7.4, 600 mM NaCl, 1 mM EDTA, 1% NP-40, 60 mM MgCl2) and substrate oligonucleotide. After incubation at 25°C, the selection mixture was passed through an affinity column specific for a tag sequence on the substrate oligonucleotide. Ligated molecules that contained the tag sequence were retained while unligated molecules were washed from the column. The selected ribozymes were then reverse transcribed and selectively PCR amplified with a primer specific for the tag sequence. The amplified molecules were then further amplified with a nested PCR primer that regenerated the T7 promoter. Following transcription with T7 RNA polymerase, the resultant RNA pool was ready to undergo further cycles of selection and amplification.
The aptazyme selection protocol was based upon the general ligase selection protocol with an additional negative selective step. Pool RNA (100 pmol) was denatured with 200 pmol 18.90a. Ligation buffer (pH 7.7) and 200 pmol substrate (S28A-biotin) were added and the mixture was incubated for 16–24 h at 25°C in the absence of ligand. The reaction was stopped by the addition of EDTA and the ligated species were removed by passing the reaction mixture through a column packed with 100 µl streptavidin–agarose (Gibco BRL, Gaithersburg, MD). The column eluant was then replenished with 100 pmol 18.90a, ligand (final concentration of 100 µM FMN or 1 mM chloramphenicol), and 200 pmol S28A-biotin. These reactions were incubated at 25°C for 2 h in the first round of positive (+) selection. Selection stringency was steadily increased over the course of seven rounds of selection by decreasing the (+) ligand incubation times to 1 h in the second round, 2.5 h in the third, 30 min in the fourth, 10 min in the fifth, 5 min in the sixth and seventh rounds. Additional stringency in the negative selection was included in the seventh round by performing two sequential (–) ligand incubations.
Ligation assays
Ligation assays were performed as previously described (1). Body-labeled ribozyme (10 pmol) and 20 pmol effector oligonucleotide were denatured for 3 min at 70°C in 5 µl water. The RNA mixture was cooled to room temperature followed by addition of ligation buffer (pH 7.7 unless otherwise indicated) and ligand [or water in place of ligand, in the case of (–) ligand samples]. After a 5 min equilibration at room temperature, reactions were initiated by the addition of 20 pmol substrate oligonucleotide and incubated at 25°C. Aliquots (4 µl) of each reaction were removed at two or three appropriate time points and the reactions terminated by the addition of 18 µl of stop mix (100 mM EDTA, 80% formamide, 0.05% bromophenol blue, 0.05% xylene cyanol). Samples were denatured for 3 min at 70°C and ligated and unligated species separated from one another on 8% polyacrylamide gels and analyzed using a Phosphorimager.
RESULTS
Identification of functional residues and structures in the L1 ligase
In order to engineer the L1 ligase to function as an allosteric ribozyme, it was first necessary to determine which residues and structures in the original ribozyme contributed to its catalytic function. To this end, we employed three strategies. First, site-directed mutagenesis and complementation analysis was used to identify residues that contributed to substrate-binding; second, selection from a doped sequence population was used to identify functional residues; and finally, deletion analysis was used to determine what residues could fold into a minimal, functional ligase.
The secondary structural model that we had developed for the L1 ligase (Fig. 1a) suggested that residues near the 3′ end of the ribozyme were involved in base pairing to the ligation substrate. To determine whether this was truly the substrate-binding site, several mutations were constructed in the substrate, and the mutant substrates were assayed for their ability to join to either the wild-type ligase or to substitution variants that restored the predicted base-pairing. The results of these experiments are shown in Figure 1b, and confirm our model of the secondary structure of the L1 ligase and the identity of the residues involved in substrate binding. While the activities of the ligases that contained compensatory interactions were smaller than the activity of the wild-type ribozyme, they were nonetheless much higher than the (unmeasurable) activities of the mismatched ribozyme:substrate pairs. An exception to these suppression studies is the G:U wobble pair at the ligation junction, which mutation analysis indicates is essential for activity (data not shown).
Figure 1.
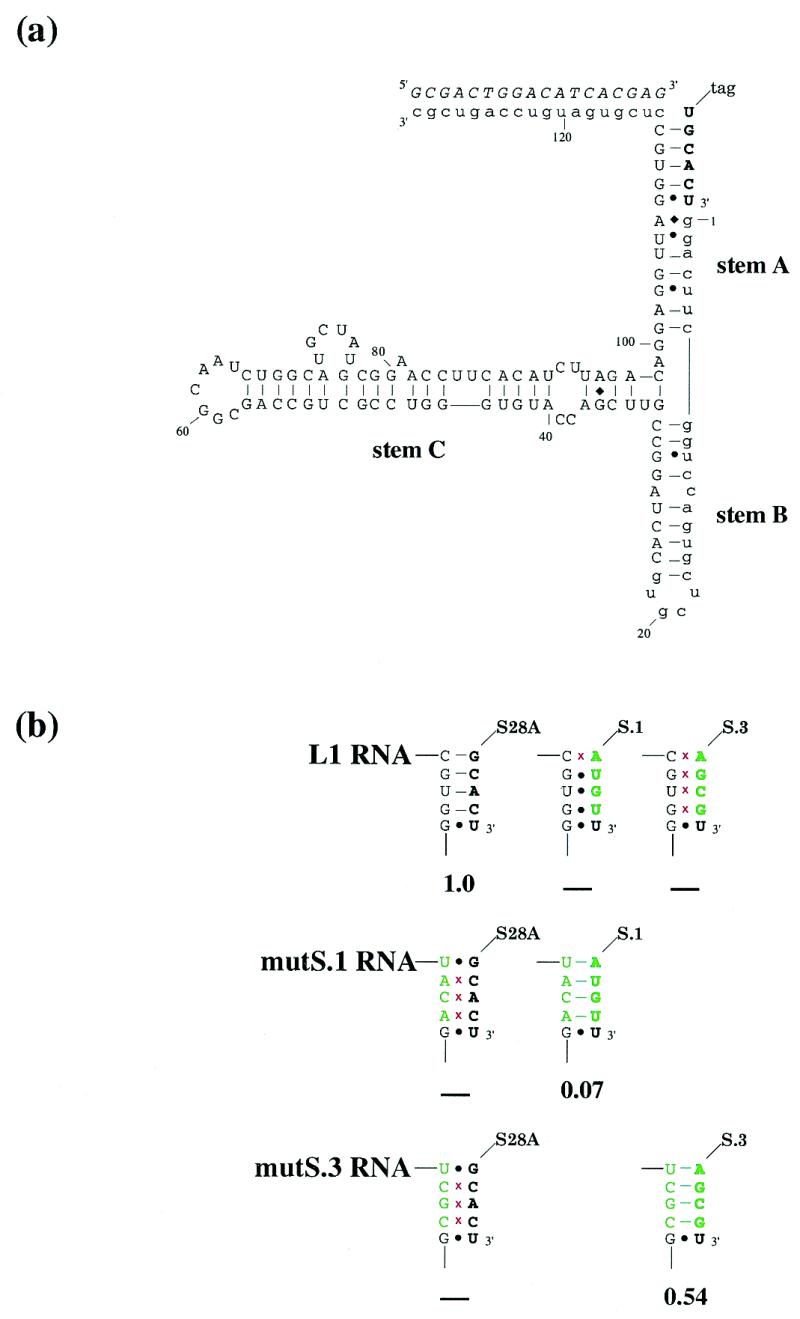
(a) Proposed secondary structure of the L1 ligase. The predicted secondary structure of the L1 ligase, based on mutational and covariation analysis. Constant regions are shown in lower case. The ligation substrate is in bold. The oligonucleotide effector is italicized. (b) Substrate binding suppression analysis. The substrate (in bold) and the region of the ribozyme involved in substrate binding are shown. Mutations relative to either the wild-type ribozyme or the wild-type substrate are shown in green; red crosses indicate disruption of Watson–Crick pairings. Ribozyme activities are shown below, relative to wild type. ‘–’ indicates no detectable activity.
A doped sequence pool centered on the wild-type L1 ligase sequence was generated. Each position in the pool contained 63% wild-type residues (e.g., 63%G at position 42) and 37% non-wild-type residues (e.g., 12.3%A, 12.3%T, 12.4C%, based on the sequences of eight unselected clones). Ribozymes were selected based on their ability to covalently append a substrate to themselves. Although the round 0 population had extremely low activity (Fig. 2a), after only four rounds of selection and amplification the pool had surpassed wild-type levels of ligation activity. Individual ribozymes were cloned and sequenced from this highly active population, and the relative degree of sequence conservation at each position was mapped onto the proposed secondary structure of the ligase (Fig. 2b). It is apparent that most of the functional residues in the ribozyme lie at or adjacent to the three helix junction. In at least two instances, C25U and A27G, sequence changes that predominate following the doped sequence selection affirm the secondary structural model.
Figure 2.

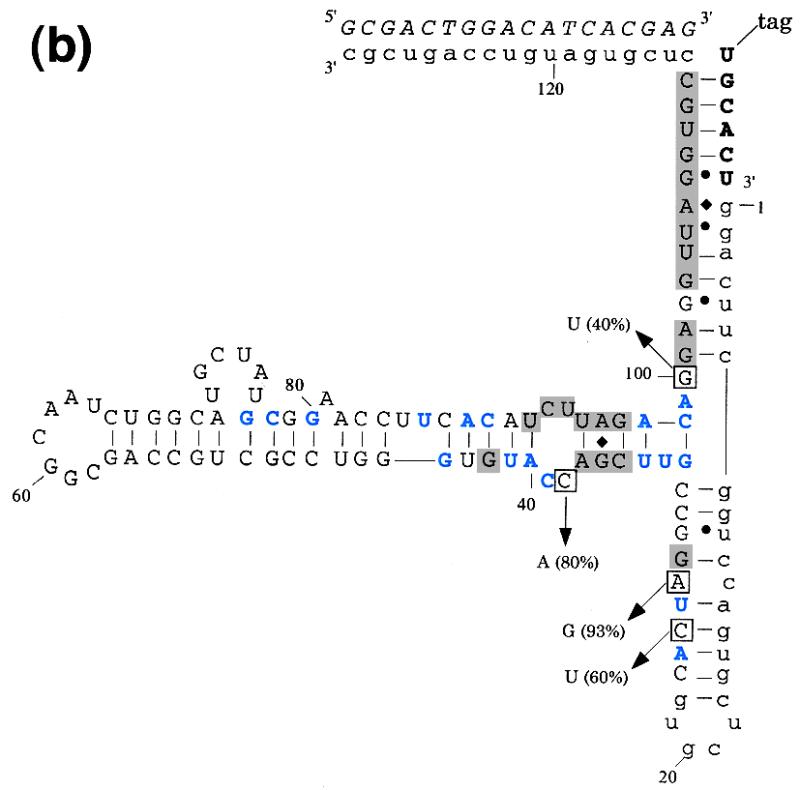
(a) Progress of the doped selection. The bar graph on the left indicates the activity of the selected population relative to the wild-type ligase as a function of ribozyme generation. The dark grey activity bars are for the population as a whole; the light grey bar in round 7 is for a ribozyme sub-population that had a small deletion in Stem C and was of a slightly different size than the major population. The table on the right shows experimental parameters during the course of the selection. Ligation activities in a standard assay are expressed relative to the wild-type ribozyme. The stringency of selection was increased throughout by decreasing the time allowed for reaction and magnesium concentration (20). (b) Secondary structure of the L1 ligase with doped selection results superimposed. The general description of the L1 ligase is as in Figure 1a. Shaded regions showed absolute sequence conservation following re-selection. Residues shown in blue were conserved in >85% of re-selected clones. Boxed positions converged to a non-wild-type residue; percentages indicate the number of clones containing the non-wild-type residue.
To validate the structural and functional model of the L1 ligase that had emerged from mutation and selection analyses, we attempted to generate a minimal ribozyme by deleting residues in outlying portions of the ribozyme that appeared to be less critical to ribozyme function. In particular, Stem C extends outward from the helical junction with mostly Watson–Crick base-pairing interrupted by a single, absolutely conserved G:A pair and a symmetrical internal loop. However, beyond the internal loop the majority of Stem C was highly variant following the doped selection, and did not form a consensus secondary structure. Consistent with the doped selection data, constructs with deletions downstream of the A40:U91 base pair retained near wild-type activity, while constructs with deletions encompassing the C38C39:C92U93 internal loop were inactive (Fig. 3).
Figure 3.

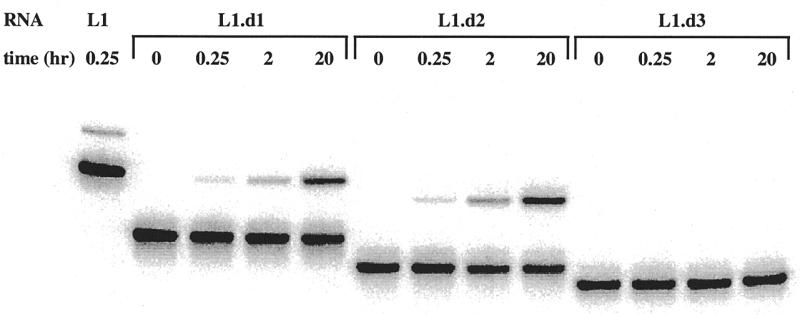
(a) Secondary structures and activities of deletion constructs. The general description of L1 ligase variants is as in Figure 1a. Residues in green have been introduced in place of a longer Stem C. (b) The activities of the three deletion constructs are shown. The upper band is ligated product, the lower band is unligated ribozyme.
Design and assay of aptazymes
Breaker and his co-workers had originally designed an effector-dependent hammerhead ribozyme by joining ribozyme residues and structures known to be important for function with aptamer residues and structures known to be aligned and stabilized by induced fit (5). Our selection and deletion analyses were successful in identifying which residues in the L1 ligase were important for function. We therefore attempted to join several different aptamers to the L1 core structure in an effort to create a hybrid ‘aptazyme’ that would exhibit effector-dependent ligation activity.
The first aptazyme construct was created by introducing an anti-adenosine aptamer (6) in place of the non-essential portion of Stem C. It was known from NMR studies that the anti-adenosine aptamer adopted a more rigid conformation upon interaction with adenosine (7). We hypothesized that upon interaction with adenosine, the connecting stem would be stabilized, would better mimic the Watson–Crick pairing normally present on Stem C, and thus would better align the catalytically important internal loop region. Since the minimal, functional structural model of the L1 ligase appeared to require the Stem C internal loop region, the minimal, functional anti-adenosine aptamer was joined to a base pair immediately adjacent to the Stem C internal loop (Fig. 4a). Our hypothesis proved to be correct, and the resultant aptazyme showed a 30-fold increase in activity in the presence of saturating (1 mM) ATP concentrations (data not shown).
Figure 4.
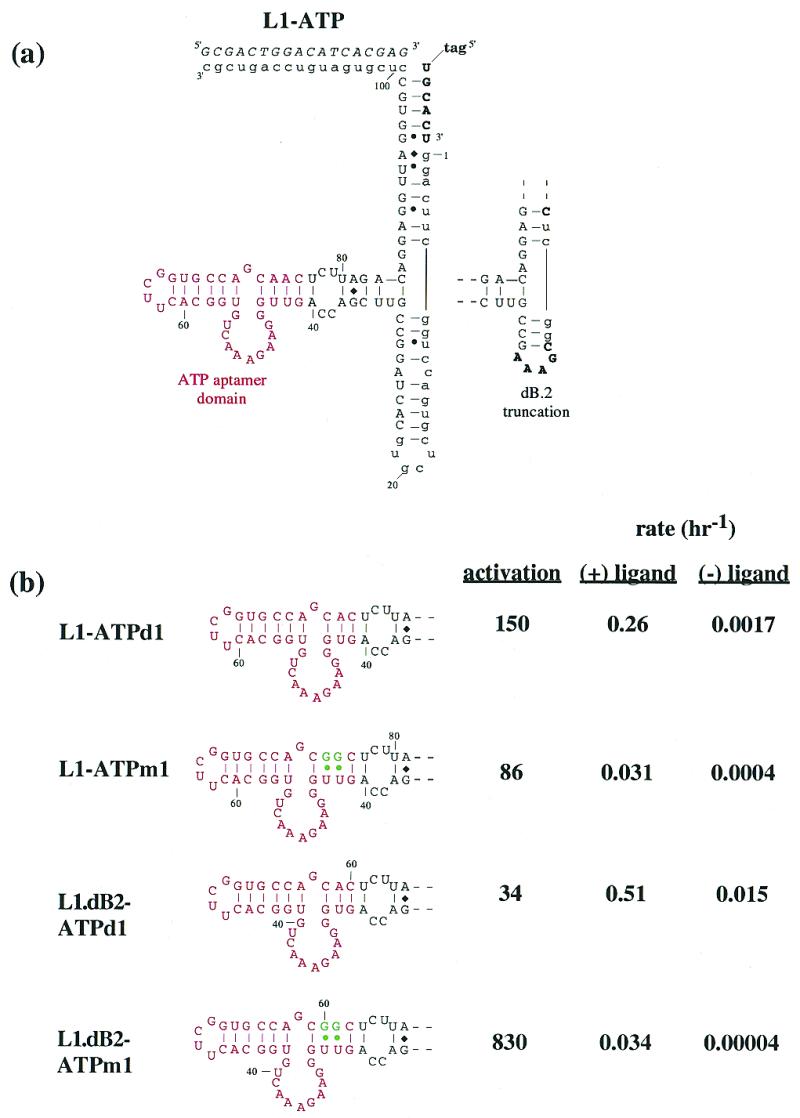

(a) Adenosine-sensing aptazyme structure. The general description of the L1 ligase aptazymes is as in Figure 1a. The anti-adenosine aptamer domain is shown in red. Mutations that are predicted to destabilize the joining region between the aptamer and the ribozyme are shown in green. (b) Aptazyme activities. Stem C variants are shown, again with the anti-adenosine aptamer in red. Aptazymes that have a ‘dB2’ designation in their title also contain the alternate Stem B shown in (a). The rate constants and relative activation in the presence of ligand (1 mM) under standard assay conditions (at pH 7.4) are shown to the right of the constructs. (c) Activity as a function of ATP concentration. Standard assays (at pH 7.7) were carried out with L1–ATPd1 [see (b)].
To the extent that the ligand-dependent stabilization of the joining region between the ribozyme and the aptamer led to ligand-dependent increases in catalysis, then destabilization of the joining region should similarly lead to an increased level of allosteric activation. We synthesized and assayed a series of constructs that rationally altered the joining region, progressively destabilizing the connecting stem structure by either introducing mismatches into or shortening the stem structure (Fig. 4b). Both destabilization approaches were successful in increasing the ATP aptazyme’s activation parameters. Shortening the stem by 1 bp (L1–ATPd1) resulted in a 5-fold further increase in activation (to 153-fold), while introducing G:U pairs in place of A:U pairs (L1–ATPm1) resulted in a 3-fold increase in activation (to 86-fold). We further reasoned that if ligase activity was a property of the global stability of the ribozyme, then altering other regions of the ribozyme might also affect global stability, and in consequence ligand-dependent re-stabilization of the global structure might increase the overall level of allosteric activation. To this end, we deleted non-essential residues in Stem B. In the context of the single base pair deletion variant, L1–ATPd1, the deletion of Stem B reduced the level of activation back to that of the parental ribozyme. However, in the context of the mutant containing wobble base pairs, L1–ATPm1, the deletion of Stem B proved to be synergistic, and led to 830-fold activation by 1 mM ATP. This level of activation exceeded the level of effector modulation previously observed with rationally designed hammerhead aptazymes, due primarily to the much slower reaction rate of the ligase in the absence of activating ligands. The reduced activity of the L1 ligase may in turn be due to the fact that it appears to be more sensitive to perturbations of its Stem C region than is the hammerhead ribozyme to perturbations of its stem II (8), indicating that the L1 ligase might be an even better vehicle for the construction of aptazymes than the hammerhead ribozyme.
We measured the responsivity of one of the adenosine-sensing aptazymes (L1–ATPd1) as a function of ATP concentration (Fig. 4c). A curve that was similar in shape to a canonical binding profile was obtained. However, the apparent Kd of the adenosine-sensing aptazyme (~97 µM) was higher than the known Kd of the anti-adenosine aptamer (~10 µM; 6,9). These results may indicate that the generic binding ability of the anti-adenosine aptamer was compromised upon conjugation to the ribozyme.
Based on the success of our ATP aptazyme, we used the same strategy to design additional aptazymes by fusing other aptamer domains onto Stem C of L1. We chose to use anti-theophylline (10) and anti-flavin aptamers (3), whose three-dimensional structures were known (11,12). In addition, both aptamers had previously proven amenable for use in hammerhead aptazymes (5,13) and thus allowed us to further compare the characteristics of self-cleaving aptazymes with ligase aptazymes. We also chose an anti-chloramphenicol aptamer (4) that had previously been extensively characterized, but whose detailed structure was unknown.
Using the results of the L1–adenosine constructs as a guide, an anti-theophylline aptamer was appended to Stem C of L1 through a 4 bp connecting stem. This initial theophylline construct showed <2-fold activation with theophylline and so a new construct was designed in which the connecting stem was destabilized by replacing a U:A base pair with a U:G wobble pair. The activation parameters of the second-generation construct were no better than the initial construct. A third-generation construct was created in which the connecting stem was shortened to 3 bp by deleting a G:C base pair (Fig. 5a). This version of the aptazyme proved to be extremely dependent on the presence of theophylline for activity. Without theophylline, the ribozyme performed self-ligations with a rate of 1.7 × 10–4 h–1. When theophylline was added to the reaction the rate improved to 0.27 h–1, a 1600-fold increase (Fig. 5b). As was the case with the adenosine-sensing aptazyme, when the activity of the theophylline-sensing aptazyme was measured as a function of theophylline concentration a characteristic response curve was observed (Fig. 5c). Again, the apparent Kd of the aptazyme (~99 µM) was much higher than the known Kd of the anti-theophylline aptamer (~300 nM).
Figure 5.
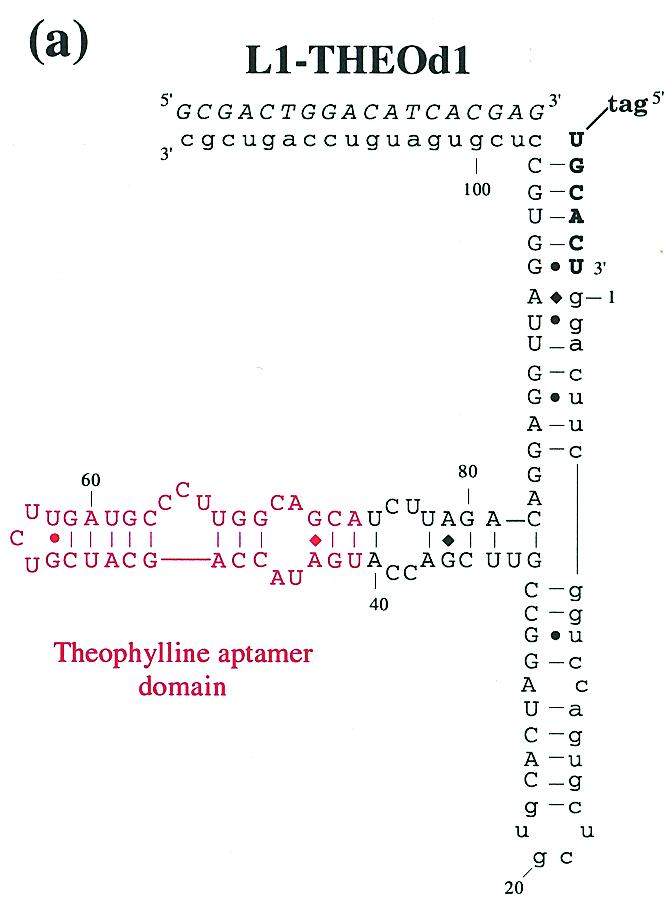
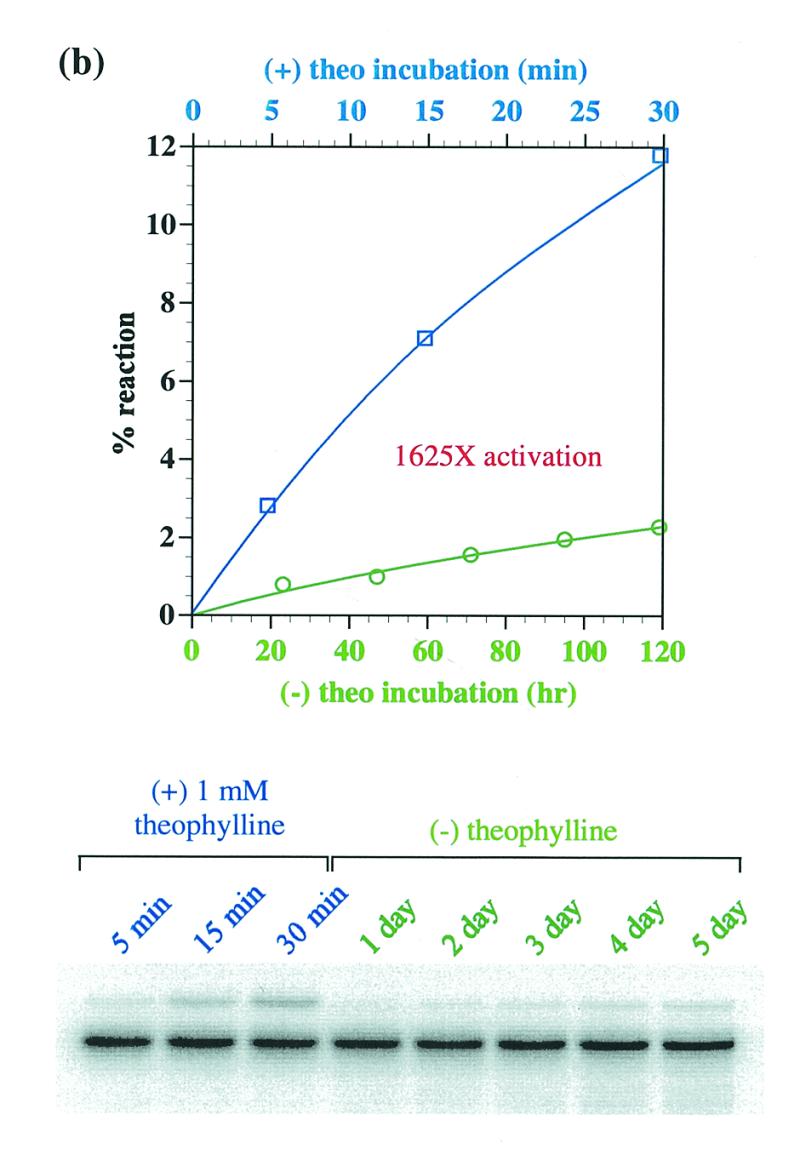

(a) Theophylline-sensing aptazyme structure. The structural description is as in Figure 1a and Figure 4a, except that the anti-theophylline aptamer domain is in red. (b) Aptazyme kinetics. The extent of ligation for the anti-theophylline aptazyme in the absence (green, x-axis in hours) and presence (blue, x-axis in minutes) of theophylline is shown as a function of time. The data for this graph were derived from the gel shown. (c) Activity as a function of theophylline concentration. Standard assays (at pH 7.7) were carried out with L1–THEOd1 [see (a)].
The anti-flavin aptamer was similarly appended to Stem C so that 4 bp connected internal loops of the aptamer and the ribozyme. Like the initial theophylline construct, the initial flavin construct (L1–FMN) showed little activation. However, in contrast to the results with the anti-adenosine and anti-theophylline aptamers, destabilizing mutations (G:U pairs, deletions) did not lead to improved activation parameters. We next attempted to insert a ‘communication module’ that had previously been shown by Soukup and Breaker (13) to greatly improve the FMN-dependent activation of a hammerhead aptazyme. However, in the context of the L1 ligase (L1–FMNm1) the hammerhead class I induction element did not yield any effector dependence; in fact, the resultant ligase construct was completely inactive (Fig. 7). Similarly, when the internal loops of the anti-chloramphenicol aptamer and the L1 ligase were joined by a 4-bp connecting stem the resultant constructs showed very little effector dependence.
Figure 7.

Cross-utilization of communication modules. Different constructs with different communication modules are shown. Aptamers are in red; communication modules or joining regions are in green. The top three constructs utilize a communication module derived from the selection for FMN-dependence and were previously shown in Figure 6b. Utilization of the hammerhead class I induction module is depicted in L1–FMNm1. The lower two constructions utilize a joining region originally designed for the theophylline-sensing aptazyme. The relative activation in the presence of ligand (1 mM, except FMN which was 100 µM) under standard assay conditions (pH 7.7, except L1–THEOd1 which is pH 7.4 indicated by asterisks) are shown to the right of the constructs.
Selection for effector dependence
In order to optimize the performance of the anti-flavin and anti-chloramphenicol aptazymes, the stem region connecting the aptamer and ribozyme domains was randomized and a selection for effector dependence was performed. The randomized pools were synthesized such that each side of the connecting region contained a random sequence mixture either 3 or 4 nt in length. It was hoped that the mixture of lengths (i.e., three residues across from three residues, three residues across from four residues, etc.) would facilitate the selection of both stem and internal loop or bulge structures that might facilitate communication between the aptamer and ribozyme. All possible sequences and combinations of lengths (102 400) were represented in the initial library.
A two-stage selection procedure was used to isolate aptazymes that achieved maximal activity only when ligand was present. The first, negative selection step involved an extended incubation with substrate in the absence of ligand. Any ribozymes capable of ligating without ligand being present were removed from the population via a biotin tag on the substrate. Ligand (flavin or chloramphenicol) was then added to the remaining RNA population and a second, positive selective step was used to isolate the best catalysts from the surviving ribozymes. The entire process was repeated using an increasingly stringent positive selective step until only the most active aptazymes remained. Over the course of seven rounds of selection for FMN activation, the activity of the selected population generally increased when FMN was present, while the activity of the same population in the absence of FMN remained low (Fig. 6a). The net result was a population of aptazymes that reacted at rates comparable to the wild-type L1 ligase when FMN was present, but ~100-fold slower in the absence of FMN. When individual clones were isolated from the selected population and assayed for activity, most variants had activations of ~100-fold with FMN in accord with the activity level of the population. However, a few variants were isolated that had superior activation parameters of up to 260-fold with FMN (Fig. 6b). While we report five joining regions that yielded superior activation, there were multiple others in the final pool, many of which showed effector-dependence similar to that observed with the pool as a whole.
Figure 6.
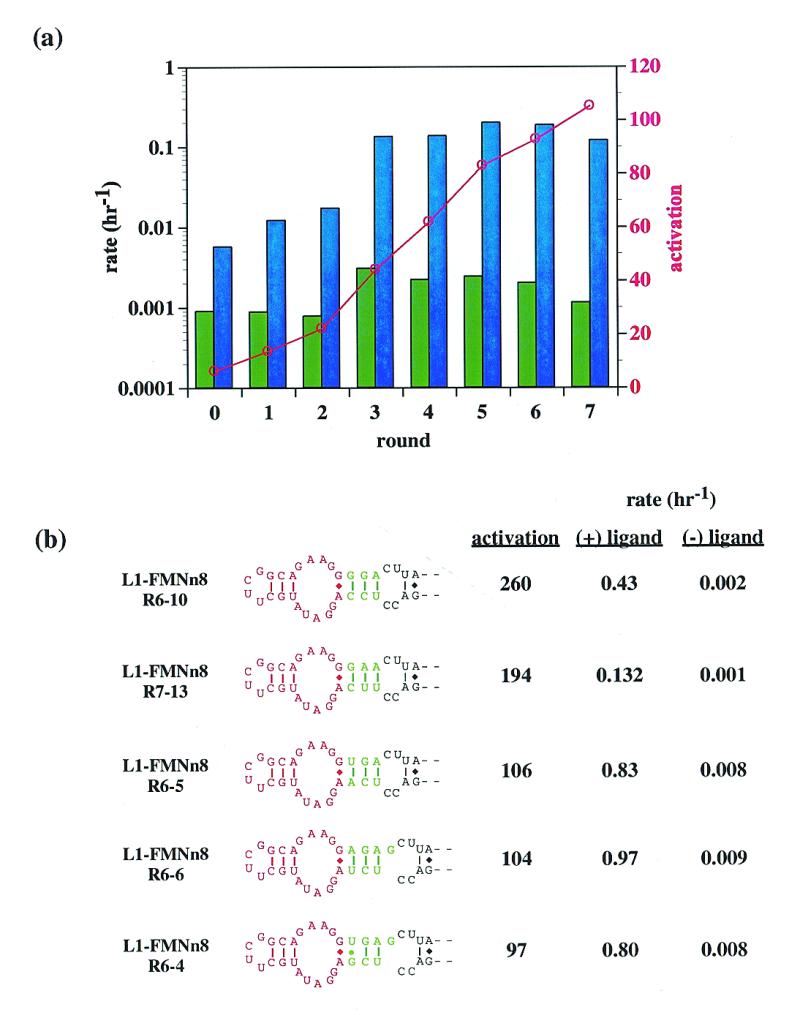
(a) Progress of the communication module selection. The rate constants of aptazyme pools in the absence (green bars) and presence (blue bars) of 100 µM FMN are shown as a function of aptazyme generation. The (logarithmic) scale for the rate data is on the left-hand y-axis. Superimposed on the rate data is the ratio of activity in the presence of FMN to the activity in the absence of FMN (red points). The scale for the ratio data is on the right-hand y-axis. (b) Sequences and activities of communication modules. The best communication modules derived from the selection are shown. The anti-flavin aptamer is in red; the communication modules selected from the N3-N4 pool are in green. The rate constants and relative activation in the presence of ligand (100 µM) under standard assay conditions (at pH 7.7) are shown to the right of the constructs.
In contrast with the selection targeting FMN, the results of the selection targeting chloramphenicol were disappointing. Over the course of four rounds of selection for chloramphenicol activation, the activity of the selected population increased to near wild-type L1 levels irrespective of the presence of chloramphenicol.
Modular effector dependence
As stated above, Soukup and Breaker have previously demonstrated (13) that randomization of the joining region between aptamer and ribozyme can yield so-called communication modules that can mediate signal transduction between a variety of aptamers and the ribozyme. In order to determine if the joining regions that arose during the selection for flavin dependence were also communication modules we substituted both the anti-theophylline and anti-adenosine aptamers for the anti-flavin aptamer in the most active construct (L1–FMNn8R6-10; Fig. 7). The anti-theophylline aptamer chimera (L1–THEOm2) showed significant (350-fold) ligand-dependent activation, though less than had been observed with the designed construct (L1–THEOd1; 1625-fold). In contrast, the anti-adenosine aptamer chimera (L1–ATPm2) not only showed no activation, but in fact had little catalytic activity at all. The ability of anti-flavin and anti-theophylline aptamers to substitute for one another, but not with the anti-adenosine aptamer, may derive from the structural similarities of both of the former aptamers including non-canonical G:A base pairs at the base of their stems, immediately adjacent to the joining region.
In order to determine the generality of the ability of anti-flavin and anti-theophylline aptamers to swap with one another we used the previously designed, highly theophylline-dependent aptazyme (L1–THEOd1) as a starting point. The anti-flavin aptamer was joined to the L1 ligase in place of the anti-theophylline aptamer, using the same designed (as opposed to selected) joining region. In accord with our expectations, the resultant anti-flavin aptamer chimera (L1–FMNm2) showed much greater ligand-dependent activation (41-fold) than had previous designed constructs, though less than that exhibited by a selected aptazyme (L1–FMNn8R6-10; 260-fold)
DISCUSSION
The L1 ligase is one of three selected ribozymes (the others being the Bartel Class I ligase and a cytidine deficient ligase; 14) that can use the 3′ hydroxyl of an oligonucleotide substrate to attack a 5′ triphosphate with concomitant displacement of pyrophosphate (data not shown). The substrate-binding domain of the ligase can be altered to accommodate other substrates, but not without a loss of activity; similar results have been observed for the Bartel Class I ligase. Its initial rate following selection, 0.71 h–1, was similar to that observed by Bartel and Szostak (2) for many of their selected ribozyme ligases, but could be improved following partial randomization and reselection. However, while the self-ligation activity of the Bartel Class I ligase was optimized by 47-fold following doping and reselection (15), the self-ligation activity of the L1 ligase was optimized by only 8-fold. The apparent difference in kinetic malleability may be due to the fact that the L1 ligase is much smaller (130 nt) than the Bartel Class I ligase (274 nt). Other small ribozymes, such as the hammerhead ribozyme, have similarly been shown to be relatively refractive to selective optimization of their kinetic parameters, especially rate (16,17).
The small size of the L1 ligase and the fact that its substrate, effector and kinetics can be readily engineered suggested that it might be developed for biotechnology applications (see below). To this end, we wished to determine whether it could be modified to recognize non-nucleic acid effectors. While the Breaker lab has previously demonstrated that hammerhead ribozymes can be converted to effector-modulated ribozymes, it was unclear whether the sequence, structure and/or mechanism of the hammerhead were in some way uniquely suited to the generation of aptazymes. By engineering a ribozyme that was similar in size, but different in sequence, structure and function, we have demonstrated that it may be possible to generate multiple different types of aptazymes.
More importantly though, by examining the comparative enzymology of allosteric ribozymes we have begun to understand which engineering tools are specific to individual ribozymes and which may be more general. First, it appears that some aptamers are more conducive to the development of aptazymes than others. The aptamer domains that conferred effector dependence on the hammerhead ribozyme, an anti-adenosine aptamer, an anti-theophylline aptamer and an anti-flavin aptamer, were the same aptamers that were found to confer effector dependence on the L1 ligase, either by rational design or selection. Other aptamer domains have so far proven less tractable. For example, Soukup and Breaker were unable to generate arginine-dependent hammerhead ribozymes via an anti-arginine aptamer (13), and we have so far been unable to generate chloramphenicol-dependent ligases via an anti-chloramphenicol aptamer. It is possible that anti-adenosine, anti-theophylline and anti-flavin aptamers may undergo more global or more relevant ligand-induced conformational changes than the anti-arginine and anti-chloramphenicol aptamers (18), and this allows them to better serve as effector domains for aptazymes. It is more likely, however, that we do not yet understand the rules for how to conjoin the anti-arginine and anti-chloramphenicol aptamers to the ribozyme in order to transduce ligand-induced conformational changes into catalysis.
Second, as was the case with many of the aptazymes identified by Breaker and his co-workers, improvements in ligand responsivity could frequently be engineered by relying on simple structural hypotheses. For the adenosine-sensing aptazyme, we hypothesized that if the conformational coupling between ligand binding and catalysis involved the joining region between the aptamer and the ribozyme, then destabilization of the join should ultimately increase responsivity. This proved to be true. Similarly, for both the adenosine- and theophylline-sensing aptazymes, we hypothesized if the ribozyme followed a two-state allosteric model in which there was a structurally less stable ‘off’ conformer and a structurally more stable ‘on’ conformer, then additional destabilization of the ribozyme as a whole should ultimately increase responsivity. This proved to be true for only some constructs; for example, the activation parameters of the adenosine-sensing aptazyme with a destabilized joining region was further increased by trimming Stem B (compare L1–ATPm1 and L1.dB2–ATPm1, Fig. 4b). However, in the absence of the destabilizing joining region, the opposite was the case (compare L1–ATPd1 and L1.dB2–ATPd1, Fig. 4b). Similarly, when the Stem B deletion was introduced into theophylline-sensing aptazymes overall responsivity to ligand was decreased (data not shown). Sequence and structural changes in different portions of the ribozyme may interact with one another to affect the overall level of effector-dependent activation.
Third, the selection of joining regions can be used for the optimization of ligand-dependent aptazymes. While in retrospect this result is manifest, prior to our experiments it was unclear whether the optimization of conformational coupling between binding and catalysis might be stymied by the nature of the ribozyme under investigation or the site of the join between the ribozyme and aptamer. Even with our successful demonstration that ribozymes other than the hammerhead can be evolutionarily engineered to function as aptazymes it is unclear whether other ribozymes will follow suit. In particular, we have encountered great difficulty in designing or selecting larger ribozymes (e.g., the Group I self-splicing intron) that can function as aptazymes (data not shown).
Finally, the concept of communication modules may be generalizable between ribozymes, but the communication modules themselves are not. The class I induction element selected for the hammerhead ribozyme (13) did not work with the L1 ligase. The communication modules selected for the hammerhead ribozyme could accommodate anti-flavin, anti-theophylline and anti-adenosine aptamers, while the communication modules that were designed or selected for the L1 ligase worked solely between the anti-flavin and anti-theophylline aptamers.
The identification of small aptazyme ligases may have particular significance for biotechnology applications. In particular, an aptazyme ligase can be viewed as a generalized reagent for transducing ligand-binding information to a variety of types of signaling. For example, immobilized aptazymes could ligate substrates conjugated with fluorophores to themselves, and transitively to the surface. Thus, transient, non-covalent interactions could be transformed into a covalent signal, potentially allowing for extremely high stringency washing of sensor surfaces. Further, since the aptazyme ligase is specific only for its oligonucleotide substrate, numerous different reporter molecules, including reporter enzymes such as β-galactosidase, could potentially be conjugated to the oligonucleotide substrate. Depending on the reporter enzyme, aptazymes could even transduce ligand binding to luminescent (luciferase) or electrochemical signals (horseradish peroxidase), rather than colorimetric signals. Coupled with the ability to carry out in vitro selection experiments to identify aptamers and ribozymes, and to optimize aptazyme function, these advantages suggest that aptazyme ligases may find increasing application in biosensor arrays (19).
REFERENCES
- 1.Robertson M.P. and Ellington,A.D. (1999) Nature Biotechnol., 17, 62–66. [DOI] [PubMed] [Google Scholar]
- 2.Bartel D.P. and Szostak,J.W. (1993) Science, 261, 1411–1418. [DOI] [PubMed] [Google Scholar]
- 3.Lauhon C.T. and Szostak,J.W. (1995) J. Am. Chem. Soc., 117, 1246–1257. [DOI] [PubMed] [Google Scholar]
- 4.Burke D.H., Hoffman,D.C., Brown,A., Hansen,M., Pardi,A. and Gold,L. (1997) Chem. Biol., 4, 833–843. [DOI] [PubMed] [Google Scholar]
- 5.Tang J. and Breaker,R.R. (1997) Chem. Biol., 4, 453–459. [DOI] [PubMed] [Google Scholar]
- 6.Sassanfar M. and Szostak,J.W. (1993) Nature, 364, 550–553. [DOI] [PubMed] [Google Scholar]
- 7.Jiang F., Kumar,R.A., Jones,R.A. and Patel,D.J. (1996) Nature, 382, 183–186. [DOI] [PubMed] [Google Scholar]
- 8.Tuschl T. and Eckstein,F. (1993) Proc. Natl Acad. Sci. USA, 90, 6991–6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dieckmann T., Suzuki,E., Nakamura,G.K. and Feigon,J. (1996) RNA, 2, 628–640. [PMC free article] [PubMed] [Google Scholar]
- 10.Jenison R.D., Gill,S.C., Pardi,A. and Polisky,B. (1994) Science, 263, 1425–1429. [DOI] [PubMed] [Google Scholar]
- 11.Zimmermann G.R., Jenison,R.D., Wick,C.L., Simorre,J.P. and Pardi,A. (1997) Nature Struct. Biol., 4, 644–649. [DOI] [PubMed] [Google Scholar]
- 12.Fan P., Suri,A.K., Fiala,R., Live,D. and Patel,D.J. (1996) J. Mol. Biol., 258, 480–500. [DOI] [PubMed] [Google Scholar]
- 13.Soukup G.A. and Breaker,R.R. (1999) Proc. Natl Acad. Sci. USA, 96, 3584–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogers J. and Joyce,G.F. (1999) Nature, 402, 323–325. [DOI] [PubMed] [Google Scholar]
- 15.Ekland E.H. and Bartel,D.P. (1995) Nucleic Acids Res., 23, 3231–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang J. and Breaker,R.R. (1997) RNA, 3, 914–925. [PMC free article] [PubMed] [Google Scholar]
- 17.Vaish N.K., Heaton,P.A. and Eckstein,F. (1997) Biochemistry, 36, 6495–6501. [DOI] [PubMed] [Google Scholar]
- 18.Patel D.J., Suri,A.K., Jiang,F., Jiang,L., Fan,P., Kumar,R.A. and Nonin,S. (1997) J. Mol. Biol., 272, 645–664. [DOI] [PubMed] [Google Scholar]
- 19.Hesselberth J., Robertson,M.P., Jhaveri,S. and Ellington,A.D. (2000) Rev. Mol. Biotech., in press. [DOI] [PubMed] [Google Scholar]
- 20.Green R. and Szostak,J.W. (1992) Science, 258, 1910–1915. [DOI] [PubMed] [Google Scholar]