


Abstract
ADP-ribose 1″,2″-cyclic phosphate (Appr>p) is produced in yeast and other eukaryotes as a consequence of tRNA splicing. This molecule is converted to ADP-ribose 1″-phosphate (Appr-1″p) by the action of the cyclic nucleotide phosphodiesterase (CPDase). Comparison of the previously cloned CPDase from Arabidopsis with proteins having related cyclic phosphodiesterase or RNA ligase activities revealed two histidine-containing tetrapeptides conserved in these enzyme families. Using the consensus phosphodiesterase signature, we have identified the yeast Saccharomyces cerevisiae open reading frame YGR247w as encoding CPDase. The bacterially expressed yeast protein, named Cpd1p, is able to hydrolyze Appr>p to Appr-1″p. Moreover, as with the previously characterized Arabidopsis and wheat CPDases, Cpd1p hydrolyzes nucleosides 2′,3′-cyclic phosphates (N>p) to nucleosides 2′-phosphates. Apparent Km values for Appr>p, A>p, U>p, C>p and G>p are 0.37, 4.97, 8.91, 12.18 and 14.29 mM, respectively. Site-directed mutagenesis of individual amino acids within the two conserved tetrapeptides showed that H40 and H150 residues are essential for CPDase activity. Deletion analysis has indicated that the CPD1 gene is not important for cellular viability. Likewise, overexpression of Cpd1p had no effect on yeast growth. These results do not implicate an important role for Appr>p or Appr-1″p in yeast cells grown under standard laboratory conditions.
INTRODUCTION
Splicing of pre-tRNAs is initiated by endonucleolytic cleavages that lead to the removal of the intron and the generation of two tRNA half molecules. In yeast and plants, the two tRNA halves are ligated via an unusual 3′,5′ phosphodiester, 2′-phosphomonoester linkage (for reviews see 1,2). In the following maturation steps, the 2′-phosphate is removed from the spliced tRNA. This reaction is catalyzed by a specific phosphotransferase which transfers the 2′ phosphate to an NAD to produce ADP-ribose 1″,2″-cyclic phosphate (Appr>p) and nicotinamide (3,4). The resulting Appr>p is then converted to ADP-ribose 1″-phosphate (Appr-1″p) by the action of a specific phosphodiesterase referred to as a cyclic phosphodiesterase (CPDase) (5,6) (Fig. 1). The tRNA splicing pathway described above is also conserved in vertebrates (1–3,7,8), although in these organisms, most of the tRNA splicing involves another RNA ligase. This ligase, to date identified only in vertebrates, joins the two tRNA exons by the regular 3′,5′-phosphodiester linkage (9,10) (Fig. 1). The finding that the 3′,5′-phosphodiester, 2′-phosphomonoester-forming pathway functions in vertebrates as a redundant route of tRNA splicing led to speculations that production of Appr>p and/or Appr-1″p might be important for the cell (2,3,5,6).
Figure 1.

tRNA splicing pathways in eukaryotes. Generation of Appr>p and its subsequent hydrolysis to Appr-1″p by the CPDase is indicated at the bottom of the 3′,5′-phosphodiester, 2′-phosphomonoester-forming pathway. For more information and references, see text.
CPDase activity has been previously identified in yeast, wheat and Arabidopsis (5,6). The cDNA encoding the CPDase in Arabidopsis has been cloned and the enzyme biochemically characterized (6); the overexpressed and purified Arabidopsis protein has similar substrate specificity to the enzyme purified previously from wheat (6,11). Both enzymes hydrolyze Appr>p and nucleoside 2′,3′-cyclic phosphates (N>p) to Appr-1″p and nucleoside 2′-phosphates (N2′p), respectively. CPDase partially purified from yeast was found to be highly specific for Appr>p: no hydrolysis of N>p to N2′p by the yeast enzyme could be observed (5). Apart from the apparent higher substrate specificity of the yeast protein, the plant and yeast enzymes share many biochemical properties, suggesting that they belong to the same family of proteins. Despite these similarities, a search of the yeast databank with the Arabidopsis protein sequence as a query did not identify a candidate gene encoding the CPDase in yeast (6).
In this work we have compared sequences of the Arabidopsis CPDase with other proteins having 2′,3′-cyclic nucleotide 3′-phosphodiesterase or related activities, and identified two histidine-containing tetrapeptides, which are highly conserved in this family of enzymes. Using the consensus phosphodiesterase signature, we have identified the yeast Saccharomyces cerevisiae YGR247w as a gene encoding CPDase. We demonstrate that the yeast enzyme has similar properties to the previously characterized plant CPDases. Site-directed mutagenesis indicated that two conserved His residues of the CPDase signature are essential for enzyme activity.
MATERIALS AND METHODS
Strains and media
The S.cerevisiae strain BMA41 (MATa/MATα, leu2-3,112, his3-11, ade2-1, ura3-1, trp1Δ, can1-100) was used. All protocols for manipulation of DNA and media are described elsewhere (12–14).
Gene deletion
Gene deletion was performed using the one-step disruption procedure (15). Two oligonucleotides, GTTTGGGAATTCG-CGATACTTCAATTTGCTAATATCAGTAACCACAGAA-AAAGATCTGAATTAATTCGGTCG and CAGTATACGT-AAATCTCTAACTAGAACATTTTCAAGTATTGACAGATATGAGATGTCGGCAAGTGCAC, were used to amplify the TRP1 gene carrying 50 nt-long flanking sequences corresponding to the 5′ and 3′ untranslated regions of YGR247w, using the pFL35 plasmid (16) as template. Yeast was transformed using the procedure of Chen et al. (17). Deletion was confirmed by PCR and Southern blotting.
Overexpression and purification of Cpd1p
The YGR247w open reading frame (ORF) was cloned in the pET28 (Novagen, Abingdon, UK) vector designed to express proteins as fusions with a His6-tag at the C-terminus. The insert was PCR-amplified, using yeast genomic DNA as a template and oligonucleotides CATGCCATGGCAATAGCATTATGGTATTGTCC and ATAAGAATGCGGCCGCAACGTCCACCCGTCCCAATACC. The product was digested by NcoI and NotI and cloned in pET28 to yield pFN207. YGR247w was also cloned in the pGEX-2T (Amersham Pharmacia Biotech, Dübendorf, Switzerland) vector designed to express proteins as fusions with GST. The oligonucleotides used were CGCGGATCCGCAATAGCATTATGGTATTGTCC and CGCGG-ATCCTTAAACGTCCACCCGTCCCAATACC. The product was digested with BamHI and cloned in pGEX-2T to yield pFN203. Both plasmids were analyzed by sequencing. Overexpression was carried out as described by Genschik et al. (6) in the Escherichia coli strain BL21. Recombinant proteins were purified in their native forms under non-denaturing conditions, using GST-resin (Amersham Pharmacia Biotech) and nickel-nitrilotriacetic acid resin (NTA) (Qiagen, Hilden, Germany), following the manufacturer’s instructions. Purified proteins were dialyzed against 20 mM Tris–HCl, pH 7.0, containing 0.1 M NaCl, 10% glycerol, 0.1 mM EDTA and 0.5 mM dithiothreitol (DTT), and stored at –70°C. Protein concentration was determined by the Bradford method (18) using bovine serum albumin as a standard. Protein purity, checked by SDS–PAGE, was >85%.
Site-directed mutagenesis
Site-directed mutagenesis was performed on the CPD1 ORF present in the plasmid pFN207 using two appropriate oligonucleotide primers, one of which carried the desired mutation. They were used to amplify the whole pFN207 plasmid as a fragment containing blunt ends. After amplification and purification, DNA was phosphorylated with T4 polynucleotide kinase, circularized in the presence of T4 DNA ligase and transformed in the E.coli DH5α strain. Positive clones were checked by sequencing the entire CPD1 ORF. Oligonucleotide W1 was used together with mutagenic oligonucleotides to create mutants P38, H39, V40, T41, V42 and T43, and oligonucleotide W2 was used to generate mutants P149, H150, V151, S152 and L15.
The following oligonucleotides were used (the mutagenized codons are underlined):
W1, TTCAAAAACTGGTGAATCTGGGAACAACG;
W2, ATGGAATTCCTCGTGCACCCAAGTGGC;
P38, GCTCATGTGACGGTAACGTCCCATTTGG;
H39, CCTGCTGTGACGGTAACGTCCCATTTGGTG;
V40, CCTCATGCGACGGTAACGTCCCATTTGGTGTGC;
T41, CCTCATGTGGCGGTAACGTCCCATTTGGTGTGC;
V42, CCTCATGTGACGGCAACGTCCCATTTGGTGTGC;
T43, CCTCATGTGACGGTAGCGTCCCATTTGGTGTGC;
P149, GCTCACGTCTCATTGCTTTATTCGG;
H150, CCTGCAGTCTCATTGCTTTATTCGGATATTC;
V151, CCTCACGCCTCATTGCTTTATTCGGATATTC;
S152, CCTCACGTCGCATTGCTTTATTCGGATATTC;
L153, CCTCACGTCTCAGCGCTTTATTCGGATATTCATCCG.
The mutant proteins were expressed in the BL21 E.coli strain and purified as described above.
Preparation of extracts
Unless described otherwise, 100 ml of cells was grown to an A600 of 1 in a complete medium (YPD: 1% yeast extract, 2% peptone, 2% glucose). Cells were harvested, washed in buffer A (0.1 M Tris–HCl, pH 6.5, 10 mM DTT) and resuspended in 2 ml of buffer B1 [20 mM Tris–HCl pH 6.5, 2 mM EDTA pH 8.0, 1 mM DTT, 1 M NaCl, 4 mM MgCl2, and protease inhibitor cocktail tablets (one tablet per 50 ml; Boehringer Mannheim, Mannheim, Germany)]. Cells were then sheared at 4°C with glass beads for 6 × 1 min. The supernatant was centrifuged at 15 000 g for 20 min, dialyzed against buffer B2 (as buffer B1 but containing 40 mM NaCl and 10% glycerol), and stored at –70°C.
Assays of cyclic nucleotide phosphodiesterase
Assays (10 or 20 µl) contained 50 mM Tris–HCl, pH 6.5, 0.01% Triton X-100, and, if not indicated otherwise, 2 mM Appr>p or 5 mM A>p. Enzyme concentrations and incubation temperatures and times are indicated in the figure legends. Sources of all nucleotides and incubations with the Arabidopsis CPDase and RNase T2 were as described by Genschik et al. (6). Appr>p was synthesized as described by Hall et al. (19). Reaction products were analyzed on TLC cellulose plates in solvent A [saturated (NH4)2SO4/3 M sodium acetate/isopropyl alcohol (80:6:2)] or polyethyleneimine–cellulose TLC glass-baked plates in solvent B (0.75 M LiCl). Nucleotide markers and reaction products were visualized under UV. To calculate kinetic and other parameters, a quantitative assay based on a measurement of inorganic phosphate (20) was used. Reactions (20 µl) containing 10 ng of enzyme were incubated for 10 min at 30°C. They were stopped by boiling for 3 min and 80 µl of 0.1 M Tris–HCl, pH 8.0, containing 0.2 U of calf intestine phosphatase (CIP; Boehringer Mannheim) was added. After incubation at 37°C for 20 min, liberated phosphate was assayed according to the protocol described by Hess and Derr (20). To determine Km and Vmax values, assays contained substrates at concentrations of 1.34–25 mM (N>p) or 0.46–5.5 mM (Appr>p). All velocities were calculated from the initial linear rates as described by Genschik et al. (6).
Computer analysis
Analyses, including pairwise comparisons, multiple alignments and amino acid shading, were performed using the GCG package of programs (Genetics Computer Group, Madison, WI). Databanks were searched using BLAST programs (21) available on the NIH server.
RESULTS
Identification of a candidate gene encoding CPDase in yeast
We have previously cloned the Arabidopsis cDNA encoding the cyclic nucleotide phosphodiesterase (AtCPDase1; Fig. 3) which hydrolyzes Appr>p and nucleoside 2′,3′-cyclic phosphates to Appr-1″p and N2′p, respectively (6). The gene encoding the related protein, with 56% identity and 66% similarity to AtCPDase1 and referred to as AtCPDase2 (Fig. 3), has been subsequently sequenced within the Arabidopsis genome project (22). Since this gene is positioned immediately downstream of the AtCPDase1 gene, within the Arabidopsis genome, it was likely formed by DNA duplication and encodes a protein with properties similar to AtCPDase1. Inspection of amino acid sequences of both Arabidopsis proteins revealed that they have two His residue-containing tetrapeptides which are also conserved, in terms of both sequence and approximate spacing, in two other families of enzymes with either 2′,3′-cyclic nucleotide 3′-phosphodiesterase or related activities (Fig. 2). The first family corresponds to RNA ligases involved in tRNA splicing. The C-terminal domains of these enzymes have phosphodiesterase activity that opens the terminal cyclic phosphate in RNA into the 2′-phosphate (1,2,23); for the tRNA splicing ligase from wheat germ, it has been demonstrated that the enzyme also inefficiently hydrolyzes 2′,3′-cyclic mononucleotides (11). To the second family belong prokaryotic RNA ligases which are able to ligate tRNA half molecules containing 2,3′-cyclic phosphate and 5′-OH termini to products containing the 2′,5′-phosphodiester linkage (24,25). Physiological substrates of these enzymes are not known. Although these enzymes have not been demonstrated to have a cyclic 3′-phosphodiesterase activity, it is likely, based on the structure of the ligation product, that the reaction they catalyze involves hydrolysis of the 2′,3′-cyclic phosphate into 2′-phosphate. Moreover, brain CNPases, which hydrolyze cyclic 2′,3′-phosphates into 2′-phosphates in mono- or oligonucleotides (26–28, and references therein), contain two conserved tetrapeptide sequences that are similar to those of the proteins described above. While the second tetrapeptide conforms with the consensus derived from other three classes, the first one deviates from the consensus in the second and fourth amino acid position (Fig. 2). The consensus signature derived from CPDases, tRNA-splicing RNA ligases and prokaryotic RNA ligases, H[ILVMF]T[LVM][X66–107]H[ILVM][TS][ILV] (where X represents any amino acid) (Fig. 2), was used to search protein sequence databanks. A putative protein from S.cerevisiae, containing two signature tetra amino acids, was detected. The gene, YGR247w, identified within the yeast genome project (29), encodes a 239-amino acid protein of 26.6 kDa with a predicted isoelectric point of 7.7. The codon bias of 0.04 (30) indicates that YGR247w is expressed at a low level. Figure 3 shows the alignment of the yeast protein and Arabidopsis CPDases. In addition to the regions corresponding to the two tetrapeptides, and amino acids adjacent to them, the Arabidopsis and yeast proteins share a few additional stretches of similar sequences.
Figure 3.

Alignment of Ygr247wp-Cpd1p with the Arabidopsis AtCPDase1 and AtCPDase2 sequences. Identical residues present in all proteins are on a black background; residues conserved in at least two proteins are on a gray background. The two conserved signature tetrapeptides are underlined.
Figure 2.

Conserved sequences in four different classes of proteins having 2′,3′-cyclic nucleotide 3′-phosphodiesterase or related activities. The sequences shown were extracted from four classes of enzymes. CPDases: S.cerevisiae (P43314:sp), A.thaliana 1 (CAA16750.1:embl) and A.thaliana 2 (CAA16751.1:embl); tRNA-splicing ligases: S.cerevisiae (P09880:sp), Schizosaccharomyces pombe (Q10313:sp) and Candida albicans (P43075:sp); prokaryotic 2′,5′ RNA ligases: E.coli (P37025:sp), Deinococcus radiodurans (BAA21329:dbj), Thermotoga maritima (AAD36922:gb), Methanococcus jannaschii (O58963:sp), Archaeoglobus fulgidus (O28125:sp), Aeropyrum pernix (BAA80795.1:dbj), Bacillus stearothermophilus (S43916:pir), Methanobacterium thermoautotrophicum (O26683:sp), Pyrococcus horikoshii (BAA29168.1:dbj), Aquifex aeolicus (2983341:gi), Pyrococcus abyssi (CAB49026.1:embl) and Bacillus subtilis (CAB14974:embl). Brain CNPases and related RICH proteins: Homo sapiens (P09543:sp), Bos taurus (P06623:sp), Mus musculus (P16330:sp), Rattus norvegicus (Q64575:sp), Gallus gallus (O57389:sp), Rana catesbeiana (O57390:sp), Carassius auratus (O42487:sp and Q90306) and Danio rerio (28). Positions of histidines in the proteins, and numbers of amino acids separating the two conserved tetrapeptides are indicated.
The yeast protein has CPDase activity
The YGR247w ORF was cloned in pGEX-2T and pET-28 vectors designed for expression of proteins as fusions with the GST and His6-tag, respectively. The purified recombinant His-tagged protein was tested for phosphodiesterase activity using different cyclic nucleotides as substrates. The yeast protein converted Appr>p to the product which chromatographs in two different TLC systems with an identical mobility to that of Appr-1″p, obtained by the hydrolysis of Appr>p by the Arabidopsis CPDase (Fig. 4A). Moreover, the product of Appr>p hydrolysis by the yeast enzyme migrated differently from Appr-2″p generated by treatment of Appr>p with RNase T2 (Fig. 4B) (5). The yeast protein also hydrolyzed all four nucleoside 2′,3′-cyclic phosphates to the corresponding 2′-phosphomonoesters; no formation of nucleoside 3′-phosphates was detected. The same results were obtained with the GST fusion protein (data not shown). These results indicate that the yeast YGR247w encodes a cyclic phosphodiesterase with substrate specificity similar to the enzyme characterized previously in wheat and Arabidopsis (5–6). Thus, the gene was renamed CPD1 (for cyclic phosphodiesterase).
Figure 4.


Hydrolysis of nucleoside 2′,3′-cyclic phosphates and Appr>p by the S.cerevisiae CPDase. (A) All indicated substrates were incubated with or without 100 ng of Cpd1p. Products were analyzed on cellulose TLC plates chromatographed in solvent A (Materials and Methods). Positions of products and other nucleotide markers are indicated. Appr-1″p and Appr-2″p are not separated in this TLC system; identification of the Appr>p hydrolysis product as Appr-1″p is based on other criteria (B). (B) Appr>p was incubated with Cpd1p, AtCPDase or RNase T2, or without any enzyme (control), and products analyzed on the PEI-cellulose TLC plate chromatographed in solvent B.
Biochemical characterization of Cpd1p
Reaction requirements of the S.cerevisiae CPDase (Cpd1p) were determined using A>p as substrate. The amount of A>p hydrolyzed was linearly dependent on enzyme concentration up to at least 30 ng per 20 µl assay (Fig. 5A). Optimal activity was found at pH 6.5 (data not shown). The rate of A>p hydrolysis was similar at 30 and 37°C, and ∼2-fold slower at 4°C (Fig. 5B). The kinetic parameters, Km and Vmax, were calculated for Appr>p and four nucleoside 2′,3′-cyclic phosphates (Table 1). As based on Vmax/Km values, the yeast CPDase has the following specificity towards cyclic nucleotides: Appr>p:A>p:C>p:U>p:G>p = 100:43:30:20:16. Thus, the enzyme has a small preference for Appr>p over N>p.
Figure 5.

Hydrolysis of A>p by the yeast CPDase. (A) Dependence on enzyme concentration. Reactions (20 µl) were incubated at 30°C for 30 min. (B) Kinetics of A>p hydrolysis. Assays (20 µl), containing 10 ng of the enzyme, were incubated at 4, 30 and 37°C (not shown, identical to 30°C). All values are averages of duplicates.
Table 1. Substrate specificity of the yeast CPDase.
| Substrate | Vmax | Km | Vmax/Km |
|---|---|---|---|
| (µmol/min/mg) | (mM) | (× 103) | |
| A>p | 353 ± 23 | 4.97 ± 0.83 | 71 ± 28 |
| G>p | 390 ± 39 | 14.29 ± 2.78 | 27 ± 14 |
| C>p | 598 ± 35 | 12.18 ± 1.46 | 49 ± 24 |
| U>p | 296 ± 40 | 8.91 ± 2.70 | 33 ± 15 |
| Appr>p | 61 ± 40 | 0.37 ± 0.09 | 164 ± 48 |
Parameters in the table are the weighted means, together with their standard errors, of at least two determinations.
Functional analysis
To find out whether the CPD1 gene is essential, a null allele was created using a one-step disruption procedure. The DNA fragment carrying the TRP1 marker flanked by 50 bp-long sequences corresponding to the 5′ and 3′ untranslated regions of CPD1, was transformed into the diploid BMA41 strain. Tryptophan prototrophs were selected and subjected to PCR and Southern blotting analysis. Of the 20 transformants analyzed, 18 showed the expected profile for the strain heterozygous for CPD1. Three of the heterozygous diploids (CPD1/cpd1Δ::TRP1) were sporulated and 40 asci dissected, most of which gave rise to complete tetrads showing 2:2 segregation of trp+:trp–. Hence, CPD1 is not essential for cellular viability.
The growth of the CPD1-disrupted and wild-type (wt) haploid strains was assessed on different standard media (complete fermentable, complete non-fermentable and minimal SD medium) at 16, 28 and 37°C. No differences could be detected. Since phosphodiesterases converting cyclic phosphodiesters into phosphatase-sensitive monoesters may represent catabolic enzymes replenishing the cellular phosphate pool, we have measured growth of the cpd1Δ::TRP1 strain on a phosphate-free medium. Two CPD1 and two independent cpd1Δ::TRP1 strains were pre-grown in a complete medium and then spotted in 10 serial dilutions onto a minimal medium with or without a phosphate. No differences in growth of the wt and mutant strains could be detected (data not shown).
We determined whether deletion of CDP1 entirely eliminates the ability of yeast extracts to hydrolyze Appr>p and A>p. Extracts were prepared from haploid cpd1Δ::TRP1 and CPD1 wt strains obtained from a complete tetrad. Only extracts of the two strains carrying the CPD1 gene hydrolyzed Appr>p (Fig. 6), suggesting that CPD1 is the only gene in S.cerevisiae to encode an Appr>p cyclic phosphodiesterase. We have found that the product of Appr>p hydrolysis, Appr-1″p, is converted during incubation with the extract to another compound, co-migrating during TLC analysis with ADP-ribose (Fig. 6 and data not shown). Thus, yeast cells are likely to contain a phosphatase removing the monoester phosphate group resulting from hydrolysis of the cyclic phosphate in Appr>p. Comparison of cpd1Δ::TRP1 and wt strain extracts for activity to hydrolyze A>p revealed that Cpd1p is not the only phosphodiesterase able to hydrolyze A>p to A2′p. Low levels of such activity were also present in extracts from CPD1 deleted strains (Fig. 6B). Significantly, however, extracts from wt strains yielded more A2′p than those from strains deleted in CPD1, further demonstrating that Cpd1p hydrolyzes not only Appr>p but also nucleoside 2′,3′-cyclic phosphates to N2′p. Interestingly, extracts from both cpd1Δ::TRP1 and wt strains also contained phosphodiesterase activity converting A>p to the product comigrating with A3′p (Fig. 6B).
Figure 6.

Hydrolysis of Appr>p (A) and A>p (B) by crude extracts prepared from haploid strains derived from one dissected CPD1 disruption tetrad. Reactions (10 µl) containing 500 ng of extract protein and 100 ng of purified Cpd1p were incubated at 30°C for 14 h and applied to the cellulose TLC plate. Control, incubation without addition of the extract. Chromatography was in solvent A.
Site-directed mutagenesis of the CPDase signature
We have investigated whether two tetrapeptide regions conserved among different classes of 2′,3′-cyclic nucleotide 3′-phosphodiesterases or related enzymes (Fig. 2) are important for the activity of Cpd1p. The amino acids that form part of the tetrapeptides or are adjacent to them (P38, H39, V40, T41, V42, T43, P149, H150, V151, S152 and L153) were individually mutagenized to alanine. Mutant proteins were overexpressed in E.coli and purified on NTA-Ni columns. They were first tested for A>p hydrolysis under limiting (10 min incubation) and exhaustive (60 min incubation) conditions (Fig. 7A and B, respectively). No hydrolysis of A>p could be detected in reactions containing mutants H39 and H150, and mutations T41 and S152 strongly decreased activity of the enzyme. Quantitative determinations, using a phosphate release assay (Materials and Methods), indicated that mutations H39 and H150 lower activity of Cpd1p at least 1000-fold, while T41 and S152 mutants have 2 and 30% of the wt protein activity, respectively (data not shown). Other mutations had no significant effect on Cpd1p activity (Fig. 7, and data not shown). When mutant proteins were tested with Appr>p as a substrate, mutants H39 and H150 were again found to be inactive. Interestingly, mutants T41 and S152 were considerably more active with Appr>p than A>p as a substrate (Fig. 7C). Quantitative determinations by the inorganic phosphate assay indicated that the T41 mutant has 49% activity of the wt protein whereas activities of S152 and a wt protein are identical when Appr>p is used as a substrate (data not shown). Hence, residues T41 and S152 are likely to play a role in substrate recognition rather than the catalysis.
Figure 7.
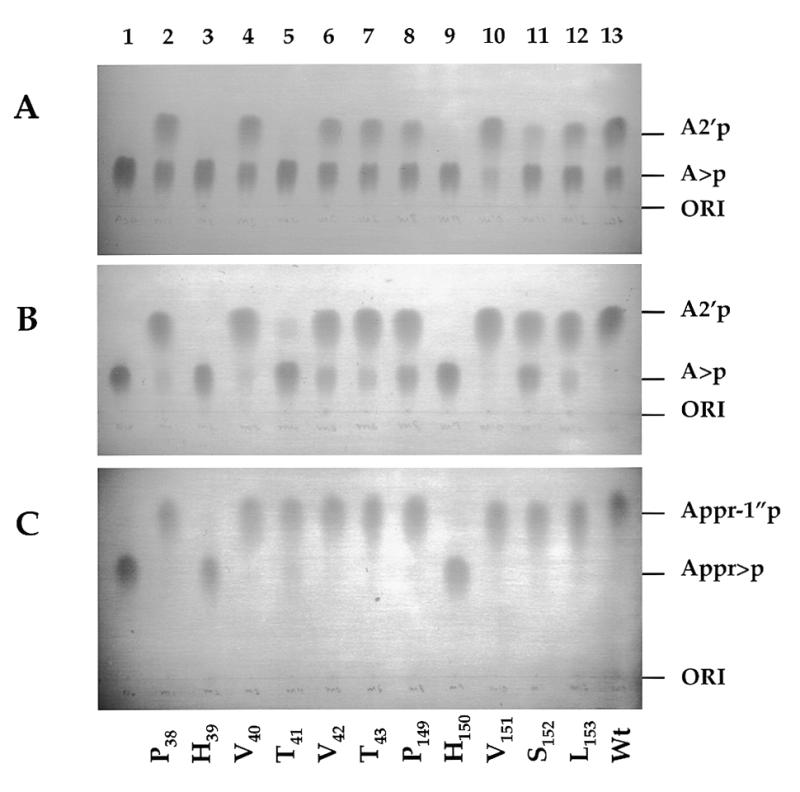
Hydrolysis of A>p and Appr>p by the wt Cpd1p and its mutants. (A) Hydrolysis of A>p under non-exhaustive conditions. Reactions (10 µl), containing 25 ng of proteins, were incubated for 10 min at 30°C. (B) Hydrolysis of A>p under exhaustive conditions. Reactions (10 µl), containing 25 ng of proteins, were incubated for 1 h at 30°C. (C) Hydrolysis of Appr>p under exhaustive conditions specified in (B). Products were analyzed on cellulose TLC plates chromatrographed in solvent A. Mutants are identified at the bottom. Lane 1, control incubations without addition of the enzyme.
DISCUSSION
In all eukaryotes, splicing of tRNAs via the 3′,5′-phosphodiester, 2′-phosphomonoester-forming pathway yields tRNA intermediates with a 2′-phosphate at the splice junction (1,2). This 2′-phosphate is removed by the specific phosphotransferase and transferred to an NAD acceptor to produce Appr>p (3). The latter product is further converted to Appr-1″p by the action of the cyclic phosphodiesterase previously identified in yeast, wheat and Arabidopsis (5,6,11). The cDNA encoding the Arabidopsis CPDase has been cloned and its product (AtCPDase1) biochemically characterized. In addition to Appr>p, the Arabidopsis CPDase, and also its wheat counterpart, hydrolyze all 2′,3′-cyclic nucleotides to the corresponding nucleoside 2′-phosphates (6). A second gene encoding a related protein to AtCPDase1 and referred to as AtCPDase2, has been subsequently sequenced within the Arabidopsis genome project.
By aligning the Arabidopsis CPDases with three other families of enzymes having either 2′,3′-cyclic nucleotide 3′-phosphodiesterase or related activities, we identified two tetrapeptide sequences, which are conserved in this class of proteins. Using the derived signature, H[ILVMF]T[LVM] [X66–107]H[ILVM][TS][ILV], as a profile to search sequence databases, we identified the gene YGR247w as a candidate gene encoding the CPDase in yeast. We found that the encoded protein indeed has cyclic phosphodiesterase activity with substrate specificity similar to that of the previously characterized Arabidopsis and wheat enzymes. When this work had been nearly completed, Martzen et al. (31) also reported identification of YGR247w as the CPDase gene by a biochemical genomics approach, based on assaying biochemical activities of all yeast ORFs expressed as GST fusions.
The bacterially overexpressed yeast protein hydrolyzed Appr>p and nucleoside 2′,3′-cyclic phosphates to Appr-1″p and N2′p, respectively. Based on relative Vmax/Km values, the specificity of the yeast CPDase towards the cyclic nucleotides is Appr>p:A>p:C>p:U>p:G>p = 100:43:30:20:16. Hence, the enzyme has a small preference for Appr>p over nucleoside 2′,3′-cyclic phosphates. Notably, the yeast, Arabidopsis, and wheat enzymes hydrolyze different nucleoside 2′,3′-cyclic phosphates with the same order of preference (6,11; this work).
The demonstration that Cpd1p utilizes both Appr>p and nucleoside 2′,3′-cyclic phosphates as substrates is in disagreement with previous findings indicating that crude or partially purified preparations of the yeast phosphodiesterase hydrolyze Appr>p but not nucleoside 2′,3′-cyclic phosphates (5). It is unlikely that the enzyme studied previously and that reported in this work represent two different proteins. We have demonstrated that extracts prepared from strains deleted for the CPD1 gene are devoid of activity to hydrolyze Appr>p. Moreover, Culver et al. (5) have previously shown that the Appr>p cyclic phosphodiesterase activity detectable in yeast extracts also appears to be due to only one protein species. We have investigated whether the apparent discrepancy between the previous and current results may result from the presence in the extract of an inhibitor preventing hydrolysis of N>p but not Appr>p. We have found that crude extracts of yeast cells indeed hydrolyze A>p to A2′p with the efficiency which, relative to the hydrolysis of Appr>p, is lower than that of the purified recombinant enzyme (Fig. 6, Table 1). However, the CPD1-gene-dependent hydrolysis of A>p to A2′p in extracts was clearly apparent (Fig. 6). It is possible that partially purified CPDase fractions investigated previously (5), prepared from the yeast strain other than that used in this work, contained inhibitory factors interfering with the hydrolysis of nucleoside 2′,3′-cyclic phosphates.
To assess the importance of the CPDase signature for enzymatic activity, all residues within the two conserved tetrapeptides of Cpd1p were individually mutated to alanine. Mutations of the two conserved histidine residues, H39 or H150, resulted in a very strong inhibition of activity of the enzyme to hydrolyze A>p and Appr>p. Mutations of T41 and S152 inactivated the enzyme partially and their effect on A>p hydrolysis was much more pronounced than on Appr>p hydrolysis. Hence, it is possible that T41 and S152 are involved in substrate recognition rather than the catalysis. Ballestro et al. (28) have recently analyzed, by mutagnesis, the sequence 330GSRAHVTLGCSAGV343 of the zebrafish zRICH phosphodiesterase, a protein homologous to mammalian brain CNPases. The investigated sequence was identified as a region conserved in all CNPases with similarity to the active site motif of β-ketoacyl synthases. Although this analysis did not provide evidence of the expected relationship, two of the point mutations analyzed, H334A and T336A, inactivated the zebrafish enzyme. Significantly, H334 and T336 represent two conserved residues of the C-terminus-proximal tetrapeptide identified in CPDases and CNPases (Fig. 2). Hence, His and Ser/Thr residues of the proposed signature may indeed be universally important for enzymatic activity of the 2′,3′-cyclic nucleotide 3′-phosphodiesterases and related enzymes discussed in this work. Interestingly, two His residues conserved in many cyclizing ribonucleases likewise play critical roles in catalysis (for reviews see 32,33).
The tRNA-splicing pathway generating the 3′,5′-phosphodiester, 2′-phosphomonoester bond and, consequently, Appr>p, also functions in vertebrates, even though most of the tRNA splicing in these organisms occurs via another pathway (Introduction). These findings led to speculations that production of Appr>p and/or Appr-1″p might be important for the cell (2,3,5,6). Other NAD or ADP-ribose derivatives, such as nicotinic acid adenine dinucleotide phosphate (NAADP) and cyclic ADP-ribose (cADPR), have been recently shown to act as intracellular messengers mobilizing Ca2+ from intracellular stores in animal and plant cells (for reviews see 34,35). Although we have reported previously that Appr>p and Appr-1″p neither affect nor modify Ca2+ release in sea urchin egg homogenates (6), cloning of CPD1 provided the possibility to investigate whether these compounds have any other biological function. We found that the CPD1 gene is not essential for cellular viability and that the cpd1Δ::TRP1 strain is at no disadvantage when grown at different temperatures on rich or minimal media, and with or without phosphate (Results). Since Cpd1p appears to be the only phosphodiesterase able to hydrolyze Appr>p in yeast cells, these results suggest that formation of Appr-1″p is not essential for yeast growth under standard laboratory conditions. We have also found that overproduction of Cpd1p in strains transformed with multicopy 2µ plasmids expressing Cpd1p from either GAL10 or its own promoter, a condition that should deplete Appr>p, has no effect on yeast growth in standard media (data not shown).
The YGR247w gene was recently identified in a screen designed to identify genes whose expression elicits the SOS response in E.coli (36). The genes of the SOS system in bacteria are generally induced by agents that damage DNA or inhibit its metabolism, or play a role in DNA metabolism and/or genome stability. We checked whether the CPD1 deletion has any effect on growth and viability of yeast cells treated with a DNA-damaging agent such as UV (254 nm) light. No effect could be detected with cells grown on YPD and treated with different doses of UV light (our unpublished results).
In summary, the current results do not implicate an important role for Appr>p or Appr-1″p in yeast cells grown under standard laboratory conditions. Other approaches, such as a search for mutations which are synthetic lethal with a CPD1 deletion, might provide more insight into the role of Cpd1p and, consequently, Appr>p and Appr-1″p, in the cell. It is possible that Cpd1p, and its counterparts in other organisms, are only responsible for catabolism of Appr>p and nucleoside 2′,3′-cyclic phosphates generated as products of tRNA splicing and endonucleolytic degradation of RNA by cyclizing ribonucleases, respectively.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Jan Hofsteenge and Vitaly Boyko for valuable discussions. F.N. was supported by a long-term fellowship from EMBO.
REFERENCES
- 1.Phizicky E.M. and Greer,C. (1993) Trends Biochem. Sci., 18, 31–34. [DOI] [PubMed] [Google Scholar]
- 2.Westaway S.K. and Abelson,J. (1995) In Soell,D. and RajBhandary,U.L (eds), tRNA: Structure, Biosynthesis and Function. American Society for Microbiology, Washington DC, pp. 79–92.
- 3.Culver G.M., McCraith,S.M., Zillmann,M., Kierzek,R., Michaud,N., LaReau,R.D., Turner,D.H. and Phizicky,E.M. (1993) Science, 261, 206–208. [DOI] [PubMed] [Google Scholar]
- 4.Culver G.M., McCraith,S.M., Consaul,S.A., Stanford,D.R. and Phizicky,E.M. (1997) J. Biol. Chem., 272, 13203–13210. [DOI] [PubMed] [Google Scholar]
- 5.Culver G.M., Consaul,S.A., Tycowski,K.T., Filipowicz,W. and Phizicky,E.M. (1994) J. Biol. Chem., 269, 24928–24934. [PubMed] [Google Scholar]
- 6.Genschik P., Hall,J. and Filipowicz,W. (1997) J. Biol. Chem., 272, 13211–13219. [DOI] [PubMed] [Google Scholar]
- 7.Zillmann M., Gorovsky,M.A. and Phizicky,E.M. (1991) Mol. Cell. Biol., 11, 5410–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zillmann M., Gorovsky,M.A. and Phizicky,E.M. (1992) J. Biol. Chem., 267, 10289–10294. [PubMed] [Google Scholar]
- 9.Filipowicz W. and Shatkin,A.J. (1983) Cell, 32, 547–557. [DOI] [PubMed] [Google Scholar]
- 10.Laski F.A., Fire,A.Z., RajBhandary,U.L. and Sharp,P.A. (1983) J. Biol. Chem., 258, 11974–11980. [PubMed] [Google Scholar]
- 11.Tyc K., Kellenberger,C. and Filipowicz,W. (1987) J. Biol. Chem., 262, 12994–13000. [PubMed] [Google Scholar]
- 12.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 13.Ausubel F., Brent,R., Kingstom,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1990) Current Protocols in Molecular Biology. Greene Publishing, New York, NY.
- 14.Kaiser C., Michaelis,S. and Mitchell,A. (1994) Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbour Laboratory Press, Cold Spring Harbor, NY.
- 15.Eberhardt I. and Hohmann,S. (1995) Curr. Genet., 27, 306–308. [DOI] [PubMed] [Google Scholar]
- 16.Bonneaud N., Ozier-Kalogeropoulos,O., Li,G., Labouesse,M., Minivielle-Sebastia,L. and Lacroute,F. (1991) Yeast, 7, 609–615. [DOI] [PubMed] [Google Scholar]
- 17.Chen D.-C., Yang,B.-C. and Kuo,T.-T. (1992) Curr. Genet., 21, 83–84. [DOI] [PubMed] [Google Scholar]
- 18.Bradford M.M. (1976) Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 19.Hall J., Genschik,P. and Filipowicz,W. (1996) Helv. Chim. Acta, 79, 1005–1010. [Google Scholar]
- 20.Hess H.H. and Derr,J.E. (1975) Anal. Biochem., 63, 607–613. [DOI] [PubMed] [Google Scholar]
- 21.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bevan M., Bancroft,I., Bent,E., Love,K., Goodman,H., Dean,C., Bergkamp,R., Dirkse,W., Van Staveren,M., Stiekema,W. et al. (1998) Nature, 391, 485–488. [DOI] [PubMed] [Google Scholar]
- 23.Xu Q., Teplow,D., Lee,T.D. and Abelson,J. (1990) Biochemistry, 29, 6132–6138. [DOI] [PubMed] [Google Scholar]
- 24.Arn E.A. and Abelson,J.N. (1996) J. Biol. Chem., 271, 31145–31153. [DOI] [PubMed] [Google Scholar]
- 25.Arn E.A. and Abelson,J.N. (1998) In Simons,R.W. and Grunberg-Manago,M. (eds), RNA Structure and Function. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 695–726.
- 26.Sprinkle T.J. (1989) CRC Crit. Rev. Neurobiol., 4, 235–401. [PubMed] [Google Scholar]
- 27.Ballestero R.P., Wilmot,G.R., Agranoff,B.W. and Uhler,M.D. (1997) J. Biol. Chem., 272, 11479–11486. [DOI] [PubMed] [Google Scholar]
- 28.Ballestero R.P., Dybowski,J.A., Levy,G., Agranoff,B.W. and Uhler,M.D. (1999) J. Neurochem., 72, 1362–1371. [DOI] [PubMed] [Google Scholar]
- 29.Tettelin H., Agostoni Carbone,M.L., Albermann,K., Albers,M., Arroyo,J., Backes,U., Barreiros,T., Bertani,I., Bjourson,A.J., Bruckner,M. et al. (1997) Nature, 387 (Suppl), 81–84. [PubMed] [Google Scholar]
- 30.Sharp P.M. and Li,W.H. (1987) Nucleic Acids Res., 15, 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martzen M.R., McGraith,S.M., Spinelli,S.L., Torres,F.M., Fields,S., Grayhack,E.J. and Phizicky,E.M. (1999) Science, 286, 1153–1155. [DOI] [PubMed] [Google Scholar]
- 32.Cuchillo C.M., Vilanova,M. and Nogués,M.V. (1997) In D’Alessio,G. and Riordan,J.F. (eds), Ribonucleases: Structures and Functions. Academic Press, San Diego, CA, pp. 271–304.
- 33.Irie M. (1997) In D’Alessio,G. and Riordan,J.F. (eds), Ribonucleases: Structures and Functions. Academic Press, San Diego, CA, pp. 101–130.
- 34.Lee H.C., Munshi,C. and Graeff,R. (1999) Mol. Cell Biochem., 193, 89–98. [DOI] [PubMed] [Google Scholar]
- 35.Lee H.C. (2000) J. Membr. Biol., 173, 1–8. [DOI] [PubMed] [Google Scholar]
- 36.Perkins E.L., Sterling,J.F., Hashem,V.I. and Resnick,M.A. (1999) Proc. Natl Acad. Sci. USA, 96, 2204–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]