

Abstract
Background:
Perturbations in the timing of puberty, with potential adverse consequences in later health, are increasingly common. The underlying neurohormonal mechanisms are unfolded, but nutritional alterations are key contributors. Efforts to unveil the basis of normal puberty and its metabolic control have focused on mechanisms controlling expression of Kiss1, the gene encoding the puberty-activating neuropeptide, kisspeptin. However, other regulatory phenomena remain ill-defined. Here, we address the putative role of the G protein-coupled-receptor kinase-2, GRK2, in GnRH neurons, as modulator of pubertal timing via repression of the actions of kisspeptin, in normal maturation and conditions of nutritional deficiency.
Methods:
Hypothalamic RNA and protein expression analyses were conducted in maturing female rats. Pharmacological studies involved central administration of GRK2 inhibitor, βARK1-I, and assessment of gonadotropin responses to kisspeptin or phenotypic and hormonal markers of puberty, under normal nutrition or early subnutrition in female rats. In addition, a mouse line with selective ablation of GRK2 in GnRH neurons, aka G-GRKO, was generated, in which hormonal responses to kisspeptin and puberty onset were monitored, in normal conditions and after nutritional deprivation.
Results:
Hypothalamic GRK2 expression increased along postnatal maturation in female rats, especially in the preoptic area, where most GnRH neurons reside, but decreased during the juvenile-to-pubertal transition. Blockade of GRK2 activity enhanced Ca+2 responses to kisspeptin in vitro, while central inhibition of GRK2 in vivo augmented gonadotropin responses to kisspeptin and advanced puberty onset. Postnatal undernutrition increased hypothalamic GRK2 expression and delayed puberty onset, the latter being partially reversed by central GRK2 inhibition. Conditional ablation of GRK2 in GnRH neurons enhanced gonadotropin responses to kisspeptin, accelerated puberty onset, and increased LH pulse frequency, while partially prevented the negative impact of subnutrition on pubertal timing and LH pulsatility in mice.
Conclusions:
Our data disclose a novel pathway whereby GRK2 negatively regulates kisspeptin actions in GnRH neurons, as major regulatory mechanism for tuning pubertal timing in nutritionally-compromised conditions.
Keywords: GRK2, G protein-coupled receptors (GPCR), GPR54, GnRH, Kisspeptin, Energy balance, Puberty, Reproduction
1. Introduction
Reproduction is controlled by sophisticated regulatory mechanisms that allow attainment and maintenance of fertility, if proper environmental and endogenous conditions concur [1,2]. Sexual and somatic maturation is completed at puberty, when reproductive capacity is achieved [1]. Worrying trends for deterioration of reproductive health have been reported worldwide that manifest in an increasing prevalence of pubertal and fertility disorders. Indeed, different epidemiological studies have revealed changes in the age of puberty, which mainly consist of advancement of pubertal onset, especially in girls, but possibly also in boys [3–7]. While the basis for such trends are yet to be disclosed, metabolic and nutritional factors are considered major contributors, with a clear parallelism between the rise of pubertal disorders and the incidence of child obesity [1,2,8]. Conditions of malnutrition (e.g., anorexia nervosa) and strenuous exercise, which are common in adolescents, are also linked to altered (delayed) puberty [1,2]. The relevance of this phenomenon goes beyond its reproductive dimension, since early or late pubertal maturation are bound to a large number of adverse health outcomes later in life, as behavioral alterations, perturbation of final height, and a number of musculoskeletal, cognitive, onco-logic, gastrointestinal and, prominently, metabolic and cardiovascular disorders [9–11].
Puberty onset is driven by the activation of the components of the hypothalamic-pituitary-gonadal (HPG) axis, whose major hierarchical element is the population of neurons producing the gonadotropin-releasing hormone (GnRH), whose neurosecretory activity becomes fully activated at puberty [12,13]. GnRH is secreted in pulses to stimulate the release of gonadotropins, LH and FSH [13], which, in turn, promote the testicular and ovarian maturation, the secretion of sex steroids and peptide hormones, and the production of gametes [14]. While full awakening of GnRH neurons is a primus event in pubertal activation, the mechanisms whereby this phenomenon takes place are yet to be fully disclosed. The pioneering work of Knobil and colleagues set the contention that a network of afferents, including excitatory and inhibitory inputs [12,15–17], converge onto GnRH neurons to define the GnRH pulse generator [18]. This system is responsible for driving the pulsatile secretion of GnRH and its awakening during the pubertal transition.
While the dynamic activation of GnRH neurons at puberty requires multiple regulatory mechanisms, substantial attention has been paid in recent years to identification of the major neuropeptide drivers of puberty, and the molecular mechanisms underlying their precise secretory control [1,2]. This is epitomized by numerous studies addressing the essential roles of kisspeptins, the products of Kiss1 gene acting via the receptor, Gpr54, in the control of puberty [1,2]. Likewise, analysis of the pubertal roles of a member of the tachykinin (TAC) family, neurokinin B (NKB), encoded by TAC3 in humans, which operates via the neurokinin receptor, NK3R, has drawn considerable interest [19,20]. Inactivating mutations of the elements of the Kiss1/Gpr54 and NKB/NK3R systems have been reported to prevent puberty in humans [21–23]. Detailed studies in sheep and rodents, later confirmed in other species, including humans, have unveiled the presence of a prominent neuronal population of Kiss1 neurons in the arcuate nucleus (ARC), which co-express NKB; they are called KNDy neurons, due to co-localization of Kisspeptin, NKB and Dynorphin [24]. In this system, NKB would operate as major activator of kisspeptin output to GnRH neurons, with a major role in pubertal activation of the gonadotropic axis.
Whereas the signals and mechanisms responsible for the control of these neuropeptide drivers have been closely scrutinized, other regulatory phenomena are likely to contribute to the fine tuning of pubertal timing, and its modulation by a plethora of signals, including metabolic and nutritional cues. Notably, most of the neuropeptides involved in the central control of GnRH secretion and puberty operate via G protein-coupled receptors (GPCR), such as NK3R and Gpr54, which activate signaling cascades in Kiss1 and GnRH neurons, respectively [1,12]. A key process in the regulation of GPCR is desensitization; a phenomenon triggered by continuous exposure to the ligand. In this phenomenon, eviction of the Gα subunit to switch on the signaling cascade, allows the GPCR kinases (GRKs) to phosphorylate intracellular serine/threonine residues of the receptor, thereby increasing its affinity for the binding of β-arrestins, which prevents receptor coupling to its Gα protein [25,26]. Among the members of the GRK family, GRK2 has emerged as major integrator of multiple signaling cascades, due to its capacity not only to interplay with β-arrestins, but also to phosphorylate multiple non-GPCR targets [27–29], and to respond to various pathophysiological stimuli. Nonetheless, information about the potential contribution GRKs in the regulation of these reproductive GPCRs remains surprisingly limited, despite initial biochemical evidence in a heterologous cell system suggesting that Gpr54 signaling may be modulated by GRK2 in vitro [30]. By a combination of expression, pharmacological and functional genomic studies, we present here evidence for a critical physiological role of GRK2 in GnRH neurons as putative modulator of kisspeptin signaling, thereby controlling puberty onset and its regulation by nutritional cues.
2. Methods
2.1. Ethics statement
All experimental procedures were approved by the University of Córdoba Ethical Committee for Animal Experimentation and were conducted in accordance with the European Union norms for the care and use of experimental animals.
2.2. Animals and drugs
Wistar rats and genetically modified C57Bl6 mice, engineered for conditional ablation of GRK2 in GnRH cells (namely, G-GRKO mice; see below), bred in the vivarium of the University of Córdoba, Spain, were used. The day the litters were born was considered as day 1 of age. The animals were maintained under constant conditions of light (14 h of light, lights on at 7:00 am) and temperature (22 °C). They were weaned at 21 days postpartum, with free access to standard rat chow and water ad libitum. In hormonal tests involving intracerebroventricular (icv) administration of drugs, rats or mice were caged individually from the day before cannula implantation. Correct positioning of the cannula was checked by visual inspection and confirmed at necropsy.
Rat/mouse kisspeptin-10 (Kp-10) was obtained from Phoenix Pharmaceuticals. Depending on the experiments, doses of 50 pmol or 1 nmol of Kp-10, as capable to sub-maximally or maximally eliciting LH and FSH secretion [31,32], were used. The NK3R agonist, Senktide, was from Sigma Chemical Co., and it was administered at a dose of 600 pmol in rats, in keeping with previous references [19,33]. The GRK2-inhibitor, methyl[(5-nitro-2-furyl)vinyl]-2-furoate (aka, βARK1-I), was from Calbiochem (La Jolla, CA, USA). Kp-10 and Senktide were diluted in physiological saline, 0.9% NaCl. βARK1-I was dissolved in DMSO and administered at a dose of 6.53 nmol, in line with previous studies [34,35]. The compounds were injected icv in a final volume of 5 μL per rat. As a general procedure, blood samples (>200 μL) were obtained by jugular venipuncture, in keeping with standard procedures routinely running in our laboratory [19,31–33], before and at different time points after administration of the various drugs. Animals injected with vehicle (0.9% NaCl, or DMSO) served as controls. Blood was centrifuged to isolate the serum, which was stored at −20 °C until hormone measurements.
2.3. Generation of a GnRH neuron-specific GRK2 null mouse line: the G-GRKO mouse
To study the potential role of GRK2 regulating on GnRH neurons, mice lacking functional GRK2 specifically in GnRH-expressing cells were generated by crossing a well-validated mouse line carrying Cre recombinase expression under the GnRH promoter, the GnRH-Cre+/− mouse [36], with mice harboring LoxP sites flanking exons 3 to 6 of the Adrbk-1 gene [37]. GnRH-Cre+/− mice were initially mated with Grk2loxP/loxP animals. The resulting genotypes, GnRH-Cre+/−;Grk2loxP/−, were self-crossed to generate all possible genotypic combinations. For experiments, only GnRH-Cre+/−;Grk2loxP/loxP (named G-GRKO) and GnRH-Cre−/−;Grk2loxP/loxP (referred as control) mice were used.
Mice were genotyped using the following combination of primers for GnRH-Cre, forward: 5′-CTG GTG TAG CTG ATG ATC CG-3′; and reverse: 5′-ATG GCT AAT CGC CAT CTT CC-3′; resulting in a 400 base-pair (bp) amplicon for the Cre allele, whose expression is under the GnRH promoter. Determination of Grk2 alleles, floxed or not, and recombination events was done using combinations of the following primers: GT2 (5′-TGA GGC TCA GGG ATA CCT GTC AT-3′); GT4 (5′-GTT AGC TCA GGC CAA CAA GCC-3′); and GT5 (5´-CAG GCA TTC CTG CTG GAC TAG-3′). Combination of GT2 and GT5 primers generates two products of amplification: a) one amplicon of 400 bp, for the floxed allele; and b) one amplicon of 341 bp, in case the allele is not floxed. On the one hand, the combination of GT4 and GT5 primers generates an amplicon of 1916 bp, when the genome is not excised by the action of Cre recombinase; however, when the Cre recombinase cuts the genome at the loxP sites of the Grk2 allele, this results in an amplicon of 350 bp. The strategy for generation and genotyping of the G-GRKO mouse line is shown in Suppl. Fig. S1A. Effective recombination and Grk2 exon excision was demonstrated by PCR in POA (see amplicon marked by an arrow), where the majority of GnRH neurons are located, while in other tissues devoid of GnRH expression, e.g., liver and ear, no evidence of Cre-mediated recombination was obtained.
2.4. Experimental designs
As general procedure, assignment of animals to the different experimental groups was done by stratified randomization (within each genotype); body weight was used as major co-variate. Due to operational reasons, groups were not blinded to the experimenters. For studies involving intervention, application of intervention/treatments was conducted in blocked random order.
In Experiment 1, expression of GRK2, at the mRNA and protein levels, was analyzed during postnatal development, in the whole hypothalamus, or in preoptic (POA) and medio-basal (MBH) hypothalamic fragments. Age-points for analyses were: neonatal (postnatal day 1, PND1), infantile (PND8), juvenile (PND21), late juvenile/early pubertal (PND28), and peripubertal (PND36). An additional group of adult female rats at diestrus was used for reference. The youngest group (neonatal or infantile, depending on the analysis) was taken as reference (100%), and other values were normalized accordingly.
In addition, the relevance of GRK2 to modulate responsiveness to major central regulators of the gonadotropic axis, acting via GPCRs, were evaluated in pubertal female rats. Thus, in Experiment 2, female rats at PND28 were icv injected twice with vehicle (VEH) or the GRK2-inhibitor, βARK1-I (6.53 nmol per injection), 13-h and 1-h before of central icv injection of a bolus of Kp-10 (1 nmol). To allow delivery of the compounds into the lateral cerebral ventricle, animals were implanted with icv cannulae lowered to a depth of 3 mm beneath the surface of the skull; the insert point was placed 1 mm posterior and 1.2 mm lateral to bregma. Hormonal tests were conducted at least 1–2 days after cannula implantation, when animals were fully recovered. Blood samples were obtained before (0 min) and at 15, and 60 min after icv injection of Kp-10. A similar approach was used in Experiment 3 to test the gonadotropin-releasing effects of the agonist of NK3R, Senktide. A bolus of 600 pmol Senktide was icv injected after twice injection of βARK1-I, and blood samples were obtained at 0-, 15 and 60-min for LH and FSH determinations.
In Experiment 4, the role of GRK2 in the regulation of Gpr54-mediated signaling was evaluated in vitro by measuring intracellular calcium responses to kisspeptin administration in the presence of βARK1-I or vehicle in a HEK293T cell line, stably transfected with a vector including the coding sequence of the human kisspeptin receptor, termed HEK293T-GPR54. Cells were exposed to an effective concentration of the GRK2 inhibitor, βARK1-I (150 μM), or vehicle, and Ca2+ mobilization responses to an effective dose of Kp-10 (10 nM) were analyzed, as described in the section Calcium Mobilization Assay below.
In Experiment 5, we evaluated the impact of chronic blockade of central GRK2 signaling on the onset of puberty in female rats. Rats were icv injected with the GRK2 inhibitor (βARK1-I; dose: 6.53 nmol) or vehicle, every 12 h, from PND25 to PND34. Similar to Experiments 2–3, the animals were implanted with icv cannulae to allow delivery of the antagonist to the lateral ventricle. During treatment, food intake (FI), body weight (BW), vaginal opening (VO) and first estrus (FE) were daily monitored. Animals were euthanized at PND34, when trunk blood, uterus and ovaries were collected, as described elsewhere [38].
Generation of a model of conditional ablation of GRK2 from GnRH neurons allowed us to conduct acute pharmacological tests, similar to those of Experiment 2, in G-GRKO mice. Thus, acute responses to a single bolus of Kp-10 (50 pmol; icv) were evaluated in Experiment 6. Mice of the two genotypes (G-GRKO and control) were used. To allow repeated sampling, adult male mice were used for this proof-of-concept study, hence avoiding limitations related with the scarcity of blood volume (adult) and the estrus cyclicity (males). To allow delivery of Kp-10 into the lateral cerebral ventricle, animals were implanted with icv cannulae lowered to a depth of 2 mm beneath the surface of the skull; the insert point was placed 1 mm posterior and 1.2 mm lateral to bregma. Hormonal tests were conducted at least 1–2 days after implantation of cannulae, when animals were fully recovered. Blood samples were obtained before (0 min) and at 15- and 60-min after icv injection of Kp-10.
Pubertal progression was monitored in G-GRKO mice and their respective controls, by recording phenotypic markers of puberty, similar to those described in Experiment 5. Thus, in Experiment 7, body weight, food intake, VO and FE were analyzed in control and KO female mice between PND24 and PND43. In addition, uterus and ovarian weights were recorded, and ovarian sections were analyzed in both genotypes at the termination of the experiments (PND43). Based on results from this experiment, in a parallel study (Experiment 8), somatic (body weight, food intake) and reproductive indices [preputial separation, as external sign of puberty, and testosterone (T) levels] were assessed in pubertal G-GRKO male mice and their age-paired controls.
Hypothalamic expression analyses of GRK2 were conducted in two rat models of delayed female puberty due to subnutrition, included in Experiment 9. First, a model of postnatal underfeeding during lactation was used. To this end, rats were reared in large litters (LL: 20 pups/litter), as model of delayed puberty [38,39]. Rats from normal-size litters (NL, 12 pups/litter) served as controls. After weaning, rats were fed ad libitum. Subsets of rats were euthanized at PND5, 15 and 35, for analysis of hypothalamic expression of GRK2. From PND25, body weight and VO were daily monitored. In all the groups, blood and hypothalamic samples were obtained at the end of the experiment, for LH measurements and GRK2 protein analyses, respectively. In addition, a model of chronic undernutrition (UN) during juvenile period was studied. A 30% reduction of daily food intake was applied from PND23 to PND37, as established model of delayed puberty [40]. Age-paired female rats fed ad libitum (NN) served as controls. Body weight and VO were daily monitored. Rats were euthanized on PND37, when blood and hypothalamic samples were obtained.
The contribution of central GRK2 signaling to the delay of puberty caused by conditions of negative energy balance was explored in Experiment 10, using a pharmacological approach in LL rats, as model of early undernutrition and delayed puberty. LL rats were icv injected with the GRK2 inhibitor (βARK1-I; dose: 6.53 nmol) or vehicle, every 12 h, from PND25 to PND37. Body weight, food intake, VO and FE were daily monitored. Histological scoring of follicular maturation and ovulation was also conducted. Animals were euthanized at PND37, when trunk blood, uterus, and ovaries were collected.
Similarly, in Experiment 11, we evaluated the impact of chronic subnutrition on pubertal progression in female G-GRKO mice. Body weight, food intake, VO and FE were daily monitored, from PND 23 to PND 50, in control female mice fed ad libitum, and in control and G-GRKO mice subjected to 20% caloric restriction from PND 22 onwards. Tail blood samples were obtained at alternative days for mean LH measurements in the experimental group during the whole study-period. Animals were euthanized at PND50, when trunk blood, uterus, and ovaries were collected, as described in Experiment 10.
2.5. Quantitative real-time PCR
Standard procedures for RNA isolation, retro-transcription and quantitative real-time PCR are described in detail in Supplemental Annex-I.
2.6. Protein analysis by Western blot
Total protein was extracted from hypothalamic blocks or specific hypothalamic areas (POA and MBH), following previously described procedures [38]. Standard procedures for Western blot analyses are described in detail in Supplemental Annex-I.
2.7. Hormone measurements by radioimmunoassay
Standard procedures for measurement of gonadotropins and testosterone, using specific radioimmuno-assays are described in detail in Supplemental Annex-I.
2.8. Analysis of LH pulsatility and LH determinations by ultrasensitive ELISA
LH secretory profiles were evaluated in young adult female control and G-GRKO mice, either fed ad libitum or subjected to chronic 25% subnutrition (UN), following validated protocols for LH pulsatility analysis [41,42], with minor modifications [43]. In brief, mice were habituated during three weeks prior to bleeding. Pulsatility test consisted in the extraction of either 4 μL (ad lib feeding) or 7 μL (undernutrition) of blood from the tail of the animals, every 5-min during a period of 3 h. Blood samples were collected in 46 or 43 μL of PBS-Tween (PBS 1× with 0.05% of Tween20) and were frozen immediately over dry ice.
LH determinations were conducted using a super-sensitive sandwich ELISA, as described in detail elsewhere [41]. In brief, 96-well high-affinity binding microplates (Corning) were coated with 50 μl per well of the capture antibody (monoclonal antibody 518B7, bovine LHβ, from Lillian E Sibley, UC Davis), diluted 1:1000 in PBS (pH 7.4), and incubated overnight at 4 °C in a humidity chamber. Thereafter, each well was incubated 2 h at room temperature with 200 μL of blocking buffer (5% milk powder in PBS-T). After standard curve preparation (1:2 serial dilutions in PBS-T of rLH-RP3 from Dr. A.F. Parlow, National Hormone and Pituitary Program, CA, USA) and washing steps, 50 μL of standards and samples were loaded into the plate and were incubated 2 h at room temperature (RT) with constant shaking. Standard curves were generated with the reference preparation, rLH-RP3 (supplied also by Dr. Parlow), used at 2-fold serial dilutions, from 1 ng/ml to 0.0078125 ng/ml in PBS-Tween. Then, 50 μl aliquots of the diluted blood samples or the each of the points of the standard curve were incubated for 2 h at room temperature. Thereafter, 50 μl of the detection, polyclonal antibody, AFP240580Rb (provided also by the National Hormone and Pituitary Program), diluted 1:1000 in PBS, were added to each well. After 1.5 h and washing steps, the secondary antibody (P-0048 Dako Cytomation polyclonal goat anti-rabbit IgG/HRP; 1:1000 diluted in 50% PBS 1× and 50% blocking buffer) was added and incubated during 1.5 h at room temperature and agitation. Finally, aliquots of 100 μl of o-Phenylenediamine (OPD; Sigma-Aldrich Corp., St. Louis, MO) were diluted in citrate buffer with 0.1% of H2O2, added to the plate after washing steps and incubated during 30 min at room temperature. Reactions were stopped by adding 50 μL of HCl 3 M. Absorbance was read at 490 nm and at 665 nm (for subtraction of the background) in a Bio-Rad iMARKTM microplate reader. LH concentrations were calculated by interpolating the OD values of unknown samples against a nonlinear regression of the LH standard curve. The assay sensitivity was 0.002 ng/ml, with intra- and inter-assay coefficients of variations of 5.8% and 10.2%.
For pulsatile analyses, a LH pulse was identified as any increase in hormone concentrations of at least 125% from the baseline nadir to peak. The basal level was determined as the mean of the 5 lowest values recorded in 3 h. This criterion is adapted from recent methodological guidelines, based on analysis of correlation between synchronized episodes of calcium activity in ARC Kiss1 neurons and LH secretory pulses, which defined this threshold [42].
2.9. Calcium mobilization assay
Standard procedures for calcium assays are described in detail in Supplemental Annex-I.
2.10. Ovarian histological analysis for precise assessment of the completion of puberty
Pubertal progression was estimated using a scoring method (Pub-Score), recently validated by one of our groups, based on ovarian histometric analyses of follicular development and development of corporal lutea [44]. For further details of standard procedures for ovarian histological analyses and pubertal scoring, please see Supplemental Annex-I.
2.11. Analysis of Grk2 and Gnrh mRNA co-localization in GnRH neurons
RNAscope® Multiplex v.2 in situ hybridization (ISH) was employed for co-detection of Grk2 and Gnrh mRNAs in GnRH neurons from preoptic area of the hypothalamus. Brains from female control and G-GRKO mice (n = 4/group) were excised immediately after euthanasia, frozen and stored at −80C. By using a freezing microtome (Leica CM1850UV), brain coronal sections of 20 μm were cut and distributed into four sets of slides containing the main hypothalamic areas were GnRH neurons are located, i.e., medial septal nucleus and medial preoptic area. Sets of brain sections were fixed for 30 min in paraformaldehyde (PFA) 4% prior to dehydration. Four representative brain sections per group were selected for RNAscope assay, together with two sections for positive/negative controls. RNAscope Multiplex v.2 protocol was followed, as recommended by the manufacturer. Briefly, cells from brain sections were permeabilized and pre-treated to unmask the target RNAs. Next, RNAscope probes were incubated for 2 h to hybridize with the corresponding target mRNA. For Gnrh mRNA hybridization, the predesigned RNAscope® Probe, Mm-Gnrh1-O1, was employed. Custom RNAscope® Probe- Mm-GRK2-C2 was designed to target mRNA nucleotides corresponding to the loxP-flanked Grk2 sequence, ablated by the Cre recombinase in the G-GRKO mice. After washing, RNAscope reagents amplify the hybridization signals by consecutive amplifiers incubations. For detection of mRNA probes, opal dye fluorophores were added. Opal dye 480 (1:350) and 570 (1:500) were employed for Gnrh and Grk2 visualization, respectively. Sections were cover slipped with Fluoroshield™ with Dapi histology mounting medium (F6057, Sigma). ISH signals were visualized on a Zeiss LSM confocal microscopy to assess co-localization, using 63× lens. Confocal images were taken and Z-stacks were condensed to maximum intensity. For quantification of Gnrh and Grk2 co-expression, the green signal detected from GnRH probe hybridization was used to delineate as the region of interest (ROI), denoting a GnRH cell. Inside ROI, the numbers of Grk2 spots were counted as co-localization events per GnRH neuron. The operator performing the raw counting was blinded to the genotype. The total number of neurons counted were (n = 11) in controls and (n = 23) in G-GRKO mice, by using the 3D object counter plug-in from ImageJ application. In addition to absolute numbers, the percentage of reduction in co-localization events in G-GRKO neurons was calculated, as quotient of (number of co-localization events in control minus number of events in G-GRKO neurons) divided by the total number of co-localization events in control neurons x 100.
2.12. Presentation of data and statistics
Data of BW, absolute organ weights, hormonal levels, and protein quantification values are expressed as the mean ± SEM. Results were analyzed using Student t-test or ANOVA followed by Student–Newman–Keuls multiple range tests (Prism GraphPad 5.0 software; GraphPad Software, Inc.). Outlier tests were conducted in the datasets using Prism GraphPad 5.0 software. Significance level was set at P ≤ 0.05, and pads or asterisks indicate statistical significance. Group sizes are indicated in the figure legends for each experiment. As general rule, the sample sizes were selected based on our previous experience with studies addressing neuroendocrine regulation of puberty in rodent models, assisted by power analyses performed using values of standard deviation that we usually obtain when measuring parameters analogous to those examined in this study, as analyses using these sample size should provide at least 80% power to detect effect sizes using the tests indicated above, with a significance level of 0.05. Note, however, that according to standard procedures, more complex molecular and histological analyses were implemented in a representative subset of randomly assigned samples from each group. Further details are provided in the Method section and the corresponding figure legends. As general rule, the investigators directly performing the experimentation involving physiological/molecular determinations were not blinded to the group allocation, but primary data analyses conducted by senior authors were conducted independently to avoid any potential bias.
2.13. Data availability
The authors declare that the data supporting the findings of this study are included in this published article and its supplementary information files. All relevant original data are available from the corresponding authors, upon reasonable request.
3. Results
3.1. Hypothalamic expression of GRK2 postnatally and modulation of kisspeptin responsiveness
Expression of GRK2, at the RNA and protein levels, was analyzed in whole hypothalamus of female rats at different stages of postnatal maturation, corresponding to the neonatal, infantile, juvenile, early-pubertal, pubertal and adult periods. Relative Grk2 mRNA levels in the hypothalamus increased steadily during postnatal maturation, with the lowest levels at the neonatal stage and peak levels at the peri-pubertal period (Fig. 1A). Similar profiles were observed for GRK2 protein content, which was low at the infantile age and increased significantly at puberty (Fig. 1B). Similar RNA expression analyses conducted in dissected hypothalamic fragments, containing specifically the rostral/preoptic area (POA) and the mediobasal hypothalamic (MBH) area, revealed grossly similar results. In the POA, peak Grk2 mRNA levels were detected during the late-juvenile to early puberty transition, whereas expression declined thereafter (Fig. 1C). In contrast, Grk2 mRNA levels in the MBH increased sharply between the infantile and the early juvenile period, and remained elevated up to puberty (Fig. 1D).
Fig. 1.
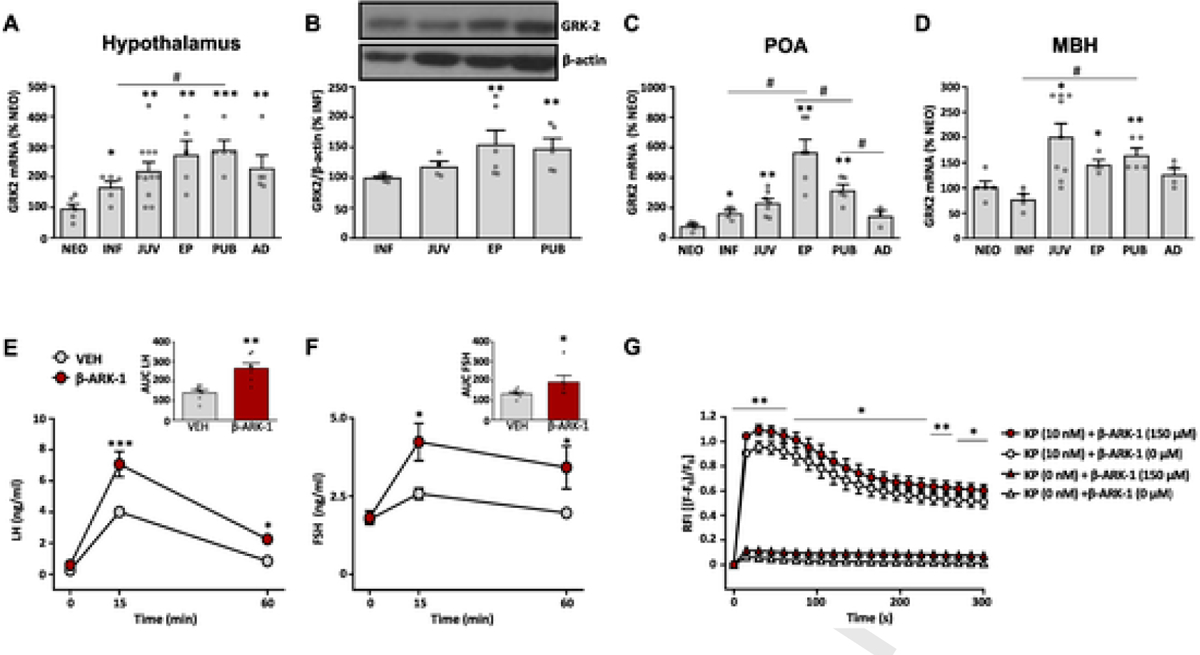
GRK2 expression in rat hypothalamus and GRK2 modulation of kisspeptin signaling. GRK2 mRNA expression (A) and protein content (B) in whole hypothalamic samples from female rats at different stages of postnatal development. Data on GRK2 mRNA expression in the preoptic area (POA; panel C) and the medio-basal hypothalamus (MBH; panel D) of female rats at different stages of postnatal development are also shown. For presentation of data, the level of expression of GRK2 mRNA in neonatal samples (NEO), or protein content in infantile samples (INF), were taken as 100%, and the other values were normalized accordingly. Data represent mean ± SEM. Group sizes are: N = 6 for NEO, INF and EP stage, n = 11 for JUV stage and n = 5 for AD stage in figs. A-D. *P < 0.05, **P < 0.01, ***P < 0.001 vs reference (NEO or INF) values; # P < 0.05, ## P < 0.01 vs the indicated paired groups (denoted by the continuous overline). Statistically significant differences were assessed by ANOVA followed by Student–Newman–Keuls multiple range tests. NEO: neonatal; INF: infantile; JUV: juvenile; EP: early-pubertal; PUB: pubertal; AD: adult. In addition, in the lower panels, functional analyses of the impact of blockade of GRK2 on kisspeptin signaling in vivo are presented. LH (E) and FSH (F) responses to icv administration of Kp-10 in female rats (N = 7/group), pretreated with the GRK2-inhibitor, βARK1-I, or vehicle, are presented. In addition to time-course profiles, integral (AUC) secretory responses are shown in the insets. Finally, in G, we show data on calcium mobilization responses to Kp-10 stimulation in a cell line stably expressing the human kisspeptin receptor (HEK293T-GPR54), pretreated with βARK1-I or vehicle. Data represent mean ± SEM of three independent experiments. Statistical significance of the differences was assessed by ANOVA followed by Student–Newman–Keuls multiple range tests (time-course hormonal data) or paired Student t-test (AUC and in vitro data). *P < 0.05, **P < 0.01, ***P < 0.001 vs. corresponding vehicle-treated groups. RFI = relative fluorescence intensity.
To address the putative relevance of central GRK2 signaling in the modulation of GnRH responses to two major activators of the gonadotropic axis acting via GPCRs, namely, kisspeptins and NKB, a pharmacological approach was used. Early-pubertal female rats were pretreated with a specific GRK2 inhibitor, βARK1-I, followed by intracerebroventricular (icv) injection of effective doses of kisspeptin-10 (Kp-10) or the agonist of NK3R, Senktide. In line with previous literature [31, 45], icv injection of 50 pmol Kp-10 evoked a significant rise of LH, and to a lesser extent, FSH levels, which peaked at 15-min and partially declined by 60-min. Blockade of GRK2 significantly augmented both LH and FSH responses, as documented by the time-course profiles and the integral (AUC) responses to Kp-10, which were increased by approx. 2-fold and 50% in case of LH and FSH, respectively (Fig. 1E–F). In clear contrast, similar tests injecting Senktide revealed that central inhibition of GRK2 failed to increase LH responses to the NK3R agonist. Consistent with previous data [46], no FSH responses to Senktide were detected in pubertal female rats, regardless of the pretreatment with vehicle or the GRK2 inhibitor, βARK1-I (Suppl. Fig. S2). Of note, Gpr54 is prominently expressed in GnRH neurons [12], whereas NK3R is mostly expressed in Kiss1 neurons in the MBH [47]. Thus, these data suggest a predominant regulatory role of GRK2 on Gpr54 in GnRH neurons (rather than on NK3R in Kiss1 cells) in the control of the gonadotropic axis.
The capacity of GRK2 to modulate Gpr54-mediated actions of kisspeptin was further attested in vitro, using a HEK293T cell line, stably expressing the human kisspeptin receptor (HEK293T-GPR54). Cells were exposed to an effective dose of the specific GRK2 inhibitor, βARK1-I (150 μM), or vehicle, in the presence or absence of Kp-10 (10 nM). Stimulation of HEK293T-GPR54 cells with Kp-10 induced a rapid and sustained increase in intracellular calcium as expected; the calcium response to Kp-10 was significantly augmented in the presence of βARK1-I. βARK1-I alone, in the absence of Kp-10, failed to significantly alter the basal pattern of calcium mobilization, indicating that pharmacological blockade of GRK2 alone did not induce intracellular calcium. These results indicate that calcium mobilization responses to Kp-10-mediated activation of Gpr54 are significantly enhanced when GRK2 activity is concurrently inhibited by βARK1-I in HEK293T-GPR54 cells (Fig. 1G).
3.2. Central blockade of GRK2 advances puberty onset in female rats
To address the physiological relevance of central GRK2 signaling in the control of puberty, as a key kisspeptin-driven phenomenon in reproductive maturation [1], prepubertal female rats were injected icv twice daily with an effective dose of the GRK2 inhibitor, βARK1-I, or vehicle, from PND25 onwards. Monitoring of pubertal progression demonstrated that treatment with the GRK2 inhibitor advanced the onset of puberty, as documented by moderately earlier vaginal opening (VO) and first estrus (FE) (Fig. 2A–B). This advancement was associated with an increase in mean LH and FSH levels (Fig. 2C–D), as well as in ovarian and uterus weights (Fig. 2E–F). In line with VO and FE data, histological analyses of the ovaries identified only a marginal advancement of the overall stage of follicular maturation, without changes in the percentage of ovulatory females at the end of the experiment (Fig. 2G–H).
Fig. 2.
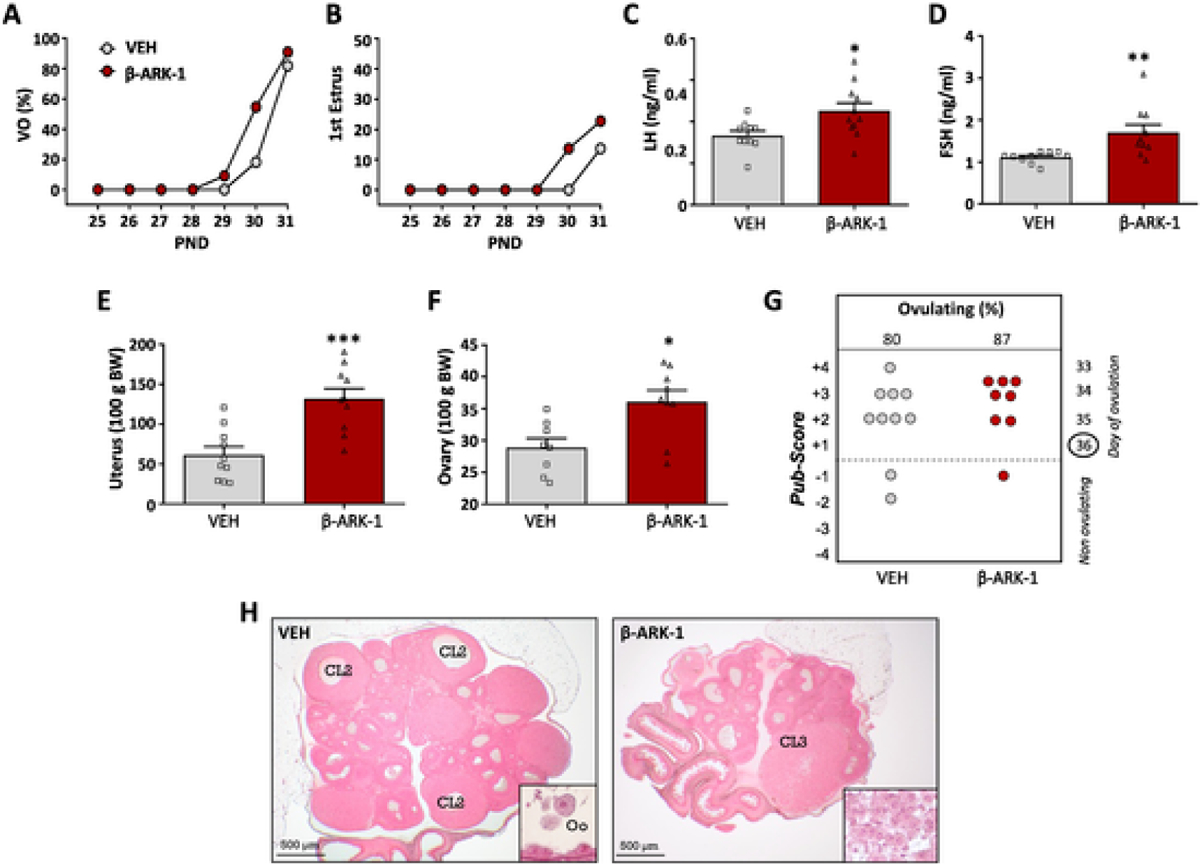
Impact of central blockade of GRK2 on the timing of puberty onset in female rats. Immature female rats were injected icv with the GRK2 inhibitor, βARK1-I (red symbols), every 12 h, from PND25 to PND34; females injected icv with vehicle (VEH; grey symbols) served as controls (N = 11 rats/group). The parameters presented are: cumulative percentage of vaginal opening (VO; panel A) and first estrus (B), LH (C) and FSH (D) levels, as well as uterus (E) and ovary (F) weights. In addition, the histological score of follicular development/ovulation (Pub-score; panel G) and representative images of ovarian maturation (H) are presented from both groups. Data represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the vehicle-treated group (Student t-test). CL: corpus luteum.
3.3. Conditional ablation of GRK2 in GnRH neurons augments Kp-10 responsiveness and advances puberty
Based on our previous data, and in order to address the physiological relevance of GRK2 signaling specifically in GnRH neurons, we generated a conditional mouse line with GnRH-specific ablation of GRK2, using Cre-loxP technology, that we termed G-GRKO, for GnRH-specific GRK2 KO. A well-validated GnRH-Cre mouse line was crossed with homozygous mice harboring modified Grk2 alleles, in which the DNA region coding exons 3 to 6 is flanked by loxP sites. This strategy is depicted in Suppl. Fig. S1A. Validation of this mouse line was conducted by PCR, which documented the presence of Grk2 recombination events, indicating excision of the floxed-region of the Grk2 gene, in the POA, where most GnRH neurons reside, but not in other tissues, such as the liver and ear (Suppl. Fig. S1B). Effective GRK2 ablation was further demonstrated by dual in situ hybridization, using RNAscope™ probes for detection of transcripts of GnRH and Grk2; the latter probe targeting the loxP-flanked region of Grk2 subjected to recombination. These analyses demonstrated abundant co-localization of Grk2 mRNA in GnRH-expressing cells in control mice, whereas in G-GRKO mice, co-expression of Grk2 in GnRH neurons was significantly decreased, with an average number of co-localization events of 18.91 ± 2.19 per GnRH neuron in control mice vs. 6.26 ± 0.55 co-localization events in GnRH neurons from G-GRKO mice (Student t-test; P < 0.0001). This represents a 66.9% suppression of co-expression in our conditional null mouse line. These data confirm that GnRH neurons normally express GKR2, but this is markedly blunted in GnRH cells from G-GRKO animals (Suppl. Fig. S1C).
Functional tests were applied to G-GRKO mice to evaluate changes in GnRH responsiveness to Kp-10 as result of conditional GRK2 ablation, which was assessed by measuring circulating levels of LH and FSH, as surrogate markers of GnRH activation. As shown in Fig. 3A–D, LH and FSH responses to icv injection of Kp-10 were significantly augmented in G-GRKO mice, which displayed also elevated basal LH levels (P = 0.056; Fig. 3A). In addition, pubertal maturation was monitored in G-GRKO mice. Analysis of phenotypic markers of puberty revealed a substantial advancement of pubertal onset, as denoted by earlier VO and FE (Fig. 3E–F), together with a significant increase in the uterus and ovarian weights, as well as a reduction of the expected mean age of the first ovulation (Fig. 3G–I). These trends were fully confirmed by histological analyses of the ovaries of G-GRKO mice, which demonstrated a significant advancement of the state of follicular maturation and an increase in the % of ovulatory animals in G-GRKO mice (Fig. 3J). These phenotypic changes occurred in the absence of detectable changes in body weight or food intake during the pubertal maturation (Suppl. Fig. S3A–B).
Fig. 3.
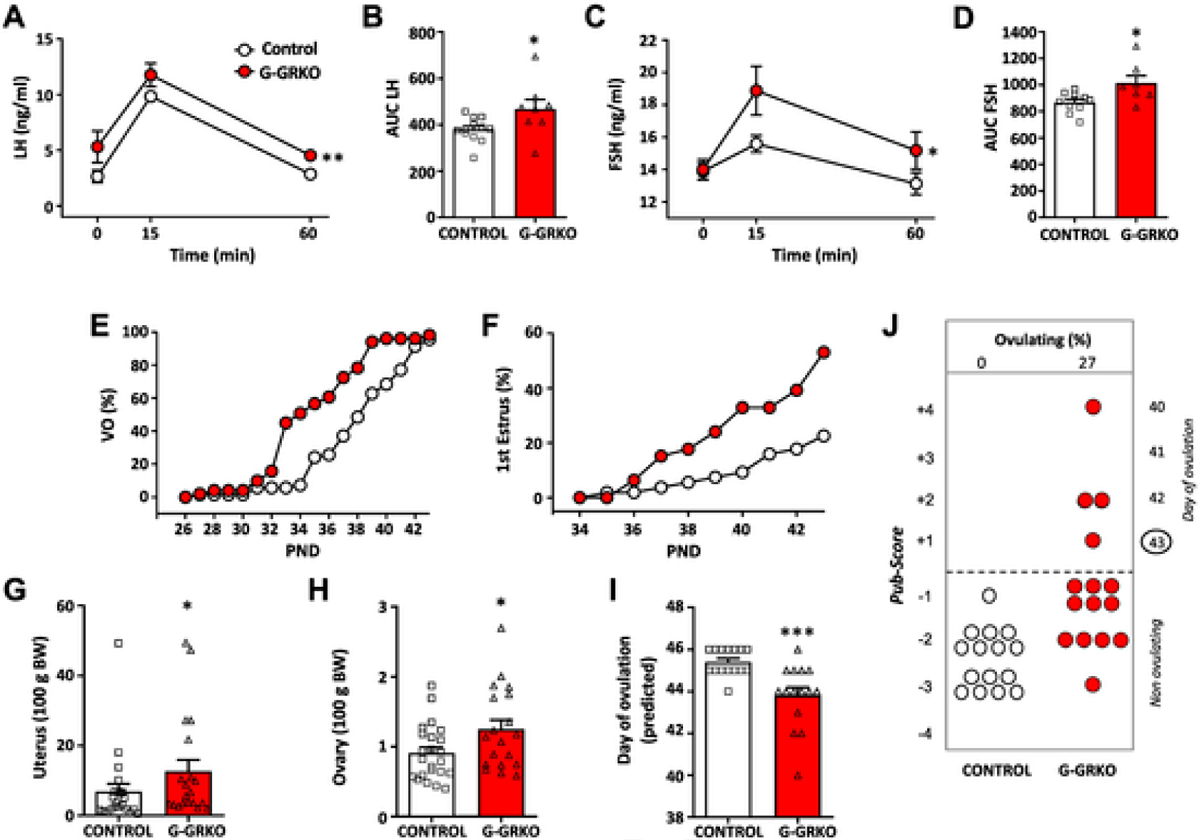
Characterization of mice with conditional ablation of GRK2 in GnRH cells (G-GRKO). In the upper panels, the patterns of LH (A) and FSH (C) responses to Kp-10 stimulation in control and G-GRKO mice are presented. Young adult male mice of both genotypes were injected icv with 50 pmol Kp-10, and hormonal determinations were conducted at 0- (basal) and 15- and 60-min after Kp-10 injection. In addition to time-course profiles, integral (AUC) LH (B) and FSH (D) secretory responses are shown. Data represent mean ± SEM. Group sizes: N = 12 for controls; N = 8 for G-GRKO mice. In addition, phenotypic characterization of puberty onset in control and G-GRKO female mice is presented in the lower panels. The parameters presented are: cumulative percentage of vaginal opening (VO; panel E) and first estrus (F), as well as uterus (G) and ovary (H) weights. In addition, the histological score of follicular development/ovulation (Pub-score; panel J) and the expected mean age of first ovulation, calculated on the basis of the stage follicular maturation (I) are presented from both groups. Data represent mean ± SEM. Group sizes: N = 23 for controls; N = 19 for G-GRKO mice. *P < 0.05, ***P < 0.001 vs. corresponding control group (Student t-test).
For comparative purposes, we also analyzed markers of puberty in male counterparts of both genotypes. In line with the female phenotype, and despite the lack of changes in body weight or food intake during the pubertal transition (Suppl. Fig. S3C–D), the age of preputial separation (as an external sign of puberty) was advanced, and the circulating levels of testosterone were increased in G-GRKO male mice, as compared to control mice (Suppl. Fig. S3E–F).
To explore the basis for such accelerated puberty onset in G-GRKO mice, we assessed the profiles of pulsatile LH secretion in G-GRKO mice, following previous references [41–43]; due to operational reasons, which hamper conduction of proper habituation and repeated blood sampling in pubertal mice, analyses were carried out in young adult females. Ablation of GRK2 in GnRH neurons significantly decreased basal LH levels, with a concomitant reduction of mean, total and integral LH secretion over the 3-h period of analysis (Fig. 4A–D). In contrast, G-GRKO mice displayed a significant increase in LH pulsatility, with double number of pulses (on average) per 3-h than the corresponding controls (Fig. 4E). Representative LH secretory profiles from control and G-GRKO mice are shown in Fig. 4F–G.
Fig. 4.
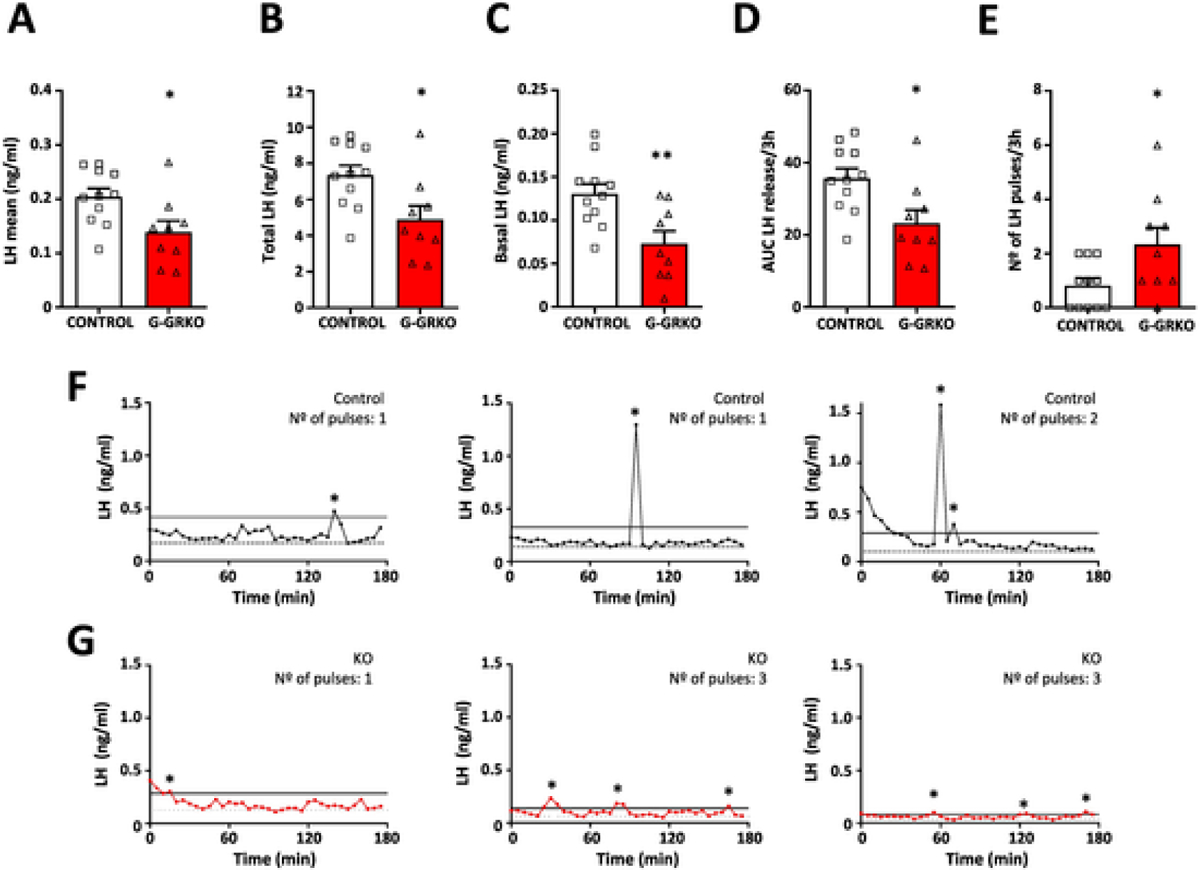
LH pulsatility in female G-GRKO mice, with conditional ablation of GRK2 in GnRH cells. LH secretory profiles were characterized in young adult, control and G-GRKO mice. The following parameters of LH secretion are presented in the upper panels: mean LH (A), total LH (B) and basal LH (C) levels; in addition, the area under the curve (AUC) of LH release (D) and the number of LH pulses (E) during the 3-h study period are shown. In the lower panels, three representative individual LH secretory profiles of control (F) and G-GRKO (G) female mice are displayed. Sample sizes: N = 11 for controls; N = 9 for G-GRKO mice. *p < 0.05, **p < 0.01 vs. corresponding control group (Student t-test for A-D panels; Mann- Whitney U test for E panel). In panels F-G, pulses are denoted by asterisks, dotted lines represent basal LH levels and continuous lines represent the 125% peak threshold level.
3.4. Functional role of GRK2 in pubertal inhibition linked to negative energy balance caused by undernutrition
Conditions of sustained negative energy balance before puberty are known to suppress pubertal progression. To address the potential contribution of central GRK2 signaling to this phenomenon, a combination of expression and functional studies was applied. Hypothalamic expression levels of GRK2 were assayed, at the RNA and protein levels, in two rat models of early-onset undernutrition, namely, postnatal and post-weaning subnutrition. The former was generated by rearing of pups in large litters (20 pups/dam; LL) until weaning, whereas the latter was produced by reducing the daily food ration to 70% of controls fed ad lib (UN). Rats reared in normal litters (NL), and fed ad libitum after weaning (NN), served as respective controls.
In line with previous reports [39], lean LL rats displayed reduced body weight and delayed VO during the peripubertal period, without overt changes in LH levels (Fig. 5A–C). This was associated with significantly increased Grk2 mRNA levels in the hypothalamus across postnatal maturation, as detected at PND5, PND15 and PND35 (Fig. 5D). Likewise, the hypothalamic content of GRK2 protein was enhanced in LL rats at PND35 (Fig. 5E). Similar profiles were observed in the second model of metabolic stress. Rats subjected to 30% UN from PND23 onwards showed lower body weight, together with reduced LH levels and delayed VO during the pubertal transition (Fig. 5F–H). This pubertal delay was associated with a significant increase in Grk2 mRNA levels in the POA, as well as a detectable rise of GRK2 protein content, in the POA and MBH, of UN female rats at PND35.
Fig. 5.
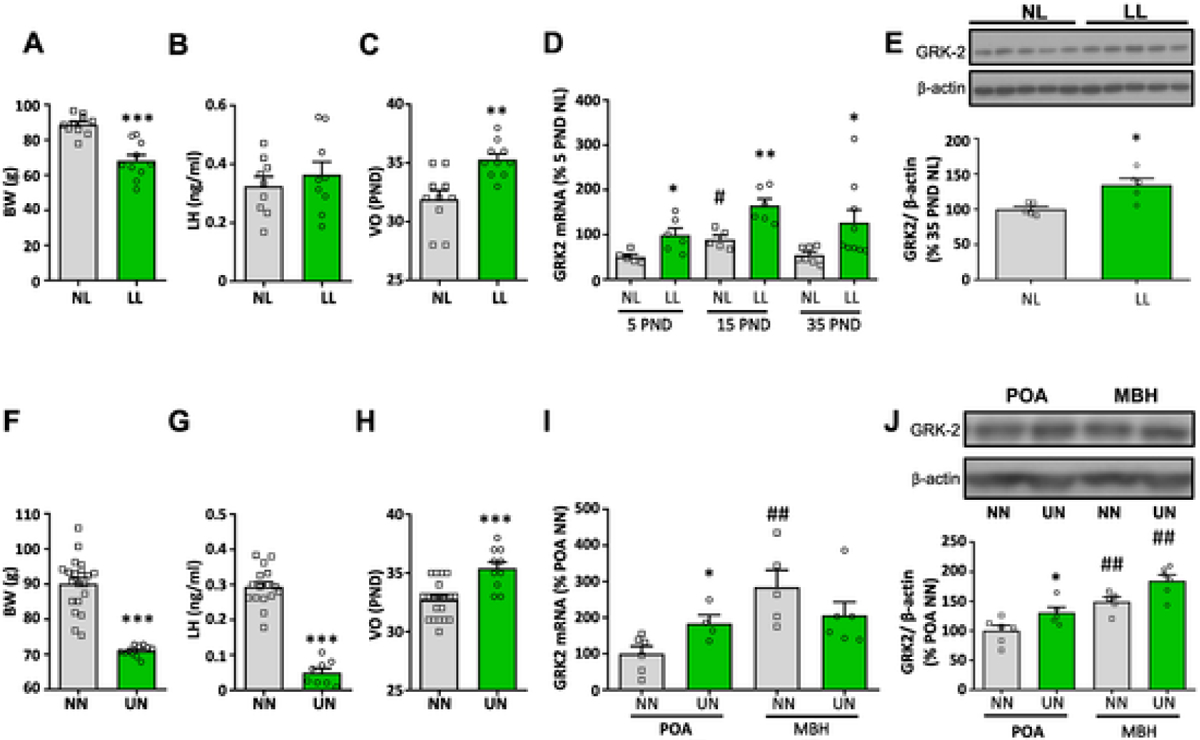
Hypothalamic expression of GRK2 in rat models of postnatal undernutrition and delayed puberty. In the upper panel, data from a model of postnatal underfeeding, generated by rearing female rats in large litters (LL: 20 pups/litter) during lactation, are presented. Female rats from normal-sized litters (NL, 12 pups/litter) served as controls. Body weight (A) and LH levels (B) at PND35, as well as the mean age of vaginal opening (VO; panel C), are shown. In addition, hypothalamic expression of GRK2 mRNA was assayed in NL and LL rats at PND5, 15 and 35 (D), while GRK2 protein content was measured at PND35 (E). In the lower panel, data from a model of chronic undernutrition (UN) of female rats during the juvenile period are presented. A 30% reduction of daily food intake was applied from PND23 to PND37, as established model of delayed puberty. Female rats that were fed ad libitum (NN) served as controls. Body weight (F) and LH levels (G) at PND37, as well as the mean age of vaginal opening (VO; panel H), are presented. In addition, hypothalamic levels of GRK2 mRNA (I) and GRK2 protein content (J) in the POA and MBH of UN and NN rats at PND37, are shown. Data represent mean ± SEM. Group sizes: N = 10 for the NL and the LL group (panels A-E); yet, mRNA expression and protein content analyses were conducted in a subset of representative samples of each group. For panels F-J, N = 20 for the NN group and n = 11 for the UN group, with RNA and protein analyses being conducted in a subset of representative samples. *P < 0.05, **P < 0.01 vs corresponding control (NL or NN) groups. #P < 0.05, ##P < 0.01 vs PND5 NL or POA NN (Student t-test or ANOVA followed by Student–Newman–Keuls multiple range tests).
To ascertain the functional relevance of the observed increase of GRK2 expression in the hypothalamus of peripubertal females rats subjected to undernutrition, which displayed also delayed puberty, functional studies involving chronic administration of the GRK2 inhibitor, βARK1-I, were applied, using a protocol similar to that conducted in pubertal females fed ad lib. As expected, LL female rats showed reduced body weight and food intake (Fig. 6A–B), together with a substantial delay in the age of VO and FE, as well as a decrease in ovarian and uterus weights, as compared to NN rats (shown in Fig. 6 as grey symbols, for reference purposes). Central blockade of GRK2 partially restored the delayed pubertal onset, in terms of VO and FE, observed in LL rats, causing an intermediate phenotype between NN and LL rats treated with vehicle (Fig. 6C–D). In addition, chronic administration of βARK1-I to lean LL rats significantly increased ovarian and uterus weights, which were fully normalized to control values (Fig. 6E–F). Likewise, histological analyses of follicular maturation revealed that the delay caused by LL was restored by repeated icv treatment with the GRK2 inhibitor (Fig. 6I–J). No changes in LH or FSH levels were detected across the experimental groups (Fig. 6G–H).
Fig. 6.
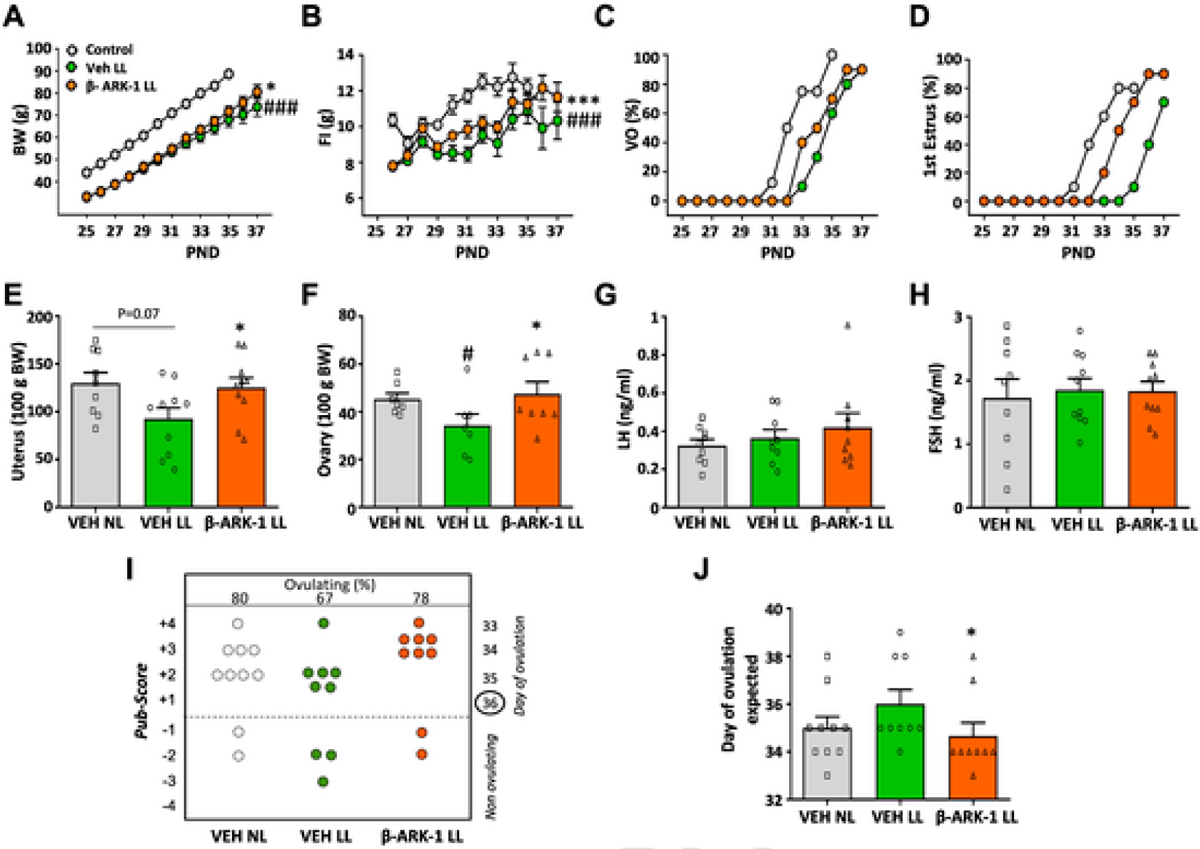
Central blockade of GRK2 partially recues pubertal delay caused by postnatal undernutrition in female rats. Female rats reared in large litters (LL) were icv injected with GRK2 inhibitor, βARK1-I, or vehicle every 12 h, from PND25 to PND37. Reference values from control (NL) females icv injected with vehicle (VEH) served as controls; these are shown as grey symbols. The parameters presented are: body weight (A), daily food intake (FI; panel B), cumulative percentage of vaginal opening (VO; panel C) and first estrus (D). In addition, uterus (E) and ovary (F) weights, as well as LH (G) and FSH (H) levels are shown, together with the histological score of follicular development/ovulation (Pub-score; panel I), and the mean age of expected first ovulation, calculated on the basis of the stage follicular maturation (J). Data represent mean ± SEM (N = 10/group). *P < 0.05 vs. the LL group treated with vehicle (ANOVA followed by Student–Newman–Keuls multiple range tests). Data represent mean ± SEM. *P < 0.05, ***P < 0.001 vs VEH LL. #P < 0.05, ##P < 0.01, ###P < 0.001 vs VEH NL.
Finally, to further address the putative contribution of GRK2 in GnRH neurons to the pubertal delay observed in conditions of subnutrition, a protocol of chronic food restriction from weaning onwards was applied to control and G-GRKO female mice, as described elsewhere [48]. Chronic UN, applied from PND23 to PND49, caused a substantial drop in body weight gain, together with lower daily food intake, as expected due to restricted food availability, which was 80% of the ad lib daily ration (Fig. 7A–B). In line with previous data [48], UN caused a delay in pubertal onset, evidenced by deferred progression and mean age of VO (Fig. 7C–D), together with delayed FE (Fig. 7E), and reduced LH levels, and uterus (P = 0.07) and ovarian weights (Fig. 7F–H). Ablation of GRK2 in GnRH cells partially rescued this pubertal delay, as denoted by partial normalization of VO progression and complete rescue of the mean age of VO, as well as uterus and ovarian weights. In addition, FE progression was moderately advanced, while LH levels remained suppressed in G-GRKO female mice subjected to UN (Fig. 7C–H). To get insight into the underlying hormonal basis for this partial rescue, pulsatile LH secretion was assessed in G-GRKO female mice subjected to UN; again, due to operational reasons, this was conducted in young adult females. As shown in Fig. 7I–L, 25% UN caused a reduction in mean, basal and integral LH secretion, as well as a suppression of the number of LH pulses, over a 3-h period in control mice. In contrast, G-GRKO female mice subjected to a similar regimen of UN displayed higher number of LH pulses than the corresponding UN controls; their pulse frequency being similar to that of controls fed ad libitum. Notwithstanding, mean, basal and integral LH secretion over the 3-h period was equally suppressed by UN in G-GRKO and control mice (Fig. 7I–L).
Fig. 7.
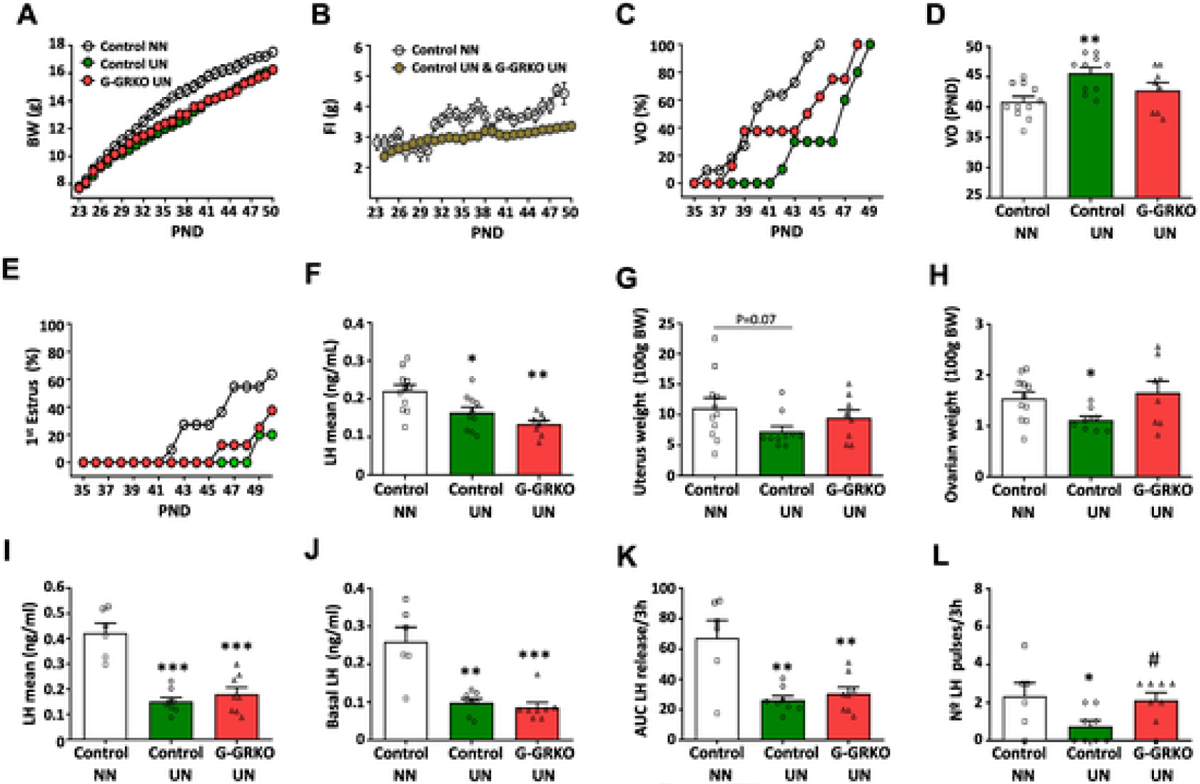
GRK2 ablation in GnRH cells partially prevents the impact of undernutrition on puberty onset and LH pulsatility. Body weight (A), daily food intake (B), cumulative percentage of vaginal opening (VO) (C), mean age of VO (D), and cumulative percentage of first estrus (E) in control and G-GRKO female mice subjected to chronic undernutrition (UN) from weaning onwards. Pair-aged, control female mice fed ad libitum (NN) served as controls. Mean LH levels (F), as well as uterus (G) and ovarian (H) weights in pubertal control and G-GRKO mice are also presented. In addition, features of LH secretory profiles of young adult female control and G-GRKO mice subjected to UN are shown; the parameters presented are: mean LH (I) and basal LH (J) levels, as well as the area under the curve (AUC) of LH release (K) and the number of LH pulses (L) during the 3-h study period. Data represent mean ± SEM. Group sizes: N = 11 for NN control group; N = 10 for UN control group; and N = 8 for UN G-GRKO group (panels A-H). In panels I-L, N = 6 for NN control group; N = 9 for UN control group; and N = 8 for UN G-GRKO group. *P < 0.05, **P < 0.01, ***p < 0.001 vs control NN group; #P < 0.05 vs control UN group.
4. Discussion
Awakening of the reproductive axis at puberty is the result of the delicate interplay of numerous signals and regulatory mechanisms, which permit the timely heightening of GnRH secretion, ultimately leading to complete gonadal and sexual maturation [1,2,12,13]. Kisspeptins, acting via Gpr54, have emerged in recent years as master regulators of GnRH neurons, with an indispensable role in the amplification of GnRH neurosecretion that is mandatory for puberty to occur [13,49]. In fact, compelling evidence has documented a sophisticated developmental program, affecting Kiss1 neurons and kisspeptin actions during pubertal maturation, which includes not only an increase in Kiss1 expression and kisspeptin content in the hypothalamus during the pubertal transition, but also plastic changes in Kiss1 neuronal populations and their projections to GnRH neurons, which substantially increase at puberty [1,13,50]. Characterization of these presynaptic components of kisspeptin signaling, and their changes during puberty onset, has drawn substantial attention in recent years and has dominated the search for the mechanisms controlling pubertal timing, and its modulation by metabolic and nutritional factors. This is epitomized by our recent studies addressing the roles of cellular energy sensors in the transcriptional control of Kiss1 in conditions of metabolic stress, such as subnutrition and obesity, bound to altered puberty [38,48]. However, initial evidence suggested that during the pubertal transition, postsynaptic changes affecting kisspeptin signaling are likely to occur also, leading to an increase in kisspeptin responsiveness [51,52], whose basis remained totally unexplored. Our current dataset discloses a novel role of GRK2 in GnRH neurons, as a key component of the kisspeptin signaling-regulatory machinery, with a physiological function in the control of puberty. GRK2 would operate as repressor signal and counterbalance for excessive activation of GnRH neurons by kisspeptins, therefore aiding to keep puberty at check and preventing precocious pubertal maturation. Considering that powerful stimulatory actions of kisspeptins on the gonadotropin axis manifest already during the infantile/juvenile period [51,52], such inhibitory mechanism appears crucial to ensure the proper timing of puberty onset.
Among the different GPCR kinases, GRK2 is the most extensively studied, with proven patho-physiological actions in multiple bodily functions, including prominently the cardiovascular and metabolic systems [28,53]. However, the putative link of GRK2 with the central mechanisms governing puberty and reproduction remained virtually unexplored, except for very few biochemical studies addressing the role of GRK2 in the modulation of GnRH receptor in vitro [54,55], which is expressed at the pituitary to elicit gonadotropin secretion. In addition, a single report had documented, using an heterologous (HEK293) cellular system, that GRK2 can interact with Gpr54 and may contribute to mediating kisspeptin-induced desensitization of the kisspeptin receptor in vitro [30]. While that biochemical study set the grounds of our current analyses, it did not provide evidence for the potential pathophysiological relevance of GRK2 in the control of kisspeptin signaling, or its putative role in the modulation of puberty. Interestingly, over-expression of GRK2 in HEK293T cells suppressed Gpr54 signaling, as measured by inositol-phosphate (IP) formation, in basal and kisspeptin-stimulated conditions, but did not affect Gpr54 surface expression or receptor internalization [30], which are hallmarks of desensitization. These data would suggest alternative modes of action of GRK2 in the tuning of Gpr54 signaling, in line with recent mounting evidence demonstrating that multiple actions of GRK2 are actually conducted independently of its GPCR-desensitization effects [28,29]. In good agreement, our current results demonstrate that either pharmacological blockade of central GRK2 or selective ablation of GRK2 from GnRH neurons result in a clear augmentation of acute responses to kisspeptin, suggesting that GRK2 conducts a repressive action on kisspeptin signaling in basal conditions, which contributes to shape the activation of GnRH secretion and, hence, the timing of puberty.
While pharmacological tests involving central administration of the GRK2 inhibitor, βARK1-I, already suggested a role of GRK2 in controlling kisspeptin signaling and puberty onset, conclusive demonstration of the primary site and physiological relevance of such GRK2 regulatory system came from our studies in the G-GRKO mouse. A combination of conditional ablation of GRK2 in GnRH-expressing cells in vivo and testing of the releasing effects of kisspeptin on LH secretion, as a surrogate marker of GnRH, demonstrated that GRK2 in GnRH neurons operates as a repressive signal that restrains the stimulatory actions of kisspeptins in this cell population. Reducing this restraint, either via genetic or pharmacological means, results in acceleration of puberty, which is especially evident when GRK2 inactivation is selectively applied in GnRH neurons. Notably, pubertal G-GRKO mice displayed elevation of LH levels in single point measurements, together with advanced puberty onset and first ovulation in females. A similar acceleration of puberty seems to take place also in males, where external signs of puberty were advanced and testosterone levels were elevated in pubertal mice. Altogether, this evidence attests a sustained inhibitory action of GRK2 in the control of GnRH neurons during the pubertal transition, at least partially via repression of kisspeptin signaling. The relevance of this repressive pathway in the control of adult reproductive axis is yet to be fully defined, although our unpublished pharmacological data strongly suggest that GnRH secretion is also restrained by GRK2 in adulthood (Avendaño, Perdices-Lopez & Tena-Sempere, unpublished). In addition, our current LH pulsatility analyses in G-GRKO mice, which were conducted in young adult mice for operational reasons, further support a sustained role of GRK2 in the regulation of adult gonadotropic axis, as denoted by changes in LH pulsatility after congenital ablation of GRK2 in GnRH neurons. Intriguingly, such elimination resulted in diminished basal and total levels but significantly increased LH pulsatility, therefore pointing out that elimination of the restrain imposed by GRK2 in GnRH neurons contribute to the physiological shaping of LH pulse frequency, as major determinant for appropriate reproductive function [56]. In this context, the putative role of GRK2 signaling in GnRH neurons in the pathogenesis of reproductive diseases involving GnRH/LH hyper-secretion, such as the polycystic ovary syndrome, one of whose hallmarks is actually increased LH pulsatility [57], is currently under investigation in our laboratory. From a mechanistic standpoint, it is tenable that this elevation of LH pulsatility is a major driving force for the accelerated puberty detected in G-GRKO female mice.
Our data not only suggest that GRK2 plays a role in the physiological control of puberty, but also identify GRK2 as a novel regulatory node for the metabolic modulation of pubertal maturation. Indeed, conditions of early-onset negative energy balance, such as postnatal undernutrition during lactation and after weaning, which cause a substantial delay of puberty onset [39,48,58], were associated with a significant increase in GRK2 expression, at mRNA and protein levels, in the hypothalamus, including the POA, where most GnRH neurons reside. Moreover, central pharmacological blockade of GRK2 partially rescued the pubertal delay imposed by early undernutrition, therefore attesting to the relevance of enhanced GRK2 signaling in the suppression of puberty caused by conditions of negative energy balance. In good agreement, mice congenitally devoid of GRK2 in GnRH neurons were partially protected from the deleterious impact of chronic subnutrition on pubertal onset, as denoted by normalization of several phenotypic and hormonal markers of pubertal initiation. Interestingly, young adult females with congenital elimination of GRK2 from GnRH neurons maintained higher LH pulsatility when subjected to subnutrition, when compared to underfed controls, although they did not display rescued LH secretory mass. These observations further attest to the key role of GRK2 in GnRH neurons in modulating LH pulsatility and strongly suggest that preserved LH pulsatility is seemingly a major contributor for preservation of pubertal progression in G-GRKO mice despite undernutrition.
Recent data from our laboratory have documented that a key component of the pubertal suppression caused by early subnutrition is the transcriptional repression of Kiss1 expression, mediated via activation of AMPK and SIRT1 in Kiss1 neurons, that results in the inhibition of the major stimulatory drive (kisspeptins) on GnRH neurons [38,48]. Our present findings disclose another, previously unnoticed regulatory layer, operating within GnRH neurons, whereby kisspeptin effects would be physiologically restrained and further inhibited in conditions of energy deficit that cause pubertal delay. These data are compatible with a distinct role of other nutrient-sensing mechanisms in GnRH neurons (e.g., AMPK), as putative transducers for at least part of the control of puberty and the reproductive axis by metabolic cues [43]. While the ubiquitous nature of GRK2 and its capacity to modulate different GPCR makes feasible that part of its suppressive effects on puberty may be Gpr54-independent, our current findings, including those in the mouse model of conditional ablation of GRK2, strongly argue in favor of a role of this kinase in the modulation of kisspeptin signaling in GnRH neurons. Considering that GRK2 has been proposed as a potential target for the treatment of various pathological conditions, our present data have translational potential, as they open up the possibility for the design of novel pharmacological strategies, targeting GRK2, for the management of pubertal disorders, and eventually other reproductive alterations, including those linked to metabolic perturbations.
Supplementary Material
Funding information
This work was supported by grants BFU2017-83934-P, PID2020-118660GB-I00 (to MTS) and SAF2017-84125-R (to FM), Agencia Estatal de Investigación, Spain; co-funded with EU funds from FEDER Program; Project PIE-00005 (to MTS) and CB16/11/00278 (to FM) - Instituto de Salud Carlos III, Ministerio de Sanidad, Spain; Project P12-FQM-01943 (to MTS) and Project PI-0370-2016 (to MSA- Junta de Andalucía, Spain); and Project REP-655232 (ReprObesity; to MTS - European Union). CIBER Fisiopatología de la Obesidad y Nutrición and CIBER Enfermedades Cardiovasculares are initiatives of Instituto de Salud Carlos III, Spain.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.metabol.2022.155141.
Declaration of competing interest
The authors have no conflict of interest to disclose in relation to the contents of this work.
References
- [1].Avendano MS, Vazquez MJ, Tena-Sempere M. Disentangling puberty: novel neuroendocrine pathways and mechanisms for the control of mammalian puberty. Hum Reprod Update 2017;23:737–63. [DOI] [PubMed] [Google Scholar]
- [2].Vazquez MJ, Velasco I, Tena-Sempere M. Novel mechanisms for the metabolic control of puberty: implications for pubertal alterations in early-onset obesity and malnutrition. J Endocrinol 2019;242:R51–65. [DOI] [PubMed] [Google Scholar]
- [3].Aksglaede L, Juul A, Olsen LW, Sorensen TI. Age at puberty and the emerging obesity epidemic. PLoS One 2009;4:e8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].De Leonibus C, Marcovecchio ML, Chiavaroli V, de Giorgis T, Chiarelli F, Mohn A. Timing of puberty and physical growth in obese children: a longitudinal study in boys and girls. Pediatr Obes 2014;9:292–9. [DOI] [PubMed] [Google Scholar]
- [5].Lee JM, Appugliese D, Kaciroti N, Corwyn RF, Bradley RH, Lumeng JC. Weight status in young girls and the onset of puberty. Pediatrics 2007;119:e624–30. [DOI] [PubMed] [Google Scholar]
- [6].Aksglaede L, Sorensen K, Petersen JH, Skakkebaek NE, Juul A. Recent decline in age at breast development: the Copenhagen puberty study. Pediatrics 2009;123: e932–9. [DOI] [PubMed] [Google Scholar]
- [7].Herman-Giddens ME, Steffes J, Harris D, Slora E, Hussey M, Dowshen SA, et al. Secondary sexual characteristics in boys: data from the pediatric research in office settings network. Pediatrics 2012;130:e1058–68. [DOI] [PubMed] [Google Scholar]
- [8].Li W, Liu Q, Deng X, Chen Y, Liu S, Story M. Association between obesity and puberty timing: a systematic review and meta-analysis. Int J Environ Res Public Health 2017;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Day FR, Elks CE, Murray A, Ong KK, Perry JR. Puberty timing associated with diabetes, cardiovascular disease and also diverse health outcomes in men and women: the UK Biobank study. Sci Rep 2015;5:11208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Day FR, Thompson DJ, Helgason H, Chasman DI, Finucane H, Sulem P, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet 2017;49:834–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kaya G, Yavas Abali Z, Bas F, Poyrazoglu S, Darendeliler F. Body mass index at the presentation of premature adrenarche is associated with components of metabolic syndrome at puberty. Eur J Pediatr 2018;177:1593–601. [DOI] [PubMed] [Google Scholar]
- [12].Pinilla L, Aguilar E, Dieguez C, Millar RP, Tena-Sempere M. Kisspeptins and reproduction: physiological roles and regulatory mechanisms. Physiol Rev 2012; 92:1235–316. [DOI] [PubMed] [Google Scholar]
- [13].Herbison AE. Control of puberty onset and fertility by gonadotropin-releasing hormone neurons. Nat Rev Endocrinol 2016;12:452–66. [DOI] [PubMed] [Google Scholar]
- [14].Tena-Sempere M, Huhtaniemi I. Gonadotropins and gonadotropin receptors. In: Fauser BCJM, editor. Reproductive medicine - molecular, cellular and genetic fundamentals. New York: Parthenon Publishing; 2003. p. 225–44. [Google Scholar]
- [15].Maeda K, Ohkura S, Uenoyama Y, Wakabayashi Y, Oka Y, Tsukamura H, et al. Neurobiological mechanisms underlying GnRH pulse generation by the hypothalamus. Brain Res 2010;1364:103–15. [DOI] [PubMed] [Google Scholar]
- [16].Ojeda SR, Lomniczi A, Sandau U. Contribution of glial-neuronal interactions to the neuroendocrine control of female puberty. Eur J Neurosci 2010;32:2003–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prevot V, Dehouck B, Sharif A, Ciofi P, Giacobini P, Clasadonte J. The versatile tanycyte: a hypothalamic integrator of reproduction and energy metabolism. Endocr Rev 2018;39:333–68. [DOI] [PubMed] [Google Scholar]
- [18].Knobil E The neuroendocrine control of the menstrual cycle. Recent Prog Horm Res 1980;36:53–88. [DOI] [PubMed] [Google Scholar]
- [19].Navarro VM, Ruiz-Pino F, Sanchez-Garrido MA, Garcia-Galiano D, Hobbs SJ, Manfredi-Lozano M, et al. Role of neurokinin B in the control of female puberty and its modulation by metabolic status. J Neurosci 2012;32:2388–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gill JC, Navarro VM, Kwong C, Noel SD, Martin C, Xu S, et al. Increased neurokinin B (Tac2) expression in the mouse arcuate nucleus is an early marker of pubertal onset with differential sensitivity to sex steroid-negative feedback than Kiss1. Endocrinology 2012;153:4883–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr, Shagoury JK, et al. The GPR54 gene as a regulator of puberty. N Engl J Med 2003;349: 1614–27. [DOI] [PubMed] [Google Scholar]
- [22].de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A 2003;100:10972–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin B in the central control of reproduction. Nat Genet 2009;41: 354–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lehman MN, Coolen LM, Goodman RL. Minireview: kisspeptin/neurokinin B/dynorphin (KNDy) cells of the arcuate nucleus: a central node in the control of gonadotropin-releasing hormone secretion. Endocrinology 2010;151:3479–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gurevich VV, Gurevich EV. GPCR signaling regulation: the role of GRKs and arrestins. Front Pharmacol 2019;10:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Black JB, Premont RT, Daaka Y. Feedback regulation of G protein-coupled receptor signaling by GRKs and arrestins. Semin Cell Dev Biol 2016;50:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Penela P, Murga C, Ribas C, Lafarga V, Mayor F Jr. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol 2010;160:821–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Murga C, Arcones AC, Cruces-Sande M, Briones AM, Salaices M, Mayor F Jr. G protein-coupled receptor kinase 2 (GRK2) as a potential therapeutic target in cardiovascular and metabolic diseases. Front Pharmacol 2019;10:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Evron T, Daigle TL, Caron MG. GRK2: multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol Sci 2012;33:154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pampillo M, Camuso N, Taylor JE, Szereszewski JM, Ahow MR, Zajac M, et al. Regulation of GPR54 signaling by GRK2 and {beta}-arrestin. Mol Endocrinol 2009; 23:2060–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Navarro VM, Castellano JM, Fernandez-Fernandez R, Tovar S, Roa J, Mayen A, et al. Characterization of the potent luteinizing hormone-releasing activity of KiSS-1 peptide, the natural ligand of GPR54. Endocrinology 2005;146:156–63. [DOI] [PubMed] [Google Scholar]
- [32].Leon S, Barroso A, Vazquez MJ, Garcia-Galiano D, Manfredi-Lozano M, Ruiz-Pino F, et al. Direct actions of kisspeptins on GnRH neurons permit attainment of fertility but are insufficient to fully preserve gonadotropic axis activity. Sci Rep 2016;6: 19206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Garcia-Galiano D, van Ingen Schenau D, Leon S, Krajnc-Franken MA, Manfredi-Lozano M, Romero-Ruiz A, et al. Kisspeptin signaling is indispensable for neurokinin B, but not glutamate, stimulation of gonadotropin secretion in mice. Endocrinology 2012;153:316–28. [DOI] [PubMed] [Google Scholar]
- [34].Taguchi K, Matsumoto T, Kamata K, Kobayashi T. Inhibitor of G protein-coupled receptor kinase 2 normalizes vascular endothelial function in type 2 diabetic mice by improving beta-arrestin 2 translocation and ameliorating Akt/eNOS signal dysfunction. Endocrinology 2012;153:2985–96. [DOI] [PubMed] [Google Scholar]
- [35].Won KA, Kim MJ, Yang KY, Park JS, Lee MK, Park MK, et al. The glial-neuronal GRK2 pathway participates in the development of trigeminal neuropathic pain in rats. J Pain 2014;15:250–61. [DOI] [PubMed] [Google Scholar]
- [36].Messina A, Langlet F, Chachlaki K, Roa J, Rasika S, Jouy N, et al. A microRNA switch regulates the rise in hypothalamic GnRH production before puberty. Nat Neurosci 2016;19:835–44. [DOI] [PubMed] [Google Scholar]
- [37].Vila-Bedmar R, Cruces-Sande M, Lucas E, Willemen HL, Heijnen CJ, Kavelaars A, et al. Reversal of diet-induced obesity and insulin resistance by inducible genetic ablation of GRK2. Sci Signal 2015;8:ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vazquez MJ, Toro CA, Castellano JM, Ruiz-Pino F, Roa J, Beiroa D, et al. SIRT1 mediates obesity- and nutrient-dependent perturbation of pubertal timing by epigenetically controlling Kiss1 expression. Nat Commun 2018;9:4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Castellano JM, Bentsen AH, Sanchez-Garrido MA, Ruiz-Pino F, Romero M, Garcia-Galiano D, et al. Early metabolic programming of puberty onset: impact of changes in postnatal feeding and rearing conditions on the timing of puberty and development of the hypothalamic kisspeptin system. Endocrinology 2011;152: 3396–408. [DOI] [PubMed] [Google Scholar]
- [40].Castellano JM, Navarro VM, Fernandez-Fernandez R, Nogueiras R, Tovar S, Roa J, et al. Changes in hypothalamic KiSS-1 system and restoration of pubertal activation of the reproductive axis by kisspeptin in undernutrition. Endocrinology 2005;146:3917–25. [DOI] [PubMed] [Google Scholar]
- [41].Steyn FJ, Wan Y, Clarkson J, Veldhuis JD, Herbison AE, Chen C. Development of a methodology for and assessment of pulsatile luteinizing hormone secretion in juvenile and adult male mice. Endocrinology 2013;154:4939–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].McQuillan HJ, Han SY, Cheong I, Herbison AE. GnRH pulse generator activity across the estrous cycle of female mice. Endocrinology 2019;160:1480–91. [DOI] [PubMed] [Google Scholar]
- [43].Franssen D, Barroso A, Ruiz-Pino F, Vazquez MJ, Garcia-Galiano D, Castellano JM, et al. AMP-activated protein kinase (AMPK) signaling in GnRH neurons links energy status and reproduction. Metabolism 2021;115:154460. [DOI] [PubMed] [Google Scholar]
- [44].Gaytan F, Morales C, Leon S, Heras V, Barroso A, Avendano MS, et al. Development and validation of a method for precise dating of female puberty in laboratory rodents: the puberty ovarian maturation score (Pub-Score). Sci Rep 2017;7:46381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Navarro VM, Castellano JM, Fernandez-Fernandez R, Tovar S, Roa J, Mayen A, et al. Effects of KiSS-1 peptide, the natural ligand of GPR54, on follicle-stimulating hormone secretion in the rat. Endocrinology 2005;146:1689–97. [DOI] [PubMed] [Google Scholar]
- [46].Ruiz-Pino F, Garcia-Galiano D, Manfredi-Lozano M, Leon S, Sanchez-Garrido MA, Roa J, et al. Effects and interactions of tachykinins and dynorphin on FSH and LH secretion in developing and adult rats. Endocrinology 2015;156:576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Navarro VM, Gottsch ML, Chavkin C, Okamura H, Clifton DK, Steiner RA. Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J Neurosci 2009;29: 11859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Roa J, Barroso A, Ruiz-Pino F, Vazquez MJ, Seoane-Collazo P, Martinez-Sanchez N, et al. Metabolic regulation of female puberty via hypothalamic AMPK-kisspeptin signaling. Proc Natl Acad Sci U S A 2018;115:E10758–E67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Clarkson J, Boon WC, Simpson ER, Herbison AE. Postnatal development of an estradiol-kisspeptin positive feedback mechanism implicated in puberty onset. Endocrinology 2009;150:3214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Clarkson J, Herbison AE. Postnatal development of kisspeptin neurons in mouse hypothalamus; sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology 2006;147:5817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Han SK, Gottsch ML, Lee KJ, Popa SM, Smith JT, Jakawich SK, et al. Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci 2005;25:11349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Castellano JM, Navarro VM, Fernandez-Fernandez R, Castano JP, Malagon MM, Aguilar E, et al. Ontogeny and mechanisms of action for the stimulatory effect of kisspeptin on gonadotropin-releasing hormone system of the rat. Mol Cell Endocrinol 2006;257–258:75–83. [DOI] [PubMed] [Google Scholar]
- [53].Cipolletta E, Gambardella J, Fiordelisi A, Del Giudice C, Di Vaia E, Ciccarelli M, et al. Antidiabetic and cardioprotective effects of pharmacological inhibition of GRK2 in db/db mice. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Neill JD, Duck LW, Musgrove LC, Sellers JC. Potential regulatory roles for G protein-coupled receptor kinases and beta-arrestins in gonadotropin-releasing hormone receptor signaling. Endocrinology 1998;139:1781–8. [DOI] [PubMed] [Google Scholar]
- [55].Neill JD, Musgrove LC, Duck LW, Sellers JC. High efficiency method for gene transfer in normal pituitary gonadotropes: adenoviral-mediated expression of G protein-coupled receptor kinase 2 suppresses luteinizing hormone secretion. Endocrinology 1999;140:2562–9. [DOI] [PubMed] [Google Scholar]
- [56].Stamatiades GA, Kaiser UB. Gonadotropin regulation by pulsatile GnRH: signaling and gene expression. Mol Cell Endocrinol 2018;463:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Moore AM, Campbell RE. Polycystic ovary syndrome: understanding the role of the brain. Front Neuroendocrinol 2017;46:1–14. [DOI] [PubMed] [Google Scholar]
- [58].Caron E, Ciofi P, Prevot V, Bouret SG. Alteration in neonatal nutrition causes perturbations in hypothalamic neural circuits controlling reproductive function. J Neurosci 2012;32:11486–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are included in this published article and its supplementary information files. All relevant original data are available from the corresponding authors, upon reasonable request.