

Summary:
Human pluripotent stem cell-derived cardiomyocytes (hPSC-CMs) offer a promising cell-based therapy for myocardial infarction. However, the presence of transitory ventricular arrhythmias, termed engraftment arrhythmias (EAs), hampers clinical applications. We hypothesized that EA results from pacemaker-like activity of hPSC-CMs associated with their developmental immaturity. We characterized ion channel expression patterns during maturation of transplanted hPSC-CMs, and used pharmacology and genome editing to identify those responsible for automaticity in vitro. Multiple engineered cell lines then were transplanted in vivo into uninjured porcine hearts. Abolishing depolarization-associated genes HCN4, CACNA1H, and SLC8A1, along with overexpressing hyperpolarization-associated KCNJ2, creates hPSC-CMs that lack automaticity but contract when externally stimulated. When transplanted in vivo, these cells engrafted and coupled electromechanically with host cardiomyocytes without causing sustained EA. This study supports the hypothesis that the immature electrophysiological prolife of hPSC-CM mechanistically underlies EA. Thus, targeting automaticity should improve the safety profile of hPSC-CMs for cardiac remuscularization.
Keywords: Human Pluripotent Stem Cell-derived Cardiomyocytes, cell therapy, heart regeneration, myocardial infarction, cardiac remuscularization, arrhythmia, engraftment arrhythmia, automaticity, pacemaker, hPSC-CM maturation
eTORC
Engraftment arrhythmia (EA) compromises the safety of hPSC-CM cell therapy. We hypothesized that spontaneous graft depolarizations are the source of EA. We used a CRISPR screen to demonstrate that targeting excitatory channels HCN4, CACNA1H, SLC8A1 and expressing the inhibitory KCNJ2 channel generates quiescent-yet-excitable cardiomyocytes that engraft without resulting in EA.
Graphical Abstract
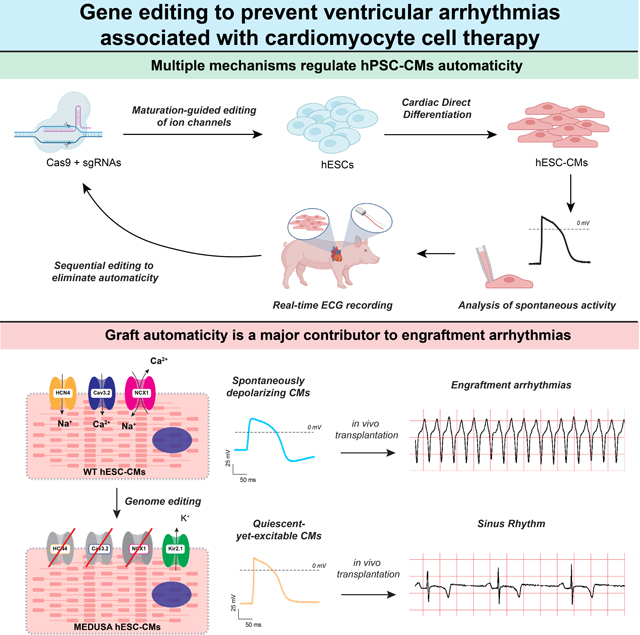
Introduction
The human heart loses its regenerative potential soon after birth1,2. After a myocardial infarction (MI), ~1 billon adult cardiomyocytes are replaced by non-contractile scar tissue; this impairs heart function and often progresses to chronic heart failure3–6. Ischemic heart disease affects over 120 million individuals per year, and it is the leading cause of death and hospitalization worldwide5,6. The discovery of pluripotent stem cells (PSCs) opened a new horizon in the treatment of MI and the prevention of heart failure7. Human PSCs, indeed, can be differentiated rapidly and at large scale into highly pure cardiomyocytes (hPSC-CMs). Intra-myocardial transplant of hPSC-CMs leads to long-lasting grafts of new myocardium in infarcted hearts8,9. These grafts form a functional syncytium with the host and are able to follow pacing from the sinoatrial node8,9. Moreover, in mouse10, rat11, guinea pig12, and non-human primate (NHP)13,14 models of subacute MI, transplantation of hPSC-CMs improves heart contractile function. For all these reasons, hPSC-CMs are being studied intensively as preclinical candidates for bona fide human heart regeneration9,15.
Compared to adult ventricular CMs (vCMs), however, hPSC-CMs exhibit automaticity, described as the ability to spontaneously and rhythmically depolarize and elicit action potentials (APs)16–18. Automaticity is a feature of all early-stage cardiomyocytes that, during normal development, becomes restricted to specialized cells in the pacemaking/conduction system. Upon transplantation of hPSC-CMs into large animal models (e.g., NHPs and pigs), whose heart rates compares to that of humans (~70 beats per minute, bpm), we and others observed transitory but severe arrhythmias, hampering the translation of this technology into the clinic13,14,19–21. We named this phenomenon “engraftment arrhythmia” (EA, see the Method section for our detailed clinical characterization of EA)20. EA typically presents as polymorphic sustained ventricular tachycardia (VT) arising after graft-host electrical coupling (~1 week after transplantation) and lasting on average 1 month13,14,19,20,22. In severe cases, the heart rate of pigs and NHPs can reach ~300 bpm13,14,19,20. Although EA is well tolerated in NHPs and gradually wanes as the grafts mature13,14, it can be lethal for arrhythmia-sensitive animals such as pigs19,20,22. Likewise, humans may not tolerate rapid EA, and it is imperative to identify strategies to prevent or at least control EA until electrical maturation of the graft20.
Electrical mapping studies in NHPs and pigs indicated that EA originates locally from the sites of cell transplant, suggesting that an abnormal impulse generation from the graft, rather than conduction defects (i.e., re-entry pathways), represents the major source of EA14,23. Indeed, overdrive pacing and cardioversion, which typically restore sinus rhythm if re-entry pathways are responsible, could not terminate EA14,23. We thus proposed that spontaneous impulse generation by the graft is a critical aspect in the etiology of EA. Compared to mature adult vCMs, hPSC-CMs have a more depolarized membrane potential and shorter AP duration18,24. This corresponds to a fetal-like gene expression profile for most ion channels24–27. We therefore hypothesized that the arrhythmogenic currents causing EA result from the presence of depolarizing fetal channels that are normally absent in adult vCMs and/or from the absence of hyperpolarizing channels that are normally present in the adult state.
In this work, we determined the expression dynamics of all ion channel genes after hPSC-CM transplantation to establish a list of candidate EA mediators. Using CRISPR/Cas9 technology, we systematically knocked out and/or overexpressed ion channel genes – singly and in combination – with the goal of generating cardiomyocytes that, like adult vCMs, have reduced automaticity but beat in response to electrical stimulation. We characterized their electrophysiological behavior in vitro and quantified the burden of EA after cell transplantation in uninjured Yucatán minipigs20. After screening over ten different cell lines, we found that simultaneous modification of four genes (knockout of HCN4 CACNA1H, and SLC8A1, coupled with knock-in of KCNJ2 under the transcriptional control of the HCN4 promoter) eliminated automaticity in vitro without affecting the ability of the cell to fire action potentials when stimulated. Importantly, compared to WT controls and the other cell lines, transplantation of this quadruple-gene edited CMs did not result in sustained EA. These modifications prevented the otherwise high morbidity and mortality and acute heart failure associated with hPSC-CM transplantation in pigs19,20,22. These results identify multiple redundant mechanisms that control automaticity in hPSC-CMs and provide evidence that pacemaker-like activity of the immature graft is the primary source of EA. Interventions that reduce graft automaticity therefore should minimize arrhythmic complications of hPSC-CM transplantation.
Results
Maturation of hPSC-CMs in vivo remodels the expression of ion channels involved in automaticity.
As shown in Fig. 1A, the AP in immature hPSC-CMs has several differences from an adult vCM. It exhibits a spontaneous diastolic depolarization (or phase 4, generally absent in adult vCMs16,17,25), a slower phase 0, the absence of a notch in phase 1, and shorter repolarization phases 2 and 3; this leads to an overall shorter AP duration compared to adult cells18,24. Maturation of hPSC-CMs strongly affects AP morphology, and this correlates with changes in ion channel expression24,25. While some degree of hPSC-CM maturation can be achieved through in vitro long-term culture, electromechanical, and/or hormonal stimulation, the gene expression profile following prolonged culture never reaches a bona fide adult stage16,17. In contrast, we have previously shown that hPSC-CMs transplanted into the infarcted rat heart mature to have adult-like myofibril isoform expression and organization28. Most importantly, this model more closely mimics the maturation milieu that would be experienced by hPSC-CMs in human subjects, and is thus more clinically relevant. To characterize the genome-wide expression dynamics of hPSC-CMs during in vivo maturation, we performed laser-capture microdissection (LCM) followed by bulk RNA-seq time-course of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) from 1 day to 12 weeks after transplantation in infarcted rat hearts (Fig. 1B). As a comparison, we analyzed hiPSC-CMs cultured long-term in vitro for up to 1 year. To identify and extract in vivo-transplanted hiPSC-CMs from the host rat heart, we transduced them with GCaMP3 prior to injection28, and isolated the GCaMP3+ (i.e., green fluorescent) grafts from tissue sections using LCM (Fig. 1C). To distinguish rat host signal from human graft signal, human-specific RNA-seq reads were then analyzed separately from those mapping ambiguously or clearly rat-specific, which were both discarded (Supplementary Fig. 1A). Dimensionality reduction of the data through Principal Component Analysis (PCA, Fig. 1D) revealed that the hiPSC-CM transcriptome was strongly altered rapidly upon early stages of engraftment (PC1), possibly a response to the associated stress. A similar magnitude of gene expression variability was explained by PC2, which separated samples by both time in culture and time in vivo. We interpreted PC2 as a maturation index. In agreement with protein expression studies previously reported by our group28, gene expression profiles showed stronger and faster maturation in vivo compared to in vitro culture, which lagged behind even after 1 year in culture (Fig. 1E, Supplementary Fig. 1B and Supplementary Table 1). For instance, fetal to adult isoform switching of myofibril-related genes (TNNI1 to TNNI3, MYH6 to MYH7, and MYL7 to MYL2) as well as upregulation of genes involved in oxidative metabolism were more strongly induced by in vivo transplantation (Fig. 1E).
Figure 1. Gene expression analysis of hiPSC-CMs during in vivo transplantation compared to 2D culture.

(A) Representative action potential traces from hESC-CMs and adult ventricular CMs (dotted line indicates 0 mV). Adult CMs adapted from Karbassi et al.16 (B) Experimental layout for RNA-seq experiments. (C) Representative hematoxylin and eosin-stained (H&E) histological analyses of rat heart engrafted with hiPSC-CMs at day 84 after injection (a-b), and adjacent unstained sections showing the graft site before (c, fluorescence image of GCaMP3 signal) and after laser capture microdissection (LCM; d, brightfield image). Scale bar = 200 μm for a, d; 50 μm for b, c. (D) Principal component analysis (PCA) of RNA-seq data set described in B. The percentage of gene expression variance expressed by each PC is indicated. Day 0 indicates hiPSC-CMs in vitro at day 18–21 as depicted in B. (E) RNA-seq expression heatmap of selected maturation-related genes (data plotted as average log2 fold-change from day 0 for three biological replicates). (F) Selected RNA-seq data for ion channels involved in AP. Red shading indicates the approximate window of engraftment arrhythmia. See also Supplementary Figs. 1C, D.
We then analyzed changes in gene expression specifically of ion channels involved in hPSC-CM APs (Fig. 1F). At early time points, corresponding to the onset of EA in large animals, we saw strong expression of HCN4 and CACNA1H (whose protein products mediate If and ICaT, responsible for Na+/K+ and Ca2+ currents, respectively, and are involved in phase 4), while we detected barely any KCNJ2 transcript (which encodes for Kir2.1, responsible for the inward rectifying K+ current, IK1, one of the major repolarizing potassium currents). By 3 months, well after EA would be resolved and corresponding to a more mature hiPSC-CM state, this relationship was inverted, with KCNJ2 expression being strongly upregulated (~1,000% more compared to early time points), while HCN4 and CACNA1H decreased by ~50% and >90%, respectively. We also evaluated the expression kinetics of the other subunits that potentially contribute to If (HCN1, HCN2 and HCN3), as well as ICaT (CACNA1G and CACNA1I). As shown in Supplementary Figures 1C, D, the expression of these subunits in hiPSC-CMs is substantially lower compared to HCN4 and CACNA1H, suggesting that in hiPSC-CMs, HCN4 and CACNA1H are the major contributors for If and ICaT, respectively. Overall, the expression patterns of HCN4, CACNA1H, and KCNJ2 correlate with the absence of phase 4 spontaneous depolarization in more mature hPSC-CMs and vCMs16.
SCN5A (mediating the INa current), CACNA1C (mediating the ICaL current), and SLC8A1 (mediating the INCX current) regulate the amount of depolarizing current after the initiation of the AP in vCMs. In hiPSC-CMs, INa and ICaL were significantly lower than in adult vCMs24–27, and, as shown in Fig. 1F (Phase 0, 2), SCN5A and CACNA1C expression levels were almost 4 times lower than SLC8A1. This is consistent with studies indicating a predominant role of INCX in the control of calcium handling in hPSC-CMs27,29,30. We also analyzed the expression of potassium channels responsible for the repolarization phase of the AP, including KCND3 (Ito), KCNQ1 (IKs) and KCNH2 (IKr). Electrophysiological studies previously reported that maturation of hPSC-CMs coincides with increased repolarizing currents as well as the presence of the characteristic notch in the AP trace, mediated by the upregulation of KCND3/Ito16,24. As shown in Fig. 1F (Phase 1, 3), as hiPSC-CMs matured in vivo the expression of KCND3, KCNQ1 and KCNH2 increased with time. All together, these results indicate that in vivo transplantation progressively affects ion channels expression towards a more mature, adult-like phenotype. This suggests that inducing a more adult-like ion channel gene expression profile in hPSC-CMs could reduce automaticity and, potentially, the burden of EA after transplantation.
Pharmacologic studies link hPSC-CMs automaticity to calcium trafficking.
The role of ion channels in automaticity has been largely studied in the context of the adult mouse sinoatrial node31–36. Whether the same mechanisms also apply to hPSC-CMs remains controversial29. Moreover, the electrophysiological properties of hPSC-CMs may differ based on the specific differentiation protocol and/or the homogeneity of the population24,26,29,37. We thus set out to understand the drivers of automaticity in human embryonic stem cell-derived cardiomyocytes (hESC-CMs) differentiated by modulating the WNT pathway with small molecules.
We began by testing a variety of pharmacological compounds using multielectrode array system (MEA). First, we studied the role of HCN4 (If), a key mediator of spontaneous phase 4 depolarization38. Inhibition of If with Ivabradine potently reduced hESC-CM beating frequency39,40. Nevertheless, Ivabradine did not prevent spontaneous AP firing, even at high doses (Supplementary Fig. 2A). Zacopride, a reported agonist for the Kir2.1 channel (KCNJ2/IK1)41, did not show measurable effects on the beat rate of hESC-CMs, likely due to the very low expression of the target ion channel in immature hPSC-CMs (Supplementary Fig. 2B). We attempted to inhibit ICaT using both ML-218 and Mibefradil (Supplementary Figs. 2C, D), reported to inhibit ICaT with moderate selectivity42,43. Mibefradil caused a dose-dependent reduction in beating rate and, at higher concentrations, reduced spike amplitude. ML-218 reduced hESC-CMs beating rate only at concentrations associated with undetectable spike amplitudes (Supplementary Fig. 2C, right panel). Since both inhibitors can block ICaL42,43 at high concentrations, we consider this evidence for ICaT in automaticity, but not definitive proof.
Arguing for an important role of ICaL for AP generation, we treated hESC-CMs with Verapamil, a class IV antiarrhythmic that blocks L-type calcium channels44. Verapamil, indeed, is indicated for the treatment of supraventricular arrhythmias, where it decreases the firing rate of nodal cells by reducing the rate of phase 4 depolarization45. In hESC-CMs, Verapamil caused a strong and progressive decrease in both the beat rate and the spike amplitude (Supplementary Fig. 2E), suggesting that the same mechanisms controlling AP firing in nodal cells might apply to hESC-CMs as well. A strong dose-response reduction in frequency and spike amplitude was observed also after the inhibition of the sodium-calcium exchanger NCX1 with the inhibitors SEA0400 and KB-R3702 (Supplementary Fig. 2F)46,47. NCX1 could also participate in AP generations because it mediates the uptake of three Na+ ions for every Ca2+ ion extruded, independent from voltage membrane. This creates an influx of positive charge per cycle that could increase the spontaneous activity of hPSC-CMs in case of Ca2+ intracellular accumulation33,48. All together, these results highlight the importance of Ca2+ trafficking in hESC-CMs automaticity.
Gene editing to suppress automaticity
Pharmacologic studies are inherently limited by the specificity of drugs. To assess definitively the role of specific ion currents, we genetically manipulated candidate genes encoding for those ion channels likely to play an important role in hESC-CMs automaticity, based on our transcriptomic and pharmacological evidence. Specifically, we sought to obtain hESC-CMs that are quiescent-yet-excitable, like mature vCMs. This required the targeting of HCN4 (HCN4), CACNA1H (Cav3.2), SLC8A1 (NCX1) and KCNJ2 (Kir2.1). Gene edits were performed individually and combinatorically to identify the minimum set of perturbations needed to abrogate automaticity and/or EA (Fig. 2A and Supplementary Fig. 3A).
Figure 2. Ablation of HCN4 and CACNA1H is not sufficient to prevent automaticity of hESC-CMs.

(A) Experimental layout for the generation of gene-edited cell lines, cardiac differentiation, and in vitro/in vivo characterization. SpCas9: Streptococcus pyogenes Cas9, sgRNA: single-guide RNA. (B, C) Patch clamp analyses of funny current (If) from HCN4 knockout (B) and T-type calcium current (C) from CACNA1H KO, compared to WT cardiomyocytes. (For If, WT: n = 7, HCN4 KO cl.1: n = 6, HCN4 KO cl. 2: n = 4; for ICaT WT: n=22, KO: n=15). Representative current traces showed on the right. Differences in ICaT vs. WT by two-way ANOVA with Sidak correction for multiple comparison (green-shaded area from −20 mV to +20 mV, p < 0.001). The traces for ICaT were recorded from quadruply edited MEDUSA hESC-CMs and reported here with the parental line for simplicity (see also Supplementary Fig. 3A for detail of gene-editing strategy). (D - F) Spontaneous electrical activity of gene-edited hESC-CMs on MEA system. Data shown as mean ± SEM of 2–3 independent experiments each with 8 technical replicates, and normalized on WT frequency (D and F) or WT spike amplitude (E). Statistical differences are reported vs. WT hESC-CMs by one-way ANOVA with Sidak correction for multiple comparisons (* p <0.05, ** p < 0.01 and *** p <0.001). (G) Quantification of engraftment arrhythmia burden (% of time each day) and heart rate after transplanting HCN4 KO hESC-CMs compared to WT hESC-CMs. Data shown as mean ± SEM for HCN4 KO (N = 2) and WT controls up to day 14 and as individual animals thereafter (N = 7 for starting cohort, then N = 2, shown as individual trace). Red-colored symbols represent animals that reached endpoints as defined in the Method section. Representative ECG traces shown on the right.
We began with knockout (KO) perturbations. First, we abrogated the expression of HCN4, either alone and/or in combination with CACNA1H (Supplementary Figs. 3A, B). Western blot analyses confirmed the absence of HCN4 protein (Supplementary Fig. 3C) and, most importantly, patch clamp measurements demonstrated functional ablation of If (Fig. 2B) and a significant reduction in ICaT (Fig. 2C), in agreement with the absence of compensatory upregulation on the other lowly expressed genes potentially contributing to these currents (Supplementary Figs. 3D, E). Confirming pharmacological observations, KO of HCN4 alone did not eliminate hESC-CM automaticity in vitro, but rather, it reduced their beating frequency by nearly half (Fig. 2D). Interestingly, we also observed a moderate increase in the spike amplitude, that was significant for only one of the two clones studied (Fig. 2E). KO of CACNA1H did not alter hESC-CMs beat rate (Fig. 2F) or spike amplitude (Supplementary Fig. 3F). The combination of HCN4/CACNA1H 2KO showed a slightly more suppressed beat rate than HCN4 KO alone, but automaticity was not completely abrogated (Fig. 2F).
To test the impact of gene edits on EA, we developed an in vivo model where 150 million hESC-CMs are transplanted into the uninjured hearts of immunosuppressed Yucatán minipigs (Supplementary Video 1 and Supplementary Table 2). We monitored heart rate and rhythm by telemetric continuous electrocardiogram (ECG) system. The number of animals treated with gene edited cells was, of necessity, limited since we wished to screen multiple genomic edits and were seeking dramatic impacts on EA. Indeed, moderate improvement of EA burden can be achieved already with pharmacologic therapy20. Moreover, although we did not utilize infarcted pigs in this work, the complication rate due to unstable EA (including sudden cardiac death and heart failure), as well as the general burden of EA, was comparable with our previous study where 500 million hESC-CMs were transplanted into infarcted pig hearts (Supplementary Figs. 3G, H)20. In all, we reasoned that gene editing would be clinically relevant only if it led to much greater safety profiles in a model where EA has the same clinical severity. Seven pigs received WT cells and served as controls. In WT-receiving hearts, EA commenced by day 4 and progressed to occupy more than ~50% of the day, especially in the first two weeks after engraftment (Fig. 2G, left panel). As described previously20, EA began as premature ventricular contractions (PVCs), progressed into non-sustained ventricular tachycardia (NSVT) and then to sustained VT. The VT was often polymorphic, with both wide- and narrow-QRS complexes. As grafts mature, EA slows down, associated with increasing duration of normal sinus rhythm. Three control subjects exited the study prematurely based on prespecified EA severity endpoints described in the Methods section: one animal was euthanized due to unstable VT and cardiogenic shock at day 6, and two required acute pharmacological intervention with anti-arrhythmic therapy at day 4 and day 14, therefore these animals were excluded from further analysis (Fig. 2G, red points). The remaining four controls survived until planned endpoints of 14 (n=2), 35 (n=1), or 49 (n=1) days (See Methods for explanation of time points).
We began analysis of gene-edited hESC-CM using HCN4 KO cells (n=2). Both animals receiving HCN4 KO CMs developed EA: although there was a trend toward a reduced arrhythmia burden (% of the day in VT) and both subjects survived to the planned endpoint of 4 weeks, the removal of HCN4 did not significantly reduce the rapid rise in heart rate (Fig. 2G, right panel). We concluded that additional reductions in EA severity were needed to increase safety.
Next, we turned to the possibility of overexpressing hyperpolarizing currents (Supplementary Fig. 3A). Since the IK1 current is virtually absent in hESC-CMs16,49 and is known to set the resting membrane potential at a more hyperpolarized, less excitable level in adult cardiomyocytes50–52, we decided to overexpress KCNJ2 (Kir2.1/IK1)49. We initially attempted to constitutively overexpress this channel through knock-in into the AAVS1 genomic safe harbor with a constitutively active promoter53. However, despite numerous attempts and the fact that a control EGFP knock-in was consistently successful, we could not isolate any correctly targeted KCNJ2-overexpressing hESCs (data not shown). This unexpected finding strongly suggested that KCNJ2 overexpression is not compatible with maintenance of pluripotency. To overcome this issue, we knocked in KCNJ2 into the HCN4 locus (Fig. 3A and Supplementary Fig. 4A). This strategy allowed us to both overexpress KCNJ2 and knockout HCN4 in a single gene-editing step and drive KCNJ2 expression only after cardiac specification (Figs. 3A, B). Indeed, KCNJ2 expression during hESC-CMs differentiation paralleled the normal pattern of HCN4, rising after the transition from progenitor to definitive cardiomyocyte (Fig. 3B). Given our earlier observation that during in vivo maturation of hiPSC-CMs HCN4 levels decrease as endogenous KCNJ2 is activated, this knock-in/knockout strategy provided the added potential benefit of providing temporally controlled KCNJ2 upregulation, bridging its expression from immature to mature cardiomyocytes. We then compared HCN4 KO/KCNJ2 KI hESC-CMs with control cells that have EGFP knocked-in to the HCN4 locus. Interestingly, even though the dual perturbation did not affect cardiac differentiation efficiency (Supplementary Fig. 4B), it significantly delayed the onset of spontaneous beating (Fig. 3C). Moreover, these dual edited hESC-CMs exhibited rapid and irregular bursts of electrical activity interposed with long quiescent pauses (Fig. 3D). This suggests that automaticity relies on the simultaneous activity of more than one or two ion channels31–34.
Figure 3. HCN4 and KCNJ2 perturbation is not sufficient to prevent automaticity or EA.

(A) Gene-editing approach to knock-in KCNJ2 under the transcriptional control of the HCN4 promoter in RUES2 hESCs. Genotyping PCR strategies for on- and off-target insertions are indicated; see also Supplementary Figure 4A. (B) Time course qRT-PCR analysis of HCN4, KCNJ2 and TNNT2 expression during cardiac differentiation of the indicated WT and gene-edited hESCs. ES: embryonic stem cell, MS: mesoderm, CP: cardiac progenitor, CM: cardiomyocyte. N = 2 differentiations per cell line. (C) Representative quantification of spontaneous beating during hESC-CM differentiation from HCN4 KO/KCNJ2 KI clones compared to WT. See Methods for details on quantification. (D) Spontaneous activity of HCN4 KO/KCNJ2 KI clones quantified by MEA, and representative traces. Given the marked irregularity of automaticity in these lines, data are reported as average beats in 5 min recording (left panel) and the corresponding % beat irregularity (right panel), calculated as standard deviation of the beat period record in 100 sec, divided by the mean of the beat period in that same period. Data are plotted as mean ± SEM of 3 independent experiments each with 8 technical replicates. (E) In vivo data showing EA burden (left panel) and heart rate of animals transplanted with HCN4 KO/KCNJ2 KI hESC-CMs compared to WT, with representative ECG traces on the right. Data shown for WT controls as described in Fig. 2G and individual traces (N = 2) for HCN4 KO/KCNJ2 KI. Red-colored symbol represents animals that reached prespecified EA endpoints leading to withdrawal from the study, as defined in the Methods section.
Intrigued and puzzled by these results, we decided to test HCN4 KO/KCNJ2 KI hESC-CMs in vivo. Both animals receiving the dual edited cells showed a delay in the onset of EA by ~3 days (Fig. 3E). Once EA commenced, however, it rapidly degenerated into unstable VT in both animals, reaching euthanasia criteria on days 10 and 11. Collectively, these data indicate a complex role of KCNJ2 in the regulation of both automaticity and EA, possibly through the interplay with other ion channels31–34.
Triple gene editing: deletion of CACNA1H reduces pacemaker activity but does not prevent EA.
Work from others has shown that, in a simplified in silico model and in a non-excitable cell, the balanced alternation of hyperpolarization-potentiated depolarizing If and depolarization-activated repolarizing IK1 is sufficient to create rhythmic oscillations in the membrane potential, thus setting the stage for AP formation40,50. However, these antagonizing currents lead to moderate oscillations in the voltage membrane that alone cannot reach the threshold for phase 0 depolarization, and an additional depolarizing current is required for triggering the AP40. In agreement with other results suggesting the existence of multiple mechanisms regulating automaticity for nodal cells29,32, our results indicate that a more complex circuit regulates hESC-CM automaticity as well. Indeed, the perturbation of HCN4 and KCNJ2 resulted in a slower and/or irregular beat rate, suggesting that the oscillation created by If and IK1 might principally set the depolarizing rhythm, whereas a second mechanism is required to maintain and stabilize automaticity. We thus focused on both the T-type calcium channel (CACNA1H) and NCX1 (SLC8A1). ICaT is activated at a more hyperpolarized voltage compared to ICaL or INa32,35,40, whereas INCX is mainly regulated by the concentration of Na+/Ca2+ across the sarcolemma, rather than membrane voltage29,31,32,35. We hypothesized that ICaT and/or INCX might participate in cooperative mechanisms of automaticity, as suggested by in silico and in vitro results33,40.
We decided first to generate hESCs lacking both depolarizing If and ICaT currents with KCNJ2 overexpression (Supplementary Figs. 3A and 4C). Triple edited hESCs maintained a normal karyotype and cardiac differentiation potential (Supplementary Figs. 4D, E). Since knockout of a gene may lead to compensatory upregulation of genes sharing sequence similarity54, we evaluated the expression of other ion channels mediating If and ICaT. As shown in Fig. 4A, knockout of HCN4 and CACNA1H did not affect the expression of gene family members. Triple edited HCN4/CACNA1H 2KO/KCNJ2 KI hESC-CMs displayed delayed and inconsistent beating during differentiation (Fig. 4B), and MEA analysis indicated either complete absence of depolarization or irregular burst activity (Fig. 4C; Supplementary Fig. 4F). Nevertheless, when transplanted in vivo triple edited hESC-CMs still elicited EA at day 6 post engraftment (Fig. 4D). Once initiated, heart rate progressively accelerated and reached euthanasia criteria by day 8 (Fig. 4D). Collectively, adding CACNA1H KO appeared to further reduce automaticity in vitro compared to the parental HCN4 KO/KCNJ2 KI cells, but these hESC-CMs still caused severe EA in vivo. This indicated that another mechanism maintains some degree of automaticity in hPSC-CMs. We hypothesized that this mechanism might be mediated by NCX1, as described in the next section.
Figure 4. Triple gene edits decrease automaticity but do not fully prevent EA.

(A) qRT-PCR gene expression analysis of HCNs, T-type ion channel genes, and KCNJ2 in HCN4/CACNA1H 2KO/KCNJ2 KI compared to WT hESC-CMs at day 14 of differentiation. Data shown as mean ± SEM of 3 independent experiments normalized on WT. Differences vs. WT by multiple paired t test (* p <0.05, ** p <0.01 and *** p <0.001). (B) Representative onset of beating during cardiac differentiation in HCN4/CACNA1H 2KO/KCNJ2 KI hESC-CMs. (C) MEA analysis of HCN4/CACNA1H 2KO/KCNJ2 KI clones compared to WT hESC-CMs. Data are shown as average beats/min recorded in 5 min ± SEM of 2 independent experiments with 8 replicates each. See also Supplementary Fig. 5F. (D) Arrhythmia burden and heart rate of one pig engrafted with HCN4/CACNA1H 2KO /KCNJ2 KI hESC-CMs compared to WT (N = 7), and representative EKG traces during EA. Data shown as described in Fig. 3E. Red-colored symbols represent animals that reached prespecified EA endpoints and withdrawn from the study as described in the Methods section. (E) Western blot of NCX1 KO clones compared to WT hESC-CMs. cTnT: cardiac troponin T. (F) Representative time course analysis of onset of beating during cardiac differentiation of SLC8A1 KO clones compared to WT. (G) Representative field potential traces and quantifications of SLC8A1 KO clones at different points of a 2 weeks culture on MEA plates. Data shown as average beats/min recorded in 5 min ± SEM of 3 independent experiments with 4 replicates each. (H) EA burden and respective heart rate of pigs transplanted with SLC8A1/HCN4 2KO/KCNJ2 KI hESC-CMs compared to WT. Data shown for WT controls as mean ± SEM up to 14 and as individual animals thereafter (N = 7 for starting cohort), and for SLC8A1/HCN4 2KO/KCNJ2 KI as individual traces to demonstrate heterogeneity (N = 3); Red-colored symbols represents animals withdrawn from the study due to death and/or EA severity, as described in Methods section. (I) Representative histological analysis of SLC8A1/HCN4 2KO/KCNJ2 KI graft 4 weeks after injection, stained with human-specific β-myosin heavy chain antibody. Scale bar = 200 μm.
An SLC8A1-dependent mechanism critically contributes to pacemaker activity and EA burden.
As mentioned above, the opening of NCX1, mediated by the concentration of Na+/Ca2+ across the sarcolemma, might contribute to the generation of AP as a cooperative mechanism for hPSC-CM automaticity. Indeed, the existence of both a “voltage-clock”, mostly mediated by If/ICaT/IK1, and a “calcium-clock”, where the main player is NCX1, has been described not only for nodal cells but also proposed for immature hPSC-CMs29,30. Although NCX1 is expressed in adult vCMs, we consider that its role might be ancillary in calcium handling in adulthood, compared to immature cardiomyocytes55,56. The calcium-induced calcium release (CICR) during excitation-contraction coupling in adult CMs is mainly regulated by L-type calcium channels, ryanodine receptors, and the Sarco-Endoplasmic Reticulum Calcium ATPase, (SERCA2A), pump55. In immature CMs, such as hPSC-CMs, CICR has not developed yet16,48; hence, NCX1 might play a predominant role in calcium cycling in immature CMs. This notion is supported by the pharmacological suppression of automaticity by NCX1 inhibition (Supplementary Fig. 2F).
All considered, we decided to knockout the gene encoding NCX1 (SLC8A1) either alone or in combination with the dual targeting of HCN4/KCNJ2 (Supplementary Figs. 3A and 4G, H). Western blot confirmed the absence of NCX1 protein in both single and triple-edited clones (Fig. 4E). SLC8A1 KO hESC-CMs showed no differences in the onset of beating during differentiation but exhibited intermittent periods of quiescence (Fig. 4F). Quiescence was accentuated in the triply edited SLC8A1/HCN4 2KO/KCNJ2 KI (Fig. 4F). Indeed, from both observation during differentiation and MEA analysis, the triply edited hESC-CMs remained mostly quiescent, with only very sporadic beating episodes (Fig. 4G).
These results encouraged us to proceed with the in vivo transplantation of SLC8A1/HCN4 2KO/KCNJ2 KI cardiomyocytes. Two out of three pigs, over the 4-weeks observation period, showed minimal EA with only episodes of PVCs (see the Method section for detailed definition of EA presentation). The third one developed unstable VT requiring euthanasia on day 9 post-transplant (Fig. 4H). Although graft size varied among animals, all pigs had readily detectable grafts (Fig. 4H, right panel; Supplementary Table 2). Notably, we had not seen the absence of EA in any animal to this point in the study. In combination with the in vitro data, we concluded that deactivation of both the calcium and voltage clocks significantly reduces automaticity in vitro and potentially reduced EA burden in vivo. This suggested that a state of total quiescence might be necessary to completely abrogate EA.
MEDUSA: a quadruple gene-edit that induces hESC-CM quiescence but maintains excitability.
Since the triply edited SLC8A1/HCN4 2KO/KCNJ2 KI CMs can still exhibit some automaticity and caused EA, we hypothesized that removal of CACNA1H might deactivate what redundant depolarization mechanisms remain. We therefore generated a clone of HCN4/CACNA1H/SLC8A1 3KO/KCNJ2 KI hESCs. Despite undergoing four sequential rounds of genome editing (Supplementary Fig. 3A), this cell line retained a normal karyotype and homogeneous expression of pluripotency markers (Supplementary Figs. 5A, B). Henceforth, we will refer to this quadruple edited cell line with the shorter acronym MEDUSA (Modification of Electrophysiological DNA to Understand and Suppress Arrhythmias). MEDUSA hESCs were still able to differentiate into cardiomyocytes (Supplementary Figs. 5C, D) that, as expected, lack expression of HCN4, CACNA1H and showed increased expression of KCNJ2 (Fig. 5A and Supplementary Figs. 5E, F). SLC8A1 mRNA did not undergo nonsense-mediated decay, yet the gene edit prevented NCX1 translation as expected (Fig. 5A, insert and Supplementary Fig. 5E). As before, also in this quadruple gene edited cell line we did not observe compensatory upregulation of the other ion channel isoforms (Supplementary Fig. 5F). MEDUSA hESC-CMs remained quiescent with sporadic beating seen only when the cells were undergoing stress (e.g., lactate selection or heat-shock, Fig. 5B). Similarly, on the MEA system (Fig. 5C), we observed minimal spontaneous activity (less than 5 beats/hour), even after 2 weeks of observation, indicating that these cells are more quiescent compared to both the WT and the other cell lines tested so far.
Figure 5. In vitro characterization of MEDUSA hESC-CMs.

(A) qRT-PCR of gene-edited ion channels in MEDUSA hESC-CMs compared to WT control. Data shown as mean ± SEM of 3 independent experiments. Statistical differences are reported vs. WT hESC-CMs by unpaired t-test (* p <0.05, ** p < 0.01 and *** p <0.001). Insert showing western blotting analysis for NCX1 in MEDUSA hESC-CMs, see also Supplementary Figs. 5E, F. (B) Onset of beating and beating rate during cardiac differentiation of MEDUSA hESC-CMs. Data shown as mean ± SEM of 2 independent batches of monolayer differentiation (12-wells/cell line per batch of differentiation). See Methods section for details on quantification. (C) Representative MEA analysis of MEDUSA hESC-CMs cultured for 2 weeks on MEA plates. Data shown as average ± SEM of total beats recorded for 5 min every hour. Spontaneous activity at day 35 is shown as continuous recording for 20 hours, panel below. Differences vs. WT hESC-CMs by two-way ANOVA with Sidak correction (*** p < 0.001). (D) Distribution of quiescent and spontaneously depolarizing cells in patch clamp preparation. (E) Quantification of patch clamp metrics under pacing condition (RMP: resting membrane potential; MDP: maximum diastolic potential). Data shown as violin plots of 16 WT and 11 MEDUSA hESC-CMs. Differences vs. WT hESC-CMs by unpaired t-test with Welch’s correction (**p<0.01, ***p<0.001). (F) AP traces of WT and MEDUSA hESC-CMs after electrical stimulation (RMP, MDP as in 5E; APD90: action potential at 90% repolarization). (G) Representative AP traces of MEDUSA hESC-CMs stimulated at 0.5 – 3 Hz, during patch clamp experiments. (H) Calcium transient analysis of WT and MEDUSA hESC-CMs during pacing at 1 Hz. Data shown as mean ± SEM of 12 individual cells/cell line. Statistical differences are shown by multiple unpaired t-test (***p<0.001). Representative traces shown in Supplementary Fig. 5H.
To gain further insight on the excitability of MEDUSA CMs, we performed patch clamp experiments in the perforated-patch configuration on individual CMs. Similar to what was observed in MEA experiments, we found that MEDUSA hESC-CMs were essentially quiescent, as only 1/12 CMs (8%) exhibited automaticity after engaging the patch electrode (in contrast with 70% of WT hESC-CMs; Fig. 5D). Consistent with our expectations for CMs that overexpress KCNJ2, we found that the resting membrane potentials of quiescent MEDUSA hESC-CMs were significantly more hyperpolarized compared to spontaneously beating WT hESC-CMs, both in terms of resting membrane potential (RMP) and maximum diastolic potential (MDP; Figs. 5E, F). Moreover, the action potential duration at 90% repolarization was not different between MEDUSA and WT-CMs (Supplementary Fig. 5G). The capacitance of MEDUSA hESC-CMs did not differ from WT hESC-CMs (Supplementary Fig. 5G), indicating that membrane surface area was comparable.
Before proceeding with in vivo transplantation, we tested whether quiescent MEDUSA hESC-CMs were still excitable. Both patch clamp analyses (Fig. 5G) and calcium transient measurements (Fig. 5H and Supplementary Fig. 5H) confirmed that MEDUSA hESC-CMs can follow electrical pacing, generate APs and Ca2+ transients, and visibly contract up to 3 Hz stimulation (Fig. 5G). Calcium transients showed a delayed decay in MEDUSA CMs, potentially due to lack of NCX1 (Fig. 5H and Supplementary Fig. 5H). We wondered whether MEDUSA cardiomyocytes might compensate for lack of NCX1 by down-regulating systolic calcium entry. Interestingly, MEDUSA hESC-CMs showed reduced ICaL currents, compared to WT control (Supplementary Fig. 5I), despite no significant changes in CACNA1C transcript levels (p = 0.82 with unpaired t-test). This suggest that, like the mouse cardiac NCX1 knockout 56, MEDUSA cardiomyocytes might reduce influx of Ca2+ via L-type Ca2+ channels to prevent calcium overload. Together, these data indicate that MEDUSA gene edits result in cardiomyocytes that are quiescent under baseline conditions but are still able to fire action potentials upon stimulation.
MEDUSA gene edits attenuate EA in vivo.
To test the efficacy of MEDUSA hESC-CMs in preventing EA, we proceeded with their transplantation into pig hearts. Remarkably, all three animals receiving 150M MEDUSA hESC-CMs showed no sustained VT for the entire period of observation (Figs. 6A, B) and reached the planned endpoints of 4 or 7 weeks (2 animals and 1 animal, respectively), without any adverse events. Pigs spent >95% of the time post-transplantation in stable sinus rhythm (Fig. 6B), with only sporadic PVCs and rare NSVT (Fig. 6A, right panel, see the Methods for detailed definitions). No sustained VT occurred at any point of the study (Fig. 6B). Indeed, MEDUSA hESC-CMs showed ~95% reduction in combined EA burden (VT, PVC, NSVT) for the first 2 weeks after transplant, compared to WT hESC-CMs (p < 0.001; Fig. 6C and Supplementary Fig. 6A). Importantly, the MEDUSA graft size was consistent with others examined at comparable endpoints (Fig. 6D and Supplementary Table 2).
Figure 6. Characterization of MEDUSA hESC-CMs in vivo.
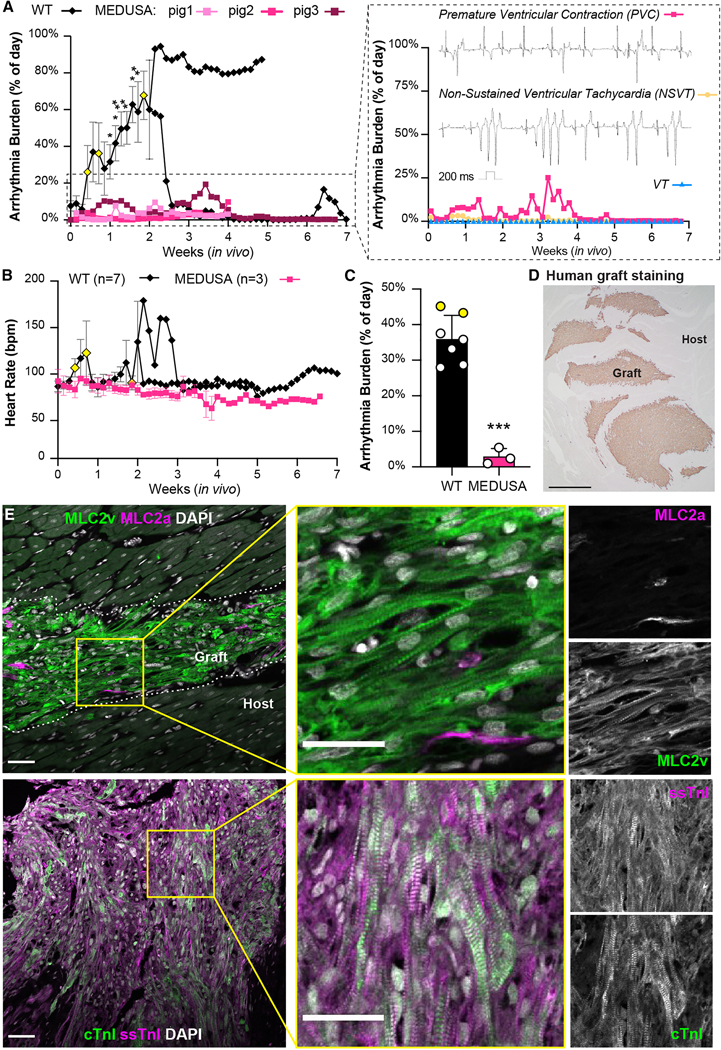
(A, B) Arrhythmia burden and heart rate of pigs receiving 150M MEDUSA CMs and monitored with telemetry up to 7 weeks post-transplantation. Data shown for WT controls as mean ± SEM as detailed in Fig. 2G. Yellow-colored symbols represent animals that reached EA severity endpoints leading to study withdrawal as defined in Methods section. Insert from panel in A shows arrhythmia from pig N.3 subdivided in isolated premature ventricular contraction (PVC), non-sustained VT (NSVT) and VT, with relative representative ECG traces. (C) Aggregate analysis of combined EA daily burden (VT, NSVT, PVC) for WT and MEDUSA hESC-CMs for the first 2 weeks after transplantation. Data shown as mean ± SEM of animals receiving either WT (N=7) or MEDUSA hESC-CMs (N=3). Yellow-colored symbols represent animals analyzed until EA severity necessitated their withdrawal from the study (day 4 and day 6, respectively; see also panel A and Supplementary Fig. 6A). Differences vs. WT by unpaired t-test (*** p <0.001). (D) Representative image of MEDUSA hESC-CMs graft 7 weeks after injection stained with human-specific β-myosin heavy chain. Scale bar = 1 mm. Note the large, multifocal graft throughout the host ventricle. (E) Low and high magnification immunofluorescence images of MLC2v/MLC2a and ssTnI/cTnI in MEDUSA-CMs grafts 4 weeks post transplantation. Single channels monochromatic images for both MLC2a/MLC2v and ssTnI/cTnI shown on the right. Dotted lines indicate host/graft interface. Scale bars = 50 μm for low magnifications; 20 μm for high magnification.
As cardiomyocytes mature, they undergo isoform switching of several myofibril proteins such as MLC2a to MLC2v and ssTnI to cTnI, providing developmental benchmarks16,17,28. Immunofluorescence imaging revealed that MEDUSA hESC-CM grafts undergo progressive maturation in vivo. Indeed, we observed signs of increased maturation of sarcomere as indicated by the isoforms switching from MLC2a→MLC2v and ssTnI→cTnI (Fig. 6E). This indicates ongoing but not yet complete maturation at 7 weeks, consistent with what we previously observed with WT CMs engrafted in rat hosts.
Encouraged by the results of these studies, we proceeded with a dose-escalation study, transplanting 500 million the MEDUSA CMs (n=2) to more rigorously study safety at a clinically-relevant dose. Our recent data in this model demonstrated that this dose leads to 100% EA penetrance, persisting for multiple weeks, and resulting 67% mortality when WT hESC-CMs are employed20. Upon high-dose engraftment of MEDUSA hESC-CMs we observed brief, self-terminating episodes of VT shortly after transplantation lasting less than 24 hours, followed by return to normal sinus rhythm until endpoint at 3 months (Figs. 7A, B and Supplementary Fig. 6B). This brief and abruptly self-terminating presentation of VT was never observed in animals receiving WT hESC-CMs at the same dose, which exhibited a gradual onset of ectopy developing into sustained VT and lasting uninterrupted for weeks until electrical maturation at 4–6 weeks after transplantation. The MEDUSA grafts were large and viable out to 3 months post-transplantation and comparable in size with the ones previously seen in the infarcted heart (Supplementary Figs. 6C, D and Supplementary Table 2), ruling out reduced durability from the multiple edits.
Figure 7. Electromechanical coupling of high-dose MEDUSA hESC-CMs.
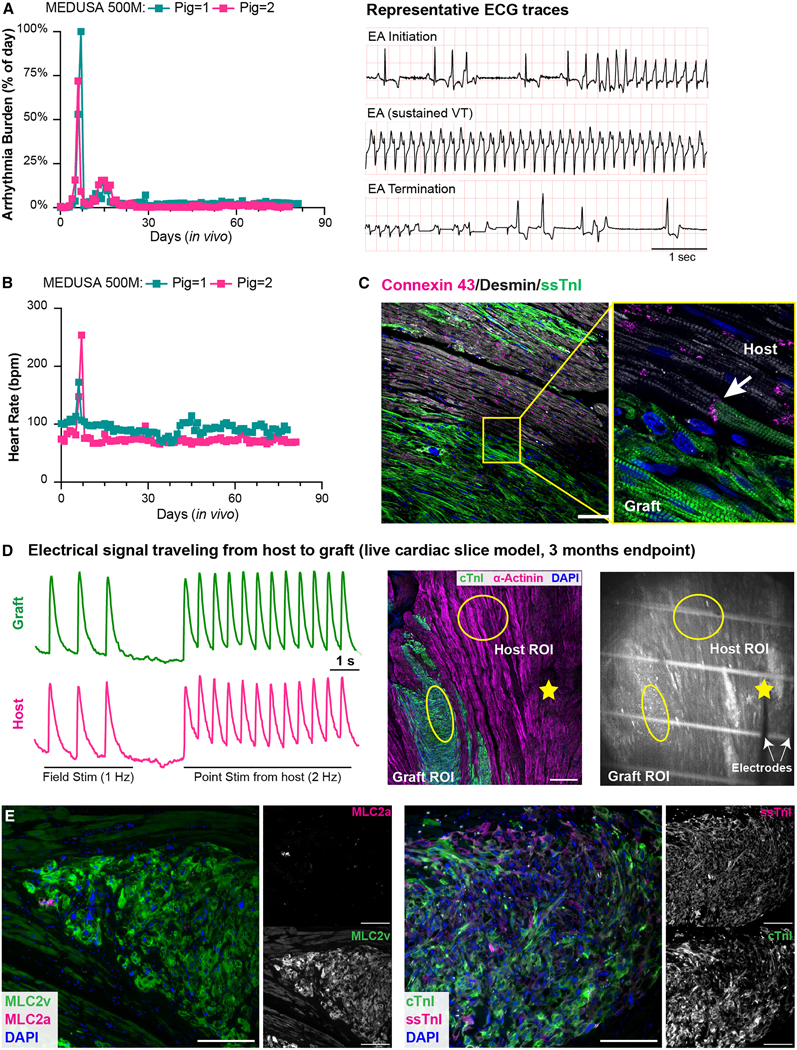
(A, B) Arrhythmia burden and heart rate of animals receiving 500M MEDUSA hESC-CMs (N = 2). Panel A shows the cumulative incidence of sustained VT, PVC, and NSVT; See also Supplementary Figs. 6B. Right panel in A showing representative ECG traces of one animal transitioning without intervention from sustained VT to sinus rhythm. (C) Representative immunofluorescence images of 3 months old MEDUSA grafts stained with Connexin 43 (white arrows indicate junctions between host, Desmin/graft, ssTnI. Scale bars = 100). (D) Cardiac slice model of 12 weeks old MEDUSA-CMs grafts loaded with Fluo-4 and paced with field and point stimulation from host region; mid panel showing immunofluorescence image indicating graft and host in the cardiac slice studied are shown. Yellow circle indicates ROI for Fluo-4 recording, yellow star indicates point stimulation location. Scale bar = 500 μm. Right panel showing one frame from Supplementary Video 2 showing parallel bipolar electrodes placement. White lines are plastic monofilament used to keep the cardiac slice in place during recording. (E) MLC2a/MLC2v (left) and ssTnI/cTnI (right) staining of 3 months-old MEDUSA grafts, with relative monochromatic images on the right side. Scale bar: 200 μm.
Histological analyses showed that MEDUSA CMs formed gap junctions with the host, demonstrating a structural basis for electrical coupling (Fig. 7C). To assess the electrical integration of MEDUSA grafts in the porcine heart, we performed calcium imaging in live cardiac slices. Through field stimulation, we first assessed that MEDUSA grafts at 3 months after transplant were still viable and able follow electrical pacing (Supplementary Video 2). Importantly, we found that MEDUSA grafts fired APs in response to electrical point stimulation of the host myocardium up to 2 Hz (the fasted rate tested, Fig. 7D). Conversely, pacing the MEDUSA grafts resulted in the synchronous depolarization of the host myocardium (Supplementary Fig. 6E). This indicates that depolarizing impulses can flow bidirectionally between graft and host. Thus, MEDUSA hESC-CM grafts show electrical coupling with host myocardium that appears identical to what we have previously reported for WT hESC-CMs12,57. Moreover, we observed increased sign of maturation at 3 months after injection; indeed, the transition from MLC2a to MLC2v that was largely complete, as well as the switch from ssTnI to cTnI (Fig. 7E).
Taken together these results demonstrate that a nearly complete suppression of spontaneous electrical activity of quadruple gene edited MEDUSA hESC-CMs in vitro is associated with potent mitigation of EA in vivo. This supports the hypothesis that a series of electrical currents underlying automaticity are a primary cause of EA. The fact that multiple ion channel manipulations are required to abrogate automaticity and EA indicates complex and partially redundant roles among the resulting currents.
Discussion
Clinical translation of hPSC-CM cell therapy for heart regeneration has been complicated by transient arrhythmias following engraftment13,14,19,20. Engraftment arrhythmias (EA) were not observed in small animal models10–12, likely being masked by theirs high heart rates at baseline, but are readily observed in large animal models, where heart rates approach that of humans58. Although NHPs tolerate EA, it can be lethal in pigs19,20. In general terms, arrhythmias arise from either a defect in conduction that leads to re-entry or from the presence of abnormal depolarization (pacemaker-like activity or after-depolarization)59. Invasive electrophysiology studies in monkeys and pigs demonstrate that EA has a focal source, cannot be extinguished by overdrive pacing or direct-current (DC) cardioversion, and cannot be induced by programmed electrical stimulation14,23; these findings favor local impulse generation rather than reentry. This led us to hypothesize that currents that normally underlie automaticity in immature hPSC-CMs are culprits in generating EA. Automaticity in hPSC-CMs is incompletely understood, and the mechanisms proposed are largely based on sinoatrial node models, with additional insights drawn from computational modeling and limited in vitro experiments29,33,36. Multiple mechanisms have been proposed depending on the differentiation protocol and/or the maturation state of the hPSC-CMs29,49,50; these models involve either the voltage clock, the calcium clock, or both32–34.
To address this hypothesis experimentally, we first analyzed ion channel mRNA expression dynamics in hPSC-CMs as they matured after engraftment (Fig. 1), arguably the most powerful and physiologically relevant model of hPSC-CM maturation available to date. We used this information to identify key ion channels in hPSC-CMs, with a goal of engineering their electrophysiology towards an adult-like phenotype. Interestingly, single, double, and triple edits could reduce the beating rate and destabilize the rhythm, but they were insufficient to eliminate automaticity in vitro or EA in vivo. We eventually discovered that a quadruple edit (KO of HCN4, CACNA1H, and SLC8A1, along with overexpression of KCNJ2, which we termed MEDUSA) generated hESC-CMs that were quiescent-yet-excitable. When transplanted into the uninjured pig hearts, MEDUSA cardiomyocytes engrafted stably for 3 months, beat synchronously with the host myocardium, and markedly attenuated ventricular arrhythmias.
These findings support the hypothesis that sarcolemmal currents involved in automaticity underlie the pathogenesis of EA. Achieving electrically quiescent cardiomyocytes required manipulation of Na+, Ca2+, and K+ channels and transporters, resulting in hyperpolarized cells that lacked diastolic depolarization currents. This indicates considerable redundancy in the circuitry underlying automaticity (perhaps not surprisingly, considering its importance to life). For example, KO of HCN4 or its pharmacologic inhibition by ivabradine reduced in vitro beating rates by ~50% but eliminated neither automaticity nor EA (Fig. 2 and Supplementary Fig. 2). Layering onto the HCN4 KO the overexpression of KCNJ2 to hyperpolarize cells induced periods of electrical quiescence (Fig. 3), interrupted by irregular bursts of beating activity. These cells also caused EA after transplantation. Further layering onto these edits the KO of depolarizing genes CACNA1H or SLC8A1 similarly failed to induce full quiescence or to prevent EA (Fig. 4). Only when we combined the KO of CACNA1H and SLC8A1 with HCN4 KO and KCNJ2 overexpression we were successful in generating quiescent-yet-excitable cells with markedly reduction in arrhythmogenicity post-transplantation (Figs. 5–7 and Supplementary Figs. 6A, B). These findings support a model whereby trans-sarcolemmal movement of Na+ and Ca2+ via the HCN4 channel, the T-Type Ca2+ channel, and the sodium-calcium exchanger triggers action potentials within the hESC-CM grafts, a process that is antagonized by hyperpolarization via the IK1 current encoded by KCNJ2. Why these pacemaking-like activities accelerate after transplantation remains unknown.
While our manuscript was in revision, a relevant paper from Selvakumar et al. appeared in preprint form22. These scientists studied EA after transplanting 750M hPSC-CMs into infarcted pig hearts. They confirmed our earlier findings20 that amiodarone plus ivabradine partially suppresses EA, and they showed that ablating grafts with radiofrequency catheters could reduce EA burden (although with time EA recurred, driven from other foci). They also showed that cell preparations with a higher percentage of atrial or pacemaker-like cells, and faster in vitro beating, caused more severe EA. These observations further link the phenotype of graft cell automaticity to EA burden.
Our path to generate the MEDUSA line was non-linear, in that it involved multiple iterations of exploratory research to arrive at the final combination of gene edits (Supplementary Fig. 3A). We performed one at a time, and clonal lines were derived, qualified, and tested electrophysiologically prior to adding the next edit. Now that this combination is known, new lines can be generated much more quickly, e.g., through multiplexed editing. For example, one could perform simultaneous KO of SLC8A1 and CACNA1H and then, using the bulk-edited cells, perform a second round of editing to knock KCNJ2 into the HCN4 locus, followed by cell cloning to derive the final lines. Alternatively, the knock-out and knock-in edits could be done simultaneously in bulk, followed by clone selection. Strategies like these should markedly reduce the time from initiation of editing to final line qualification.
It is interesting to speculate how MEDUSA cardiomyocytes would compare functionally to host cardiomyocytes after maturation in situ. After transplantation, MEDUSA cells retained their ability to engraft long term, to undergo myofibril developmental maturation, and to form gap junctions and beat synchronously with host myocardium. The KO of HCN4 and CACNA1H should not affect function, since these genes are downregulated in mature cardiomyocytes16,17. Similarly, our strategy to overexpress KCNJ2 should not affect mature cell function, since the transgene locus (inside HCN4) should naturally be downregulated with maturation, simultaneously with activation of the endogenous KCNJ2 loci25,26. The impact of deleting SLC8A1 is less clear, because the NCX1 channel is the adult cardiomyocyte’s principal route of Ca2+ extrusion. Although the global deletion of Slc8a1 is embryonically lethal in mice60, mice with cardiac-specific deletion of Slc8a1 are viable, grow to adulthood, and are fertile56. These animals exhibit modestly reduced systolic function but appear to have adapted to the absence of NCX1 by reducing the amount of Ca2+ entering the cell during the AP. This suggests that adult cardiomyocytes may be less dependent on NCX1 for function48,55, and that hPSC-CMs lacking NCX1 may still be efficacious and healthy. Indeed, MEDUSA CMs are still responsive to pacing and exhibit calcium transients even after long-term engraftment (Supplementary Videos 2 and 3), suggesting the absence of calcium overload. Further studies are needed to test whether MEDUSA hESC-CMs are efficacious after transplantation into infarcted hearts.
It should be noted that MEDUSA cells are not perfectly quiescent. We occasionally observed spontaneous beating when the cells were stressed, e.g., during lactate selection or heat shock. While we do not know the source of the depolarizing current in these situations, one could imagine that stresses related to transplantation such as ischemia or inflammation could induce similar beating activity in vivo. This could underlie the brief, self-limited episodes of EA we observed when we implanted high doses of MEDUSA cells (Figs. 7A, B). Although this does not exclude other mechanisms of automaticity or arrhythmogenicity (i.e., macro-re-entry), MEDUSA cardiomyocytes have a substantially improved safety profile compared to their WT counterpart. Combining less arrhythmogenic cardiomyocytes with anti-arrhythmic pharmacology, such as amiodarone/ivabradine, may offer additive safety benefit for clinical trials.
In conclusion, these results provide new insights into the mechanisms behind automaticity of hESC-CMs and strongly support the hypothesis that EA results from pacemaker-like activity in the graft that wanes as ion channels mature toward an adult state. Although additional studies are needed to determine whether MEDUSA cardiomyocytes have retained the ability to restore systolic function, these cells represent an advance toward safely remuscularizing the injured heart.
Limitations of the study
The principal aim of this study was to understand the mechanism of engraftment arrhythmia by identifying the culprit ion channels involved in its pathogenesis. Because there is currently no in vitro or small animal model for EA, we used an in vivo pig model, transplanting cells into the normal heart to test the efficacy of our gene edits. This was an expensive and labor-intensive approach (typically with n=2 pigs/group), which limited the screen’s statistical robustness to identifying only large treatment effects. As such, the negative predictive power for arrhythmia is less than its positive predictive power in this screen. Once the MEDUSA hit was identified, we increased our animal numbers and cell doses to have greater statistical confidence in the findings. Moreover, assessing the efficacy of MEDUSA hPSC-CMs for functional remuscularization will require properly controlled and adequately powered experiments in infarcted hearts with clinically relevant cell doses.
STAR Methods text
RESOURCE AVAILABILITY
Lead contact
Further information and requests about reagents and other resources used in this work should be directed to and will be fulfilled by the lead contact: Charles E. Murry (murry@uw.edu).
Materials availability
Plasmids and cell lines generated in this study are available upon request to the lead contact and with a complete Materials Transfer Agreement.
Data and code availability
Bulk-RNA-seq data have been deposited at GEO and are publicly available as of the date of publication (accession number can be found in the key resource table). Original western blot images are included in the supplementary material as specified in the figure legends. Microscopy data reported in this paper will be shared by the lead contact upon request.
The paper does not report any original code.
Any further information required to reanalyzed the data reported in this paper is available from the lead contact upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies (working concentration, application*) | ||
| NCX1 Mouse monoclonal (dil. 1:200, WB) | Swant | RRID: AB_2716744 |
| Cardiac Troponin T Antibody Rabbit polyclonal (dil. 1:1000, WB) | Abcam | RRID: AB_956386 |
| HCN4 Mouse monoclonal (S114/10) (dil. 1:200, WB) | NovusBio | RRID: AB_2935644 |
| GAPDH Mouse monoclonal (dil. 1:3000, WB) | Abcam | RRID: AB_2107448 |
| Cardiac Troponin T-APC REA400 (dil. 1:100, FC) | Miltenyi Biotec | RRID: AB_2783887 |
| REA293-APC (dil. 1:100, FC) | Miltenyi Biotec | RRID: AB_2733446 |
| Mouse monoclonal anti-CD56-PE (dil. 1:5, FC) | BD Bioscience | RRID: AB_395906 |
| Mouse monoclonal anti-PDGFRα-APC (dil. 1:10, FC) | R&D System | RRID: AB_883910 |
| Mouse monoclonal IgG1-PE (dil. 1:5, FC) | BD Biosceince | RRID: AB_396091 |
| Mouse monoclonal IgG1-APC (dil. 1:10, FC) | R&D System | RRID: AB_398576 |
| Rabbit monoclonal anti NKX2.5 (dil. 1:200, FC) | Cell Signaling | RRID: AB_2935645 |
| Rabbit IgG1 (dil. 1:500, FC) | Cell Signaling | RRID: AB_1031062 |
| Slow Skeletal Troponin I Mouse monoclonal (dil. 1:200, IHC/IF) | Novus Biological | RRID: AB_2935646 |
| β-Myosin Heavy chain Mouse A4.951 (dil. 1:1, IHC/IF) | Developmental Studies Hybridoma Bank | RRID: AB_528385 |
| Biotin-SP-conjugated AffiniPure Goat anti-Mouse IgG (dil. 1:500, IHC) | Jackson Immunoresearch | RRID: AB_2338557 |
| Cardiac Troponin I Rabbit Monoclonal (dil. 1:100, IF) | Abcam | RRID: AB_869983 |
| MLC2a Mouse monoclonal (dil. 1:500, IF) | BD Life Science | RRID: AB_2739265 |
| MLC2v Rabbit polyclonal (dil. 1:100, IF) | Proteintech | RRID: AB_2147453 |
| Connexin43 Rabbit polyclonal (dil 1:200, IF) | Merck | RRID: AB_476857 |
| Desmin goat polyclonal (dil. 1:100) | Origene | RRID: AB_2935647 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 (dil. 1:1000, IF) | Life Technologies | RRID: AB_141436 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (dil. 1:1000, IF/FC) | Life Technologies | RRID: AB_143165 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 (dil. 1:1000, IF) | Life Technologies | RRID: AB_141844 |
| *WB: Western blotting, FC: flow cytometry; IF: Immunofluorescence, IHC: Immunohistochemistry | ||
| Bacterial and virus strains | ||
| 𝛼-Select Gold Efficiency Chemically competent cells | BIOLINE | Cat # BIO-85027 |
| Chemicals, peptides, and recombinant proteins | ||
| Fast Digest Bpil | Thermo Scientific | Cat # FD1014 |
| Fast Digest Mph1103I (NsiI) | Thermo Scientific | Cat # FD0734 |
| Fast Digest Pfi23II (BsiWI) | Thermo Scientific | Cat # FD0854 |
| Fast Digest SgsI (AscI) | Thermo Scientific | Cat # FD1894 |
| Fast Digest NotI | Thermo Scientific | Cat # FD0593 |
| Fast Digest ScaI | Thermo Scientific | Cat # FD0434 |
| Fast Digest BamHI | Thermo Scientific | Cat # FD0054 |
| Fast Digest Bsu15I (ClaI) | Thermo Scientific | Cat # FD0143 |
| Fast Digest NcoI | Thermo Scientific | Cat # FD0575 |
| T4 DNA Ligase Reaction Buffer | New England Bio-Labs | Cat # B0202S |
| Quick Ligation Kit | New England Bio-Labs | Cat # M2200S |
| T4 Polynucleotide Kinase | New England Bio-Labs | Cat # M0201S |
| Ampicillin sodium salt | Sigma-Aldrich | Cat # A0166 |
| LB Agar, Miller (Luria-Bertani) | BD Life Sciences | Cat # 244520 |
| LB Broth, Miller (Luria-Bertani) | BD Life Sciences | Cat # 244620 |
| Granulated Agar | BD Life Sciences | Cat # 214530 |
| OPTI-MEM Reduced Serum Medium | Gibco | Cat # 31985062 |
| GeneJuice Transfection Reagent | Sigma-Aldrich | Cat # 70967–3 |
| DPBS, no calcium, no magnesium | Gibco | Cat # 14190–250 |
| mTeSR Plus | Stem Cell Technologies | Cat # 100–0276 |
| Essential 8™ Medium | Thermo Scientific | Cat # A1517001 |
| Matrigel Growth Factor Reduced (GFR), Phenol Red-free, LDEV-free | Corning | Cat # 356231 |
| rLaminin-521 (human) | Corning | Cat # 354221 |
| Y-27632 dihydrochloride | Tocris | Cat # 1254–50 |
| Versene Solution | Gibco | Cat # 15040066 |
| TrypLE Select | Gibco | Cat # A12177–02 |
| RPMI 1640 medium | Gibco | Cat # 11875119 |
| RPMI 1640 medium, no glucose | Life Technologies | Cat # 11879020 |
| RPMI 1640 medium, no phenol red | Life Technologies | Cat # 11835055 |
| Bovine Serum Albumin, suitable for cell culture | Sigma-Aldrich | Cat # A9418–50G |
| L-Ascorbic Acid 2-phosphate sesquimagnesium salt hydrate | Sigma-Aldrich | Cat # A8960–5G |
| CHIR99021 | Cayman | Cat # 13122 |
| Wnt-C59 | Selleck Chemicals | Cat # S7037 |
| Sodium L-lactate | Sigma-Aldrich | Cat # L7022–10G |
| B-27 Supplement (50X), serum free | Gibco | Cat # 17504044 |
| CryoStor cell cryopreservation media - CS10 | Sigma-Aldrich | Cat # C2874 |
| Q5 High-Fidelity 2X Master Mix | New England Bio- Labs | Cat # M0492S |
| M-MLV Reverse Transcriptase | Invitrogen | Cat # 28025013 |
| RNaseOUT Recombinant Ribonuclease Inhibitor | Invitrogen | Cat # 10777019 |
| SYBR Select Master Mix | Applied Biosystems | Cat # 4472913 |
| SpCas9 2NLS Nuclease | Synthego | NA |
| 4–15% Mini-PROTEAN TGX Precast Protein Gel | Biorad | Cat # 4561084 |
| Immobilon-P membrane (0.45 μm) | Millipore | Cat # IPVH00010 |
| Hematoxylin Solution (Mayer’s, Modified) | Abcam | Cat # ab220365 |
| Hoechst 33342 Solution | Thermo Scientific | Cat # 62249 |
| DNase I | Millipore | Cat # 260913 |
| Fluo-4 AM | Invitrogen | Cat # 14201 |
| Ivabradine hydrochloride | Tocris | Cat # 6542 |
| ML-218 hydrochloride | Tocris | Cat # 4507 |
| Mibefradil dihydrochloride | Tocris | Cat # 2198 |
| Zacopride hydrochloride | Tocris | Cat # 1795 |
| SEA0400 | Tocris | Cat # 6164 |
| KB-R7943 mesylate | Tocris | Cat # 1244 |
| Verapamil hydrochloride | Tocris | Cat # 0654 |
| Cyclosporine A | Cayman | Cat # 12088 |
| Buprenorphine SR-Lab | ZooPharm | NA |
| Euthasol | Virbac | NA |
| Abatacept (CTLA4-Ig) | Bristol-Myers Squibb | NA |
| 4% Paraformaldehyde | Santa Cruz Biotechnology | Cat # sc-281692 |
| Membrane-coated slides | Leica Microsystem | Cat #11600289 |
| VECTASTAIN Elite ABC HRP kit | Vector Laboratories | Cat # PK-6100 |
| SIGMAFAST™ 3,3′-Diaminobenzidine tablets | Sigma-Aldrich | Cat # D4168 |
| HEPES | Fisher | Cat # BP310–500 |
| D-(+)-Glucose | Sigma-Aldrich | Cat # G5767 |
| EGTA | Sigma-Aldrich | Cat # E3889 |
| ATP Magnesium Salt | Sigma-Aldrich | Cat # A9187 |
| Amphotericin B | Sigma-Aldrich | Cat # A9528 |
| Tetraethylammonium Chloride | Sigma-Aldrich | Cat # T2265 |
| 4-Aminopyridine | Sigma-Aldrich | Cat # 275875 |
| Tetraethylammonium Hydroxide | Sigma-Aldrich | Cat # 177806 |
| Cesium Chloride | Sigma-Aldrich | Cat # 289329 |
| GTP Sodium Salt Hydrate | Sigma-Aldrich | Cat # G8877 |
| Cesium Hydroxide Monohydrate | Sigma-Aldrich | Cat # 516988 |
| Critical commercial assays | ||
| QIAEX II Gel Extraction Kit | Qiagen | Cat # 20021 |
| QIAprep Miniprep Kit | Qiagen | Cat # 27104 |
| QIAfilter Plasmid Midiprep Kit | Qiagen | Cat # 12243 |
| BD StemflowTM Human and Mouse Pluripotent Stem Cell Analysis Kit | BD Life Sciences | Cat # 560477 |
| RNAesy Mini kit | Qiagen | Cat # 74106 |
| PicoPure® RNA Isolation Kit | Applied Biosystems | Cat # KIT0204 |
| Leica Laser Microdissection (LMD) System | Leica Microsystem | Cat # 8118616 |
| Pierce BCA Protein assay kit | Thermo Scientific | Cat # 23225 |
| NEBuilder HiFi DNA Assembly Cloning kit | New England Bio- Labs | Cat # E5520S |
| Maestro Pro multiwell microelectrode array (MEA) | Axion Biosystems | Cat #CP-MCV48W-UPF |
| Deposited data | ||
| RNA-seq data set | This paper | GSE190758 |
| Experimental models: Cell lines | ||
| Human induced-pluripotent Stem Cells – 253G1 -Camp3 | Kyoto University | CVCL_B51828 |
| Human Embryonic Stem Cells – RUESe002-A | Rockefeller University | CVCL_C1W369 |
| Experimental models: Organisms/strains | ||
| Athymic male Sprague Dawley rats (Hsd:RH-Foxn1rnu) | Envigo | RGD_5508395 |
| Yucatan minipigs | Premier BioSource | NA |
| Oligonucleotides (see also Methods Tables 1, 2) | ||
| HCN4_gRNA1 TCGTGAAGCGGACAATGCGC | This paper | NA |
| CACNA1H_gRNA1 GGATTTCTTCATCGTCGTGG | This paper | NA |
| CACNA1H_gRNA2 GACCATCTCCACCGCAAAAA | This paper | NA |
| HCN4_gRNA1_KI CAGCTTGTCCATGGCGCCAG | This paper | NA |
| HCN4_gRNA2_KI GGCAGCTTGTCCATGGCGCC | This paper | NA |
| SLC8A1_gRNA1 GGATCATATTACTGTAAGAA | Synthego | NA |
| SLC8A1_gRNA2 CAGCAATTACATGGTCCACA | Synthego | NA |
| SLC8A1_gRNA3 TGAAATCCCATTGAAAAGGT | Synthego | NA |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (pX459) V2.0 | Addgene | Addgene_6298870 |
| AAV_CAGGS_EGFP | Addgene | Addgene_2221271 |
| MV-PGK-Puro-TK | Hera BioLabs | Cat # SGK-0062 |
| KCNJ2 cDNA cloning fragment (RefSeq: NM_000891.3) | IDT | NA |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | Addgene_42230 |
| Software and algorithms and equipment | ||
| Guidescan (https://guidescan.com/) | Ventura lab | 72 |
| Cas-OFFinder (http://www.rgenome.net/cas-offinder/) | Kim lab | 73 |
| Axis Navigator 2.0.3 | Axion Biosystem | NA |
| Prism 8.4.2 | GraphPad | NA |
| FlowJo V9 | BD Life Sciences | NA |
| SnapGene 5.0.8 | SnapGene | NA |
| ImageJ | Fiji | NA |
| ecgAUTO 3.3.5.10 | EMKA Technologies | NA |
| Graft quantification analysis | This paper | 20 |
| Calcium analysis Fluo4 MatLab | Sniadecki Lab | 62 |
| pClamp 11.1 | Molecular Devices | NA |
| Multiclamp 700B | Molecular Devices | NA |
| Digidata 1550B | Molecular Devices | NA |
| Patchmaster v2×73 | HEKA Elektronik | NA |
| Fitmaster v2×91 | HEKA Elektronik | NA |
| Other | ||
| Karyotype analysis | Diagnostic Cytogenetics, Seattle WA | NA |
| ChemiDoc Imaging system | Biorad | NA |
| Vibratome 7000smz-2 | Camden Instrument | NA |
| NOGA-MyoStar platform | BioSense Webster | NA |
| PBS-0.5 MAG Single-Use Vessel | PBS Biotech | Cat #IA-0.5-D-001 |
| PBS-Mini Mag Drive Bioreactor Base Unit | PBS Biotech | Cat #FA-UNI-B-501 |
| Neon Transfection System | Life Technologies | Cat # MPK5000 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
hPSC-CMs culture and differentiation (monolayer and suspension)
Human induced pluripotent stem cells (hiPSCs, 253G1-Camp3, Kyoto University, JP) and human embryonic stem cells (hESCs, RUES2e002-A, Rockefeller University, USA) were maintained and differentiated into cardiomyocytes as previously described61. Briefly, hiPSCs and hESCs were cultured in mTeSR Plus on Matrigel-coated plates (0.17 mg/mL) and passaged with 10μM Y-27632 when ~70% confluency was reached using Versene. For monolayer cardiac differentiation, ~90%-confluent hiPSCs/hESCs were primed with mTeSR Plus supplemented with 1 μM CHIR99021 for 24 hours. On day 0 of differentiation, mesoderm was induced using a cell line-optimized concentration of CHIR99021 (range: 3–5 μM) in RPMI 1640 supplemented with 213 μg/mL ascorbic acid and 500 μg/mL bovine serum albumin (RBA media). After 48 hours, on day 2 of differentiation, cells were washed with DPBS and cardiac progenitors were induced with 2 μM Wnt-C59 in RBA. After 48 hours, on day 4, cells were washed again with DPBS and incubated with plain RBA media for an additional 48 hours. From day 6 to day 10 cardiomyocytes were maintained in RPMI 1640 supplemented with Penicillin-Streptomycin and B-27 (RPMI-B27 media), performing media changes every 48 hours. Lactate selection was performed from day 10 through 14 by culturing cells in RPMI without glucose supplemented with 4 mM sodium L-lactate, with a media change after 48 hours. Media was changed with RPMI B-27 media until heat shock. The day before cryopreservation (Day 16–20), hPSC-CMs were heat-shocked for 30 min at 42°C. The following day, hPSC-CMs were dissociated using 5X TrypLE in Versene, washed in RPMI B-27, frozen in Cryostor CS10 at cell density of 3 × 107 cells/mL, and stored in liquid nitrogen.
As listed in Supplementary Table 2, WT controls, HCN4 KO, HCN4 KO/KCNJ2 KI, HCN4/CACNA1H 2KO/KCNJ2 KI hESC-CMs were differentiated and cryopreserved in suspension cultures, as previously described20. This same procedure was followed to generate MEDUSA hESC-CMs for high-dose animals described in Fig. 7. Briefly, pluripotent aggregates were expanded in suspension culture using Essential 8 media; cardiomyocytes differentiation was induced on day 0 using cell-line optimized CHIR99021 (range: 5–6 μM) for 24 hours as described in14. WNT pathway inhibition was performed at timing optimized empirically for each cell line. When cardiac state was reached (~ day 6 of differentiation), suspension cultures were fed using RPMI 1640, supplemented with B-27; purified between day 10 – 14 using lactate selection media (as described for monolayer differentiation) and heat-shocked the day before harvesting. HCN4 KO/KCNJ2 KI and HCN4/CACNA1H 2KO/KCNJ2 KI hESC-CMs were differentiated by the same method until the fourth day of differentiation, at which point aggregates were dissociated with TrypLE, plated on rhLaminin-521 (BioLamina), and maintained as adherent cultures for the remainder of the differentiation protocol.All cells were harvested enzymatically 17–22 days after initiating differentiation and cryopreserved in CryoStor CS10.
Athymic Sprague Dawley rats (Hsd:RH-Foxn1 rnu )
All protocols were approved and conducted in accordance with the University of Washington (UW) Office of Animal Welfare and the Institutional Animal Care and Use Committee. Eight-ten weeks old male athymic rats (240–300 g) were housed and monitored by the Department of Comparative Medicine at UW, received ad libitum water and food. For all surgical procedures, rats were anesthetized by intraperitoneal injection of 68.2 mg/kg ketamine and 4.4 mg/kg xylazine (first thoracotomy) or isoflurane by inhalation (second thoracotomy), intubated and mechanically ventilated. Rats received analgesic (buprenorphine 0.05 mg/kg) twice daily for 48 hours after each surgery. Cyclosporine A (5 mg/kg/day) was given for 7 days beginning the day before transplantation to prevent graft cell death and euthanized by intravenous Euthasol.
Yucatán mini pigs
All protocols were approved and conducted in accordance with the University of Washington (UW) Office of Animal Welfare and the Institutional Animal Care and Use Committee. Castrated male, Yucatán minipigs between 30–40 kg were used in this study. Animals received ad libitum water and were fed twice a day (Lab Diet-5084 Laboratory Porcine Grower Diet). For surgical procedures, anesthesia was induced with a combination of intramuscular butorphanol, acepromazine and ketamine. Animals were intubated and mechanically ventilated using isoflurane and oxygen to maintain a surgical plane of anesthesia. Vital signs were monitored continuously throughout each procedure. All animals received subcutaneous Buprenorphine SR-Lab for post-operative analgesia and were euthanized by intravenous Euthasol. All post-mortem examinations were performed by a blinded board-certified veterinary pathologist. Immunosuppression regimen consisted of oral cyclosporine, which was titrated to a goal serum trough of 300 ng/mL for the duration of the study. Two days before transplant, oral methylprednisolone was started at 1.5 mg/kg for 2 weeks, and then was decreased to 1 mg/kg for the remained of the study. On the day of cell transplantation, Abatacept (CTLA4-Ig, Bristol-Myers Squibb), 12.5 mg/kg, was administered intravenously and dosed every 2 weeks thereafter. Prophylactic cephalexin (500 mg PO BID) was initiated at the time of central venous catheter placement and sulfamethoxazole/trimethoprim (50 mg/kg PO daily), valganciclovir (450 mg PO daily) and probiotics five days prior to transplant.
METHOD DETAILS
In vitro experiments for hPSCs editing and hPSC-CMs characterization.
Flow cytometry assessment of hPSC-CM differentiation and purity.
Flow cytometry analysis to assess pluripotency of hESCs (expressed as OCT-4 %) was performed after every round of gene-editing and at day 0 of cardiac differentiation using from BD StemflowTM Human and Mouse Pluripotent Stem Cell Analysis Kit, according to manufacturers’ instruction. For mesoderm markers (CD56 and PDGFRα), an aliquot of hESCs at day 4–5 of cardiac differentiation were resuspend in DBPS supplemented with 5% bovine serum albumin (blocking buffer) and incubated with APC-/PE-conjugated antibodies or isotype controls diluted in blocking buffer for 1 hour at room temperature. Cells were then washed with DPSB and resuspend in blocking buffer before flow cytometry analysis. For cardiac markers (cardiac troponin T [cTnT] and NKX2.5), hESC-CMs were fixed in BD Cytofix (BD Life Science) and permeabilized in BD Wash/Perm Buffer (BD Life Science) supplemented with 0.1% Triton-X100 for 5 minutes at room temperature. HESC-CMs were then incubated with NXK2.5 primary antibody/isotype control diluted in BD Wash/Perm Buffer for 1 hour at room temperature. Cells were then wash twice with DPBS and resuspended in BD Wash/Perm Buffer containing APC-conjugated cTnT antibody or isotype control and Alexa Fluor anti-rabbit 488. After 1-hour incubation, hESC-CMs were resupsended in BD Wash/Perm Buffer and analyzed by flow cytometry.
Gene-editing for HCN4 and CACNA1H KO.
For HCN4 and CACNA1H KO, single gRNAs targeting key coding exons were cloned in pX459V2 plasmid (which includes the SpCas9 coding sequence driven by the CMV enhancer, see Supplementary Table 3 for gRNAs sequence) according to the Zhang lab’s protocol and verified by Sanger sequencing. Plasmids for gene editing were prepared using QIGEN Midi prep kit. RUES2 hESCs were seeded at 15,000 cells/cm2 in mTeSR Plus supplemented with 10 μM Y-27632, and transfected using 6 μL of GeneJuice and 2 μg of pX459V2_gRNA plasmid in 100 μL of OPTI-MEM. After 24 hours, cells were washed with DPBS and incubated with mTeSR Plus supplemented with 5 μM Y-27632 and 0.5 μg/mL puromycin for 48 hours. Cells were subsequently washed with DPBS and maintained as described above. Single clones were isolated using a limiting dilution protocol by seeding single cells at 0.5 cell/well in 96-well cell culture plates. Genotype analysis was performed on clonal lines using Sanger sequencing of PCR amplicons obtained using the primer listed in Methods Table 1 and Q5 High-Fidelity Master Mix. When a mixed sequencing trace was observed, amplicons were TOPO-cloned and Sanger sequencing was performed on at least 48 individual bacterial clones to determine edits on individual alleles. Standard G-banding analysis was performed on undifferentiated cells to confirm absence of karyotype abnormalities. CRISPR/Cas9 off-target analysis was performed using Cas-OFFinder software (cut-off: up to 4 mismatches with 1 bulge on either DNA or RNA sequence) and potential off-targets in an exon of a gene expressed in hESC-CMs were selected for genotyping by Sanger sequencing to exclude the presence of indels.
Gene-editing for KCNJ2 KI in HCN4 locus
Knockin of KCNJ2 in the HCN4 locus was performed through CRISPR/Cas9 homology-directed repair (HDR). The donor vector was generated using the NEBuilder Hifi DNA Assembly kit. 3’ and 5’ HCN4 homology arms (3’/5’-HAs) were amplified by PCR from hESCs RUES2 genomic DNA, while an SV40 polyA fragment was amplified from AAVS1_CAGGS_EGFP (all using primers listed in Supplementary Table 3). The Puro-T2A-TK cassette was isolated from on MV-PGK-Puro-TK plasmid through restriction digestion using NsiI and BsiWI. The same MV-PGK-Puro-TK plasmid was digested with NotI and AscI to isolate a backbone vector containing transposon inverted tandem repeats and ampicillin resistance genes. The donor plasmid resulting from this assembly (pbHCN4) and a synthetic fragment carrying the KCNJ2 cDNA were then digested with ClaI and NcoI restriction enzymes and ligated using Quick Ligation kit. To generate a control vector, the same procedure was performed but using EGFP amplified from AAVS1_CAGGS_EGFP. The resulting targeting plasmids (pbHCN4_KCNJ2 or pbHCN4_GFP) were amplified as described above. CRISPR/Cas9 plasmids designed to induce double strand breaks necessary to drive HDR were obtained by cloning two gRNAs targeting the HCN4 promoter (HCN4_gRNA1_KI and HCN4_gRNA2_KI, see Supplementary Table 3) in pX330-U6-Chimeric_BB-CBh-hSpCas9, all according to Zhang lab’s protocol and amplified as described above. RUES2 hESCs were then transfected with 1 μg of donor plasmid (either pbHCN4_KCNJ2 or pbHCN4_GFP) and 0.5 μg for each of pX330_HCN4gRNA1_KI and pX330_HCN4gRNA2_KI using GeneJuice as described above. After puromycin selection, single clones were obtained through limiting dilution and genotyped through PCR. On site integration of the transgene cassette downstream of the HCN4 locus was confirmed using primers mapping on the cassette and on the locus but outside of the homology arm; the number of gene edited loci was determined by amplifying the wild-type allele (resulting in loss of amplification in homozygous edited cells); absence of random integrations of the targeting plasmids were confirmed using primers mapping to the insert and plasmid backbone. Karyotype analysis was performed to confirm absence of abnormalities, and CRISPR/Cas9 offtargets were excluded as described above.
Gene-editing for SLC8A1 KO series
To induce SLC8A1 KO, hESCs were electroporated with CRISPR/Cas9 ribonucleoprotein complexes targeting exon 1 of SLC8A1. Briefly, 500,000 RUES2 hESCs were resuspended in a mixture of 60 pmol of SpCas9 2xNLS nuclease (Synthego) and 60 pmol of SLC8A1 gRNAs mix (20 pmol/gRNA, Supplementary Table 3) and 300 μL of Neon Buffer R. Cells were then electroporated (1 pulse at 1,300 V for 30 milliseconds) using a Neon electroporation system, and immediately replated on Matrigel-coated 6-well plate in mTeSR1 supplemented with 10 μM Y-27632. SLC8A1 KO clones were isolated through limiting dilution and genotyped by PCR and Sanger sequencing. Karyotype analysis was performed to confirm absence of abnormalities, and CRISPR/Cas9 off-targets were excluded as described above.
Gene expression analysis via quantitative real-time PCR
Total RNA was extracted from day 14 RUES2 CMs from the different cell lines using RNAesy Mini kit according to manufacturer’s instruction. Single-stranded cDNA was obtained by reverse transcription using M-MLV RT kit, and quantitative real-time reverse transcription PCR (RT-qPCR) was performed with SYBR Select Master Mix using 10 ng of cDNA and 400 nM forward and reverse primers (Supplementary Table 4). Reactions were run on a CFX384 RealTime System (Biorad), and data was analyzed using the ΔΔCt method using HPRT1 as the housekeeping gene. Primers were designed using PrimerBlast and confirmed to amplify a single product.
Western Blotting
Day 14 hESC-CMs WT and SLC81A KO clone series and MEDUSA hESC-CMs clones were incubated with ice-cold RIPA Buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris·HCl, 1X protease inhibitors) for 30 minutes at 4°C. Protein lysates were cleared by centrifugation at 21,000 rcf for 15 minutes at 4 °C and quantified by BCA assay. 30 μg of protein per sample were incubated in 1X non-reducing sample buffer for 30 minutes at 37 °C, run on a 4–20% Mini-Protean gel and transferred on a PVDF membrane. Membranes were incubated with 5% BSA in TBS buffer supplemented with 0.1% Tween-20 (blocking buffer) for 1 hour at room temperature. Primary antibodies were incubated in blocking buffer for 2 hours at room temperature; membranes were then washed three times in blocking buffer and incubated for 1 hour with fluorophore-conjugated secondary antibodies. Fluorescent signals were acquired using with a GelDoc Imager.
Spontaneous electrical activity measurements with MEA system and during differentiation
For MEA, CytoView MEA 48 well plates were coated with 0.17 mg/mL of Matrigel for 1 hour at 37 °C. 50,000 hESC-CMs were resuspended in 6 μL and plated on each MEA well, as previously described5. Media was changed with RPMI-1640 supplemented with B-27 every other day for 1 week. For spontaneous beat recording, spontaneous electrical activity was recorded for 5 minutes using Maestro Pro system at 37 °C with 5% CO2. Considering the irregular beat rate of the cell lines bearing KCNJ2 KI, the data are shown as total number of beats recorded per experiment, divided by the total time of recording (average beats/min). For pharmacological studies, the different compounds were diluted in DMSO and incubated directly on MEA plates for 5 minutes at 37°C. Effects on spontaneous beat rate was determined by recording for 15 minutes after drug incubation. A non-linear regression curve was calculated as Y=100/(1+10^((LogIC50-X)*HillSlope))). Voltage was acquired simultaneously for all the electrodes at 12.5 kHz, with a low-pass digital filter of 2 kHz for noise reduction. Quantification of beat rate, spike amplitude and beat period irregularity was performed using Axis Navigator (beat detector threshold: 30–100 μV; minimum beat period: 200–500 milliseconds; maximum beat period: 30 seconds).
For quantification of spontaneous electrical activity/onset of beating during monolayer differentiation, 12-wells plates with hESCs were monitored from day 0 to day 18 or 35 of cardiac differentiation; spontaneous contraction was quantified as percentage of beating wells observed for 5 minutes.
Patch-clamp Electrophysiology
Single hESC-CMs (Day 14 – 21) were seeded on laminin-coated glass coverslips at a density of 13–18.5k cells/cm2. Perforated-patch action potential (AP) recordings were conducted in current-clamp mode using borosilicate glass pipettes with typical resistances of 2 – 4 MΩ. Data were acquired at 5 kHz and filtered at 2 kHz using a Multiclamp 700B amplifier, Digidata 1550 digitizer, and Clampex 11 software. Experiments were performed 35 ± 1 °C under continuous perfusion of Tyrode’s solution containing (in mM): 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose and 10 HEPES, with the pH adjusted to 7.4 with NaOH. The pipette solution contained (in mM): 150 KCl, 5 NaCl, 5 MgATP, 10 HEPES, 5 EGTA, 2 CaCl2, and 240 μg/mL of amphotericin B, with the pH adjusted to 7.2 with KOH. Evoked action potentials were recorded using 5 milliseconds current injection pulses (0.3 – 0.5 nA) at a frequency of 1Hz. Spontaneous action potential parameters were determined from 30 seconds recordings, while stimulated AP parameters were determined from 10 second recordings. APs were analyzed using Clampfit 11 software.
For If, currents were recorded in the modified Tyrode solution (with 0.2 mmol/L CdCl2) at room temperature. From the holding potential of −40 mV, If currents were elicited by 2-s test pulses from −120 to −50 mV at 10-mV increments. If currents were defined as 0.5 mmol/L Ba2+-insensitive and 5 mmol/L Cs+-sensitive currents. Modified Tyrode solution was composed of (mM):120 NaCl, 20 KCl, 10 HEPES, 10 Glucose, 2 CaCl2, and 1 MgCl2, pH adjusted to 7.4 with NaOH. The pipette solution contained (mM): 100 KCl, 10 NaCl, 14 EGTA, 10 HEPES, 5 MgATP, 1 CaCl2; pH adjusted to 7.2 with KOH.
To record ICaT, whole-cell Ca2+ currents were recorded using pipettes with resistances between 1.5 and 3 MΩ. Data was acquired at 20 kHz and filtered at 1kHz using a HEKA EPC10 amplifier and Patchmaster v2×73 software. Experiments were performed at room temperature in extracellular solution containing (in mM): 130 TEA-Cl, 2 CaCl2, 1 MgCl2, 10 4-aminopyridine, and 25 HEPES, with the pH adjusted to 7.4 with TEA-OH. The pipette solution contained (in mM): 130 CsCl, 10 EGTA, 25 HEPES, 3 Mg-ATP, and 0.4 NaGTP, with the pH adjusted to 7.2 with CsOH. Total Ca2+ current (ICa) was elicited by 100 milliseconds depolarizing voltage steps between −80 and +70 mV, in +10 mV increments, following 3 seconds at a holding potential of −90 mV. L-type Ca2+ current (ICaL) was elicited by 100 milliseconds depolarizing voltage steps from −80 to +70 mV following 3 seconds at a holding potential of −50 mV. ICaT was calculated by subtracting ICaL from ICa. Series resistance was compensated at least 60%. Voltages were corrected for a −8 mV liquid junction potential. Data analysis was performed using Fitmaster v2×91 software.
Calcium transient analysis
HESC-CMs were seeded at 5,000 cells/cm2 in Matrigel-coated 6-well plates with RPMI-1640 supplemented with B-27 and penicillin-streptomycin. Cells were fed every other day for 1 week before incubation with 1 μM Fluo-4 AM for 30 min at 37 °C. Calcium transients were acquired while the culture was electrically paced at 1 Hz (0.025 seconds pulse duration, 40 mV) using an IonOptix pacing system and analyzed with custom MATLAB code62.
Rodents and pig surgeries and in vivo experiments.
Cell injection preparation (rodent surgery)
hiPSC-CMs used in rodent surgery were cryopreserved on day 18–20 of differentiation, and thawed immediately prior to cell injection, all following our previously described protocol28,57,63. One day prior to cryopreservation, cells were heat shocked for 30 min at 42°C. Prior to enzymatic dispersion, the ROCK inhibitor Y-27632 (10 μM) was added to culture medium for 1 hour, and cells were dispersed by incubation with Versene followed by 0.05% Trypsin in EDTA. hiPSC-CMs were re-suspended in CryoStor cell preservation media and frozen in cryovials in a controlled rate freezer to −80°C before being stored in liquid nitrogen. To thaw cryopreserved cells, cryovials were thawed briefly at 37°C followed by addition of RPMI+B27+insulin with Y-27632 (10 μM). Cells were washed with PBS and re-suspended in an RPMI-based pro-survival cocktail63 containing 50% growth factor-reduced Matrigel, 100μM ZVAD (benzyloxycarbonyl-Val-Ala-Asp(O-methyl)-fluoromethyl ketone, Calbiochem), 50 nM Bcl-XL BH4 (cell-permeant TAT peptide, Calbiochem), 200 nM cyclosporine A (Novaritis), 100 ng/mL IGF-1 (Peprotech), and 50 μM pinacidil (Sigma).
Rodent surgery and laser-capture microscopy
Eight-ten weeks old male athymic rats (240–300 g) were anesthetized as described in “Experimental model and subject details” section. A thoracotomy exposed the heart and LAD was occluded for 60 minutes, reperfused, and the chest was closed. Four days after ischemia/reperfusion injury, rats were anesthetized by isoflurane inhalation, and mechanically ventilated. The heart was exposed via a second thoracotomy and 10 × 106 hiPSC-CMs were injected to the center of infarcted left ventricle wall (3–4 injections, 30 μl each).
RNA-seq preparation and sequencing
For in vitro samples, total RNA was isolated using the RNeasy Mini Kit according to the manufacturer’s protocol, including DNase treatment. For in vivo samples, rat hearts were harvested, sliced, immediately embedded in TissueTek® O.C.T. Compound (VWR), and stored at −80 °C. Tissues were sectioned at 10 μm thickness using a cryostat (Leica), and serial sections were mounted with 4ʹ, 6-diamidino-2-phenylindole (DAPI) to visualize the graft area through green fluorescent protein autofluorescence. The graft areas were captured from unstained unfixed specimens attached to membrane-coated using a laser capture microdissection system. Total RNA was extracted from the graft areas in rat heart tissues using the Arcturus PicoPure RNA isolation kit. RNA integrity (RNA integrity number equivalent ≥ 7) and quantity were determined using an Agilent 4200 Tapestation (Agilent Technologies). cDNA was synthesized using the SMART-Seq v4 ultra low input RNA kit for sequencing (Takara Bio). Library preparations were conducted using Nextera XT DNA Library Preparation Kit (Illumina) according to manufacturer’s instructions. RNA-seq libraries were sequenced on an Illumina Next-Seq 500 with single end configuration (72 bp). Reads were mapped to a custom library, a hybrid of human (hg38) and rat (Rnor_6.0) genomes using STAR aligner64. Reads that mapped specifically and uniquely to the human genome were used for downstream analysis using Cufflinks65. Reads that mapped uniquely to the rat or to both rat and human genomes were not used. Gene ontology analysis was performed using topGO R package66.
Histology (rodent heart)
Rat hearts of each time point were collected, sliced as described above, and immediately embedded in an OCT-embedding compound and stored at −80 °C. Tissues were sectioned at a thickness of 10 μm using a Cryostat (Leica), fixed in 4% paraformaldehyde for 5 minutes and stained with hematoxylin and eosin.
Cell injection preparation (pig surgery)
The day of cell transplant, hESC-CMs were thawed in RPMI 1640 without phenol red supplemented with 500 μg/mL bovine serum albumin and 100 KU/mL of DNAse I. Thawed hESC-CMs were collected by centrifugation at 400 g for 5 min at 4°C. hESC-CMs were washed twice with RPMI 1640 without phenol red and resuspended at 0.2 × 106 hESC-CMs/μL for percutaneous trans-endocardial transplant or 5 syringes for direct epicardial injection20. Syringes were individually packed in sterile containers and kept on ice until the moment of injection.
Pig surgery
As listed in Supplementary Table 2, WT controls, HCN4 KO, HCN4 KO/KCNJ2 KI and HCN4/CACNA1H 2KO/KCNJ2 KI hESC-CMs recipients underwent cell transplantation via percutaneous trans-endocardial injection using the NOGA-MyoStar platform, as previously described without myocardial infarction20. Briefly, percutaneous transplantation was performed by first mapping the left ventricle and then deliver five discrete endocardial injections of 150 μL each for total dose of 150 × 106 hESC-CMs to the anterior wall. Injections were only performed with excellent location and loop stability, ST-segment elevation and presence of premature ventricular contraction (PVC) with needle insertion in an appropriate location by electroanatomical map.
Cell transplantation for the remaining edits, namely SLC8A1/HCN4 2KO/KCNJ2 KI and MEDUSA hESC-CMs, were performed by direct transepicardial surgical injection as previously described without myocardial infarction20 due to discontinuation of the NOGA-MyoStar device by the manufacturer or availability of a suitable alternative platform for percutaneous delivery. Briefly, transepicardial injection via partial median sternotomy was performed to expose the anterior left ventricle. Purse-string sutures were preplaced at five discrete locations subtended by the LAD. After cinching the purse-string tightly around the needle, three injections of 150 μL each were performed by partial withdrawal and lateral repositioning, for a total of 15 injections to deliver total dose of 150 × 106 hESC-CMs (most experiments) or 500 × 106 hESC-CMs for high-dose experiments with MEDUSA hESC-CMs. The pericardium was closed by primary repair and the sternum and chest reapproximated using standard surgical technique.
Telemetry characterization for EA and endpoint description
Telemetric ECG was continuously monitored in real-time from the time of transplant to detect arrythmia as previously described20. Briefly, semi-automated quantification of heart rate and arrhythmia was performed by a board-certified cardiologist using the ecgAUTO 3.5.5.28 software package (EMKA Technologies). EA was defined as sustained ventricular tachycardia lasting ≥ 30 seconds. Premature ventricular contractions (PVCs) were defined as 1 or 2 ectopic ventricular contractions and non-sustained ventricular tachycardia (NSVT) as ≥ 3 consecutive PVCs but < 30 seconds in duration. The original endpoint screening was set at day 14 after injection; to ensure absence of EA recrudescence, we extended the period of observation to 4–7 weeks (for 150 × 106 hESC-CMs dose) and 3 months (for 500 × 106 hESC-CMs dose). For control animals, the endpoints at 35- and 49-days post injection were chosen to support ex vivo electrophysiology studies of EA. Animals were removed from further study when they met prespecified endpoints defined as either spontaneous death (from arrhythmia or heart failure), clinically directed rescue antiarrhythmic therapy with amiodarone/lidocaine or euthanasia necessitated by sustained tachycardia > 350 bpm or signs of heart failure.
Histology (pig heart)
Whole heart was harvested and processed as previously described20. Briefly, the left ventricle was isolated from the whole heart, weighed, and embedded in 2% agar for slicing. Sections of 2.5 mm were obtained and fixed over-night in 4% paraformaldehyde at 4°C. Sections were then dehydrated in 70% ethanol before embedding in paraffin. Sections were then cut at 4 μm and placed on positively charged microscopy slides. Before staining, slides were progressively deparaffinized and re-hydrated by subsequent washes in xylene and ethanol at different concentration. After quenching in a mixture of hydrogen peroxide and methanol (1:20), heat-induced antigen retrieval treatment was performed using citrate buffer (or proteinase K digestion for MLC2a and MLC2v staining). Slides then were incubated in 1.5% normal horse serum diluted in DPBS (blocking buffer) for 1 hour at room temperature. Primary antibodies were diluted in blocking buffer and incubated over-night at 4°C. Secondary antibodies were diluted in blocking buffer and incubated for 1 hour at room temperature. For avidin-biotin complex (ABC) detection, slides were incubated with ABC Peroxidase for 30 min at room temperature before visualization with 3,3’ diaminobenzidine. Graft was quantified using a custom code on whole-slide images and normalized on both area of block and LV weight (see Supplementary Table 2)20. For immunofluorescence, after secondary antibody incubation, slides were imaged with a 20X and 60X oil objective on a Nikon Eclipse microscope with Yokogawa W1 spinning disk head and Nikon A1R confocal microscope. Fiji software was used to process the images.
Cardiac slice model for electrical coupling
For cardiac slices, the animal was maintained under mechanical ventilation and isoflurane anesthesia while a sternotomy was performed to access the heart. The whole heart was perfused by cannulating the aorta to allow retrograde perfusion using St. Thomas cardioplegic solution until cardiac arrest (St. Thomas solution: 110mM NaCl, 16mM KCl, 16mM MgCl2, 1.2mM CaCl2, 10mM NaHCO3). The whole heart was then harvested and kept at 4°C in perfusion buffer until slicing (4 mm sections as described above). The 4 mm slices were stored in Custodial solution (15 mM NaCl, 0.015 mM CaCl2*2H2O, 9 mM KCl, 4 mM MgCl2, 18 mM Histidine HCl, 180 mM Histidine, 2 mM Tryptophan, 0.03 mM Mannitol, 0.001 mM Ketoglutarate, pH 7.4) at 4°C. A vibratome was used to generate ultra-thin (300 μm) living cardiac slices (LCS), which performed well when stored at 4°C for up to 24 h from the heart isolation. The LCS were generated with an adaptation of the previously described protocols67,68. Briefly, the 2.5 mm slice was trimmed to select the portion with the grafted cells, which was glued to the specimen holder and sliced into 300 μm LCS with a high-precision vibratome (7000smz-2, Camden Instrument) in slicing Tyrode’s solution (10 mM BDM, 140 mM NaCl, 9mM KCl, 1 mM Glucose, 10 mM HEPES, 1mM MgCl2, 0.9 mM CaCl2, pH 7.4). The LCS were then loaded for 20 minutes (in Medium199) at 37°C with calcium (Fluo-4 AM 5 μg/ml) or voltage (FluoVolt - 1:1000 dilution as per manufacturer’s instruction) indicators and used for optical mapping. An upright Nikon ECLIPSE FN1 microscope with a Kinetix sCMOS camera was used to record videos at 200 frames per second. The experiments were performed at 37°C in Recording Tyrode’s solution (10 mM BDM, 140 mM NaCl, 5 mM KCl, 1 mM Glucose, 10 mM HEPES, 1 mM MgCl2, 1.8 mM CaCl2, pH 7.4) and monofilament anchors were positioned on top to keep the LCS flat and facilitate the live imaging. The LCS were electrically paced by field stimulation with two custom-made platinum electrodes connected to an IonOptix MyoPacer at 20V (5 milliseconds duration). A parallel bipolar microelectrode was used to electrically pace (1–2V and 5 milliseconds duration) a small portion of the LCS, and therefore apply a point stimulation either to the host tissue or to the graft. Calcium and voltage traces and kinetics were analyzed with Labchart8. After analysis, LCS were then fixed in 4% paraformaldehyde and processed for staining as described above.
QUANTIFICATION AND STATISTICAL ANALYSIS
For in vitro studies, the number of biological replicates (intended as independent batches of hESC-CMs differentiated and harvested at different days) is detailed in the corresponding figure legends. For each experiment, a minimum of three technical replicates were used. Statistical testing was performed using an unpaired t-test for two-group comparisons. For multiple-group comparison, one-way or two-way ANOVA with a post-hoc Sidak correction was used. Differences between groups were considered statistically significant when p value was < 0.05. All analyses were performed using GraphPad Prism software (Version 9.3.1). All results are expressed as mean ± SEM, unless otherwise stated.
Supplementary Material
Supplementary Video 1. Injection of hPSC-CMs into uninjured pig heart through direct thoracotomy; related to Methods section – Pig surgery.
Supplementary Video 2. Point stimulation of host myocardium and calcium transient elicited in graft (related to Figure 7D).
Supplementary Video 3. Point stimulation of graft and calcium transient elicited in host myocardium (related to Supplementary Fig. 6E).
HIGHLIGHTS:
Ion channel gene expression changes extensively as hPSC-CM grafts mature in vivo.
Targeting individual ion channels was insufficient to prevent engraftment arrhythmia.
Automaticity of hPSC-CMs relies on calcium and voltage clocks.
Graft automaticity is a major contributor to arrhythmias post-hPSC-CM engraftment.
Acknowledgments
We thank the UW Department of Comparative Medicine for their exceptional veterinary care and consultation for the animal experiments. We also thank Dr. Naoto Muraoka, Ms. Elisabeth Mahen and Ms. Cathy Sanford for their help and experimental support. This research was assisted by the University of Washington Cell Analysis Facility (Department of Immunology), the Lynn and Mike Garvey Cell Imaging Core and the Tom and Sue Ellison Stem Cell Core from the Institute for Stem Cell and Regenerative Medicine) and Specialty VETPATH for histology services. These studies were supported by the UW Medicine Heart Regeneration Program, the Washington Research Foundation, a gift from Mike and Lynn Garvey, and a sponsored research agreement from Sana Biotechnology (all Seattle, WA). This work also was supported in part by NIH grants R01HL128368, R01HL146868, and R01HL148081 (to C.E.M.), a grant from the Fondation Leducq Transatlantic Network of Excellence (Boston, MA; to C.E.M.), and the Bruce-Laughlin Research Fellowship (Seattle, WA; to K.N.). S.M. was supported in part by a fellowship from the UW Institute for Stem Cell and Regenerative Medicine.
Footnotes
Declaration of interests
K.N., B. C. K., and W. R. M. were advisors to Sana Biotechnology. M. L., A. J., F. A. K., A. F.T., S. D., H. T., L. P., S. K., R.S. T., and C. E. M. were employees while part of this study was performed and continue to hold equity in Sana Biotechnology. S. M., H. R., A. B., and C. E. M. have submitted a provisional patent application pertaining to the work in this manuscript (PCT/US2022/027382). The remaining authors have nothing to disclose.
Inclusion and Diversity statement
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, et al. (2009). Evidence for cardiomyocyte renewal in humans. Science 324, 98–102. 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marchiano S, Bertero A, and Murry CE (2019). Learn from Your Elders: Developmental Biology Lessons to Guide Maturation of Stem Cell-Derived Cardiomyocytes. Pediatr Cardiol 40, 1367–1387. 10.1007/s00246-019-02165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Velagaleti RS, Pencina MJ, Murabito JM, Wang TJ, Parikh NI, D’Agostino RB, Levy D, Kannel WB, and Vasan RS (2008). Long-term trends in the incidence of heart failure after myocardial infarction. Circulation 118, 2057–2062. 10.1161/CIRCULATIONAHA.108.784215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prabhu SD, and Frangogiannis NG (2016). The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res 119, 91–112. 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diseases GBD, and Injuries C (2020). Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222. 10.1016/S0140-6736(20)30925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ, Benjamin EJ, Benziger CP, et al. (2020). Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J Am Coll Cardiol 76, 2982–3021. 10.1016/j.jacc.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eschenhagen T, Bolli R, Braun T, Field LJ, Fleischmann BK, Frisen J, Giacca M, Hare JM, Houser S, Lee RT, et al. (2017). Cardiomyocyte Regeneration: A Consensus Statement. Circulation 136, 680–686. 10.1161/CIRCULATIONAHA.117.029343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura K, and Murry CE (2019). Function Follows Form- A Review of Cardiac Cell Therapy. Circ J 83, 2399–2412. 10.1253/circj.CJ-19-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertero A, and Murry CE (2018). Hallmarks of cardiac regeneration. Nat Rev Cardiol 15, 579580. 10.1038/s41569-018-0079-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Laake LW, Passier R, Monshouwer-Kloots J, Verkleij AJ, Lips DJ, Freund C, den Ouden K, Ward-van Oostwaard D, Korving J, Tertoolen LG, et al. (2007). Human embryonic stem cell-derived cardiomyocytes survive and mature in the mouse heart and transiently improve function after myocardial infarction. Stem Cell Res 1, 9–24. 10.1016/j.scr.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Caspi O, Huber I, Kehat I, Habib M, Arbel G, Gepstein A, Yankelson L, Aronson D, Beyar R, and Gepstein L (2007). Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J Am Coll Cardiol 50, 1884–1893. 10.1016/j.jacc.2007.07.054. [DOI] [PubMed] [Google Scholar]
- 12.Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J, Palpant NJ, Gantz J, Moyes KW, Reinecke H, et al. (2012). Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature 489, 322–325. 10.1038/nature11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiba Y, Gomibuchi T, Seto T, Wada Y, Ichimura H, Tanaka Y, Ogasawara T, Okada K, Shiba N, Sakamoto K, et al. (2016). Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 538, 388–391. 10.1038/nature19815. [DOI] [PubMed] [Google Scholar]
- 14.Liu YW, Chen B, Yang X, Fugate JA, Kalucki FA, Futakuchi-Tsuchida A, Couture L, Vogel KW, Astley CA, Baldessari A, et al. (2018). Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat Biotechnol 36, 597–605. 10.1038/nbt.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selvakumar D, Clayton ZE, and Chong JJH (2020). Robust Cardiac Regeneration: Fulfilling the Promise of Cardiac Cell Therapy. Clin Ther 42, 1857–1879. 10.1016/j.clinthera.2020.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Karbassi E, Fenix A, Marchiano S, Muraoka N, Nakamura K, Yang X, and Murry CE (2020). Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat Rev Cardiol 17, 341–359. 10.1038/s41569-019-0331-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y, and Pu WT (2020). Cardiomyocyte Maturation: New Phase in Development. Circ Res 126, 1086–1106. 10.1161/CIRCRESAHA.119.315862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peinkofer G, Burkert K, Urban K, Krausgrill B, Hescheler J, Saric T, and Halbach M (2016). From Early Embryonic to Adult Stage: Comparative Study of Action Potentials of Native and Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Dev 25, 1397–1406. 10.1089/scd.2016.0073. [DOI] [PubMed] [Google Scholar]
- 19.Romagnuolo R, Masoudpour H, Porta-Sanchez A, Qiang B, Barry J, Laskary A, Qi X, Masse S, Magtibay K, Kawajiri H, et al. (2019). Human Embryonic Stem Cell-Derived Cardiomyocytes Regenerate the Infarcted Pig Heart but Induce Ventricular Tachyarrhythmias. Stem Cell Reports 12, 967–981. 10.1016/j.stemcr.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura K, Neidig LE, Yang X, Weber GJ, El-Nachef D, Tsuchida H, Dupras S, Kalucki FA, Jayabalu A, Futakuchi-Tsuchida A, et al. (2021). Pharmacologic therapy for engraftment arrhythmia induced by transplantation of human cardiomyocytes. Stem Cell Reports 16, 2473–2487. 10.1016/j.stemcr.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu JK, Franceschi W, Huang Q, Pashakhanloo F, Boyle PM, and Trayanova NA (2019). A comprehensive, multiscale framework for evaluation of arrhythmias arising from cell therapy in the whole post-myocardial infarcted heart. Sci Rep 9, 9238. 10.1038/s41598-01945684-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selvakumar D, Clayton ZE, Prowse A, Dingwall S, George J, Shah H, Chen S, Hume RD, Tjahjadi L, Igoor S, et al. (2022). Cellular Heterogeneity of Pluripotent Stem Cell Derived Cardiomyocyte Grafts is Mechanistically Linked to Treatable Arrhythmias. bioRxiv, 2022.2009.2015.500719. 10.1101/2022.09.15.500719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filice D, Dhahri W, Solan JL, Lampe PD, Steele E, Milani N, Van Biber B, Zhu WZ, Valdman TS, Romagnuolo R, et al. (2020). Optical mapping of human embryonic stem cell-derived cardiomyocyte graft electrical activity in injured hearts. Stem Cell Res Ther 11, 417. 10.1186/s13287-020-01919-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ, and January CT (2011). High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol 301, H2006–2017. 10.1152/ajpheart.00694.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Wada Y, Ballan N, Schmeckpeper J, Huang J, Rau CD, Wang Y, Gepstein L, and Knollmann BC (2021). Triiodothyronine and dexamethasone alter potassium channel expression and promote electrophysiological maturation of human-induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 161, 130–138. 10.1016/j.yjmcc.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Z, Lan H, El-Battrawy I, Li X, Buljubasic F, Sattler K, Yucel G, Lang S, Tiburcy M, Zimmermann WH, et al. (2018). Ion Channel Expression and Characterization in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Int 2018, 6067096. 10.1155/2018/6067096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ivashchenko CY, Pipes GC, Lozinskaya IM, Lin Z, Xiaoping X, Needle S, Grygielko ET, Hu E, Toomey JR, Lepore JJ, and Willette RN (2013). Human-induced pluripotent stem cell-derived cardiomyocytes exhibit temporal changes in phenotype. Am J Physiol Heart Circ Physiol 305, H913–922. 10.1152/ajpheart.00819.2012. [DOI] [PubMed] [Google Scholar]
- 28.Kadota S, Pabon L, Reinecke H, and Murry CE (2017). In Vivo Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Neonatal and Adult Rat Hearts. Stem Cell Reports 8, 278–289. 10.1016/j.stemcr.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JJ, Yang L, Lin B, Zhu X, Sun B, Kaplan AD, Bett GC, Rasmusson RL, London B, and Salama G (2015). Mechanism of automaticity in cardiomyocytes derived from human induced pluripotent stem cells. J Mol Cell Cardiol 81, 81–93. 10.1016/j.yjmcc.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang XH, and Morad M (2020). Ca(2+) signaling of human pluripotent stem cells-derived cardiomyocytes as compared to adult mammalian cardiomyocytes. Cell Calcium 90, 102244. 10.1016/j.ceca.2020.102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsutsui K, Monfredi OJ, Sirenko-Tagirova SG, Maltseva LA, Bychkov R, Kim MS, Ziman BD, Tarasov KV, Tarasova YS, Zhang J, et al. (2018). A coupled-clock system drives the automaticity of human sinoatrial nodal pacemaker cells. Sci Signal 11. 10.1126/scisignal.aap7608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lakatta EG, Maltsev VA, and Vinogradova TM (2010). A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res 106, 659–673. 10.1161/CIRCRESAHA.109.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyashkov AE, Behar J, Lakatta EG, Yaniv Y, and Maltsev VA (2018). Positive Feedback Mechanisms among Local Ca Releases, NCX, and ICaL Ignite Pacemaker Action Potentials. Biophys J 114, 1176–1189. 10.1016/j.bpj.2017.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maltsev VA, and Lakatta EG (2012). The funny current in the context of the coupled-clock pacemaker cell system. Heart Rhythm 9, 302–307. 10.1016/j.hrthm.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mesirca P, Torrente AG, and Mangoni ME (2015). Functional role of voltage gated Ca(2+) channels in heart automaticity. Front Physiol 6, 19. 10.3389/fphys.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verkerk AO, van Ginneken AC, and Wilders R (2009). Pacemaker activity of the human sinoatrial node: role of the hyperpolarization-activated current, I(f). Int J Cardiol 132, 318–336. 10.1016/j.ijcard.2008.12.196. [DOI] [PubMed] [Google Scholar]
- 37.Kernik DC, Morotti S, Wu H, Garg P, Duff HJ, Kurokawa J, Jalife J, Wu JC, Grandi E, and Clancy CE (2019). A computational model of induced pluripotent stem-cell derived cardiomyocytes incorporating experimental variability from multiple data sources. J Physiol 597, 4533–4564. 10.1113/JP277724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DiFrancesco D (2010). The role of the funny current in pacemaker activity. Circ Res 106, 434–446. 10.1161/CIRCRESAHA.109.208041. [DOI] [PubMed] [Google Scholar]
- 39.Niehoff J, Matzkies M, Nguemo F, Hescheler J, and Reppel M (2019). The Effect of Antiarrhythmic Drugs on the Beat Rate Variability of Human Embryonic and Human Induced Pluripotent Stem Cell Derived Cardiomyocytes. Sci Rep 9, 14106. 10.1038/s41598-019-50557-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen K, Zuo D, Wang SY, and Chen H (2018). Kir2 inward rectification-controlled precise and dynamic balances between Kir2 and HCN currents initiate pacemaking activity. FASEB J 32, 3047–3057. 10.1096/fj.201701260R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu QH, Li XL, Xu YW, Lin YY, Cao JM, and Wu BW (2012). A novel discovery of IK1 channel agonist: zacopride selectively enhances IK1 current and suppresses triggered arrhythmias in the rat. J Cardiovasc Pharmacol 59, 37–48. 10.1097/FJC.0b013e3182350bcc. [DOI] [PubMed] [Google Scholar]
- 42.Xiang Z, Thompson AD, Brogan JT, Schulte ML, Melancon BJ, Mi D, Lewis LM, Zou B, Yang L, Morrison R, et al. (2011). The Discovery and Characterization of ML218: A Novel, Centrally Active T-Type Calcium Channel Inhibitor with Robust Effects in STN Neurons and in a Rodent Model of Parkinson’s Disease. ACS Chem Neurosci 2, 730–742. 10.1021/cn200090z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leuranguer V, Mangoni ME, Nargeot J, and Richard S (2001). Inhibition of T-type and L-type calcium channels by mibefradil: physiologic and pharmacologic bases of cardiovascular effects. J Cardiovasc Pharmacol 37, 649–661. 10.1097/00005344-200106000-00002. [DOI] [PubMed] [Google Scholar]
- 44.Godfraind T (2017). Discovery and Development of Calcium Channel Blockers. Front Pharmacol 8, 286. 10.3389/fphar.2017.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vohra J (1982). Verapamil in cardiac arrhythmias: an overview. Clin Exp Pharmacol Physiol Suppl 6, 129–134. [PubMed] [Google Scholar]
- 46.Watanabe Y, Koide Y, and Kimura J (2006). Topics on the Na+/Ca2+ exchanger: pharmacological characterization of Na+/Ca2+ exchanger inhibitors. J Pharmacol Sci 102, 7–16. . [DOI] [PubMed] [Google Scholar]
- 47.Tanaka H, Nishimaru K, Aikawa T, Hirayama W, Tanaka Y, and Shigenobu K (2002). Effect of SEA0400, a novel inhibitor of sodium-calcium exchanger, on myocardial ionic currents. Br J Pharmacol 135, 1096–1100. 10.1038/sj.bjp.0704574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li S, Chen G, and Li RA (2013). Calcium signalling of human pluripotent stem cell-derived cardiomyocytes. J Physiol 591, 5279–5290. 10.1113/jphysiol.2013.256495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lieu DK, Fu JD, Chiamvimonvat N, Tung KC, McNerney GP, Huser T, Keller G, Kong CW, and Li RA (2013). Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol 6, 191–201. 10.1161/CIRCEP.111.973420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun Y, Timofeyev V, Dennis A, Bektik E, Wan X, Laurita KR, Deschenes I, Li RA, and Fu JD (2017). A Singular Role of IK1 Promoting the Development of Cardiac Automaticity during Cardiomyocyte Differentiation by IK1 -Induced Activation of Pacemaker Current. Stem Cell Rev Rep 13, 631–643. 10.1007/s12015-017-9745-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, and Kurachi Y (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90, 291–366. 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 52.Costa ADS, Mortensen P, Hortigon-Vinagre MP, van der Heyden MAG, Burton FL, Gao H, Simitev RD, and Smith GL (2021). Electrophysiology of hiPSC-Cardiomyocytes Co-Cultured with HEK Cells Expressing the Inward Rectifier Channel. Int J Mol Sci 22. 10.3390/ijms22126621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bertero A, Pawlowski M, Ortmann D, Snijders K, Yiangou L, Cardoso de Brito M, Brown S, Bernard WG, Cooper JD, Giacomelli E, et al. (2016). Optimized inducible shRNA and CRISPR/Cas9 platforms for in vitro studies of human development using hPSCs. Development 143, 4405–4418. 10.1242/dev.138081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Gunther S, Fukuda N, Kikhi K, Boezio GLM, Takacs CM, Lai SL, et al. (2019). Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197. 10.1038/s41586-019-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bers DM (2008). Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70, 23–49. 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 56.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, Chantawansri C, Ritter MR, Friedlander M, Nicoll DA, et al. (2004). Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ Res 95, 604–611. 10.1161/01.RES.0000142316.08250.68. [DOI] [PubMed] [Google Scholar]
- 57.Gerbin KA, Yang X, Murry CE, and Coulombe KL (2015). Enhanced Electrical Integration of Engineered Human Myocardium via Intramyocardial versus Epicardial Delivery in Infarcted Rat Hearts. PLoS One 10, e0131446. 10.1371/journal.pone.0131446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clauss S, Bleyer C, Schuttler D, Tomsits P, Renner S, Klymiuk N, Wakili R, Massberg S, Wolf E, and Kaab S (2019). Animal models of arrhythmia: classic electrophysiology to genetically modified large animals. Nat Rev Cardiol 16, 457–475. 10.1038/s41569-019-0179-0. [DOI] [PubMed] [Google Scholar]
- 59.Antzelevitch C, and Burashnikov A (2011). Overview of Basic Mechanisms of Cardiac Arrhythmia. Card Electrophysiol Clin 3, 23–45. 10.1016/j.ccep.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reuter H, Henderson SA, Han T, Mottino GA, Frank JS, Ross RS, Goldhaber JI, and Philipson KD (2003). Cardiac excitation-contraction coupling in the absence of Na(+) - Ca2+ exchange. Cell Calcium 34, 19–26. 10.1016/s0143-4160(03)00018-6. [DOI] [PubMed] [Google Scholar]
- 61.Palpant NJ, Pabon L, Friedman CE, Roberts M, Hadland B, Zaunbrecher RJ, Bernstein I, Zheng Y, and Murry CE (2017). Generating high-purity cardiac and endothelial derivatives from patterned mesoderm using human pluripotent stem cells. Nat Protoc 12, 15–31. 10.1038/nprot.2016.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bertero A, Fields PA, Smith AST, Leonard A, Beussman K, Sniadecki NJ, Kim DH, Tse HF, Pabon L, Shendure J, et al. (2019). Chromatin compartment dynamics in a haploinsufficient model of cardiac laminopathy. J Cell Biol 218, 2919–2944. 10.1083/jcb.201902117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, et al. (2007). Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol 25, 1015–1024. 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 64.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alexa A, Rahnenfuhrer J, and Lengauer T (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607. 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- 67.Wen Q, Gandhi K, Capel RA, Hao G, O’Shea C, Neagu G, Pearcey S, Pavlovic D, Terrar DA, Wu J, et al. (2018). Transverse cardiac slicing and optical imaging for analysis of transmural gradients in membrane potential and Ca(2+) transients in murine heart. J Physiol 596, 3951–3965. 10.1113/JP276239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watson SA, Scigliano M, Bardi I, Ascione R, Terracciano CM, and Perbellini F (2017). Preparation of viable adult ventricular myocardial slices from large and small mammals. Nat Protoc 12, 2623–2639. 10.1038/nprot.2017.139. [DOI] [PubMed] [Google Scholar]
- 69.Lacoste A, Berenshteyn F, and Brivanlou AH (2009). An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell 5, 332–342. 10.1016/j.stem.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 70.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27, 851–857. 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, and Ventura A (2017). GuideScan software for improved single and paired CRISPR guide RNA design. Nat Biotechnol 35, 347–349. 10.1038/nbt.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bae S, Park J, and Kim JS (2014). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1. Injection of hPSC-CMs into uninjured pig heart through direct thoracotomy; related to Methods section – Pig surgery.
Supplementary Video 2. Point stimulation of host myocardium and calcium transient elicited in graft (related to Figure 7D).
Supplementary Video 3. Point stimulation of graft and calcium transient elicited in host myocardium (related to Supplementary Fig. 6E).
Data Availability Statement
Bulk-RNA-seq data have been deposited at GEO and are publicly available as of the date of publication (accession number can be found in the key resource table). Original western blot images are included in the supplementary material as specified in the figure legends. Microscopy data reported in this paper will be shared by the lead contact upon request.
The paper does not report any original code.
Any further information required to reanalyzed the data reported in this paper is available from the lead contact upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies (working concentration, application*) | ||
| NCX1 Mouse monoclonal (dil. 1:200, WB) | Swant | RRID: AB_2716744 |
| Cardiac Troponin T Antibody Rabbit polyclonal (dil. 1:1000, WB) | Abcam | RRID: AB_956386 |
| HCN4 Mouse monoclonal (S114/10) (dil. 1:200, WB) | NovusBio | RRID: AB_2935644 |
| GAPDH Mouse monoclonal (dil. 1:3000, WB) | Abcam | RRID: AB_2107448 |
| Cardiac Troponin T-APC REA400 (dil. 1:100, FC) | Miltenyi Biotec | RRID: AB_2783887 |
| REA293-APC (dil. 1:100, FC) | Miltenyi Biotec | RRID: AB_2733446 |
| Mouse monoclonal anti-CD56-PE (dil. 1:5, FC) | BD Bioscience | RRID: AB_395906 |
| Mouse monoclonal anti-PDGFRα-APC (dil. 1:10, FC) | R&D System | RRID: AB_883910 |
| Mouse monoclonal IgG1-PE (dil. 1:5, FC) | BD Biosceince | RRID: AB_396091 |
| Mouse monoclonal IgG1-APC (dil. 1:10, FC) | R&D System | RRID: AB_398576 |
| Rabbit monoclonal anti NKX2.5 (dil. 1:200, FC) | Cell Signaling | RRID: AB_2935645 |
| Rabbit IgG1 (dil. 1:500, FC) | Cell Signaling | RRID: AB_1031062 |
| Slow Skeletal Troponin I Mouse monoclonal (dil. 1:200, IHC/IF) | Novus Biological | RRID: AB_2935646 |
| β-Myosin Heavy chain Mouse A4.951 (dil. 1:1, IHC/IF) | Developmental Studies Hybridoma Bank | RRID: AB_528385 |
| Biotin-SP-conjugated AffiniPure Goat anti-Mouse IgG (dil. 1:500, IHC) | Jackson Immunoresearch | RRID: AB_2338557 |
| Cardiac Troponin I Rabbit Monoclonal (dil. 1:100, IF) | Abcam | RRID: AB_869983 |
| MLC2a Mouse monoclonal (dil. 1:500, IF) | BD Life Science | RRID: AB_2739265 |
| MLC2v Rabbit polyclonal (dil. 1:100, IF) | Proteintech | RRID: AB_2147453 |
| Connexin43 Rabbit polyclonal (dil 1:200, IF) | Merck | RRID: AB_476857 |
| Desmin goat polyclonal (dil. 1:100) | Origene | RRID: AB_2935647 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 (dil. 1:1000, IF) | Life Technologies | RRID: AB_141436 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (dil. 1:1000, IF/FC) | Life Technologies | RRID: AB_143165 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 (dil. 1:1000, IF) | Life Technologies | RRID: AB_141844 |
| *WB: Western blotting, FC: flow cytometry; IF: Immunofluorescence, IHC: Immunohistochemistry | ||
| Bacterial and virus strains | ||
| 𝛼-Select Gold Efficiency Chemically competent cells | BIOLINE | Cat # BIO-85027 |
| Chemicals, peptides, and recombinant proteins | ||
| Fast Digest Bpil | Thermo Scientific | Cat # FD1014 |
| Fast Digest Mph1103I (NsiI) | Thermo Scientific | Cat # FD0734 |
| Fast Digest Pfi23II (BsiWI) | Thermo Scientific | Cat # FD0854 |
| Fast Digest SgsI (AscI) | Thermo Scientific | Cat # FD1894 |
| Fast Digest NotI | Thermo Scientific | Cat # FD0593 |
| Fast Digest ScaI | Thermo Scientific | Cat # FD0434 |
| Fast Digest BamHI | Thermo Scientific | Cat # FD0054 |
| Fast Digest Bsu15I (ClaI) | Thermo Scientific | Cat # FD0143 |
| Fast Digest NcoI | Thermo Scientific | Cat # FD0575 |
| T4 DNA Ligase Reaction Buffer | New England Bio-Labs | Cat # B0202S |
| Quick Ligation Kit | New England Bio-Labs | Cat # M2200S |
| T4 Polynucleotide Kinase | New England Bio-Labs | Cat # M0201S |
| Ampicillin sodium salt | Sigma-Aldrich | Cat # A0166 |
| LB Agar, Miller (Luria-Bertani) | BD Life Sciences | Cat # 244520 |
| LB Broth, Miller (Luria-Bertani) | BD Life Sciences | Cat # 244620 |
| Granulated Agar | BD Life Sciences | Cat # 214530 |
| OPTI-MEM Reduced Serum Medium | Gibco | Cat # 31985062 |
| GeneJuice Transfection Reagent | Sigma-Aldrich | Cat # 70967–3 |
| DPBS, no calcium, no magnesium | Gibco | Cat # 14190–250 |
| mTeSR Plus | Stem Cell Technologies | Cat # 100–0276 |
| Essential 8™ Medium | Thermo Scientific | Cat # A1517001 |
| Matrigel Growth Factor Reduced (GFR), Phenol Red-free, LDEV-free | Corning | Cat # 356231 |
| rLaminin-521 (human) | Corning | Cat # 354221 |
| Y-27632 dihydrochloride | Tocris | Cat # 1254–50 |
| Versene Solution | Gibco | Cat # 15040066 |
| TrypLE Select | Gibco | Cat # A12177–02 |
| RPMI 1640 medium | Gibco | Cat # 11875119 |
| RPMI 1640 medium, no glucose | Life Technologies | Cat # 11879020 |
| RPMI 1640 medium, no phenol red | Life Technologies | Cat # 11835055 |
| Bovine Serum Albumin, suitable for cell culture | Sigma-Aldrich | Cat # A9418–50G |
| L-Ascorbic Acid 2-phosphate sesquimagnesium salt hydrate | Sigma-Aldrich | Cat # A8960–5G |
| CHIR99021 | Cayman | Cat # 13122 |
| Wnt-C59 | Selleck Chemicals | Cat # S7037 |
| Sodium L-lactate | Sigma-Aldrich | Cat # L7022–10G |
| B-27 Supplement (50X), serum free | Gibco | Cat # 17504044 |
| CryoStor cell cryopreservation media - CS10 | Sigma-Aldrich | Cat # C2874 |
| Q5 High-Fidelity 2X Master Mix | New England Bio- Labs | Cat # M0492S |
| M-MLV Reverse Transcriptase | Invitrogen | Cat # 28025013 |
| RNaseOUT Recombinant Ribonuclease Inhibitor | Invitrogen | Cat # 10777019 |
| SYBR Select Master Mix | Applied Biosystems | Cat # 4472913 |
| SpCas9 2NLS Nuclease | Synthego | NA |
| 4–15% Mini-PROTEAN TGX Precast Protein Gel | Biorad | Cat # 4561084 |
| Immobilon-P membrane (0.45 μm) | Millipore | Cat # IPVH00010 |
| Hematoxylin Solution (Mayer’s, Modified) | Abcam | Cat # ab220365 |
| Hoechst 33342 Solution | Thermo Scientific | Cat # 62249 |
| DNase I | Millipore | Cat # 260913 |
| Fluo-4 AM | Invitrogen | Cat # 14201 |
| Ivabradine hydrochloride | Tocris | Cat # 6542 |
| ML-218 hydrochloride | Tocris | Cat # 4507 |
| Mibefradil dihydrochloride | Tocris | Cat # 2198 |
| Zacopride hydrochloride | Tocris | Cat # 1795 |
| SEA0400 | Tocris | Cat # 6164 |
| KB-R7943 mesylate | Tocris | Cat # 1244 |
| Verapamil hydrochloride | Tocris | Cat # 0654 |
| Cyclosporine A | Cayman | Cat # 12088 |
| Buprenorphine SR-Lab | ZooPharm | NA |
| Euthasol | Virbac | NA |
| Abatacept (CTLA4-Ig) | Bristol-Myers Squibb | NA |
| 4% Paraformaldehyde | Santa Cruz Biotechnology | Cat # sc-281692 |
| Membrane-coated slides | Leica Microsystem | Cat #11600289 |
| VECTASTAIN Elite ABC HRP kit | Vector Laboratories | Cat # PK-6100 |
| SIGMAFAST™ 3,3′-Diaminobenzidine tablets | Sigma-Aldrich | Cat # D4168 |
| HEPES | Fisher | Cat # BP310–500 |
| D-(+)-Glucose | Sigma-Aldrich | Cat # G5767 |
| EGTA | Sigma-Aldrich | Cat # E3889 |
| ATP Magnesium Salt | Sigma-Aldrich | Cat # A9187 |
| Amphotericin B | Sigma-Aldrich | Cat # A9528 |
| Tetraethylammonium Chloride | Sigma-Aldrich | Cat # T2265 |
| 4-Aminopyridine | Sigma-Aldrich | Cat # 275875 |
| Tetraethylammonium Hydroxide | Sigma-Aldrich | Cat # 177806 |
| Cesium Chloride | Sigma-Aldrich | Cat # 289329 |
| GTP Sodium Salt Hydrate | Sigma-Aldrich | Cat # G8877 |
| Cesium Hydroxide Monohydrate | Sigma-Aldrich | Cat # 516988 |
| Critical commercial assays | ||
| QIAEX II Gel Extraction Kit | Qiagen | Cat # 20021 |
| QIAprep Miniprep Kit | Qiagen | Cat # 27104 |
| QIAfilter Plasmid Midiprep Kit | Qiagen | Cat # 12243 |
| BD StemflowTM Human and Mouse Pluripotent Stem Cell Analysis Kit | BD Life Sciences | Cat # 560477 |
| RNAesy Mini kit | Qiagen | Cat # 74106 |
| PicoPure® RNA Isolation Kit | Applied Biosystems | Cat # KIT0204 |
| Leica Laser Microdissection (LMD) System | Leica Microsystem | Cat # 8118616 |
| Pierce BCA Protein assay kit | Thermo Scientific | Cat # 23225 |
| NEBuilder HiFi DNA Assembly Cloning kit | New England Bio- Labs | Cat # E5520S |
| Maestro Pro multiwell microelectrode array (MEA) | Axion Biosystems | Cat #CP-MCV48W-UPF |
| Deposited data | ||
| RNA-seq data set | This paper | GSE190758 |
| Experimental models: Cell lines | ||
| Human induced-pluripotent Stem Cells – 253G1 -Camp3 | Kyoto University | CVCL_B51828 |
| Human Embryonic Stem Cells – RUESe002-A | Rockefeller University | CVCL_C1W369 |
| Experimental models: Organisms/strains | ||
| Athymic male Sprague Dawley rats (Hsd:RH-Foxn1rnu) | Envigo | RGD_5508395 |
| Yucatan minipigs | Premier BioSource | NA |
| Oligonucleotides (see also Methods Tables 1, 2) | ||
| HCN4_gRNA1 TCGTGAAGCGGACAATGCGC | This paper | NA |
| CACNA1H_gRNA1 GGATTTCTTCATCGTCGTGG | This paper | NA |
| CACNA1H_gRNA2 GACCATCTCCACCGCAAAAA | This paper | NA |
| HCN4_gRNA1_KI CAGCTTGTCCATGGCGCCAG | This paper | NA |
| HCN4_gRNA2_KI GGCAGCTTGTCCATGGCGCC | This paper | NA |
| SLC8A1_gRNA1 GGATCATATTACTGTAAGAA | Synthego | NA |
| SLC8A1_gRNA2 CAGCAATTACATGGTCCACA | Synthego | NA |
| SLC8A1_gRNA3 TGAAATCCCATTGAAAAGGT | Synthego | NA |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (pX459) V2.0 | Addgene | Addgene_6298870 |
| AAV_CAGGS_EGFP | Addgene | Addgene_2221271 |
| MV-PGK-Puro-TK | Hera BioLabs | Cat # SGK-0062 |
| KCNJ2 cDNA cloning fragment (RefSeq: NM_000891.3) | IDT | NA |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | Addgene_42230 |
| Software and algorithms and equipment | ||
| Guidescan (https://guidescan.com/) | Ventura lab | 72 |
| Cas-OFFinder (http://www.rgenome.net/cas-offinder/) | Kim lab | 73 |
| Axis Navigator 2.0.3 | Axion Biosystem | NA |
| Prism 8.4.2 | GraphPad | NA |
| FlowJo V9 | BD Life Sciences | NA |
| SnapGene 5.0.8 | SnapGene | NA |
| ImageJ | Fiji | NA |
| ecgAUTO 3.3.5.10 | EMKA Technologies | NA |
| Graft quantification analysis | This paper | 20 |
| Calcium analysis Fluo4 MatLab | Sniadecki Lab | 62 |
| pClamp 11.1 | Molecular Devices | NA |
| Multiclamp 700B | Molecular Devices | NA |
| Digidata 1550B | Molecular Devices | NA |
| Patchmaster v2×73 | HEKA Elektronik | NA |
| Fitmaster v2×91 | HEKA Elektronik | NA |
| Other | ||
| Karyotype analysis | Diagnostic Cytogenetics, Seattle WA | NA |
| ChemiDoc Imaging system | Biorad | NA |
| Vibratome 7000smz-2 | Camden Instrument | NA |
| NOGA-MyoStar platform | BioSense Webster | NA |
| PBS-0.5 MAG Single-Use Vessel | PBS Biotech | Cat #IA-0.5-D-001 |
| PBS-Mini Mag Drive Bioreactor Base Unit | PBS Biotech | Cat #FA-UNI-B-501 |
| Neon Transfection System | Life Technologies | Cat # MPK5000 |