

This randomized clinical trial investigates the safety and efficacy of ApTOLL as a neuroprotectant drug for patients with ischemic stroke when given in combination with endovascular treatment.
Key Points
Question
What is the safety and efficacy as a neuroprotectant drug of ApTOLL in patients with ischemic stroke in combination with endovascular treatment?
Findings
In this randomized clinical trial including patients with acute stroke, the mortality at 90 days was significantly lower with ApTOLL compared with placebo.
Meaning
In patients with acute stroke and anterior circulation large vessel occlusion, the administration of ApTOLL immediately before endovascular treatment may have a neuroprotective effect.
Abstract
Importance
ApTOLL is a TLR4 antagonist with proven preclinical neuroprotective effect and a safe profile in healthy volunteers.
Objective
To assess the safety and efficacy of ApTOLL in combination with endovascular treatment (EVT) for patients with ischemic stroke.
Design, Setting, and Participants
This phase 1b/2a, double-blind, randomized, placebo-controlled study was conducted at 15 sites in Spain and France from 2020 to 2022. Participants included patients aged 18 to 90 years who had ischemic stroke due to large vessel occlusion and were seen within 6 hours after stroke onset; other criteria were an Alberta Stroke Program Early CT Score of 6 to 10, estimated infarct core volume on baseline computed tomography perfusion of 5 to 70 mL, and the intention to undergo EVT. During the study period, 4174 patients underwent EVT.
Interventions
In phase 1b, 0.025, 0.05, 0.1, or 0.2 mg/kg of ApTOLL or placebo; in phase 2a, 0.05 or 0.2 mg/kg of ApTOLL or placebo; and in both phases, treatment with EVT and intravenous thrombolysis if indicated.
Main Outcomes and Measures
The primary end point was the safety of ApTOLL based on death, symptomatic intracranial hemorrhage (sICH), malignant stroke, and recurrent stroke. Secondary efficacy end points included final infarct volume (via MRI at 72 hours), NIHSS score at 72 hours, and disability at 90 days (modified Rankin Scale [mRS] score).
Results
In phase Ib, 32 patients were allocated evenly to the 4 dose groups. After phase 1b was completed with no safety concerns, 2 doses were selected for phase 2a; these 119 patients were randomized to receive ApTOLL, 0.05 mg/kg (n = 36); ApTOLL, 0.2 mg/kg (n = 36), or placebo (n = 47) in a 1:1:√2 ratio. The pooled population of 139 patients had a mean (SD) age of 70 (12) years, 81 patients (58%) were male, and 58 (42%) were female. The primary end point occurred in 16 of 55 patients (29%) receiving placebo (10 deaths [18.2%], 4 sICH [7.3%], 4 malignant strokes [7.3%], and 2 recurrent strokes [3.6%]); in 15 of 42 patients (36%) receiving ApTOLL, 0.05 mg/kg (11 deaths [26.2%], 3 sICH [7.2%], 2 malignant strokes [4.8%], and 2 recurrent strokes [4.8%]); and in 6 of 42 patients (14%) receiving ApTOLL, 0.2 mg/kg (2 deaths [4.8%], 2 sICH [4.8%], and 3 recurrent strokes [7.1%]). ApTOLL, 0.2 mg/kg, was associated with lower NIHSS score at 72 hours (mean difference log-transformed vs placebo, −45%; 95% CI, −67% to −10%), smaller final infarct volume (mean difference log-transformed vs placebo, −42%; 95% CI, −66% to 1%), and lower degrees of disability at 90 days (common odds ratio for a better outcome vs placebo, 2.44; 95% CI, 1.76 to 5.00).
Conclusions and Relevance
In acute ischemic stroke, 0.2 mg/kg of ApTOLL administered within 6 hours of onset in combination with EVT was safe and associated with a potential meaningful clinical effect, reducing mortality and disability at 90 days compared with placebo. These preliminary findings await confirmation from larger pivotal trials.
Trial Registration
ClinicalTrials.gov Identifier: NCT04734548
Introduction
Reperfusion treatments have substantially improved acute stroke care over the last 2 decades. Tissue plasminogen activator was the only approved specific treatment for acute ischemic stroke1 from 1995 until 2015, when most clinical guidelines also recommended mechanical thrombectomy as a first-line treatment in ischemic strokes due to a large vessel occlusion.2 In recent years, the number of patients benefiting from endovascular treatment (EVT) has been increasing worldwide, and its indications are expanding.3,4,5 Despite this remarkable widespread use, only approximately 15% to 25% of patients with stroke are eligible for reperfusion treatments. Moreover, even though EVT has been shown to consistently achieve substantial recanalization in 85% to 90% of cases,6 more than 50% of treated patients develop a moderate to severe disability.7,8
In this context, new drug candidates with potential neuroprotective effects are being developed to improve stroke outcomes in parallel with reperfusion treatments.6 ApTOLL, a DNA aptamer, is an antagonist at toll-like receptor 4 (TLR4), a receptor directly involved in innate immune responses and also suggested to play a role in a large number of diseases such as ischemic stroke.9 Specifically, the inflammatory component triggered in the acute phase of an ischemic stroke is considered a relevant drug target to improve recovery. ApTOLL offers a potentially strong therapeutic effect in this area, as deemed by the efficacy observed across a range of preclinical stroke models10,11,12 that followed STAIR recommendations.13 In fact, the efficacy of ApTOLL has been demonstrated in experimental models of myocardial14 and cerebral ischemia.15,16 In addition, a first-in-human study in healthy volunteers recently demonstrated that ApTOLL was safe and well tolerated and further defined the pharmacokinetics of ApTOLL.17 The current study is a phase 1b/2a clinical trial (APRIL) designed to assess the safety of ApTOLL and its biological effects in patients with acute ischemic stroke who are eligible for EVT within 6 hours from symptom onset.
Methods
Study Design and Participants
APRIL is a double-blind, randomized, multicenter, placebo-controlled, phase 1b/2a clinical study designed to assess if the administration of ApTOLL is safe and well tolerated and shows any favorable therapeutic effect in patients with stroke and confirmed anterior circulation large vessel occlusion who are candidates to receive EVT with or without intravenous thrombolysis. The trial was performed at 15 comprehensive stroke centers in Spain (12 sites) and France (3 sites). It was approved both by the ethics committee at each site and by the national regulatory authorities. Signed informed consent was obtained from the patients or their legally authorized representative. The trial is registered at EudraCT (2020-002059-38) and ClinicalTrials.gov (NCT04734548). The study protocol is available in Supplement 1, statistical analysis plan in Supplement 2, and pharmacokinetic analysis in Supplement 3.18
Eligible patients were men and nonpregnant women aged 18 to 90 years with a disabling ischemic stroke at the time of randomization (baseline National Institutes of Health Stroke Scale [NIHSS] score 5-25; range 0-42, with higher scores indicating greater stroke severity)19; they were functionally independent before the stroke, defined as a modified Rankin Scale (mRS) score between 0 and 2, with scores ranging from 0 (no symptoms) to 6 (death)20; and they presented in time for ApTOLL to be administered within 6 hours from symptom onset. For patients with a wake-up stroke, the time of onset was considered the time when symptoms were first discovered, as safety was not expected to be challenged if the study drug was administered beyond 6 hours from actual stroke onset.
Patients with a single large vessel occlusion at the level of the terminal internal carotid artery or the M1 or M2 segments of the middle cerebral artery on noninvasive vascular imaging who were considered to undergo EVT, based on noncontrast computed tomography (CT) findings (Alberta Stroke Program Early CT Score [ASPECTS]21 of 6-10; range 0-10, with 1 point subtracted for any evidence of early ischemic change in each defined region on the CT scan), were evaluated as candidates for the APRIL study. To maximize the chances of identifying a biological effect of the study drug, the identification of a specific CT perfusion profile as previously defined22 was also required for enrollment. A predicted infarct core volume on CT perfusion defined as relative cerebral blood flow less than 30% between 5 and 70 mL had to be identified with previously validated automated software (RAPID, iSchemaView) for the patient to be eligible for the APRIL study.
Qualifying imaging was planned to be acquired at the endovascular center; in a minority of cases (n = 2), qualifying imaging was performed at a primary stroke center before transfer to the thrombectomy center. No screening log was kept. A full list of eligibility criteria is available in the eAppendix in Supplement 4.
Enrollment, Randomization, and Masking
The APRIL clinical trial was divided into 2 parts. In phase 1b, 32 patients were allocated (6 to receive ApTOLL, 2 to receive placebo at each level) to 4 ascending dose groups (0.025, 0.05, 0.1 and 0.2 mg/kg over a 30-minute intravenous infusion). A staggered dosing regimen was used to maximize patient safety. After the results of the 72-hour follow-up visit were evaluated by the data and safety monitoring board (DSMB), the next dose level was approved, if deemed appropriate. The safety outcomes evaluated were any suspected serious adverse reaction, serious adverse event (SAE), or adverse event (AE) that could be related to the drug administration and blood biochemistry parameters.
In both phase 1b and phase 2a, the masked clinical adjudication committee analyzed all AEs and SAEs to determine their potential relation to the study medication. With this information, the masked DSMB reviewed all reported AEs and SAEs. After completion of phase 1b, the DSMB was instructed to select the 2 doses to be tested in phase 2a according to initial safety results. Patients treated with either placebo or 1 of the 2 selected doses in phase 1b were further analyzed together with patients enrolled into phase 2a. In phase 2a, 3 arms were studied and patients were randomized to receive 1 of the 2 selected doses of ApTOLL or placebo in a 1:1:√2 ratio, which in turn yields probabilities of assignment of 0.293, 0.293, and 0.414, respectively. ApTOLL and placebo were stored as a powder in refrigerated vials (−20 °C). These vials were visually identical except for a unique vial number so that all trial personnel and patients were fully masked to treatment allocation. Vials were reconstituted with saline into colorless solutions before administration.
In both study phases, patients were randomly assigned by using a real-time internet-based system. This process was automated from study startup, which allowed for complete concealment of the sequence of allocation. The randomization system was originally planned to execute stratified allocation according to 3 strata: extent of predicted infarct core (<35 vs ≥35 mL) on admission CT perfusion, patient age (<70 vs ≥70 years), and admission NIHSS score (<15 vs ≥15). However, because of an error in the system that went unnoticed until study termination, stratification was not applied and a simple randomization algorithm was used. To account for the potential bias introduced by this error, post hoc analyses were performed.
During phase 2a, after 100 patients had completed the 72-hour follow-up, the DSMB performed a preplanned interim analysis and decided not to adopt any changes in the protocol for the remaining patients. Figure 1 contains the flow diagram for the APRIL study.
Figure 1. Study Flow Chart.
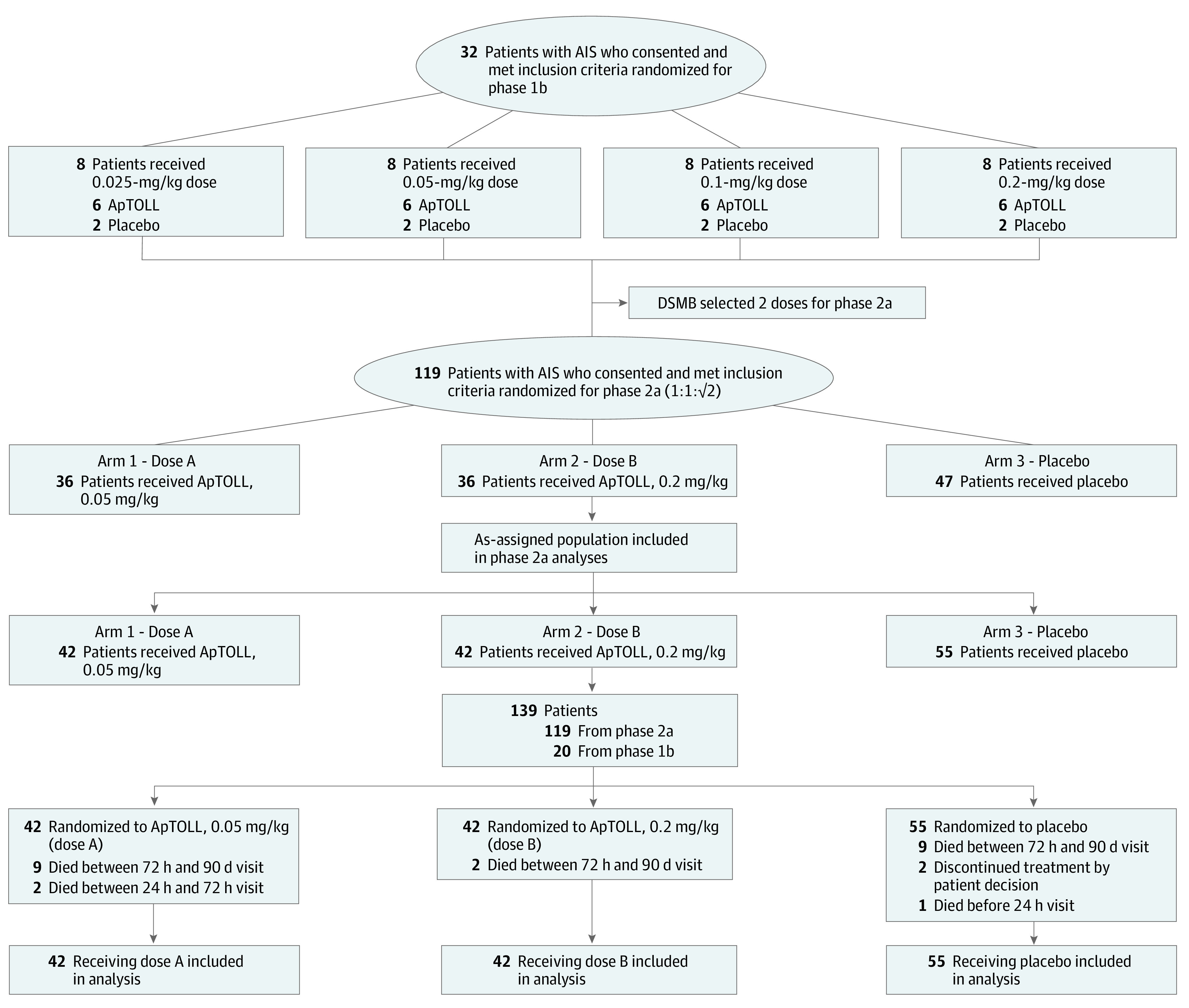
AIS indicates acute ischemic stroke; DSMB, data and safety monitoring board.
Procedures
After qualifying imaging, all patients received EVT and intravenous thrombolysis if indicated (before or during EVT, at a primary hospital before transfer, or at the thrombectomy-capable center) and were treated according to local institutional protocols and national and European Stroke Organization guidelines.23 Patients had to meet inclusion and exclusion criteria at the thrombectomy-capable center. The trial drug (ApTOLL or placebo) was administered as a single dose as soon as possible after randomization.18 Investigators were asked to ensure that the drug infusion (drug diluted in 100 mL of saline infused over 30 minutes) was started after imaging acquisition and before initiation of the EVT (arterial puncture). Sites were expected to adhere to national guidelines for stroke unit, stroke rehabilitation, and stroke prevention care.24 During the study period, 4174 patients underwent EVT.
All patients had standard assessments of demographic characteristics, medical history, laboratory values, and stroke severity (NIHSS score). The quality of reperfusion after EVT was assessed with the expanded Thrombolysis in Cerebral Ischemia (eTICI) scale.25 Follow-up brain imaging was performed at 24 hours with CT and at 72 hours with magnetic resonance imaging (MRI), if possible. Clinical follow-up was obtained at 90 days from randomization, if possible in person, to assess the degree of disability as determined by the mRS.19 Where in-person follow-up was not possible, videoconferencing or telephone follow-up was obtained.26 In all patients in phase 1b, up to 6 consecutive blood samples (baseline and 1, 6, 24, 48, and 72 hours after randomization) were drawn before and after dosing for pharmacokinetic analysis of ApTOLL concentrations and determination of plasma concentration of different proinflammatory biomarkers linked to acute stroke response. In phase 2a, blood biomarkers were determined at 5 time points (baseline and 6, 24, 48, and 72 hours after randomization). Details can be found in the study protocol (Supplement 1). Imaging interpretation was blinded and performed at a central core laboratory (UCLA, Los Angeles, California). Clinical data were verified by independent monitors (Anagram-ESIC, Barcelona, Spain).
Outcomes
The primary objective of the study was to evaluate whether the administration of ApTOLL intravenously at different doses was safe and well tolerated, as compared with placebo, when administered with EVT and intravenous fibrinolysis when indicated. The safety of ApTOLL was determined by monitoring adverse events that occurred during the study, identified on physical examination, by laboratory tests, or by neuroimaging. The primary end point was defined as the presence of any of the following events: death of any cause; intracranial hemorrhage resulting in new symptoms or in worsening of the existing ones27 (symptomatic intracranial hemorrhage [sICH]); malignant stroke resulting in herniation, neurological worsening, or death; and recurrent stroke.
Even though APRIL was a unique study, the secondary objectives were different in the 2 study parts. Phase 1b was used to also determine the pharmacokinetic profile of ApTOLL in patients with acute ischemic stroke, evaluated by the determination of ApTOLL levels in plasma and urine. Phase 1b was used to select the 2 doses to be administered in phase 2a according to their respective safety profiles and to provide an initial estimate of the therapeutic effect of ApTOLL on the final infarct volume (measured by MRI fluid-attenuated inversion recovery [MRI-FLAIR] at a mean [SD] time of 72 [24] hours) and on proinflammatory biomarkers (baseline [predose] and at the end of the infusion [up to 1 hour], 6, 24, 48, and 72 hours postdose).
Phase 2a was used to also assess the therapeutic and clinical effect of ApTOLL on (1) the final infarct volume (measured by MRI-FLAIR at a mean [SD] time of 72 [24] hours); (2) proinflammatory plasma biomarkers (predose and 6, 24, 48, and 72 hours postdose), (3) early clinical course (NIHSS score at 72 hours or discharge, whatever occurs first), and (4) long-term functional outcome (mRS score at 90 days after the stroke).
In those cases where 1 or more NIHSS measurements were missing, the last measure after study drug administration was considered. Deceased patients were included in the as-assigned population with an mRS score of 6. For patients known to be alive at 3 months postrandomization in whom follow-up evaluations were not possible, the discharge mRS score was carried forward. In cases where the MRI at 72 hours could not be obtained, the final infarct volume was determined in the last CT available after ApTOLL administration. If no follow-up image was available, final infarct volume was considered missing.
Statistical Analysis
Statistical analyses were performed by an independent external statistical consulting group. All analyses were performed on the as-assigned population, defined as all patients randomly allocated in the trial, regardless of treatment received. The total sample size was defined to be 151 patients: 32 patients in phase 1b and 119 patients in phase 2a. Because of the exploratory nature of study, there was no estimate of the statistical power. The analyses assumed a progressive relationship (ie, any dose above an unsafe dose will also be considered unsafe). Patients enrolled in phase 1b who received either placebo or 1 of the 2 selected doses of ApTOLL were combined with the phase 2a patients for the analyses. To successfully combine the patients from both study phases, the design, follow-up, and data collection were strictly identical in phase 1b and phase 2a of the trial, with the only exception being that sequential blood and urine sampling for pharmacokinetic analyses purposes were performed only in phase 1b.
Categorical variables were summarized by means of counts and percentages. Continuous variables were summarized by mean (SD) or median (IQR). Where informative, these summaries were reported by treatment and visit. Infarct volumes showed a skewed distribution and were reported as median (IQR).
The primary outcome of the study was compared between groups based on the absolute difference in proportions and asymptotic Wald confidence intervals at 95%. When both proportions to compare were zero, a generalized linear model was used. For the confidence interval of the odds ratio, asymptotic Wald confidence limits at 95% based on a log transformation of the odds ratio were used. Between-group differences in continuous secondary outcomes (final infarct volume and NIHSS score at 72 hours) were evaluated in a 1-way analysis of variance after log-transformation of the dependent variable and multiple-comparison adjustment by the Scheffe method; mean difference and 95% CIs between placebo and ApTOLL doses are reported. To evaluate the shift analysis on the mRS score at 90 days, a proportional odds ordinal logistic regression model was built, with placebo as the reference category, to estimate the unadjusted common odds ratio and 95% CIs for a better outcome at 90 days. The mRS scores 5 and 6 were collapsed in the same category for analyses. The proportional odds assumption was tested with a Brant test.
To assess assumptions related to the randomization process and baseline covariate imbalance, we conducted several statistical tests as post hoc analyses. Outcomes evaluated were death at 90 days (binary), NIHSS score at 72 hours (log-transformed), final infarct volume (log-transformed), and mRS score at 90 days (ordinal). First, permutation tests were used to assess the statistical significance of the observed differences between treatment groups. Second, we fitted linear models (for continuous outcomes) and binomial and ordered logistic regression models (for binary and ordered outcomes, respectively) to estimate the treatment effect and its 95% CIs for each outcome. To account for covariate imbalance, regression analyses were adjusted for baseline infarct core volume, hypoperfusion volume (time to maximum of 6 seconds), age, baseline NIHSS score, and time from onset to drug administration. Third, we constructed simple mediation models to explore whether the treatment effect of ApTOLL on clinical outcomes was partially or fully explained by its effect on final infarct volume. Clinical outcomes evaluated in mediation models were death at 90 days, the log-transformed NIHSS score at 72 hours, and good functional outcome at 90 days (mRS score of 0-2). Detailed methods are provided in the eMethods in Supplement 4.
Analyses were not adjusted for multiplicity of hypotheses so they should be interpreted with caution. A fully specified statistical analysis plan was reported before completing the study (Supplement 2). Analyses were performed with SAS software (SAS Institute) and R version 4.1 (R Foundation for Statistical Computing).
Results
Study Population
Between November 2020 and June 2021, 32 patients were enrolled in phase 1b and randomly assigned to receive ApTOLL or placebo (Figure 1) to complete the 4 dose escalation groups. ApTOLL plasma concentrations were obtained from 22 patients in phase 1b. Pharmacokinetic curves are shown in Supplement 3. The DSMB did not find any safety concerns and selected the 0.05-mg/kg and 0.2-mg/kg doses to be used in phase 2a. The 0.05-mg/kg dose was selected because it should be sufficient to produce some benefit, and all patients achieved sufficient concentrations of ApTOLL in the pharmacokinetic analysis. The 0.2-mg/kg dose was expected to achieve the maximum therapeutic benefit because this dose reached the maximum bioavailability in the first-in-human study.17 Additionally, there did not appear to be any toxicity risks limiting this choice.
From July 2021 to April 2022, 119 patients were assigned to receive either ApTOLL, 0.05 mg/kg (n = 36); ApTOLL, 0.2 mg/kg (n = 36); or placebo (n = 47). The pooled phase 1b/2a population was 42 patients allocated to receive ApTOLL, 0.05 mg/kg; 42 patients allocated to receive ApTOLL, 0.2 mg/kg; and 55 patients allocated to receive placebo (Figure 1). Baseline characteristics were similar between groups (Table 1). Briefly, 81 of 139 patients (58.3%) were male, and the mean (SD) age of the pooled population was 70 (12) years. The study design and patient flow are outlined in Figure 1. Major protocol deviations occurred in 10 patients (eTable 1 in Supplement 4).
Table 1. Characteristics of the Pooled Phase 1b/2a Population by Group.
| Characteristic | No. (%) | ||
|---|---|---|---|
| ApTOLL, 0.05 mg/kg (n = 42) | ApTOLL, 0.2 mg/kg (n = 42) | Placebo (n = 55) | |
| Age, mean (SD), y | 71.29 (10.97) | 68.33 (12.51) | 71.13 (12.86) |
| Sex | |||
| Male | 24 (57.14) | 24 (57.14) | 33 (60.00) |
| Female | 18 (42.86) | 18 (42.86) | 22 (40.00) |
| Prestroke mRS score, median (IQR) | 0 (0-1) | 0 (0-0) | 0 (0-1) |
| Hypertension | 29 (69.05) | 30 (71.43) | 36 (65.45) |
| Hyperlipidemia | 23 (54.76) | 23 (54.76) | 25 (45.45) |
| Diabetes | 10 (23.81) | 10 (23.81) | 9 (16.36) |
| Atrial fibrillation | 11 (26.19) | 14 (33.33) | 16 (29.09) |
| Ischemic heart disease | 4 (9.52) | 8 (19.05) | 7 (12.73) |
| Peripheral vascular disease | 3 (7.14) | 1 (2.38) | 2 (3.64) |
| Previous stroke | 5 (11.90) | 2 (4.76) | 5 (9.09) |
| Active smoker | 7 (16.67) | 9 (21.43) | 7 (12.73) |
| Systolic BP, mean (SD), mm Hg | 150.00 (19.82) | 143.98 (19.57) | 148.60 (19.46) |
| Diastolic BP, mean (SD), mm Hg | 79.76 (13.03) | 80.45 (11.84) | 79.64 (14.38) |
| Admission NIHSS score, median (IQR) | 19 (17-21) | 17 (12-21) | 18 (13-20) |
| Intravenous thrombolysis | 29 (69.05) | 27 (64.29) | 29 (52.73) |
| Time from LTSW/onset, mean (SD), h | |||
| To fibrinolysis | 2.03 (1.53-2.75) | 2.07 (1.78-2.25) | 1.92 (1.17-2.15) |
| To study randomization | 2.82 (2.02-4.05) | 2.46 (2.04-3.64) | 2.9 (2.28-4.05) |
| To study drug administration | 3.28 (2.75-4.25) | 2.78 (2.27-3.8) | 3.37 (2.55-4.67) |
| CT perfusion on admission (vs MRI) | 41 (97.62) | 41 (97.62) | 55 (100.0) |
| Predicted infarct core volume on admission, mL | |||
| Mean (SD) | 23.22 (17.71) | 20.25 (15.90) | 25.47 (17.40) |
| Median (IQR) | 16 (10-32) | 15 (7.5-26.5) | 23 (9-41) |
| Tmax >6 s on admission perfusion imaging, mL | |||
| Mean (SD) | 131.85 (50.00) | 114.00 (60.30) | 142.27 (53.75) |
| Median (IQR) | 138 (86-169) | 105 (62-166) | |
| ASPECTS, median (IQR) | 8 (7-9) | 9 (8-10) | 8 (7-8) |
| Occlusion location on admission imaging | |||
| Extracranial ICA | 8 (19.05) | 3 (7.32) | 12 (21.82) |
| TICA | 7 (16.67) | 1 (2.44) | 7 (12.73) |
| M1-MCA | 27 (64.29) | 27 (65.85) | 42 (76.36) |
| M2-MCA | 8 (19.05) | 15 (36.59) | 10 (18.18) |
| Other | 3 (7.14) | 3 (7.32) | 2 (3.64) |
| Time from LTSW to arterial puncture, median (IQR), min | 205 (147-277) | 189 (144-277) | 230 (172-324) |
| Duration of EVT procedure, median (IQR), min | 52 (29-79) | 35 (25-55) | 47 (29-73) |
| General anesthesia | 3 (7.32) | 8 (19.51) | 10 (19.23) |
| Final eTICI score | |||
| Categories 2c and 3 | 26 (61.9) | 29 (69) | 31 (56.3) |
| Categories 2b and 3 | 39 (92.3) | 38 (90.3) | 41 (82) |
Abbreviations: BP, blood pressure; CT, computed tomography; eTICI, expanded Thrombolysis in Cerebral Ischemia; EVT, endovascular treatment; mRS, modified Rankin Scale; NIHSS, National Institutes of Health Stroke Scale; LTSW, last time seen well; ASPECTS, Alberta Stroke Program Early CT Score; TICA, terminal internal carotid artery; Tmax, time to maximum; MCA, middle cerebral artery; MRI, magnetic resonance imaging.
Of the 151 patients in the pooled phase 1b/2a population, all received the allocated intervention (either active dose or placebo), and no crossovers occurred. All patients received the assigned intervention, although 3 patients received the incorrect volume or duration: 2 (4.8%) with ApTOLL, 0.05 mg/kg, and 1 (1.82%) with placebo. All patients except 1 in the ApTOLL, 0.2 mg/kg, group (99.3%) underwent an attempted EVT; intravenous thrombolysis was administered in 29 patients (69.1%) in the group receiving ApTOLL, 0.05 mg/kg; 27 patients (64.3%) receiving ApTOLL, 0.2 mg/kg; and 29 patients (52.7%) in the placebo group. Other concomitant medications are listed in eTable 2 in Supplement 4. The overall workflows (onset to randomization, onset to study drug administration, and study drug to reperfusion) and quality of reperfusion (on the eTICI scale) were similar in all groups (Table 1).
Outcomes
At 90 days, primary safety outcome data were missing for 2 patients (1.3%; 2 patients withdrew consent) (Figure 1). The primary end point occurred in 16 of 55 patients (29%) receiving placebo; 15 of 42 patients (36%) receiving ApTOLL, 0.05 mg/kg; and 6 of 42 patients (14%) receiving ApTOLL, 0.2 mg/kg. Death of any cause occurred in 10 patients (18.2%) in the placebo group, in 11 patients (26.2%) receiving ApTOLL, 0.05 mg/kg (absolute difference vs placebo, 8%; 95% CI, −9% to 25%), and in 2 patients (4.8%) receiving ApTOLL, 0.2 mg/kg (absolute difference vs placebo, −13%; 95% CI, −25% to −1%). In the placebo group, 4 of 55 patients (7.3%) had sICH, 4 (7.3%) had malignant strokes, and 2 (3.6%) had recurrent strokes. In those who received ApTOLL, 0.05 mg/kg, 3 of 42 patients (7.2%) had sICH, 2 (4.8%) had malignant strokes, and 2 (4.8%) had recurrent strokes. In patients receiving ApTOLL, 0.2 mg/kg, 2 of 42 patients (4.8%) had sICH, and 3 (7.1%) had recurrent strokes.
Table 2 and eFigure 1 in Supplement 4 show the results of the primary and secondary end points in the study groups. A complete list of all reported adverse events is available in eTables 3 and 4 in Supplement 4.
Table 2. Primary and Secondary Outcomes.
| Outcome | No. (%) | Difference, % (95% CI) | |||
|---|---|---|---|---|---|
| ApTOLL, 0.05 mg/kg (n = 42) | ApTOLL, 0.2 mg/kg (n = 42) | Placebo (n = 55) | ApTOLL, 0.05 mg/kg, vs placebo | ApTOLL, 0.2 mg/kg, vs placebo | |
| Primary outcome measures a | |||||
| Death of any cause | 11 (26.2) | 2 (4.8) | 10 (18.2) | 8 (−9 to 25) | −13 (−25 to −1) |
| Symptomatic intracranial hemorrhage | 3 (7.1) | 2 (4.8) | 4 (7.3) | 0 (−11 to 10) | −3 (−12 to 7) |
| Malignant stroke | 2 (4.76) | 1 (2.38) | 4 (7.3) | −3 (−12 to 7) | −5 (−13 to 3) |
| Recurrent stroke | 2 (4.76) | 3 (7.14) | 2 (3.6) | 1 (−7 to 9) | 4% (−6 to 13) |
| Secondary outcome measures | |||||
| Final infarct volume, median (IQR)b | 46.00 (18.00 to 100.00) | 23.50 (13.00 to 42.00) | 44.00 (26.00 to 89.00) | −12 (−49 to 35) | −42 (−66 to 1) |
| NIHSS at 72 h, median (IQR)b | 8.00 (3.00 to 17.00) | 3.00 (0.00 to 11.00) | 7.00 (3.00 to 17.00) | −1 (−41 to 40) | −45 (−67 to −10) |
| mRS score, median (IQR)c | 3.00 (2.00 to 5.00) | 2.00 (1.00 to 3.00) | 3.00 (1.00 to 4.00) | OR, 0.76 (0.37 to 1.56) | OR, 2.44 (1.76 to 5.00) |
| mRS score of 0-2, No. (%)a | 15 (37.5) | 27 (64.29) | 25 (47.17) | −10 (−29 to 10) | 17 (−2 to 37) |
| mRS score of 0-1, No. (%)a | 9 (21.4) | 19 (45.2) | 14 (25.5) | −4 (−21 to 12) | 20 (0.5 to 38) |
Abbreviations: mRS, modified Rankin Scale; NIHSS, National Institutes of Health Stroke Scale; OR, odds ratio.
Effect estimate is absolute difference (95% CI).
Effect estimate is mean difference (95% CI).
Effect estimate is common odds ratio (95% CI).
The final median (IQR) infarct volume was 44 mL (26-89) in patients allocated to receive placebo, 46 mL (18-100) in patients allocated to receive ApTOLL, 0.05 mg/kg (mean difference of log-transformed final infarct volume vs placebo, −12%; 95% CI, −49% to 35%), and 23.5 mL (13-42) in patients allocated to receive ApTOLL, 0.2 mg/kg (mean difference of log-transformed final infarct volume vs placebo, −42%; 95% CI, −66% to 1%) (eFigure 2 in Supplement 4). The median (IQR) NIHSS score assessed at 72 hours was 7 (3-17) in patients allocated to receive placebo, 8 (3-17) in patients allocated to receive ApTOLL, 0.05 mg/kg (mean difference of log-transformed 72-hour NIHSS score vs placebo, −1%; 95% CI, −41% to 40%), and 3 (0-11) in patients allocated to receive ApTOLL, 0.2 mg/kg (mean difference of log-transformed 72-hour NIHSS score vs placebo, −45%; 95% CI, −67% to −10%). The rate of patients with an mRS score of 0 through 2 at 90 days was 47.1% in patients allocated to receive placebo, 37.5% in patients allocated to receive ApTOLL, 0.05 mg/kg (common odds ratio for a better outcome vs placebo, 0.76; 95% CI, 0.37 to 1.56), and 64.3% in patients allocated to receive ApTOLL, 0.2 mg/kg (common odds ratio for a better outcome vs placebo, 2.44; 95% CI, 1.76 to 5.00). The analyses of the multiple blood biomarkers were not significantly different among the studied groups (eFigure 3 in Supplement 4). Figure 2 and Figure 3 show a graphical representation of secondary efficacy outcomes.
Figure 2. Secondary Efficacy Outcomes Based on Final Infarct Volume and National Institutes of Health Stroke Scale (NIHSS) Score at 72 Hours.
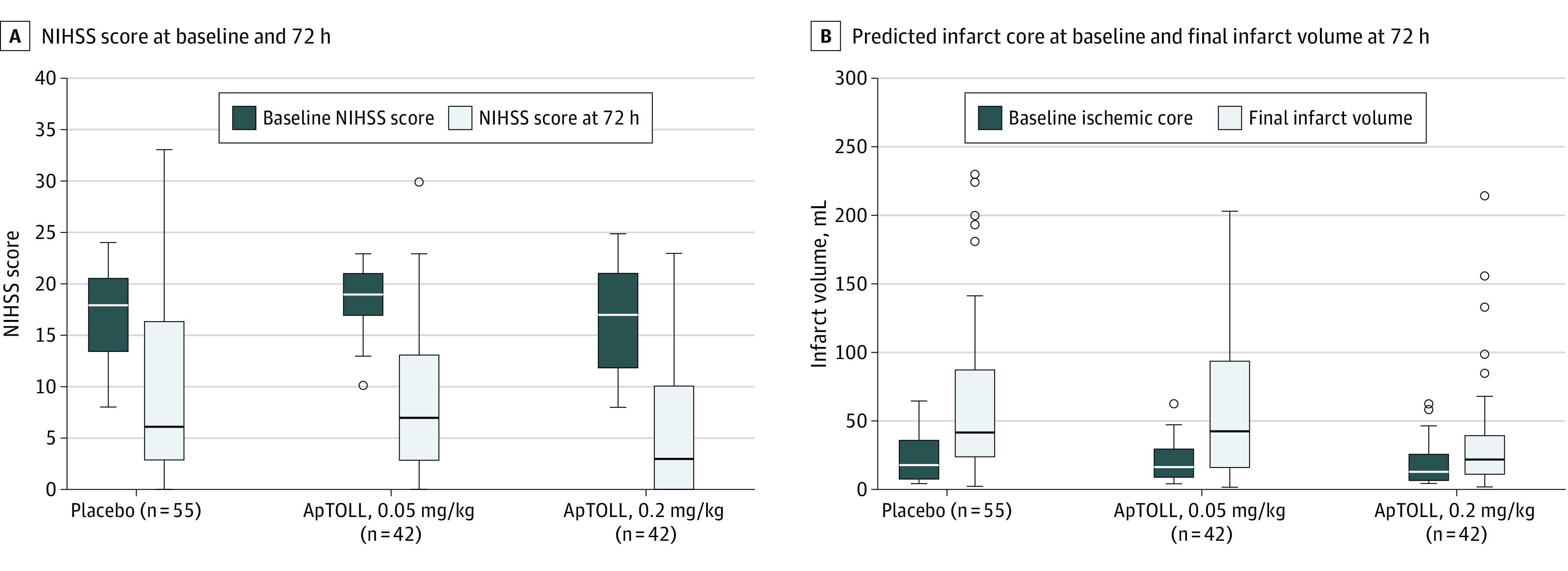
Boxplots representing baseline and 72-hour NIHSS score according to treatment allocation (A) and baseline predicted infarct core on computed tomography perfusion and final infarct volume (magnetic resonance imaging at 72 hours) according to treatment allocation (B). The horizontal line inside each box indicates the median value; upper and lower bound of each box, IQR; whiskers, range of values; circles, outliers.
Figure 3. Distribution of Global Disability at 90 Days According to Treatment Allocation.
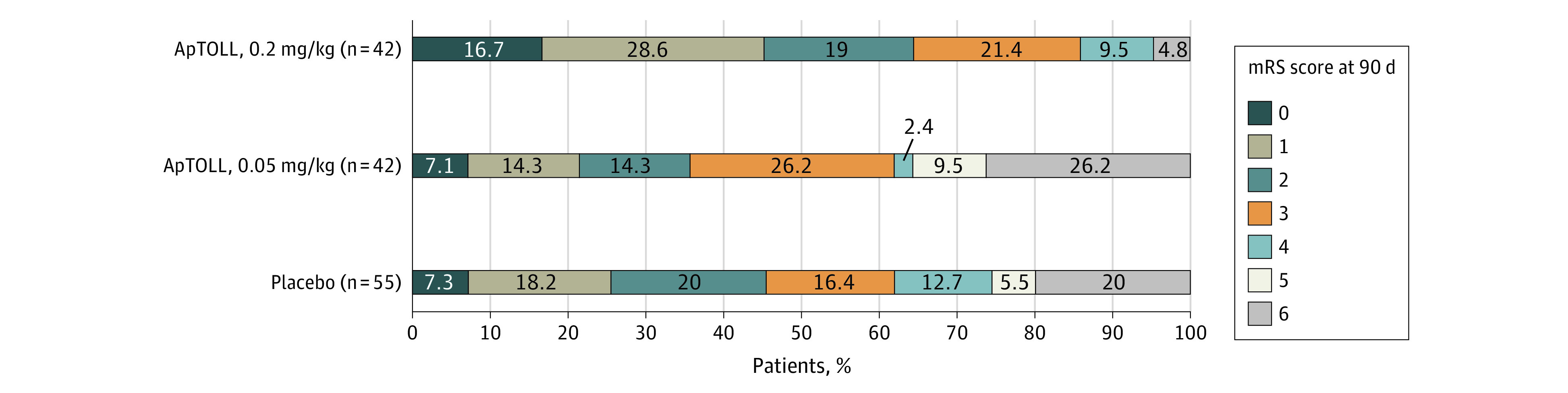
Stacked bar plots representing the distribution of the modified Rankin Scale (mRS) score at 90 days according to treatment allocation.
Post Hoc Analyses
The results of the permutation tests were consistent with the prespecified outcome models results, with adjusted P values ranging from .007 to .05 for post hoc comparisons between placebo and ApTOLL, 0.2 mg/kg. The adjusted P values for the comparison between placebo and ApTOLL, 0.05 mg/kg, ranged from .50 to .89 (eTable 5 in Supplement 4). Regression models showed a significant treatment effect of ApTOLL, 0.2 mg/kg, as compared with placebo, except for the model with death as the dependent variable (adjusted odds ratio, 0.24; 95% CI, 0.03 to 1.08) (eTable 6 in Supplement 4). Mediation models showed a significant indirect effect of ApTOLL, 0.2 mg/kg, through final infarct volume on NIHSS score at 72 hours (β = −0.265; 95% CI, −0.537 to −0.05) and good functional outcome at 90 days (β = 0.079; 95% CI, 0.012 to 0.181), but not with death (β = −0.001, 95% CI, −0.038 to 0.051). Mediation model results and sensitivity analysis are presented in eFigures 4 and 5 in Supplement 4.
Discussion
The APRIL study demonstrated that the infusion of ApTOLL, 0.2 mg/kg, in combination with EVT in selected patients with stroke is safe and reduces mortality at 90 days. Moreover, the efficacy of ApTOLL as a neuroprotectant drug in acute cerebral ischemia is supported by consistent positive results in most predefined secondary outcome measures, including final infarct volume, early neurological impairment, and long-term disability. While baseline covariate imbalance may have led to an overestimation of the treatment effect of ApTOLL, 0.2 mg/kg, post hoc statistical analyses demonstrated the robustness of the results under varying assumptions.
The high rates of recanalization currently achieved with EVT provide a promising human ischemia-reperfusion model to test novel neuroprotective agents. If administered in combination with EVT, the potential neuroprotective drug may (1) effectively reach the tissue at risk, exerting a protective effect directly in the area of ischemic penumbra and potentially extending the therapeutic window of reperfusion therapies, and (2) limit the phenomena of hemorrhagic transformation and reperfusion damage that may occur after recanalization.28,29,30,31 To increase the chances of depiction of a potential neuroprotective effect, the inclusion criteria of the APRIL trial were designed to exclude those patients with a high likelihood of achieving either an excellent or a very poor outcome with the standard-of-care treatment. In these patients, a floor/ceiling effect in the infarct volume reduction or clinical improvement could dilute the treatment effect of the studied drug.22 ApTOLL may still be effective in these patients, but higher sample sizes would be expected to be required to confirm the benefits.
A key strength of the trial is that the design largely matched the preclinical ischemia/reperfusion models of transient middle-cerebral artery occlusion. Patients had a median duration of stroke onset to study drug administration of 210 minutes and had high reperfusion rates after EVT (final eTICI 2b-3 score, 87%). The observed pharmacokinetic curves suggest that single doses of ApTOLL could be exerting neuroprotective effects, at least up to 12 hours after infusion, including the initial period of ischemia and the hours after reperfusion. The median time from drug infusion to recanalization (when achieved) was approximately 50 minutes, corresponding to the potential neuroprotective time during ischemia.
The subgroup analyses suggest a similar treatment effect in patients who received the drug in the early time window (<3 hours from symptom onset) and those who were treated in the later window (3-6 hours) (eFigure 6 in Supplement 4). Whether the efficacy of ApTOLL is also maintained later (>6 hours from symptom onset) should be investigated in future studies.
Limitations
Our study has several limitations. The study was not powered to achieve definitive conclusions regarding the efficacy of ApTOLL in improving outcomes for patients with acute stroke but to identify a safe dose in these patients. Nevertheless, the promising results observed in the present phase encourage the development of future larger trials to clearly characterize the potential neuroprotective effects. Whether higher ApTOLL doses could induce additional benefits without safety concerns may be also investigated in future studies. Initially, the study intended to stratify patients based on significant outcome predictors, such as age, NIHSS score, or predicted infarct core on inclusion. However, stratification was ultimately omitted, resulting in some degree of covariate imbalance between the treatment groups. To ensure the reliability of the study’s findings, various post hoc analyses were conducted.
Conclusion
This study found that for acute ischemic stroke, 0.2 mg/kg of ApTOLL administered within 6 hours of onset in combination with EVT was safe and associated with a potential meaningful clinical effect, reducing mortality and disability at 90 days as compared with placebo. These preliminary findings await confirmation from larger pivotal trials.
Trial protocol
Statistical analysis plan
Pharmacokinetic analysis
eAppendix. Eligibility criteria
eMethods.
eTable 1. Major protocol deviations during phase Ib and IIa
eTable 2. Concomitant medications in phase Ib and IIa
eTable 3. Summary of adverse events during phase Ib
eTable 4. Summary of adverse events during phase IIa
eTable 5. Permutation tests
eTable 6. Post-hoc analysis based on multivariate generalized linear models with and without adjustment for covariates
eFigure 1. Distribution of primary endpoints in the study groups
eFigure 2. Line plots for individual comparison of baseline ischemic core and final infarct volume
eFigure 3. Temporal profile of proinflammatory biomarkers in APRIL study
eFigure 4. Graphical representation of mediation analysis
eFigure 5. Graphical representation of the sensitivity analysis of the mediation model
eFigure 6. Secondary efficacy endpoints according to time from symptom onset to treatment administration
Data sharing statement
References
- 1.National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group . Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333(24):1581-1587. doi: 10.1056/NEJM199512143332401 [DOI] [PubMed] [Google Scholar]
- 2.Goyal M, Menon BK, van Zwam WH, et al. ; HERMES Collaborators . Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet. 2016;387(10029):1723-1731. doi: 10.1016/S0140-6736(16)00163-X [DOI] [PubMed] [Google Scholar]
- 3.Jovin TG, Nogueira RG; DAWN Investigators . Thrombectomy 6 to 24 hours after stroke. N Engl J Med. 2018;378(12):1161-1162. doi: 10.1056/NEJMc1801530 [DOI] [PubMed] [Google Scholar]
- 4.Albers GW, Marks MP, Kemp S, et al. ; DEFUSE 3 Investigators . Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. N Engl J Med. 2018;378(8):708-718. doi: 10.1056/NEJMoa1713973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshimura S, Sakai N, Yamagami H, et al. Endovascular therapy for acute stroke with a large ischemic region. N Engl J Med. 2022;386(14):1303-1313. doi: 10.1056/NEJMoa2118191 [DOI] [PubMed] [Google Scholar]
- 6.Hill MD, Goyal M, Menon BK, et al. ; ESCAPE-NA1 Investigators . Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): a multicentre, double-blind, randomised controlled trial. Lancet. 2020;395(10227):878-887. doi: 10.1016/S0140-6736(20)30258-0 [DOI] [PubMed] [Google Scholar]
- 7.Jovin TG, Chamorro A, Cobo E, et al. ; REVASCAT Trial Investigators . Thrombectomy within 8 hours after symptom onset in ischemic stroke. N Engl J Med. 2015;372(24):2296-2306. doi: 10.1056/NEJMoa1503780 [DOI] [PubMed] [Google Scholar]
- 8.Berkhemer OA, Fransen PSS, Beumer D, et al. ; MR CLEAN Investigators . A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372(1):11-20. doi: 10.1056/NEJMoa1411587 [DOI] [PubMed] [Google Scholar]
- 9.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115(12):1599-1608. doi: 10.1161/CIRCULATIONAHA.106.603431 [DOI] [PubMed] [Google Scholar]
- 10.Caso JR, Pradillo JM, Hurtado O, Leza JC, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in subacute stress-induced neuroinflammation and in the worsening of experimental stroke. Stroke. 2008;39(4):1314-1320. doi: 10.1161/STROKEAHA.107.498212 [DOI] [PubMed] [Google Scholar]
- 11.García-Culebras A, Palma-Tortosa S, Moraga A, et al. Toll-like receptor 4 mediates hemorrhagic transformation after delayed tissue plasminogen activator administration in in situ thromboembolic stroke. Stroke. 2017;48(6):1695-1699. doi: 10.1161/STROKEAHA.116.015956 [DOI] [PubMed] [Google Scholar]
- 12.Brea D, Blanco M, Ramos-Cabrer P, et al. Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J Cereb Blood Flow Metab. 2011;31(6):1424-1431. doi: 10.1038/jcbfm.2010.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savitz SI, Baron J-C, Fisher M; STAIR X Consortium . Stroke treatment academic industry roundtable X: brain cytoprotection therapies in the reperfusion era. Stroke. 2019;50(4):1026-1031. doi: 10.1161/STROKEAHA.118.023927 [DOI] [PubMed] [Google Scholar]
- 14.Oyama J, Blais C Jr, Liu X, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109(6):784-789. doi: 10.1161/01.CIR.0000112575.66565.84 [DOI] [PubMed] [Google Scholar]
- 15.Fernández G, Moraga A, Cuartero MI, et al. TLR4-binding DNA aptamers show a protective effect against acute stroke in animal models. Mol Ther. 2018;26(8):2047-2059. doi: 10.1016/j.ymthe.2018.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramirez-Carracedo R, Tesoro L, Hernandez I, et al. Targeting TLR4 with ApTOLL improves heart function in response to coronary ischemia reperfusion in pigs undergoing acute myocardial infarction. Biomolecules. 2020;10(8):1167. doi: 10.3390/biom10081167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernández-Jiménez M, Martín-Vílchez S, Ochoa D, et al. First-in-human phase I clinical trial of a TLR4-binding DNA aptamer, ApTOLL: safety and pharmacokinetics in healthy volunteers. Mol Ther Nucleic Acids. 2022;28:124-135. doi: 10.1016/j.omtn.2022.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernández-Jiménez M, Abad-Santos F, Cotgreave I, et al. APRIL: a double-blind, placebo-controlled, randomized, phase Ib/IIa clinical study of ApTOLL for the treatment of acute ischemic stroke. Front Neurol. 2023;14:1127585. doi: 10.3389/fneur.2023.1127585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyden P, Brott T, Tilley B, et al. ; NINDS TPA Stroke Study Group . Improved reliability of the NIH Stroke Scale using video training. Stroke. 1994;25(11):2220-2226. doi: 10.1161/01.STR.25.11.2220 [DOI] [PubMed] [Google Scholar]
- 20.van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke. 1988;19(5):604-607. doi: 10.1161/01.STR.19.5.604 [DOI] [PubMed] [Google Scholar]
- 21.Pexman JH, Barber PA, Hill MD, et al. Use of the Alberta Stroke Program Early CT Score (ASPECTS) for assessing CT scans in patients with acute stroke. AJNR Am J Neuroradiol. 2001;22(8):1534-1542. [PMC free article] [PubMed] [Google Scholar]
- 22.Olivé-Gadea M, Requena M, Campos D, et al. Defining a target population to effectively test a neuroprotective drug. Stroke. 2021;52(2):505-510. doi: 10.1161/STROKEAHA.120.032025 [DOI] [PubMed] [Google Scholar]
- 23.Turc G, Bhogal P, Fischer U, et al. European Stroke Organisation (ESO)- European Society for Minimally Invasive Neurological Therapy (ESMINT) guidelines on mechanical thrombectomy in acute ischemic stroke. J Neurointerv Surg. 2019;11(6):535-538. doi: 10.1136/neurintsurg-2018-014568 [DOI] [PubMed] [Google Scholar]
- 24.Dawson J, Béjot Y, Christensen LM, et al. European Stroke Organisation (ESO) guideline on pharmacological interventions for long-term secondary prevention after ischaemic stroke or transient ischaemic attack. Eur Stroke J. 2022;7(3):I-II. doi: 10.1177/23969873221100032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liebeskind DS, Bracard S, Guillemin F, et al. ; HERMES Collaborators . eTICI reperfusion: defining success in endovascular stroke therapy. J Neurointerv Surg. 2019;11(5):433-438. doi: 10.1136/neurintsurg-2018-014127 [DOI] [PubMed] [Google Scholar]
- 26.Fernández Sanz A, Ruíz Serrano J, Tejada Meza H, Marta Moreno J. Validation of the Spanish-language version of the simplified modified Rankin Scale telephone questionnaire. Neurologia (Engl Ed). 2022;37(4):271-276. doi: 10.1016/j.nrleng.2019.03.019 [DOI] [PubMed] [Google Scholar]
- 27.Wahlgren N, Ahmed N, Dávalos A, et al. ; SITS-MOST Investigators . Thrombolysis with alteplase for acute ischaemic stroke in the Safe Implementation of Thrombolysis in Stroke-Monitoring Study (SITS-MOST): an observational study. Lancet. 2007;369(9558):275-282. doi: 10.1016/S0140-6736(07)60149-4 [DOI] [PubMed] [Google Scholar]
- 28.Minnerup J, Sutherland BA, Buchan AM, Kleinschnitz C. Neuroprotection for stroke: current status and future perspectives. Int J Mol Sci. 2012;13(9):11753-11772. doi: 10.3390/ijms130911753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tymianski M. Combining neuroprotection with endovascular treatment of acute stroke: is there hope? Stroke. 2017;48(6):1700-1705. doi: 10.1161/STROKEAHA.117.017040 [DOI] [PubMed] [Google Scholar]
- 30.Savitz SI, Baron J-C, Yenari MA, Sanossian N, Fisher M. Reconsidering neuroprotection in the reperfusion era. Stroke. 2017;48(12):3413-3419. doi: 10.1161/STROKEAHA.117.017283 [DOI] [PubMed] [Google Scholar]
- 31.Bosetti F, Koenig JI, Ayata C, et al. Translational stroke research: vision and opportunities. Stroke. 2017;48(9):2632-2637. doi: 10.1161/STROKEAHA.117.017112 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial protocol
Statistical analysis plan
Pharmacokinetic analysis
eAppendix. Eligibility criteria
eMethods.
eTable 1. Major protocol deviations during phase Ib and IIa
eTable 2. Concomitant medications in phase Ib and IIa
eTable 3. Summary of adverse events during phase Ib
eTable 4. Summary of adverse events during phase IIa
eTable 5. Permutation tests
eTable 6. Post-hoc analysis based on multivariate generalized linear models with and without adjustment for covariates
eFigure 1. Distribution of primary endpoints in the study groups
eFigure 2. Line plots for individual comparison of baseline ischemic core and final infarct volume
eFigure 3. Temporal profile of proinflammatory biomarkers in APRIL study
eFigure 4. Graphical representation of mediation analysis
eFigure 5. Graphical representation of the sensitivity analysis of the mediation model
eFigure 6. Secondary efficacy endpoints according to time from symptom onset to treatment administration
Data sharing statement