


Abstract
Semi-random libraries of DNA 6mers and RNase H digestion were applied to search for sites accessible to hybridization on the genomic and antigenomic HDV ribozymes and their 3′ truncated derivatives. An approach was proposed to correlate the cleavage sites and most likely sequences of oligomers, members of the oligonucleotide libraries, which were engaged in the formation of RNA–DNA hybrids. The predicted positions of oligomers hybridizing to the genomic ribozyme were compared with the fold of polynucleotide chain in the ribozyme crystal structure. The data exemplified the crucial role of target RNA structural features in the binding of antisense oligonucleotides. It turned out that cleavages were induced if the bound oligomer could adapt an ordered helical conformation even when it required partial penetration of an adjacent double-stranded region. The major features of RNA structure disfavoring hybridization and/or RNase H hydrolysis were sharp turns of the polynucleotide chain and breaks in stacking interactions of bases. Based on the predicted positions of oligomers hybridizing to the antigenomic ribozyme we chose and synthesized four antisense DNA 6mers which were shown to direct hydrolysis in the desired, earlier predicted regions of the molecule.
INTRODUCTION
Short oligonucleotides are useful biochemical tools and promising therapeutic agents in the antisense and ribozyme strategies (for recent reviews see 1–6). Their effective base-pairing interactions with exposed regions of target molecules are of crucial importance to these approaches. In RNA molecules, target sequences are often chosen as single-stranded stretches in computer predicted RNA secondary structures. This approach has been shown, however, to be unsatisfactory. On the one hand, methods of predicting RNA secondary structures are imperfect (7–9), on the other, there is not sufficient knowledge about the effects of the structure of an RNA target on the efficiency of the formation of nucleic acid heteroduplexes (10,11).
An empirical approach to searching for antisense and ribozyme target sequences, which relies on testing several oligonucleotides chosen by a ‘walking method’, is more reliable than computer predictions. This approach requires individual synthesis and evaluation of a large number of oligomers, and is therefore rather laborious and not always successful in finding oligomers with the desired high antisense activities (12–15). Recently, a group of methods that use oligonucleotide libraries have been proposed for mapping RNA regions accessible to hybridization. In particular, the use of libraries composed of short DNA oligomers combined with RNase H digestion of RNA–DNA hybrids greatly simplifies the procedure (16–22).
Oligonucleotide libraries may be particularly useful in searching for target regions on highly structured RNA molecules, which are rather inaccessible to hybridization. Targeting such RNAs could substantially widen the applicability of the antisense and ribozyme strategies. In some cases, the binding of an oligomer to its target may involve, besides the standard Watson–Crick base pairs, also tertiary interactions. This would allow the use of very short oligomers which nevertheless interact with their targets with high efficiency and specificity. This idea originally derives from the studies on the binding of hexanucleotides to the catalytic core of a group I ribozyme, in which tertiary interactions enhance the binding by several orders of magnitude (23–25).
Recently, Mir and Southern (11) have studied hybridization of tRNAPhe to a complete set of complementary oligonucleotides immobilized on a glass plate. They analyzed the influence of a number of specific features of tRNA structure that contribute to heteroduplex stability, like helical order in single-stranded regions, sharp turns of the RNA chain, coaxial stacking of helices and stable tertiary interactions. The results pointed out that major determinants of hybridization efficiency lie in the structure of the RNA and, in the authors’ opinion, understanding of the influence of specific RNA features is likely to be improved by further studies of RNAs of known structure (11).
Here we applied oligodeoxyribonucleotide libraries and RNase H hydrolysis (Fig. 1) to search for sites accessible to hybridization on the genomic and antigenomic HDV ribozymes and their 3′ truncated derivatives. The details of the tertiary folding of a genomic ribozyme have been revealed by X-ray analysis (26) and the secondary structure of its 3′ truncate in solution has been well defined in our recent studies (27). The antigenomic ribozyme, despite a different nucleotide sequence, shares very similar secondary and, most likely, also tertiary structure with its genomic counterpart (28–31). These ribozymes and their truncates seem to be, therefore, particularly good models for a better evaluation of the factors influencing the binding of short oligomers to compact RNA structures. For identification of RNA–DNA hybrids we used RNase H, the enzyme that is thought to be involved in the antisense effects of oligonucleotides in vivo (32–34).
Figure 1.

(A) Sequences of the semi-random oligonucleotide libraries a, g, c and t are deoxyribonucleotides, and n denotes their random mixture. (B) Preferred sites of RNase H digestion on the heteroduplex substrates; N–n denotes a base pair of the Watson–Crick type between any ribo- and deoxyribonucleotide, and Y–x between a ribonucleotide in the RNA target and a deoxyribonucleotide in the third position of the bound oligonucleotide (see the text for further details).
MATERIALS AND METHODS
DNA preparation
All oligodeoxyribonucleotides were synthesized in 0.2 µmol scale and deprotected after synthesis. Oligonucleotide libraries were precipitated twice with ethanol and dissolved in TE buffer. Oligomers for preparation of DNA templates as well as DNA 6mers of defined sequences (oligomers 1–4) were purified by electrophoresis on denaturing 12% (w/v) polyacrylamide gels. DNA bands were excised, eluted with 0.3 M sodium acetate pH 5.2, 1 mM EDTA, precipitated with ethanol, DNA was recovered by centrifugation and dissolved in TE buffer.
The DNA templates for in vitro transcription of the genomic and antigenomic HDV ribozymes (3′ cleavage products) and their 3′ truncated derivatives were prepared as follows (27,35). In the case of the genomic ribozyme, two DNA oligomers were synthesized: A1: 5′-GTCCCATTCGCCATTACCGAGGGG-ACGGTCCCCTCGGAATGTTGCCCAGCCGGCGCCAG-CGAGGAGGCTGGGACCATGCCGGCC-3′ and B1: 5′-TA-ATACGACTCACTATAGGCCGGCATGGTCC-3′ (letters in italics mark the T7 RNA polymerase promotor; complementary sequences are underlined). For the antigenomic ribozyme, two other oligomers were synthesized: A2: 5′-CTCCCTTAGCC-ATCCGAGTGGACGTGCGTCCTCCTTCGGATGCCCAGGTCGGACCGCGAGGAGGTGGAGATGCCATGCCGAC-CC-3′ and B2: 5′-TAATACGACTCACTATAGGGTCGGCATGGCA-3′. Equimolar amounts of both oligomers (A1, B1 or A2, B2) were annealed and double-stranded DNA templates were generated by PCR. The reaction mixtures contained 1 µM of both DNA oligomers, 10 mM Tris–HCl pH 8.3, 15 mM MgCl2, 50 mM KCl, 200 µM each dNTP and 1.25 U of AmpliTaq polymerase (Biometra). The reactions were performed on Biometra UNO II thermocycler in seven cycles, 30 s at 94°C, 30 s at 55°C and 2 min at 72°C. The mixtures were extracted with phenol/chloroform (1:1), the reaction products precipitated with ethanol and dissolved in TE buffer. These double-stranded DNA were also used in subsequent PCR reactions for preparation of DNA templates for ribozyme truncates. The following primers were used: the 5′ primer, 5′-TAATACGACTCACTATAG-3′ corresponding to the T7 promoter sequence and the 3′ primers complementary to the 3′ terminal sequences of the truncates, 5′-CATTACCGAGGGGAC-3′ for the 1-73 truncate of genomic ribozyme and 5′-CATCCGAGTGGACGTGC-3′ for the 1-74 truncate of antigenomic ribozyme. The PCR conditions were as described above, with 0.1 µM concentration of the double-stranded DNA and 30 cycles used. The amplified DNA templates were purified, dissolved in TE buffer and used in transcription reactions.
RNA preparation
The in vitro transcription reactions contained 0.4 µM DNA template, 40 mM Tris–HCl pH 8.0, 10 mM MgCl2, 2 mM spermidine, 5 mM DTT, 1 mM each NTP, 2.5 mM guanosine, 750 U/ml ribonuclease inhibitor and 3000 U/ml T7 RNA polymerase (MBI Fermentas). Following incubation of the mixtures at 37°C for 4 h, the RNA transcripts were purified on 8% denaturing polyacrylamide gels. The RNAs were labeled at their 5′-ends using [γ-32P]ATP (Amersham) and T4 polynucleotide kinase (MBI Fermentas) in standard conditions.
RNA mapping experiments
Prior to digestion with Escherichia coli RNase H (MBI Fermentas) the 5′-32P-labeled RNA was renatured in the buffer 40 mM Tris–HCl pH 8.0, 40 mM KCl, 10 mM MgCl2, 1 mM DTT and 0.1 mM EDTA by heating at 65°C for 2 min and slow cooling to room temperature. Subsequently, RNase H was added to the final concentration of 250 U/ml. The cleavage reactions were initiated by adding separately DNA 6mer libraries (the final concentration of 200 µM) or oligomers 1–4 (the final concentration of 10 µM) to four RNA samples (20 000 c.p.m., 0.1–0.2 pmol of RNA in 10 µl reaction volume). The mixtures were incubated at 37°C for 10 or 30 min. In the experiment shown in Figure 5C, in which differences in cleavage efficiency with oligomers 1–4 were quantified as a function of oligomer concentration, 1 nM RNA was used. The reactions were quenched with equal volumes of 20 mM EDTA/7 M urea mixture and frozen on dry ice. The digestion products were analyzed by electrophoresis on 12% polyacrylamide, 7 M urea gels. Autoradiography was performed at –70°C with an intensifying screen. For quantitative analysis radioactive bands were cut off and counted in Beckman LS5000TA counter.
Figure 5.

Mapping of accessible sites on the antigenomic HDV ribozyme with RNase H and oligonucleotide 6mers 1–4 of defined sequences. (A) Autoradiogram of RNA cleavage fragments is shown, (B) fraction of RNA cleaved at the specific site after 10 min incubation was determined as function of oligomer concentration, and (C) cleavage sites as well as the sequences of complementary DNA 6mers are displayed in the ribozyme secondary structure model. Symbols used in the Figure are identical to those described in the legend to Figure 2.
Determination of cleavage sites
Products of RNase H digestion were run along with the products of alkaline RNA hydrolysis and limited T1 ribonuclease (Pharmacia) digestion of the same RNA. An alkaline hydrolysis ladder was generated by incubation of the 5′-32P-labeled RNA (dissolved in water) with formamide (1:5; v/v) at 100°C for 10 min. Limited T1 digestion was performed in 50 mM sodium citrate pH 5.3, 7 M urea with 0.1 U of the enzyme at 55°C for 30 min.
RESULTS AND DISCUSSION
Four oligoribonucleotides, derivatives of the genomic and antigenomic HDV ribozymes, were synthesized by in vitro transcription of synthetic DNA templates with phage T7 RNA polymerase (Figs 2 and 4, C and D). The first two oligomers corresponded to the ribozyme 3′ cleavage products, the next two were their truncates lacking 3′ terminal sequences of 11 nt in the genomic and 10 nt in the antigenomic variant. The 32P-end-labeled oligomers were subjected to a standard denaturation–renaturation procedure (Materials and Methods). Subsequently, semi-random oligonucleotide libraries: a, g, c and t (Fig. 1A) were added separately to four RNA samples and RNA–DNA hybrids were digested with E.coli ribonuclease H. Figures 2 and 4 show representative autoradiograms of the cleavage patterns of genomic and antigenomic ribozymes as well as their 3′ truncates. The cleavage sites are displayed in the secondary structure models of the ribozymes of a pseudoknot type (26,28,29,31). The secondary structures of 3′ truncates derive from those of full-length molecules by simple removal of RNA stretches not present in the truncates. The only difference concerns the arrangement of the central hairpin in the genomic sequence which is folded into another hairpin structure as found recently (27).
Figure 2.


Autoradiograms of RNA fragments showing cleavage of the genomic HDV ribozyme (A) and its 3′ truncate (B) with RNase H in the presence of oligonucleotide libraries. 5′-32P-end-labeled RNAs were used and reaction products were analyzed on 12% polyacrylamide gels. L, formamide ladder, T1, limited hydrolysis by RNase T1. Guanine residues are labeled on the right and for the truncate short and long run of the gel is shown. Cleavage sites are displayed on the RNA secondary structure models of the ribozyme (C) and the truncate (D). Base-paired segments are denoted P1–P4 and single-stranded regions as J1/2, J1/4 and J4/2. Boxed nucleotides interact in the ribozyme crystal structure: 2-bp helix P1.1 and an A–G base pair are formed. Cleavage sites occurring in the presence of libraries a, c, g and t are marked by filled triangles. Gray lines along the RNA sequences show the most probable positions of oligonucleotides hybridizing to the RNA targets (Fig. 1B). Underlined letters in italics denote cleavages to which the corresponding oligonucleotides could not be assigned.
Figure 4.


Autoradiograms of RNA fragments showing cleavage of the antigenomic HDV ribozyme (A) and its 3′ truncate (B) with RNase H in the presence of oligonucleotide libraries. For the ribozyme short and long run of the gel is shown. Closed diamonds denote cleavages used in selecting four antisense DNA 6mers (oligomers 1–4), which direct hydrolysis in the desired, earlier predicted regions of the molecule (see the text for further details). Cleavage sites are displayed on the RNA secondary structure models of the ribozyme (C) and the truncate (D). All symbols are specified in the legend to Figure 2.
We have attempted to correlate the RNase H cleavage sites with the most likely positions of DNA 6mers, members of the oligonucleotide libraries, hybridizing to the RNA targets. Earlier studies on RNase H cleavage of model RNA–DNA duplexes have shown that the 5′-most cleavage site was no less than 5 nt downstream of the 3′ end of the hybridizing DNA oligomer (36), which is consistent with other observations that the enzyme requires 5–6 bp to effect cleavage (37,38). Digestion also occurred outside the heteroduplex region, in single strand RNA extending 3′ from an RNA–DNA duplex. The greatest number of four cleavages was observed for the substrate containing three guanosine residues immediately adjacent to the heteroduplex, and every ‘single-stranded’ phospodiester bond was cleaved in that region. In the substrate with two guanosines the third ‘single-stranded’ phospodiester bond was the 3′-most cleavage site. In other substrates, however, cleavages did not extend beyond the second phosphodiester bond in single-strand RNA adjacent to RNA–DNA duplex regions (36). In each of our semi-random libraries, the third position was fixed, and in an RNA–DNA duplex this deoxyribonucleotide must pair with a corresponding nucleotide in the RNA strand (Fig. 1B; x–Y base pair). In the Figure, the three most likely RNase H cleavages are shown, based on the results of RNase H hydrolysis of model RNA–DNA duplexes. In our analysis we assumed that in the presence of a given DNA 6mer one of these three cleavages was induced with preference I, II, III, as shown in Figure 1B. These constraints, i.e. the third nucleotides of semi-random DNA libraries and preferred cleavage sites of RNA–DNA hybrids, were used to predict the most likely positions of complementary DNA 6mers hybridizing to the RNA targets (Figs 2 and 4, C and D).
Genomic HDV ribozyme
In the genomic HDV ribozyme cleavages occur at G6, C13, C15, U27, G40, A42, A43, G59, U79 and G81 (Fig. 2A and C; cleavages are 3′ of these nucleotides and the enzyme leaves 3′-OH and 5′-phosphate groups). The corresponding DNA 6mers bind to single-stranded RNA stretches J1/2, J1/4, J4/2 and to loop L4. In most cases, the adjacent double-stranded RNA regions are also engaged in binding. The presence of cleavages at C13, C15, U79 and G81 implicates partial unwinding of helix P2, and the cleavage at G40 of helix P1. Unexpectedly, the cleavages at G6 show that DNA 6mers hybridize efficiently to the G1–G6 and G2–C7 regions of the P1 helix. Interestingly, hybridization and/or RNase H digestion is not possible to the other side of the helix. The recently solved crystal structure of the ribozyme (26) has revealed its uniquely compact tertiary fold. Relatively few RNase H cleavages that we have found with DNA 6mer libraries suggest that the ribozyme is also highly structured in solution.
Figure 3 shows the spatial arrangement of the ribozyme polynucleotide chain determined by X-ray analysis as well as the secondary structure of the molecule modified in such a way as to allow a better visualization of base pairs and stacking interactions found in the crystals (26). The P4 stem, which contained an engineered crystallization module, is not shown, but it does not influence the ribozyme architecture (26,30). In the Figure, we marked regions accessible to hybridization with short antisense oligomers based on the results shown in Figure 2. This enables an analysis of the influence of specific structural factors on the efficiency of the binding of antisense oligomers to the RNA target. The first accessible region G1–G7 is involved in the formation of helix P1 that comprises 6 G–C bp and one terminal G–U interaction. The accessibility of this region to hybridization suggests that also in other RNAs some double-stranded stretches might be targeted with antisense oligonucleotides. However, the fact that the other side of the helix is not accessible shows that in each individual case such regions have to be found experimentally. The binding region A8–C14 consists of 3 nt of a single-stranded region J1/2 and 4 nt of helix P2. The phosphodiester chain of J1/2 makes a turn connecting two parallel stacks: one stack—P1, P1.1 and P4, and the other—P2 and P3. Oligomers that bind to J1/2 propagate into adjacent helical regions only in one direction, into helix P2. Simultaneous pairing with bases of J1/2, P1 and P2 seems to be highly disfavored, most likely because of a turn of the polynucleotide chain within J1/2. The next binding region C22–U27 maps nicely to the fragment of loop L3 connecting two continuous stacks in the spatial ribozyme structure. Bases of that region do not participate in either of those two stacks, except C22, which interacts with G38. As shown in Figure 3, moving the bound oligomer in either direction would cause its substantial turning. The binding region C36–C44 comprises the entire J1/4 region, with G38 and G39 taking part in the formation of P1.1, 2 adjacent nt of P1, and 2 nt of helix P4. Although this region consists of different elements of the secondary structure, the polynucleotide chain is arranged into a regular helical structure in the spatial ribozyme fold. Similarly, the chain of the next binding region C75–G81 is also regularly folded. A sharp turn occurs between G74 and C75, and the oligomers propagate into helix P2 and not P4.
Figure 3.
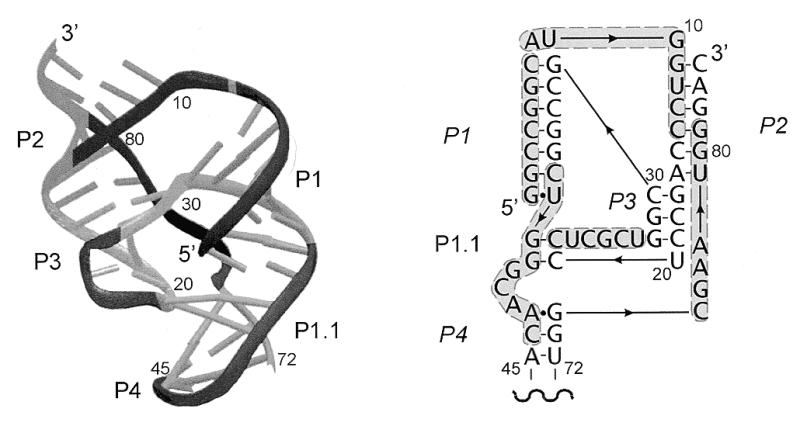
Schematic representation of the crystal structure of the genomic HDV ribozyme (left), and its secondary structure model, rearranged to reflect aspects of the three-dimensional structure (right). Regions of the polynucletide chain accessible to oligonucleotide hybridization, derived from the data shown in Figure 2, are dark. The U1A protein-binding domain engineered into helix P4, which does not participate in the ribozyme structure, is not shown (adapted from 26).
Summarizing, the data exemplify the crucial role of target RNA structural features in the binding of antisense oligomers, which is not restricted to single-stranded regions and oligomers also bind to some double-stranded RNA stretches. If binding requires partial unwinding of an adjacent helix, the oligomer propagates into that direction in which a heteroduplex forms a regular helical structure. Structural distortions of RNA targets, like turns of a polynucleotide chain and breaks in continuous stacking interactions of bases, strongly disfavor oligonucleotide binding or propagation of the bound oligomer in that direction.
The above observations correlate well with the results of analysis of the hybridization of yeast tRNAPhe to an immobilized library of complementary oligonucleotides (11). Some specific points need, however, additional comments. In yeast tRNAPhe, for instance, no significant yield of hybridization was seen from those oligomers that would require the oligonucleotide to partially penetrate a stem. The only exception was an 8mer bound to the anticodon loop in the presence of magnesium ions and penetrating partially the anticodon stem (11). In the work described here we use very short oligonucleotides, 6mers. In most cases, they are too short to bind to an unstructured RNA region initiating nucleation and to include a whole stem in an RNA–DNA hybrid (nucleation processes leading to the formation of RNA–DNA hybrids are discussed in detail in 11). The spatial fold of the ribozyme also constrains oligonucleotide binding as discussed earlier in the text. We observe, in turn, partial penetration of stem regions in the target RNA. It remains, however, to be better evaluated how beneficial is binding that includes a whole instead of a part of a stem and how its efficiency is influenced by hybridization conditions. Secondly, in yeast tRNAPhe no loop by itself gives significant binding (11). Specific conformational features of the loops like U-turns, their involvement in tertiary interactions as well as the presence of modified nucleotides hinder heteroduplex propagation. The separation of the strands of the stem in order to accommodate the loop in a duplex was also considered as an unfavorable factor (11). This seems to be, however, less detrimental since we show that short complementary oligomers bind to even very small loop regions in the HDV ribozymes and their truncates. Finally, efficient binding to the P1 helix of the genomic ribozyme observed in this work is of particular interest. This element of the ribozyme secondary structure has been well characterized with several biochemical methods in solution as a stable double-stranded region (27–30). The binding of a complementary oligonucleotide occurs most likely due to breathing of the helix allowing nucleation to take place followed by strand displacement and the formation of an RNA–DNA hybrid. The G–U pair at the end of the helix may facilitate nucleation and suggests the direction of duplex propagation from this putative nucleation site. Interestingly, in yeast tRNAPhe a 6mer oligonucleotide binds to the C27–mC32 region which consists of only 1 nt of the anticodon loop, mC32, and the entire one side of the anticodon stem (11). Binding to double-stranded RNA regions occurs, therefore, in some cases, but it requires the presence of an adjacent unpaired nucleotide or a weak base pair at the helix end to facilitate nucleation. A proper spatial fold of the RNA target also seems to be very important and it may determine the binding efficiency. Contrary to the approach used by Mir and Southern (11) the RNase H cleavage method does not allow for a quantitative analysis of binding efficiency to different regions of the RNA target. However, we determined RNase cleavage efficiency of the antigenomic sequence in the presence of four antisense DNA 6mers (see Fig. 5 and discussion later in the text). It turns out that at 0.3 µM oligomer concentration, the cleavage efficiency with oligomer 2, which partially penetrates helix P1, is only 4-fold lower than with oligomer 3, which binds to single-stranded region J1/2, and 2-fold lower than with oligomer 4, which binds to helix P1.
The most remarkable difference in the RNase H digestion patterns of the ribozyme and its 3′ truncate concerns cleavages in the J1/4 region, which are much stronger in the truncate (Fig. 2B and D). In the ribozyme crystal structure (26) the G38G39 sequence of J1/4 pairs with C21C22 of loop L3, and two adenosine residues A77 and A78 interact with base pairs G28–C19 and G29–C18 forming base triplets. In the truncate, the region corresponding to the P3/L3 hairpin folds into another hairpin structure (27). This rearrangement is fully consistent with the results of this work. Instead of cleavage at U27 in the ribozyme, cleavage at C24 occurs in the truncate, mapping nicely in both cases DNA 6mers to the loop regions of the hairpins. Strong cleavages in the J1/4 region of the truncate suggest, on the other hand, that the region is no longer involved in interactions with the central hairpin. Lastly, the digestion patterns of the full-length molecule and the truncate differ within their L4 loop regions. Although the corresponding DNA binding regions differ only slightly, the reason for the observed differences is not clear at present.
Antigenomic HDV ribozyme
The antigenomic ribozyme undergoes more intensive digestion with RNase H than its genomic counterpart in the presence of all the four DNA libraries (Fig. 4A and C). Instead of single cleavages present in the genomic ribozyme, a series of cuts are induced in the regions G6–A9 and C13–C16 in J1/2, G41–G42 in J1/4, A44–U45 and G52–G62 in stem/loop P4, G82–A83 in the ribozyme 3′ terminus, and finally, a single cleavage occurs at G29 in loop L3. Strong cleavages occurring in the J1/2 region map the oligomers to single-stranded J1/2 and to the helical region P1. High accessibility of J1/2 to hybridization and digestion is undoubtedly a consequence of the presence of 6 instead of 3 nt found in the genomic variant. In loop L3 a DNA 6mer maps to the 3′ side of the loop, similarly as in the genomic ribozyme. The J1/4 region is digested relatively weakly. It has only 3 nt, thus hybridizing DNA 6mers must partially penetrate either the P1 or P4 helix. In the P4 stem, as expected, DNA 6mers map to the loop region generating cleavages at G58, A60 and G62 as well as to the internal loop, on its side with 4 unpaired nt, generating cleavages at G52, A53 and G55. Unexpectedly, cleavages are also induced in the presence of DNA libraries of the a and g type, which require the presence of U and C or U residues in the RNA target. Since that region is composed exclusively of A and G residues, the presence of RNase H cleavages cannot be explained, unless we assume that the enzyme tolerates A–G and G–A mismatches within RNA–DNA hybrids. Although this assumption is reasonable, it has to be experimentally verified in a direct way.
The cleavage pattern of the ribozyme 3′ truncate is similar to that observed for the full-length molecule (Fig. 4B and D). Interestingly, in the C16–U32 region only a single cleavage occurs at G28. It suggests a similar structure of the central hairpin to that found in the ribozyme but with only 3 bp in the stem. The lack of RNase H cleavages in the presumably single-stranded region C16–C19 may be caused by its interactions with some other parts of the truncate. It is likely that the region interacts with the polypurine stretch G48–A56, in which almost no cleavages are observed. The G48–A56 region is susceptible to hybridization and digestion in the full-length molecule. Interestingly, the structure of loop L4 seems to be very similar in the truncate and in the full-length molecule. In the J1/4 region, essentially no changes are seen while substantial changes were observed in this region in the genomic variant. These changes were caused by an inability of J1/4 to interact with the central hairpin region rearranged into another hairpin in the truncate (27; see also the previous section). Relatively weak cleavages in J1/4 of the antigenomic ribozyme and its 3′ truncate may result from the presence of only 3 nt in that region. Alternatively, the interactions between J1/4 and L3 that have been recently proposed for the antigenomic ribozyme (31) might also be present in the truncate. Finally, low accessibility of J1/4 to hybridization and/or digestion might be caused by additional structural constraints introduced by interactions between C16–C19 and G48–A56 (discussed earlier in the text).
Cleavages in antigenomic HDV ribozyme in the presence of selected antisense DNA 6mers
We chose four cleavages from the pattern obtained with the antigenomic ribozyme in the presence of DNA libraries (Fig. 4A; the cleavages are denoted by closed diamonds). Three single cleavages were chosen from line a while the fourth cleavage site was from line g, and two cuts were present in that region of the pattern. We synthesized four DNA 6mers: 1, ccatgc; 2, tggacc; 3, atgcca; and 4, cgaccc, which were predicted to induce cleavages at the chosen sites of the ribozyme. These oligomers were used instead of oligonucleotide libraries in the digestion reactions with RNase H (Fig. 5). Since in the pattern obtained with oligonucleotide libraries, intensities of the chosen cleavages were different, reactions in the presence of oligomers 3 and 4 were performed with 5-fold lower concentration of the enzyme than in the presence of oligomers 1 and 2. In the presence of all the oligomers, RNase H cleaved the RNA chain exactly at the predicted sites. Although in the presence of oligomer 3 one major cleavage was generated, with higher concentration of the enzyme two cuts were observed (data not shown). In the case of oligomer 4, which is complementary to the region G1–G6, hydrolysis occurred at C8. This is the third ‘single strand’ phosphodiester bond, one bond further than the last 3′ cut of the three most likely cleavages predicted to occur with RNase H (Fig. 1B). However, in the presence of library a, to which oligomer 4 belongs, hydrolysis occurs exactly at the same site. Differences in cleavage efficiency with oligomers 1–4 were quantified as a function of oligomer concentration (Fig. 5C), and they followed those observed at the corresponding sites with oligonucleotide libraries. The largest difference, >10-fold, was observed for oligomers 1 and 3, at oligomer concentration of 0.3 µM. It seems that oligonucleotide libraries are useful in selecting antisense DNA oligomers which generate cleavages not only in accessible RNA regions but also occurring with relatively high efficacy.
CONCLUSIONS
Semi-random oligonucleotide libraries composed of DNA 6mers and RNase H hydrolysis were used to map RNA-accessible sites on the genomic and antigenomic HDV ribozymes. We proposed a simple way of correlating the cleavage sites and most likely sequences of oligomers, members of the oligonucleotide libraries, which were engaged in the formation of RNA–DNA hybrids.
We showed that even for highly structured HDV ribozymes and their 3′ truncates, short antisense oligomers could be found that effectively hybridize to these RNA targets. We compared the predicted positions of oligomers hybridizing to the genomic ribozyme and the fold of polynucleotide chain in the ribozyme crystal structure. It turned out that cleavages were induced if the bound oligomer could adapt an ordered helical conformation even when it required partial penetration of a double-stranded region. The major features of RNA structure disfavoring hybridization and/or RNase H hydrolysis were sharp turns of the polynucleotide chain and breaks in stacking interactions of bases.
The usefulness of the approach described here to mapping the accessibility of highly structured RNAs to hybridization and RNase H digestion was confirmed by showing that four DNA 6mers targeting the antigenomic HDV ribozyme directed hydrolysis in the earlier predicted, desired regions of the molecule.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Marcin Marciniak for performing some initial experiments and Barbara Smólska for her excellent technical assistance. The laboratory of Prof. W. T. Markiewicz of our Institute is thanked for synthesis of oligodeoxyribonucleotides. This work was supported by the Polish Committee for Scientific Research, Grants No. 6 P04A 001 12 and No. 6 P04A 006 18.
REFERENCES
- 1.Crooke S.T. (1998) Antisense Nucleic Acid Drug Dev., 8, 115–122. [DOI] [PubMed] [Google Scholar]
- 2.Crooke S.T. (1998) Biotechnol. Genet. Eng. Rev., 15, 121–157. [DOI] [PubMed] [Google Scholar]
- 3.Branch A.D. (1998) Trends Biochem. Sci., 23, 45–50. [DOI] [PubMed] [Google Scholar]
- 4.Bramlage B., Luzi,E. and Eckstein,F. (1998) Trends Biotechnol., 16, 434–438. [DOI] [PubMed] [Google Scholar]
- 5.Cook P.D. (1999) Nucl. Nucl., 18, 1141–1162. [DOI] [PubMed] [Google Scholar]
- 6.Rossi J.J. (1999) Chem. Biol., 6, R33–R37. [DOI] [PubMed] [Google Scholar]
- 7.Jaeger J.A., Turner,D.H. and Zuker,M. (1990) Methods Enzymol., 183, 281–306. [DOI] [PubMed] [Google Scholar]
- 8.Zuker M. (1994) Methods Mol. Biol., 25, 267–294. [DOI] [PubMed] [Google Scholar]
- 9.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 10.Lima W.F., Monia,B.P., Ecker,D.J. and Freier,S.M. (1992) Biochemistry, 31, 12055–12061. [DOI] [PubMed] [Google Scholar]
- 11.Mir K.U. and Southern,E.M. (1999) Nat. Biotechnol., 17, 788–792. [DOI] [PubMed] [Google Scholar]
- 12.Dean N.M., McKay,R., Condon,T.P. and Bennett,C.F. (1994) J. Biol. Chem., 269, 16416–16424. [PubMed] [Google Scholar]
- 13.Cioffi C.L., Garay,M., Johnston,J.F., McGraw,K., Boggs,R.T., Hreniuk,D. and Monia,B.P. (1997) Mol. Pharmacol., 51, 383–389. [PubMed] [Google Scholar]
- 14.Peyman A., Helsberg,M., Kretzschmar,G., Mag,M., Grabley,S. and Uhlmann,E. (1995) Biol. Chem., 367, 195–198. [DOI] [PubMed] [Google Scholar]
- 15.Monia B.P., Johnston,J.F., Geiger,T., Muller,M. and Fabbro,D. (1996) Nat. Med., 2, 668–675. [DOI] [PubMed] [Google Scholar]
- 16.Cload S.T. and Schepartz,A. (1994) J. Am. Chem. Soc., 116, 437–442. [Google Scholar]
- 17.Ho S.P., Britton,D.H, Stone,B.A., Behrens,D.L., Leffet,L.M., Hobbs,F.W., Miller,J.A. and Trainor,G.L. (1996) Nucleic Acids Res., 24, 1901–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lima W.F., Brown-Driver,V., Fox,M., Hanecak,R. and Bruice,T.W. (1997) J. Biol. Chem., 272, 626–638. [PubMed] [Google Scholar]
- 19.Bruice T.W. and Lima,W.F. (1997) Biochemistry, 36, 5004–5019. [DOI] [PubMed] [Google Scholar]
- 20.Birikh K.R., Berlin,Y.A., Soreq,H. and Eckstein,F. (1997) RNA, 3, 429–437. [PMC free article] [PubMed] [Google Scholar]
- 21.Matveeva O., Felden,B., Audlin,S., Gesteland,R.F. and Atkins,J.F. (1997) Nucleic Acids Res., 25, 5010–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scherr M. and Rossi,J.J. (1998) Nucleic Acids Res., 26, 5079–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bevilacqua P.C. and Turner,D.H. (1991) Biochemistry, 30, 10632–10640. [DOI] [PubMed] [Google Scholar]
- 24.Testa S.M., Haidaris,C.G., Gigliotti,F. and Turner,D.H. (1997) Biochemistry, 36, 15303–15314. [DOI] [PubMed] [Google Scholar]
- 25.Testa S.M., Gryaznov,S.M. and Turner,D.H. (1998) Biochemistry, 37, 9379–9385. [DOI] [PubMed] [Google Scholar]
- 26.Ferre-D’Amare A.R., Zhou,K. and Doudna,J.A. (1998) Nature, 395, 567–574. [DOI] [PubMed] [Google Scholar]
- 27.Matysiak M., Wrzesinski,J. and Ciesioka,J. (1999) J. Mol. Biol., 291, 283–294. [DOI] [PubMed] [Google Scholar]
- 28.Perrotta A.T. and Been,M.D. (1991) Nature, 350, 434–436. [DOI] [PubMed] [Google Scholar]
- 29.Rosenstein S.P. and Been,M.D. (1991) Nucleic Acids Res., 19, 5409–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Been M.D. and Wickham,G.S. (1997) Eur. J. Biochem., 247, 741–753. [DOI] [PubMed] [Google Scholar]
- 31.Wadkins T.S., Perrotta,A.T., Ferre-D’Amare,A.R., Doudna,J.A. and Been,M.D. (1999) RNA, 5, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boiziau C., Moreau,S. and Toulme,J.J. (1994) FEBS Lett., 340, 236–240. [DOI] [PubMed] [Google Scholar]
- 33.Monia B.P., Lesnik,E.A., Gonzalez,C., Lima,W.F., McGee,D., Guinosso,C.J., Kawasaki,A.M., Cook,P.D. and Freier,S.M. (1993) J. Biol. Chem., 268, 14514–14522. [PubMed] [Google Scholar]
- 34.Crooke S.T. (1998) Antisense Nucleic Acid Drug Dev., 8, 133–134. [DOI] [PubMed] [Google Scholar]
- 35.Ciesioka J., Michaowski,D., Wrzesiñski,J., Krajewski,J. and Krzy¿osiak,W.J. (1998) J. Mol. Biol., 275, 211–220. [DOI] [PubMed] [Google Scholar]
- 36.Lima W.F. and Crooke,S.T. (1997) J. Biol. Chem., 272, 27513–27516. [DOI] [PubMed] [Google Scholar]
- 37.Crooke S.T., Lemonidis,K.M., Neilson,L., Griffey,R., Lesnik,E.A. and Monia,B.P. (1995) Biochem. J., 312, 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lima W.F., Mohan,V. and Crooke,S.T. (1997) J. Biol. Chem., 272, 18191–18199. [DOI] [PubMed] [Google Scholar]